

Abstract
Synaptodendritic damage correlates with cognitive decline in many neurodegenerative diseases, including human immunodeficiency virus-1 (HIV-1)-associated neurocognitive disorders (HAND). Because HIV-1 does not infect neurons, viral-mediated toxicity is indirect, resulting from released neurotoxins such as the HIV-1 protein transactivator of transcription (Tat). We compared the effects of Tat on inhibitory and excitatory synaptic connections between rat hippocampal neurons using an imaging-based assay that quantified clusters of the scaffolding proteins gephyrin or PSD95 fused to GFP. Tat (24 h) increased the number of GFP–gephyrin puncta and decreased the number of PSD95–GFP puncta. The effects of Tat on inhibitory and excitatory synapse number were mediated via the low-density lipoprotein receptor-related protein and subsequent Ca2+ influx through GluN2A-containing NMDA receptors (NMDARs). The effects of Tat on synapse number required cell-autonomous activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). Ca2+ buffering experiments suggested that loss of excitatory synapses required activation of CaMKII in close apposition to the NMDAR, whereas the increase in inhibitory synapses required Ca2+ diffusion to a more distal site. The increase in inhibitory synapses was prevented by inhibiting the insertion of GABAA receptors into the membrane. Synaptic changes induced by Tat (16 h) were reversed by blocking either GluN2B-containing NMDARs or neuronal nitric oxide synthase, indicating changing roles for pathways activated by NMDAR subtypes during the neurotoxic process. Compensatory changes in the number of inhibitory and excitatory synapses may serve as a novel mechanism to reduce network excitability in the presence of HIV-1 neurotoxins; these changes may inform the development of treatments for HAND.
Introduction
Human immunodeficiency virus-1 (HIV-1), the causative agent of acquired immune deficiency syndrome (AIDS), affects >30 million people worldwide. HIV-associated neurocognitive disorders (HAND) are a major complication of HIV infiltrating the CNS. Cognitive decline in patients with HAND correlates with dendritic pruning and excitatory synapse loss (Sá et al., 2004). With the advent of combined antiretroviral therapy, the incidence of HIV-associated dementia has declined. However, the prevalence of mild forms of cognitive impairment is increasing, in part attributable to the prolonged lifespan of HIV-infected patients (Heaton et al., 2011). HAND pathogenesis is characterized by activation of infected monocytes and subsequent release of viral proteins, cytokines, and neurotoxins that injure neurons (Genis et al., 1992; Speth et al., 2001). The HIV transactivator of transcription (Tat) is a viral protein shed by infected cells (Nath et al., 1999; King et al., 2006). Tat-induced neuronal injuries include the following: dendritic pruning and decreased spine density (Liu et al., 2000; Fitting et al., 2010), excitatory synapse loss (Kim et al., 2008a), and neuronal death (Kruman et al., 1998; Eugenin et al., 2007). Tat expression in vivo produces neuropathologies similar to those observed in HAND (Fitting et al., 2013). Tat is elevated in brain extracts from HAND patients (Del Valle et al., 2000), suggesting that Tat contributes to HAND.
Tat-induced loss of excitatory synapses occurs via a mechanism distinct from that leading to neuronal death (Kim et al., 2008a). Tat is internalized by the low-density lipoprotein receptor-related protein (LRP) (Liu et al., 2000); it then potentiates NMDA receptors (NMDARs) (Haughey et al., 2001), causing increased intracellular Ca2+ (Bonavia et al., 2001) and triggering loss of excitatory synapses via the ubiquitin–proteasome pathway (Kim et al., 2008a) or cell death via the neuronal nitric oxide synthase (nNOS) pathway (Eugenin et al., 2007). Tat-induced synapse loss is reversible (Kim et al., 2008a; Shin et al., 2012). We hypothesize that excitatory synapse loss after Tat is a coping mechanism that protects neurons from increased NMDAR activity. This Tat-induced plasticity is reminiscent of homeostatic scaling, a physiological mechanism to reduce overall network excitability (Marder and Goaillard, 2006). Increased inhibitory tone provides another mechanism to reduce network activity (Turrigiano, 1999). How Tat affects inhibitory synapses is unclear.
Upregulation of GABAergic relative to glutamatergic neurotransmission has been observed in neurodegenerative diseases with an excitotoxic component, including Alzheimer's disease (AD; Rissman et al., 2007). Gephyrin immunoreactivity, a marker for inhibitory synapses, increased in transgenic mice expressing Tat (Fitting et al., 2013). Here, we investigated the effects of Tat on the number of inhibitory and excitatory synaptic connections between hippocampal neurons in culture using an imaging-based assay that detected GFP fused to gephyrin or PSD95, respectively. Tat exposure increased the number of inhibitory synapses concurrent with a loss of excitatory synapses via an LRP and NMDAR-dependent mechanism. Tat-induced alterations in synaptic number required Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity and were reversible. Thus, Tat elicits opposing changes in the number of inhibitory compared with excitatory synapses, suggesting that inhibitory synapses may increase as a compensatory mechanism to reduce Tat-induced increases in neuronal excitability.
Materials and Methods
Materials.
Materials were obtained from the following sources: the expression vector for PSD95 fused to enhanced green fluorescent protein (pGWI–CMV–PSD95–GFP) was kindly provided by Donald B. Arnold (University of Southern California, Los Angeles, CA; Arnold and Clapham, 1999); the expression vector for gephyrin fused to enhanced green fluorescent protein (pGFP–C2–gephyrin) was kindly provided by Heinrich Betz (Max Plank Institute, Frankfurt/Main, Germany; Fuhrmann et al., 2002); the expression vector for autocamtide inhibitory peptide fused to green fluorescent protein (AIP–GFP) was kindly provided by Steven H. Green (University of Iowa, Iowa City, IA), and the AIP insert was subsequently cloned into the ECFP–C1 expression vector from Clontech to produce CFP–AIP; the expression vector for Discosoma red 2 (pDsRed2–N1) was from Clontech; the expression vector for γ2 peptide fused to cyan fluorescent protein (γ2–CFP) was produced by cloning oligonucleotides encoding GABAAR γ2 peptide (RTGAWRHGRIHIRIAKMD) into the ECFP–N1 vector from Clontech between Pst1 and EcoR1 (Kawaguchi and Hirano, 2007); the expression vector for scrambled γ2 peptide fused to cyan fluorescent protein was produced similarly by cloning randomized oligonucleotides encoding a scrambled γ2 peptide (AAKGHRIMWGRIRTDHI) into the ECFP–N1 vector; DMEM, fetal bovine serum, and horse serum were from Invitrogen; bicuculline and tetrodotoxin (TTX) were from Ascent Scientific; nutlin-3 was from EMD Millipore; recombinant rat receptor-associated protein (RAP) was from Fitzgerald Industries International; ifenprodil hemitartate, TCN201, KN-62, and FK506 were from Tocris Bioscience; BAPTA-AM and EGTA-AM were from Millipore; and penicillin/streptomycin, dizocilpine (MK801), and all other reagents were from Sigma.
HIV-1 Tat (Clade B, recombinant) was obtained from Prospec Tany TechnoGene and obtained through the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases from Dr. John Brady. For control experiments, Tat was heat-inactivated by incubation at 85°C for 30 min.
Cell culture.
All animal use was reviewed and approved by the Institutional Animal Care and Use Committee of the University of Minnesota. Rat hippocampal neurons were grown in primary culture as described previously (Shen and Thayer, 1998) with minor modifications. Fetuses (both male and female) were removed on embryonic day 17 from maternal rats killed with CO2 inhalation. Hippocampi were dissected and placed in Ca2+ and Mg2+-free HEPES-buffered HBSS (HHSS), pH 7.45. HHSS was composed of the following (in mm): 20 HEPES, 137 NaCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 5.0 KCl, 0.4 KH2PO4, 3.0 Na2HPO4, and 5.6 glucose. Cells were dissociated by trituration through flame-narrowed Pasteur pipettes of decreasing aperture and resuspended in DMEM without glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively). Dissociated cells were then plated at a density of 80,000–120,000 cells per dish onto a 25 mm round cover glass (#1) glued to cover a 19-mm-diameter opening drilled through the bottom of a 35 mm Petri dish. The coverglass was precoated with Matrigel (200 μl, 0.2 mg/ml; BD Biosciences). Neurons were grown in a humidified atmosphere of 10% CO2 and 90% air, pH 7.4, at 37°C, and fed on days 1 and 6 by exchange of 75% of the media with DMEM, supplemented with 10% horse serum and penicillin/streptomycin. Cells used in these experiments were cultured without mitotic inhibitors for a minimum of 12 d, resulting in a mixed glial–neuronal culture. Immunocytochemistry experiments demonstrated that these cultures were composed of 18 ± 2% neurons, 70 ± 3% astrocytes, and 9 ± 3% microglia (Kim et al., 2011). Experiments were performed on pyramidal-shaped neurons, although neuronal classification cannot be made on morphology alone.
Transfection.
Rat hippocampal neurons were transfected on 11 or 12 d in vitro (DIV) using a modification of a protocol described previously (Waataja et al., 2008). Briefly, hippocampal cultures were incubated for at least 20 min in DMEM supplemented with 1 mm kynurenic acid, 10 mm MgCl2, and 5 mm HEPES to reduce neurotoxicity. A DNA/calcium phosphate precipitate containing 1 μg of plasmid DNA per well was prepared, allowed to form for 30 min at room temperature, and added to the culture. After a 90 min incubation period, cells were washed once with DMEM supplemented with MgCl2 and HEPES and then returned to conditioned media, saved at the beginning of the procedure. Cells were transfected on 11 or 12 DIV, and image acquisition was initiated on 14 DIV (48–72 h after transfection). Transfection efficiency ranged from 1 to 5%.
Confocal imaging.
Modified Petri dishes containing transfected neurons were sealed with Parafilm, transferred to the stage of an inverted confocal microscope (Olympus Fluoview 300), and viewed through a 60× oil-immersion objective (numerical aperture 1.40). Digital micrometers attached to the stage of the microscope were used to identify the location of neurons being imaged and enabled repeated imaging of the same neuron over 24 h. Optical sections spanning 8 μm in the z-dimension were collected (1 μm steps) and combined through the z-axis into a compressed z-stack. GFP was excited at 488 nm with an argon ion laser, and emission was collected at 530 nm (10 nm bandpass). DsRed2 was excited at 543 nm with a green helium–neon laser, and emission was collected at >605 nm. The cell culture dish was returned to the CO2 incubator between image collection.
Image processing.
To count and label PSD95–GFP and GFP–gephyrin puncta, an automated algorithm was created using MetaMorph 6.2 image processing software described previously (Waataja et al., 2008). Briefly, maximum z-projection images were created from the DsRed2 and GFP image stacks. Next, a threshold set 1 SD above the image mean was applied to the DsRed2 image. This created a 1 bit image, which was used as a mask via a logical AND function with the GFP maximum z-projection. A top-hat filter (80 pixels) was applied to the masked GFP image. A threshold set 1.5 SD above the mean intensity inside the mask was then applied to the contrast enhanced image. Structures between 8 and 80 pixels (∼0.37–3.12 μm in diameter) were counted as PSD95–GFP or GFP–gephyrin puncta. These structures were then dilated and superimposed on the DsRed2 maximum z-projection for visualization. GFP–gephyrin expresses as distinct puncta and has been shown previously to also form large, somatic aggregates that do not represent functional synapses (Fuhrmann et al., 2002). Thus, the somatic region of neurons expressing GFP–gephyrin was excluded from image processing to establish threshold.
Puncta counts are presented as mean ± SEM in which n is the number of cells, each from a separate cover glass. All experiments were performed using cells from at least three separate cultures from at least three separate animals. Student's t test was used for single statistical comparisons. For multiple statistical comparisons, data were first tested for homoscedasticity using Levene's squared test; all samples were found to be of equal variance. For each dataset, a two-way ANOVA was performed; Tat treatment exerted a significant interaction with the other treatment groups. Thus, separate one-way ANOVAs with Bonferroni's post hoc test were performed for the Tat and untreated (control) groups.
Immunocytochemistry.
Rat hippocampal neurons were labeled using a protocol described previously (Kim et al., 2008a). Briefly, neurons transfected with GFP–gephyrin were fixed and permeabilized with ice-cold methanol for 10 min at −20°C, washed with PBS, blocked with 10% bovine serum albumin (BSA), and incubated with a primary antibody for mouse vesicular GABA transporter (VGAT; 1:500; Synaptic Systems) for 16 h at 4°C. Cells were then washed with PBS, and labeled proteins were visualized with tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-mouse antibody (1:1000; Millipore) in 10% BSA. Coverslips were mounted with Fluoromount (Southern Biotech) and imaged on an inverted confocal microscope (Olympus Fluoview 300) using a 60× oil-immersion objective (numerical aperture 1.4). TRITC was excited at 543 nm, and emission was collected at >605 nm.
Electrophysiology.
Spontaneous miniature IPSCs (mIPSCs) were recorded in the whole-cell voltage-clamp configuration from 14 DIV hippocampal neurons in an extracellular solution containing the following (in mm): 147 NaCl, 2.1 KCl, 8.8 HEPES, 8.8 d-glucose, 1.1 CaCl2, 1.1 MgCl2, and 2.6 2-oxopropanoic acid (pyruvic acid), with a pH of 7.35 (osmolarity of 310 mOsm at room temperature; Bannai et al., 2009). d-AP-5 at 50 μm5, 50 μm NBQX, and 1 μm TTX were added to the extracellular solution to block glutamate receptor-mediated synaptic currents and action potentials, respectively, before recording mIPSCs. Extracellular solutions were applied via a gravity-fed superfusion system. Electrodes were prepared using a horizontal micropipette puller (P-87; Sutter Instruments) from glass capillaries (Narishige). Pipettes had resistances of 3–5 MΩ when filled with the following internal solution (in mm): 144 CsCl2, 10 HEPES, 5 Na2ATP, 1.1 EGTA, 0.1 CaCl2, and 5 MgCl2, with a pH of 7.35 (osmolarity of 310 mOsm; Bannai et al., 2009). The membrane potential was voltage clamped at −70 mV during recording. Resistance and capacitance were compensated for each neuron, and cells were included in the analysis if access resistance was ≤15 MΩ and changed <20% during recording. Whole-cell currents were recorded with an Axopatch 200B (Molecular Devices), low-pass filtered at 2 kHz, and digitized at 100 kHz with a Digidata 1322A digitizer with pClamp software (Molecular Devices). Data were stored and analysis performed offline using Mini Analysis software (Synaptosoft).
Results
GFP–gephyrin labels functional inhibitory postsynaptic sites
We expressed synaptic proteins with fluorescent tags to follow dynamic changes in synapse number during exposure to the HIV-1 protein Tat. Cultured hippocampal neurons were cotransfected with expression vectors for DsRed2 and either GFP–gephyrin to label inhibitory synapses or PSD95–GFP to label excitatory synapses. The fusion constructs for GFP–gephyrin and PSD95–GFP both expressed as discrete puncta that contrasted well from diffuse green fluorescence in the cell (Fig. 1A,D). DsRed2 expression filled the soma and processes and was used to track morphological changes, monitor cell viability based on retention of fluorescence, and served as a mask for image processing. Neurons expressing DsRed2 and either GFP–gephyrin or PSD95–GFP were visualized using confocal laser scanning microscopy and processed using a previously described image processing algorithm (Waataja et al., 2008). The algorithm identified and counted puncta by locating intensity peaks of the appropriate size and in contact with the DsRed2 mask (Fig. 1A,D, Processed). GFP–gephyrin fluorescent puncta colocalized with functional neurotransmitter release sites as well as GABAAR immunoreactivity, indicating that they represent functional inhibitory postsynaptic sites (Fuhrmann et al., 2002). Consistent with previous reports, in some neurons, expressed GFP–gephyrin formed large aggregates in the soma; these aggregates do not represent functional synapses (Colin et al., 1996). To control for these large, somatically located GFP–gephyrin aggregates (>2× median puncta size), the soma was excluded from image processing.
Figure 1.
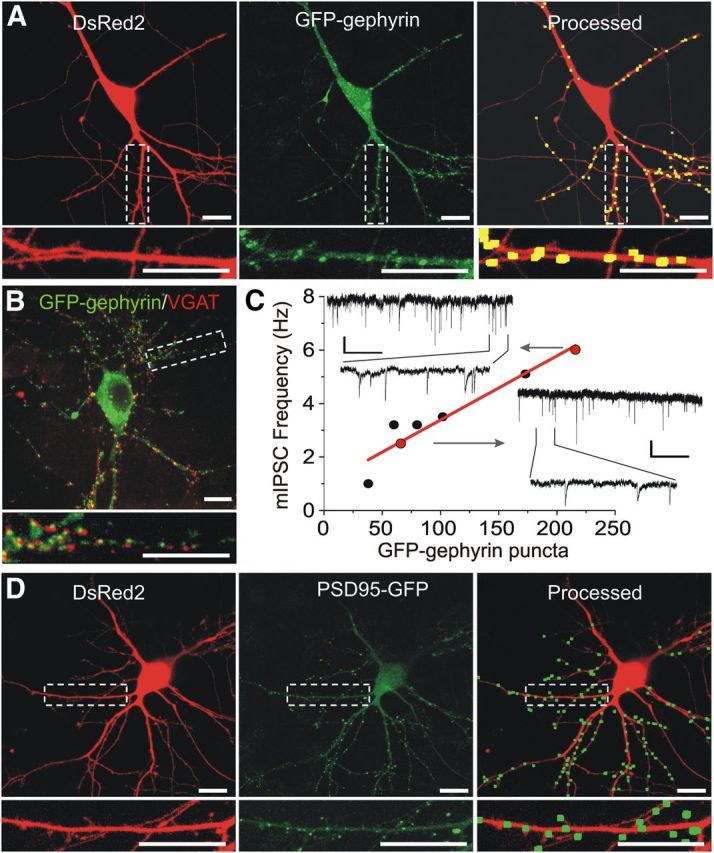
GFP–gephyrin labels functional inhibitory postsynaptic sites. Laser scanning confocal microscopy was used to image a cultured hippocampal neuron expressing DsRed2 (A, D, left) and either GFP–gephyrin (A, middle) or PSD95–GFP (D, middle). Maximum z-projections from eight 1 μm steps were created from DsRed2 z-series and GFP–gephyrin or PSD95–GFP z-series. After enhancing contrast, puncta reaching appropriate size and intensity criteria and in contact with a DsRed2 mask were enlarged and superimposed on the DsRed2 image (A, D, Processed). A more detailed description of image processing is described in Materials and Methods. The insets are enlarged images of the boxed region. Scale bars, 10 μm. B, Neurons expressing GFP–gephyrin (green) were fixed and labeled with an antibody for VGAT (red, 1:500) as described in Materials and Methods. The inset displays an enlarged image of the boxed region. Scale bars, 10 μm. C, mIPSCs were recorded from neurons expressing GFP–gephyrin as described previously, and their mIPSC frequency was plotted as a function of GFP–gephyrin puncta number. The red line is a linear regression of points demonstrating correlation between number of puncta and the frequency of inhibitory synaptic activity (r2 = 0.91, n = 7, p < 0.001). Insets show examples of mIPSC recordings from the cells indicated (red circles), demonstrating an increase in frequency between low and high puncta counts. Calibration: 25 pA, 1 or 0.1 s (for expanded trace).
To confirm that GFP–gephyrin labeled inhibitory synapses in our cultures, neurons expressing GFP–gephyrin were fixed and labeled with an antibody for VGAT. As shown in Figure 1B, GFP–gephyrin puncta colocalized with presynaptic VGAT immunoreactivity, confirming its location at inhibitory synapses. To evaluate the functional relationship of GFP–gephyrin puncta number to inhibitory synaptic transmission, neurons expressing GFP–gephyrin were imaged, and then mIPSCs were recorded (Fig. 1C). The number of GFP–gephyrin puncta was proportional to mIPSC frequency as shown by the linear regression in Figure 1C (r2 = 0.91, n = 7, p < 0.001). Thus, GFP–gephyrin puncta provide a suitable marker for functional inhibitory synapses.
Previous work from our laboratory demonstrated that fluorescent PSD95 puncta colocalized with functional neurotransmitter release sites, synaptically evoked Ca2+ increases, and NMDAR immunoreactivity, indicating that they represent functional excitatory postsynaptic sites (Waataja et al., 2008). The algorithm identified PSD95 puncta on both dendritic spines and shafts; in a previous study, we reported that 87 ± 5% of spines were labeled with PSD95–GFP puncta (Kim et al., 2008b).
We confirmed that our model system was sensitive to dynamic changes in synapse number by challenging cultured hippocampal neurons expressing DsRed2 and either GFP–gephyrin or PSD95–GFP with stimuli known to block inhibitory or excitatory synaptic transmission and then assessing changes in synapse number. The number of GFP–gephyrin or PSD95–GFP puncta was calculated for a single neuron over multiple time points to assess changes in synapse number. In untreated neurons, the number of inhibitory synaptic sites as indicated by GFP–gephyrin puncta increased by 16 ± 2% (n = 99) over 24 h (Fig. 2A,C). The number of excitatory synapses as indicated by PSD95–GFP puncta increased by 31 ± 4% (n = 87) over the same period (Fig. 2B,C). Chronic blockade of excitatory synaptic activity between cultured hippocampal neurons upregulates excitatory synapses (Turrigiano, 1999), and, similarly, blockade of inhibitory synaptic activity upregulates inhibitory synapses to maintain network homeostasis (Rannals and Kapur, 2011). In Figure 2, we show that treatment with the GABAAR antagonist bicuculline (10 μm) induced the predicted increase in inhibitory synapse number (41 ± 10%, n = 20, p < 0.01) by 24 h compared with controls. Blocking inhibitory synaptic transmission induced a slight decrease in excitatory synapse number (−1 ± 8%, n = 17, p < 0.05), which was significantly different from untreated controls, which increased. Treatment with TTX (1 μm), a voltage-gated sodium channel antagonist that potently blocks excitatory synaptic transmission, induced effects opposite to those evoked by bicuculline. TTX treatment for 24 h induced a 23 ± 10% decrease in inhibitory synapse number (n = 13, p < 0.01; Fig. 2) and a corresponding increase (58 ± 14%, n = 14, p < 0.05; Fig. 2) in excitatory synapse number. These findings demonstrate that GFP–gephyrin or PSD95–GFP puncta respond to stimuli shown previously to induce homeostatic scaling of synapse number, further validating this approach for tracking dynamic synaptic changes.
Figure 2.
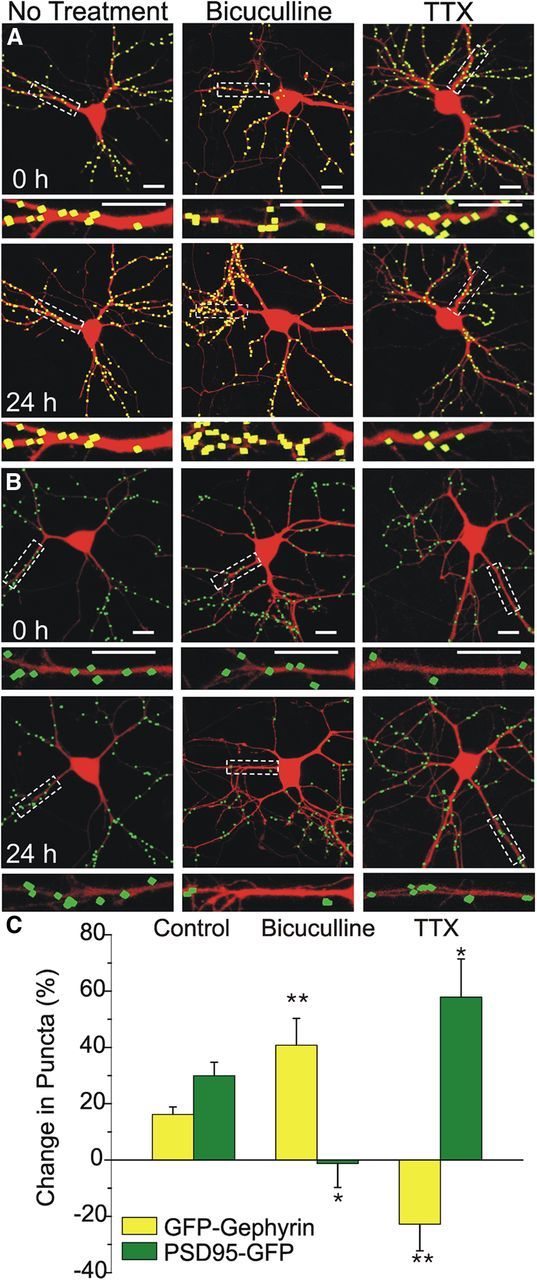
Pharmacological modulation of synaptic transmission alters GFP–gephyrin and PSD95–GFP puncta counts. A, B, Processed confocal images of neurons expressing DsRed2 and either GFP–gephyrin (A, yellow) or PSD95–GFP (B, green) before (0 h) and 24 h after treatment with 10 μm bicuculline or 1 μm TTX as indicated. Insets are enlarged images of the boxed regions. Scale bars, 10 μm. C, Bar graph summarizes the effects of no treatment or 24 h treatment with bicuculline or TTX on changes in GFP–gephyrin (yellow) or PSD95–GFP (green) puncta counts. Data are mean ± SEM. *p < 0.05, **p < 0.01 relative to corresponding control group (ANOVA with Bonferroni's post hoc test).
HIV-1 Tat exposure increases the number of inhibitory synapses
We showed previously that 24 h treatment with HIV-1 Tat induced loss of PSD95–GFP puncta via a mechanism distinct from that leading to neuronal death (Kim et al., 2008a). The effects of Tat on excitatory synapse number were concentration dependent (EC50 = 6 ± 2 ng/ml) with a maximally effective concentration of 50 ng/ml and peaked at 24 h without affecting cell survival (Kim et al., 2008a). Here, we confirmed that result: 24 h exposure to Tat (50 ng/ml) induced a 21 ± 2% decrease in PSD95–GFP puncta (corresponding to 107 ± 6 PSD95–GFP puncta before the addition of Tat, reduced to 83 ± 5 puncta after Tat; n = 87, p < 0.01; Fig. 3B,C), indicating a loss of excitatory postsynaptic sites. In contrast to its effects on excitatory synapses, treatment with Tat induced a marked increase in GFP–gephyrin puncta, indicating an increase in inhibitory synapse number. Treatment with Tat induced a 38 ± 3% increase in the number of GFP–gephyrin puncta (corresponding to 101 ± 4 GFP–gephyrin puncta before the addition of Tat, increased to 135 ± 6 puncta after Tat; n = 99, p < 0.01; Fig. 3A,C). Treatment with heat-inactivated Tat for 24 h did not alter the number of GFP–gephyrin puncta (11 ± 4% increase with heat-inactivated Tat, n = 9 vs 38 ± 3% increase with active Tat, n = 99, p < 0.05) or PSD95–GFP puncta (23 ± 8% increase with heat-inactivated Tat, n = 6 vs 21 ± 2% decrease with active Tat, n = 87, p < 0.01) compared with cells treated with active Tat. Together, these data demonstrate that Tat exerts opposing effects on excitatory and inhibitory synapse number, consistent with the idea that synaptic changes are a mechanism to cope with excess excitatory synaptic activity. What remains unclear is whether the Tat-induced increase in inhibitory synapse number is downstream of the Tat-induced increase in NMDAR activity or whether these two phenomena are mediated through distinct pathways.
Figure 3.
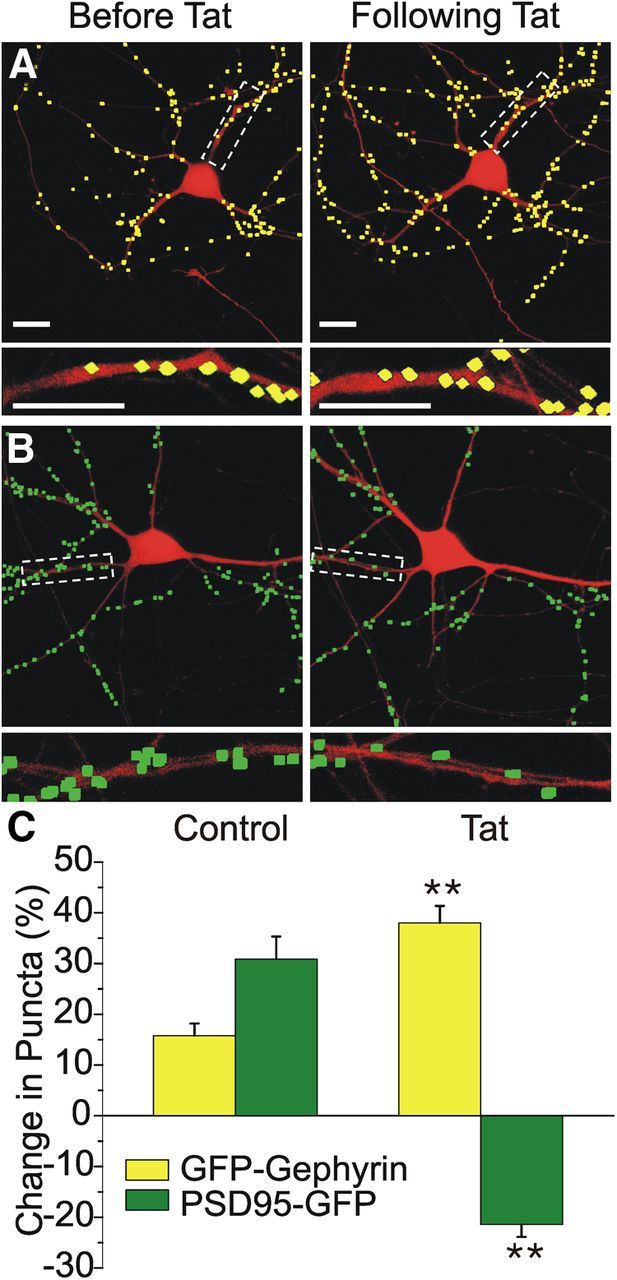
HIV-1 Tat induced concurrent upregulation of GFP–gephyrin puncta and loss of PSD95–GFP puncta. A, B, Processed confocal images of neurons expressing DsRed2 and either GFP–gephyrin (A, yellow) or PSD95–GFP (B, green) before and 24 h after Tat treatment (50 ng/ml). Insets are enlarged images of the boxed regions. Scale bars, 10 μm. C, Bar graph summarizes the effects of 24 h treatment with HIV-1 Tat or no treatment (control) on changes in GFP–gephyrin (yellow bars) or PSD95–GFP (green bars) expression. Data are mean ± SEM. **p < 0.01 relative to corresponding control group (Student's t test).
Tat-induced increases in inhibitory synapse number are dependent on binding LRP and activation of NMDAR but not ubiquitination of PSD95
The pathway by which Tat exposure induces loss of excitatory synapses is well characterized. Tat binds LRP (Liu et al., 2000), leading to activation of NMDARs (Haughey et al., 2001) and increased Ca2+ influx; the increase in [Ca2+]i activates an ubiquitin E3 ligase (Kim et al., 2008a) with subsequent degradation of PSD95 via the ubiquitin–proteasome pathway. We sought to determine whether the Tat-induced increase in inhibitory synapses was mediated through a similar pathway. First, to determine whether binding of Tat to LRP was also necessary for the Tat-induced increase in inhibitory synapses, we pretreated cultures with the chaperone protein RAP. RAP has been shown to bind and antagonize LRP (Bu, 2001), thus preventing uptake of exogenous Tat. We found that pretreatment with RAP (50 nm, 30 min) inhibited the Tat-induced increase in inhibitory synapses. Treatment with Tat in the presence of RAP yielded a 17 ± 7% increase in GFP–gephyrin puncta, significantly smaller than the 38 ± 3% increase observed with Tat alone (n = 15, p < 0.05; Fig. 4A). In a parallel experiment, RAP pretreatment abolished the Tat-induced loss of excitatory synapses (21 ± 2% decrease with Tat alone, n = 87 vs 31 ± 10% increase with RAP then Tat, n = 9, p < 0.01; Fig. 4B) as described previously (Kim et al., 2008a).
Figure 4.
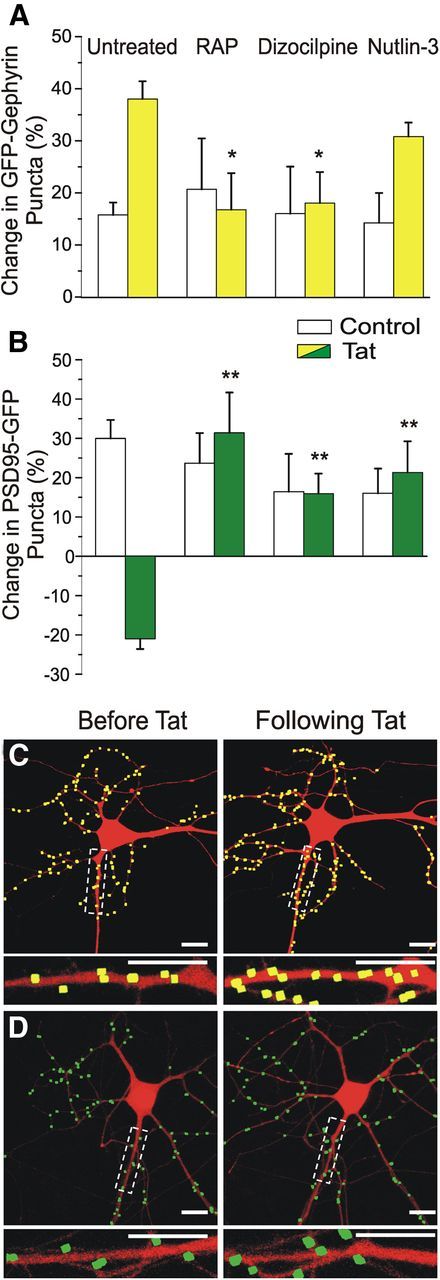
Tat-induced upregulation of GFP–gephyrin puncta is dependent on NMDA and LRP but not MDM2. Bar graphs summarize the changes in GFP–gephyrin (A) or PSD95–GFP (B) puncta in the absence (open bars) or presence (filled bars) of 50 ng/ml HIV-1 Tat for 24 h. Cells were untreated or pretreated (30 min) with 50 nm RAP, 10 μm dizocilpine, or 1 μm nutlin-3 as indicated. Data are mean ± SEM. *p < 0.05, **p < 0.01 relative to corresponding untreated + Tat groups (ANOVA with Bonferroni's post hoc test). C, D, Processed confocal images of neurons expressing DsRed2 and either GFP–gephyrin (C) or PSD95–GFP (D) before (left) and 24 h after (right) Tat (50 ng/ml) application. Cells were pretreated for 30 min with 1 μm nutlin-3. Note that nutlin-3 failed to affect the Tat-induced increase in GFP–gephyrin puncta count, whereas it blocked the effects of Tat on the number of PSD95–GFP puncta. Insets are enlarged images of the boxed regions. Scale bars, 10 μm.
Because activation of the NMDAR and subsequent Ca2+ influx is necessary for Tat-induced loss of PSD95 (Kim et al., 2008a), we next examined the possibility that the Tat-induced gephyrin increase might also be initiated through this pathway. Pretreatment with the NMDAR antagonist dizocilpine (10 μm, 30 min) significantly inhibited the Tat-induced increase in inhibitory synapses, yielding an 18 ± 6% increase in GFP–gephyrin puncta compared with a 38 ± 3% increase with Tat alone (n = 18, p < 0.05; Fig. 4A). Pretreatment with dizocilpine also prevented the Tat-induced loss of excitatory synapses, in agreement with previous studies (21 ± 2% decrease with Tat alone, n = 87 vs 16 ± 5% increase with dizocilpine and then Tat, n = 16, p < 0.01; Fig. 4B; Kim et al., 2008a). Together, these data demonstrate that the Tat-induced increase in inhibitory synapses, as well as loss of excitatory synapses, requires both LRP and NMDAR activation.
We next examined the relationship between Tat-induced alterations in excitatory and inhibitory synapses by testing the possibility that Tat-induced increases in inhibitory synapses required loss of excitatory synapses. Tat induces the loss of excitatory synapses by activating the ubiquitin–proteasome pathway. Inhibiting the pathway pharmacologically with nutlin-3, an antagonist for the ubiquitin E3 ligase murine double minute 2 (MDM2) or genetically by expressing the MDM2 inhibitor alternative reading frame polypeptide both prevented Tat-induced loss of PSD95–GFP puncta by preventing ubiquitination of PSD95 (Kim et al., 2008a). As shown in Figure 4, A and C, nutlin-3 (1 μm) had no effect on the Tat-induced increases in inhibitory synapses; GFP–gephyrin puncta increased by 31 ± 3% after 24 h exposure to Tat in the presence of nutlin-3 compared with a 38 ± 3% increase with Tat alone (n = 14). In contrast to the effects seen at inhibitory synapses, Tat-induced changes in excitatory synapses were blocked by pretreatment with nutlin-3 as described previously (21 ± 2% decrease with Tat alone, n = 87 vs 21 ± 8% increase with nutlin-3 and then Tat, n = 13, p < 0.01; Fig. 4B,D; Kim et al., 2008a). Thus, the Tat-induced increase in inhibitory synapse number is not secondary to changes in the number of excitatory synapses, indicating that a distinct mechanism mediates the increase in inhibitory synapses.
Tat-induced increases in inhibitory synapses are dependent on GluN2A-containing NMDARs
Tat-induced increases in inhibitory synapses and decreases in excitatory synapses are dependent on NMDAR activation and can be blocked by dizocilpine. In light of the distinct roles for NMDAR subtypes in Tat-induced loss of excitatory synapses (Shin et al., 2012), we examined the effects of subtype-selective NMDAR antagonists on inhibitory synapses. Micromolar concentrations of TCN201 selectively inhibit GluN2A-containing NMDARs by inhibiting the binding of glycine (Bettini et al., 2010). Ifenprodil selectively inhibits GluN2B-containing NMDARs at micromolar concentrations (Williams, 1993; Avenet et al., 1996). Pretreatment with TCN201 (10 μm, 30 min) prevented Tat-induced increases in inhibitory synapses (38 ± 3% with Tat alone, n = 99 vs 15 ± 6% with TCN201 then Tat, n = 12, p < 0.05; Fig. 5A), whereas the GluN2B-containing NMDAR antagonist ifenprodil (10 μm, 30 min) had no effect (38 ± 3% with Tat alone, n = 99 vs 35 ± 9% with ifenprodil and then Tat, n = 11; Fig. 5A). In agreement with previous findings, TCN201 also prevented the Tat-induced decrease in excitatory synapses (21 ± 2% decrease with Tat alone, n = 87 vs 20 ± 7% increase with TCN201 and then Tat, n = 12, p < 0.01; Fig. 5B), whereas ifenprodil had no effect (21 ± 2% decrease with Tat alone, n = 87 vs 29 ± 5% decrease with ifenprodil and then Tat, n = 11; Fig. 5B). These results suggest that the effects of Tat on inhibitory synapse dynamics are mediated through GluN2A-containing NMDARs in a manner similar to the effects seen at excitatory synapses.
Figure 5.
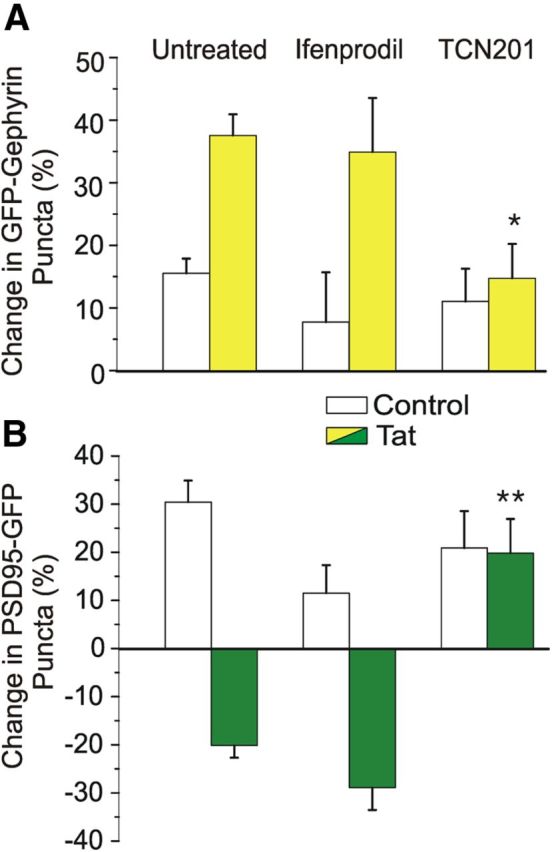
Tat-induced upregulation of GFP–gephyrin puncta is dependent on GluN2A-containing NMDARs. A, B, Bar graphs summarize the changes in GFP–gephyrin (A) or PSD95–GFP (B) puncta in the absence (control, open bars) or presence (filled bars) of 50 ng/ml HIV-1 Tat for 24 h. Cells were untreated or pretreated for 30 min with 10 μm ifenprodil or 10 μm TCN201, as indicated. Data are mean ± SEM. *p < 0.05, **p < 0.01 relative to corresponding untreated + Tat groups (ANOVA with Bonferroni's post hoc test).
Chelating intracellular calcium prevented HIV-1 Tat-induced increases in inhibitory synapses
Because NMDARs are calcium permeable, we tested the hypothesis that increased [Ca2+]i is required for Tat-induced increases in inhibitory synapse number. Loading neurons with the membrane-permeant calcium chelator BAPTA-AM (10 μm) for 30 min before exposure to Tat prevented the Tat-induced increase of gephyrin puncta (38 ± 3% with Tat alone, n = 99 vs 8 ± 5% with BAPTA-AM and then Tat, n = 8, p < 0.05; Fig. 6A,C). In agreement with previous findings, BAPTA-AM also prevented the loss of PSD95–GFP puncta (21 ± 2% decrease with Tat alone, n = 87 vs 19 ± 9% increase with BAPTA-AM and then Tat, n = 9, p < 0.01; Fig. 6D,F; Kim et al., 2008a). These findings demonstrate that Ca2+ influx through NMDARs is required for changes in both the number of inhibitory and excitatory synapses.
Figure 6.

Chelating intracellular calcium prevents HIV-1 Tat-induced increases in inhibitory synapses. A, B, Processed confocal images of neurons expressing DsRed2 and GFP–gephyrin before (left) and 24 h after (right) Tat application (50 ng/ml). Cells were pretreated for 30 min with 10 μm BAPTA-AM (A) or 10 μm EGTA-AM (B). Scale bars, 10 μm. C, Bar graph summarizes the changes in GFP–gephyrin puncta in the absence (control, open bars) or presence (filled bars) of 50 ng/ml Tat for 24 h. Cells were untreated or pretreated for 30 min with 10 μm BAPTA-AM or 10 μm EGTA-AM, as indicated. D, E, Processed confocal images of neurons expressing DsRed2 and PSD95–GFP before (left) and 24 h after (right) Tat application (50 ng/ml). Cells were pretreated for 30 min with 10 μm BAPTA-AM (D) or 10 μm EGTA-AM (E). Scale bars, 10 μm. F, Bar graph summarizes the changes in PSD95–GFP puncta in the absence (control, open bars) or presence (filled bars) of 50 ng/ml Tat for 24 h. Cells were untreated or pretreated for 30 min with 10 μm BAPTA-AM or 10 μm EGTA-AM, as indicated. Data are mean ± SEM. *p < 0.05, **p < 0.01 relative to corresponding untreated + Tat (ANOVA with Bonferroni's post hoc test).
Loss of PSD95–GFP puncta in response to direct NMDAR activation by glutamate is readily prevented by the Ca2+ chelator BAPTA-AM but not by the Ca2+ chelator EGTA-AM (Waataja et al., 2008). EGTA has an affinity for Ca2+ that is similar to BAPTA but differs in that it possesses slower binding kinetics (Tymianski et al., 1994). Thus, BAPTA is more effective than EGTA in collapsing [Ca2+]i gradients near the mouth of the NMDAR channel, preventing the activation of nearby Ca2+-sensitive targets. Loading neurons with EGTA-AM (10 μm) for 30 min before Tat exposure prevented the Tat-induced increase in inhibitory synapse number (38 ± 3% with Tat alone, n = 99 vs 8 ± 6% with EGTA-AM and then Tat, n = 13, p < 0.05; Fig. 6B,C). In contrast, pretreatment with EGTA-AM failed to prevent the Tat-induced loss of excitatory synapses (n = 8; Fig. 6E,F). PSD95 is located predominately in dendritic spines (Hering and Sheng, 2001) in close apposition to NMDARs, allowing BAPTA but not EGTA to prevent activation of Ca2+-sensitive effectors attached to the PSD scaffolding. Inhibitory postsynaptic synapses anchored by gephyrin are predominately located on the dendritic shaft (Moss and Smart, 2001) farther away from NMDARs, in good agreement with the ability of EGTA to prevent Tat-induced changes in inhibitory but not excitatory synapse number.
CaMKII is required for HIV-1 Tat-induced increases in inhibitory synapses
Many forms of synaptic plasticity are mediated through insertion and removal of glutamate (Malenka and Bear, 2004) and GABA (Michels and Moss, 2007) receptors at the postsynaptic membrane. This dynamic cycling of both excitatory (Barria et al., 1997) and inhibitory (Wei et al., 2004) synapses can be modulated via activation of CaMKII signaling pathways. Interestingly, activation of NMDARs can induce translocation of CaMKII to both excitatory (Shen and Meyer, 1999) and inhibitory (Marsden et al., 2007) synapses. We sought to determine whether the effects of Tat on inhibitory and excitatory synapses were mediated through the CaMKII signaling pathway. Pretreatment with the CaMKII inhibitor KN-62 (10 μm, 30 min) prevented both the Tat-induced increase in inhibitory synapse number (38 ± 3% with Tat alone, n = 99 vs 14 ± 4% with KN-62 and then Tat, n = 11, p < 0.05; Fig. 7A), as well as the Tat-induced loss of excitatory synapses (21 ± 2% decrease with Tat alone, n = 87 vs 22 ± 8% increase with KN-62 then Tat, n = 7, p < 0.01; Fig. 7A), demonstrating that the effects of Tat on inhibitory and excitatory synapse dynamics are CaMKII-dependent. Inhibitory synaptic organization can also be modulated by activation of the Ca2+-dependent protein phosphatase calcineurin (Marsden et al., 2010). In contrast to the effects seen with KN-62, pretreatment with the calcineurin inhibitor FK506 (10 μm) failed to affect the Tat-induced increase in inhibitory synapses (n = 7; Fig. 7A) or the Tat-induced loss of excitatory synapses (n = 5; Fig. 7A), suggesting that calcineurin activity was not required for Tat-induced synaptic reorganization.
Figure 7.
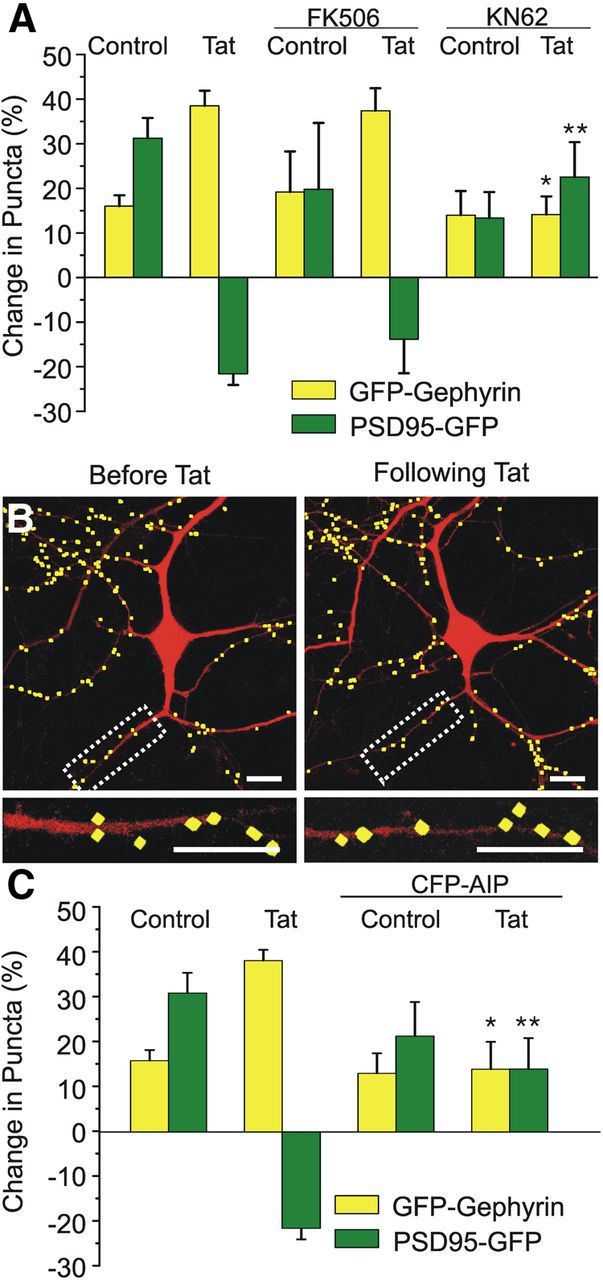
CaMKII activity is required for Tat-induced increases in inhibitory synapses. A, Bar graph summarizes the changes in GFP–gephyrin (yellow bars) or PSD95–GFP (green bars) puncta after the indicated treatments. Cells were untreated (control) or treated with Tat (50 ng/ml; 24 h), FK506 (10 μm), or KN-62 (10 μm) as indicated. Drugs were applied 30 min before addition of Tat. B, Processed representative confocal images of neurons expressing DsRed2, GFP–gephyrin, and CFP–AIP before (left) and 24 h after Tat (50 ng/ml; right). Insets are enlarged images of the boxed region. Scale bars, 10 μm. C, Bar graph summarizes the changes in GFP–gephyrin (yellow bars) or PSD95–GFP (green bars) puncta in the absence or presence of expressed CFP–AIP. Cells were either untreated (control) or treated with Tat (50 ng/ml, 24 h), as indicated. Data are mean ± SEM. *p < 0.05, **p < 0.01 relative to corresponding Tat groups (ANOVA with Bonferroni's post hoc test).
Pharmacological blockade of CaMKII activity with KN-62 would presumably affect all neurons within the hippocampal culture. To determine whether the role of CaMKII in Tat-mediated changes in synaptic number were secondary to global activation of CaMKII within the culture or a local activation of CaMKII within the dendrite, we used a genetically encoded inhibitor of CaMKII, AIP fused to CFP (CFP–AIP; Zha et al., 2009). Neuronal cultures were cotransfected with expression vectors for DsRed2, CFP–AIP, and either GFP–gephyrin or PSD95–GFP to label inhibitory or excitatory synapses, respectively. Expression of CFP–AIP prevented the Tat-induced increase in inhibitory synapse number (14 ± 6% increase in GFP–gephyrin puncta in Tat-treated cells expressing CFP–AIP, n = 10 vs 38 ± 3% increase in Tat-treated cells not expressing CFP–AIP, n = 99, p < 0.05; Fig. 7B,C). Expression of CFP–AIP also prevented the Tat-induced decrease in excitatory synapse number (14 ± 7% increase in PSD95–GFP puncta in Tat-treated cells expressing CFP–AIP, n = 12 vs 21 ± 2% decrease in Tat-treated cells not expressing CFP–AIP, n = 87, p < 0.01; Fig. 7C). These data suggest that, after Tat exposure, CaMKII acts in a cell-autonomous manner to modulate both inhibitory and excitatory synaptic reorganization.
Disruption of gephyrin-mediated insertion of GABAARs into the plasma membrane prevented HIV-1 Tat-induced increases in inhibitory synapses
Dynamic expression and stabilization of GABAARs at the membrane is influenced by a number of accessory proteins, including gephyrin and the GABAAR-associated protein (GABARAP). GABARAP links the γ2 subunit of GABAARs to tubulin (Wang et al., 1999; Nymann-Andersen et al., 2002), enabling trafficking of GABAARs to the plasma membrane (Leil et al., 2004), and is the only known protein that interacts with both gephyrin and GABAARs (Kneussel et al., 2000). Neuronal activity and subsequent CaMKII activation induces a structural change in GABARAP that alters GABAAR cell surface expression; binding of structurally altered GABARAP to GABAARs links CaMKII activation to some forms of LTP (Kawaguchi and Hirano, 2007). To further elucidate the mechanism by which CaMKII activation induces increases in inhibitory synapse number, we developed a construct to express a peptide derived from the γ2 subunit of the GABAAR fused to ECFP (γ2-CFP). The γ2 peptide prevents the interaction of the GABAAR γ2 subunit with GABARAP disrupting the trafficking of GABAARs to the postsynaptic plasma membrane (Kawaguchi and Hirano, 2007). Neuronal cultures were cotransfected with expression vectors for DsRed2, γ2–CFP, and either GFP–gephyrin or PSD95–GFP to label inhibitory or excitatory synapses, respectively. Expression of γ2–CFP prevented the Tat-induced increase in inhibitory synapse number, whereas a control peptide with a scrambled amino acid sequence did not (Tat induced a 39 ± 5% increase in GFP–gephyrin puncta in cells expressing scrambled γ2–CFP, n = 12 vs 11 ± 2% increase in puncta in Tat-treated cells expressing γ2–CFP, n = 15, p < 0.05; Fig. 8). In contrast to the effects seen at inhibitory synapses, expression of γ2–CFP did not affect the Tat-induced decrease in excitatory synapse number relative to cells expressing the scrambled peptide (Tat induced a 25 ± 4% decrease in PSD95–GFP puncta in cells expressing γ2–CFP, n = 12 and a 26 ± 4% decrease in PSD95–GFP puncta in cells expressing scrambled γ2–CFP, n = 12; Fig. 8B). These data suggest that Tat activates a CaMKII pathway at inhibitory synapses that leads to structural changes in GABARAP, resulting in increased membrane insertion of GABAARs.
Figure 8.
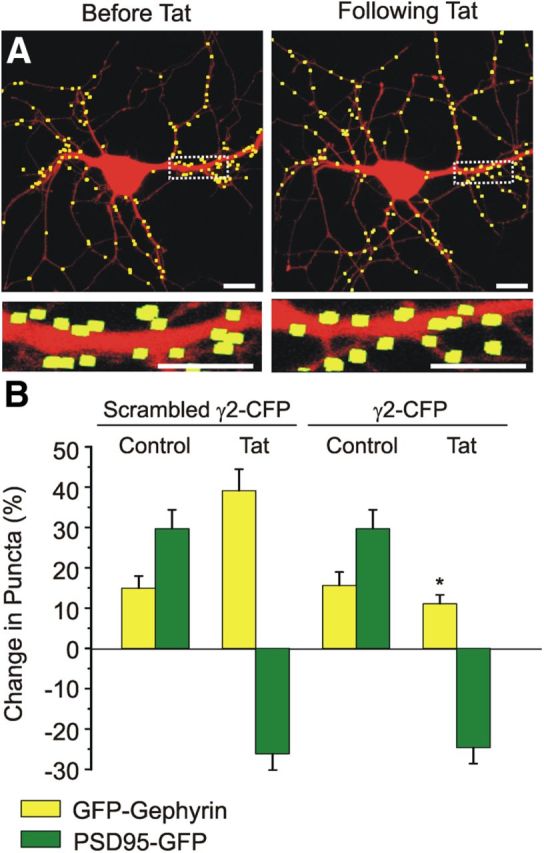
Inhibiting gephyrin-mediated insertion of GABAARs into the membrane prevents Tat-induced increases in inhibitory synapses. A, Processed representative confocal images of neurons expressing DsRed2, GFP–gephyrin, and γ2–CFP before (left) and 24 h after treatment with Tat (50 ng/ml, right). Insets are enlarged images of the boxed region. Scale bars, 10 μm. B, Bar graph summarizes the changes in GFP–gephyrin (yellow bars) or PSD95–GFP (green bars) puncta when cotransfected with scrambled γ2–CFP or active γ2–CFP. Cells were either untreated (control) or treated with Tat (50 ng/ml, 24 h), as indicated. Data are mean ± SEM. *p < 0.05 relative to corresponding Tat + scrambled γ2–CFP group (ANOVA with Bonferroni's post hoc test).
Inhibition of GluN2B-containing NMDARs restores synaptic balance after Tat exposure via reduced activity of nNOS
The loss of excitatory synapses after exposure to Tat is reversible (Kim et al., 2008a); treatment with ifenprodil, an antagonist of GluN2B-containing NMDARs, induced recovery of PSD95–GFP puncta after Tat-induced loss (Shin et al., 2012). Therefore, we decided to test the possibility that Tat-induced increases in the number of inhibitory synapses could be returned to basal levels by inhibiting GluN2B-containing NMDARs. Neuronal cultures were treated with Tat for 16 h to evoke an increase in the number of inhibitory synapses. After 16 h exposure to Tat, the neuron was imaged again, and ifenprodil (10 μm) was added to the culture media. After 8 h of ifenprodil treatment, the neuron was imaged a final time (24 h). We found that ifenprodil, added 16 h after the application of and in the continued presence of Tat, was able to restore inhibitory synapse number to basal levels, a significant decrease compared with those not receiving ifenprodil (12 ± 5% increase, n = 13 with ifenprodil vs 47 ± 5% increase, n = 19 without ifenprodil, p < 0.05; Fig. 9A). In agreement with previous findings, we found that adding ifenprodil after 16 h in the presence of Tat induced the recovery of excitatory synapse numbers to original levels (10 ± 6% increase with ifenprodil, n = 18 vs 36 ± 3% loss, n = 18 without ifenprodil, p < 0.05; Fig. 9B; Shin et al., 2012). In contrast to ifenprodil, TCN201 was unable to restore inhibitory or excitatory synapse numbers to control levels in the continued presence of Tat (Fig. 9A,B). These results demonstrate that blocking GluN2B-containing NMDARs in the presence of Tat restores balance to the synaptic network, whereas blocking GluN2A-containing NMDARs was unable to reverse the effects of Tat. These results suggest different roles for NMDAR subtypes: GluN2A-containing NMDARs mediate Tat-induced increases in inhibitory synapse number and loss of excitatory synapse number, whereas GluN2B-containing NMDARs suppress return of inhibitory and excitatory synapse number to control levels after Tat.
Figure 9.
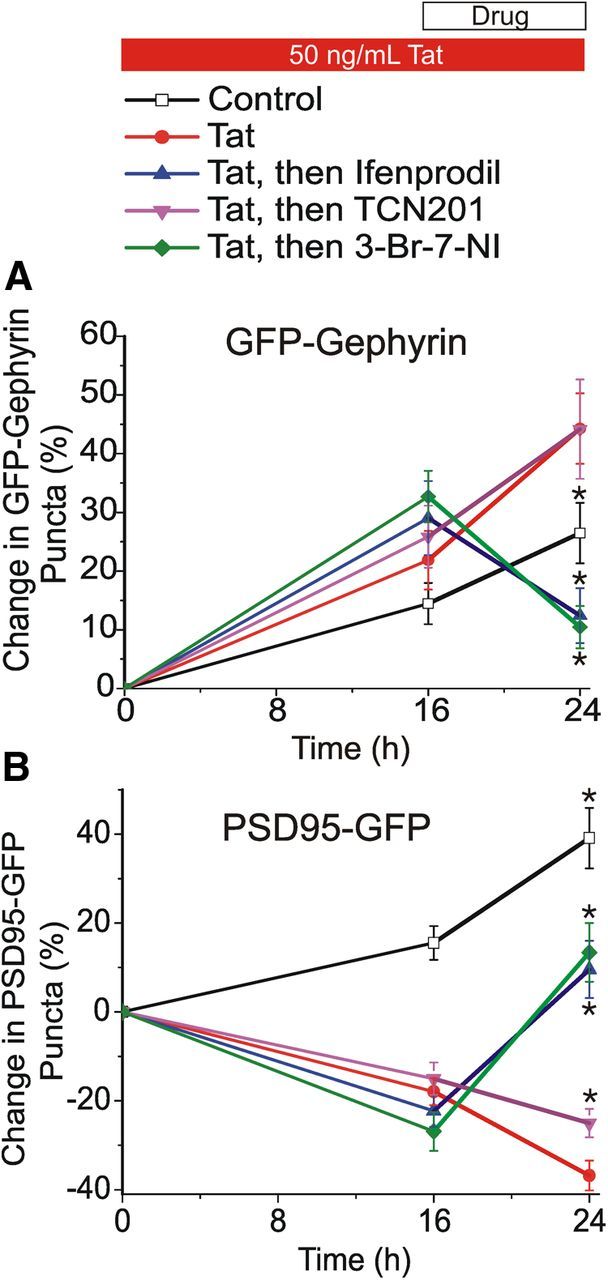
Inhibiting GluN2B-containing NMDARs or nNOS restores the balance of inhibitory synapses after Tat. Graph summarizes changes in GFP–gephyrin (A) or PSD95–GFP (B) puncta in the absence (control, open squares) or presence (24 h, filled symbols) of 50 ng/ml Tat. After 16 h exposure to Tat, cells were left untreated (red circles) or treated with 10 μm ifenprodil (blue triangles), 10 μm TCN201 (purple inverted triangles), or 10 μm 3-Br-7-NI (green diamonds). Data are expressed at mean ± SEM. *p < 0.05 relative to Tat (ANOVA with Bonferroni's post hoc test).
GluN2A- and GluN2B-containing NMDARs play distinct roles in synaptic plasticity and excitotoxicity (Liu et al., 2004, 2007). We showed previously that exposure to Tat initiates two divergent pathways, leading to either synaptic modification via the proteasome pathway or cell death via the NOS pathway (Kim et al., 2008a). Treatment with the NOS-inhibitor N-ω-nitro-l-arginine methyl ester induces recovery of excitatory synapses after Tat in a manner similar to treatment with the GluN2B-selective antagonist ifenprodil (A. Shin and S. Thayer, unpublished observations), consistent with reports suggesting that GluN2B-containing NMDARs couple to nNOS (Martel et al., 2012). To determine whether the distinct roles of GluN2A- versus GluN2B-containing NMDARs in mediating Tat-induced synaptic changes resulted from differential coupling to the nNOS pathway, neuronal cultures were treated with the nNOS-selective inhibitor 3-Br-7-NI (Wu et al., 1997). Treatment with 3-Br-7-NI (10 μm) 16 h after the application of and in the continued presence of Tat restored inhibitory synapse number to basal levels by 24 h; the number of GFP–gephyrin puncta decreased significantly in 3-Br-7-NI-treated cells compared with untreated cells (10 ± 3% increase, n = 10 with 3-Br-7-NI vs 47 ± 5% increase, n = 19 without 3-Br-7-NI, p < 0.05; Fig. 9A). Adding 3-Br-7-NI after 16 h in the presence of Tat induced the recovery of excitatory synapse numbers to original levels (13 ± 7% increase with 3-Br-7-NI, n = 11 vs 36 ± 3% loss, n = 18 without 3-Br-7-NI, p < 0.05; Fig. 9B). In contrast, pretreatment with 3-Br-7-NI (30 min) before Tat application was unable to prevent the Tat-induced increase in inhibitory synapses (n = 11) or the Tat-induced loss of excitatory synapses (n = 10). Together, these data demonstrate that 3-Br-7-NI has an efficacy profile similar to the GluN2B-containing NMDAR antagonist ifenprodil: the drug was without effect in protecting from Tat-induced increases in inhibitory synapses or decreases in excitatory synapses when applied before Tat but induced recovery of inhibitory and excitatory synapses to basal levels when applied after Tat. These data indicate that activation of GluN2B-containing NMDARs suppresses recovery of synapses via the nNOS pathway and that inhibiting this pathway restores synapse number.
Discussion
The HIV-1 protein Tat increased the number of inhibitory and decreased the number of excitatory synaptic connections between rat hippocampal neurons in culture. These results recapitulate the loss of excitatory synapses observed in HAND patients and identify a novel effect on inhibitory synapses. We suggest that Tat-evoked synaptic changes are not symptoms of the demise of the neuron but instead represent a coping mechanism to counteract excess excitatory synaptic activity. Changes in both inhibitory and excitatory synapses were initiated by Tat binding to LRP, followed by Ca2+ influx via GluN2A-containing NMDARs and subsequent activation of CaMKII (Fig. 10). The synaptic changes induced by Tat were reversed by the GluN2B-selective antagonist ifenprodil, which was intriguing because ifenprodil was without effect when applied before Tat. Thus, the pharmacology changes during exposure to Tat. Inhibition of GluN2B signaling shows promise for reversing what may be a neuroprotective mechanism gone awry.
Figure 10.

Hypothetical mechanism for HIV-1 Tat-induced increase in inhibitory synapses and recovery.
HIV-1 does not infect neurons; thus, the neurotoxicity associated with HAND is indirect and results from the release of neurotoxins such as Tat from infected cells with subsequent endocytosis by neighboring cells (Nath et al., 1999; King et al., 2006). Tat is a transcription-promoting protein, but its neurotoxic properties are unrelated to its role in viral gene expression (Ensoli et al., 1993). Extensive evidence supports the contribution of Tat to the pathogenesis of HAND. Tat protein levels are elevated in brain extracts of HAND patients (Del Valle et al., 2000), and injecting Tat into the brains of rats produces neuropathology similar to HAND (Maragos et al., 2003; Li et al., 2004). Tat dysregulates astrocyte signaling (Henderson et al., 2012), and its expression in the CNS produces gliosis (Fitting et al., 2013). Tat transgenic mice exhibit impaired learning and memory and reduced CA1 neuronal spine density (Fitting et al., 2013).
We used in vitro imaging of fluorescently tagged synaptic proteins to track changes in the number of inhibitory and excitatory synapses between hippocampal neurons in culture. PSD95–GFP puncta label functional excitatory synapses as demonstrated previously (Waataja et al., 2008). GFP–gephyrin puncta label functional inhibitory synapses as indicated by colocalization with VGAT immunoreactivity and electrophysiological experiments that correlated puncta number with mIPSC frequency. A key advantage of this imaging-based approach is that it is non-invasive, enabling longitudinal monitoring of synaptic connections. This assay detected a rapid scaling of inhibitory and excitatory synapses in response to TTX or bicuculline that was reminiscent of homeostatic plasticity (Turrigiano, 1999). Acute treatment with Tat increases mEPSC frequency, consistent with its classification as an excitotoxin (Brailoiu, 2008). Indeed, Tat evoked synaptic changes comparable with those evoked by an excitatory stimulus (Marder and Goaillard, 2006), suggesting that physiological processes responsible for maintaining network homeostasis may contribute to early synaptic changes in HAND.
The Tat-induced increases in GFP–gephyrin puncta seen here are consistent with previous findings noting increased gephyrin immunoreactivity in hippocampal lysates from Tat-expressing transgenic mice (Fitting et al., 2013). Tat-induced synaptic changes required binding to the endocytic LRP receptor (Liu et al., 2000), consistent with the LRP antagonist RAP blocking Tat-induced changes in synapse number. LRP participates in other neurodegenerative processes; it binds apolipoprotein ε4, a genetic risk factor for AD (Schellenberg et al., 2000), and LRP mRNA levels increase in AD patients with dementia (Akram et al., 2012). Tat binding to LRP increases [Ca2+]i via activation of NMDARs (Backsai et al., 2000), possibly by forming a complex with LRP, PSD95, and NMDAR (Eugenin et al., 2007).
NMDAR-mediated rises in [Ca2+]i elicit many effects, including pro-survival gene expression, synaptic remodeling, and neuronal death (Albensi, 2007). These seemingly contradictory functions can be reconciled by attributing specific roles to different NMDAR subtypes. In hippocampal neurons, activation of GluN2A-containing NMDARs stimulates pro-survival gene expression (Hardingham et al., 2002) and induces excitatory synapse loss but fails to trigger neuronal death (Shin et al., 2012). TCN201, a drug selective for GluN2A-containing NMDARs, prevented the Tat-induced increase in inhibitory synapses. The pro-survival role for this NMDAR subtype is consistent with our assertion that the increase in inhibitory synapses is a neuroprotective mechanism. Activation of GluN2B-containing NMDARs triggers neuronal death (Martel et al., 2012; Shin et al., 2012); the GluN2B-selective drug ifenprodil had no effect on Tat-induced increases in GFP–gephyrin puncta.
Tat-induced Ca2+ influx through NMDARs acted on CaMKII to alter inhibitory and excitatory synapse number. The CaMKII antagonist KN-62 blocked all Tat-induced synaptic changes. CaMKII plays a central role in synaptic plasticity (Lisman et al., 2012), and its activation can potentiate or depress glutamatergic synaptic transmission depending on experimental conditions (Wang, 2008). CaMKII activation also strengthens GABAergic synaptic transmission (Wei et al., 2004). Presynaptic CaMKII activity modulates synaptic strength in some plasticity paradigms (Ninan and Arancio, 2004). However, the actions of CaMKII described here were cell autonomous because postsynaptic expression of AIP blocked Tat-induced synaptic changes. CaMKII activation initiates insertion of GABAARs into the postsynaptic membrane (Marsden et al., 2010) via regulation of GABARAP (Kawaguchi and Hirano, 2007). The Tat-induced, CaMKII-mediated increase in inhibitory synapses was blocked by expression of the γ2 peptide, suggesting a similar mechanism. Tat-induced loss of glutamatergic synapses was not affected by the γ2 peptide. Loss of excitatory synapses was inhibited by nutlin-3, an inhibitor of the ubiquitin E3 ligase MDM2. The α isoform of CaMKII is highly enriched in dendritic spines of rat hippocampal neurons (Ding et al., 2013) and recruits the proteasome into spines (Bingol et al., 2010), suggesting that CaMKIIα may link Tat-induced increases in [Ca2+]i to PSD95 degradation via the ubiquitin–proteasome pathway (Kim et al., 2008a). The Tat-induced increase in inhibitory synapse number was not sensitive to nutlin-3, indicating that the pathways regulating inhibitory and excitatory synapse diverge downstream of CaMKII.
This bifurcation in the pathway may result in part from the spatial arrangement of CaMKII and its substrates. Chelating [Ca2+]i with BAPTA prevented the Tat-induced changes in synapse composition, indicating that Ca2+entry via NMDARs is necessary for the effects of Tat on inhibitory and excitatory synapses. However, chelating [Ca2+]i with EGTA, which binds Ca2+ with comparable affinity to BAPTA but >10-fold slower kinetics, prevented Tat-induced changes in inhibitory but not excitatory synapses. These results suggest that the CaMKII mediating excitatory synapse loss is localized in close apposition to the mouth of the NMDAR where the [Ca2+]i gradient is steep and relatively insensitive to buffering by EGTA. As shown in Figure 10, we hypothesize that the CaMKII activity that initiates formation of GFP–gephyrin puncta is located farther from the NMDAR Ca2+ source, where it would be sensitive to EGTA, presumably on the dendritic shaft. This arrangement is consistent with the purported higher density of GluN2A-containing NMDARs at synapses relative to GluN2B-containing NMDARs, which are primarily extrasynaptic (Hardingham et al., 2002).
The recovery of synaptic balance produced by ifenprodil and 3-Br-7-NI, drugs that were without effect when given before Tat, suggests that the pharmacology that controls synapse stability and formation changes during exposure to Tat. We showed previously that Tat activates separate pathways, leading to either synapse modification via the proteasome pathway or cell death via the nNOS pathway (Kim et al., 2008a). GluN2B-containing NMDARs couple to nNOS (Martel et al., 2012), suggesting that NO signaling might prevent recovery of excitatory and inhibitory synapses back to original levels. Our data support the idea that distinct signaling cascades downstream of NMDARs determine the actions of Tat; treatment with an nNOS-specific inhibitor induced recovery of excitatory and inhibitory synapses after Tat treatment but failed to prevent initial Tat-induced synaptic changes in a manner similar to the GluN2B-selective antagonist ifenprodil. GluN2B-containing NMDARs are a promising target for neuroprotective drugs because overactivation of the nNOS pathway has been linked to NMDAR-mediated cell death, and, as shown here, blocking this NMDAR subtype or nNOS restores balance to synaptic networks. Memantine, which preferentially blocks extrasynaptic NMDARs (Xia et al., 2010), improves cognition in patients with early-stage AD (Reisberg et al., 2003), suggesting that this might prove to be an efficacious clinical approach. Memantine was well tolerated in HAND patients, although larger studies of longer duration will be required to address questions of efficacy (Zhao et al., 2010).
Tat-induced changes in synapse number may be an adaptive mechanism to counteract increases in network excitability. Are these benign changes that maintain homeostasis or, when pushed to extremes during neurodegenerative disease, do they underlie impaired function? Loss of excitatory synapses correlates with cognitive decline in many neurodegenerative diseases, suggesting that there is a limit to synaptic loss, beyond which function will be impaired. Upregulation of GABAergic relative to glutamatergic signaling has been noted in other models of neurodegenerative diseases with an excitotoxic component, including AD (Rissman et al., 2007) and amyotrophic lateral sclerosis (Martin and Chang, 2012), presumably as a mechanism to counteract aberrant glutamatergic transmission. GABAergic tone is upregulated in the peri-infarct zone after stroke, and blocking this adaptation promotes recovery (Clarkson et al., 2010), suggesting that, in some situations, increases in GABAergic signaling impair function. This study recapitulates the loss of excitatory synapses observed in HAND and raises the possibility that increases in inhibitory synapses occur concurrently, suggesting that changes in inhibitory neurotransmission may be an important consideration in the treatment of HAND.
Footnotes
This work was supported by National Institutes of Health Grants DA07304 (S.A.T.) and T32DA007234 and F32DA034997 (N.J.H.).
The authors declare no competing financial interests.
References
- Akram A, Schmeidler J, Katsel P, Hof PR, Haroutunian V. Association of ApoE and LRP mRNA levels with dementia and AD neuropathology. Neurobiol Aging. 2012;33:628.e1–628.e14. doi: 10.1016/j.neurobiolaging.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albensi BC. The NMDA receptor/ion channel complex: a drug target for modulating synaptic plasticity and excitotoxicity. Curr Pharm Des. 2007;13:3185–3194. doi: 10.2174/138161207782341321. [DOI] [PubMed] [Google Scholar]
- Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron. 1999;23:149–157. doi: 10.1016/S0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Avenet P, Léonardon J, Besnard F, Graham D, Frost J, Depoortere H, Langer SZ, Scatton B. Antagonist properties of the sterioisomers of ifenprodil at NR1A/NR2A and NR1A/NR2B subtypes of the NMDA receptor expressed in Xenopus oocytes. Eur J Pharmacol. 1996;296:209–213. doi: 10.1016/0014-2999(95)00700-8. [DOI] [PubMed] [Google Scholar]
- Bacsksai BJ, Xia MQ, Strickland DK, Rebeck GW, Hyman BT. The endocytic receptor protein LRP also mediates neuronal calcium signaling via N-methyl-d-aspartate receptors. Proc Natl Acad Sci U S A. 2000;97:11551–11556. doi: 10.1073/pnas.200238297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai H, Lévi S, Schweizer C, Inoue T, Launey T, Racine V, Sibarita JB, Mikoshiba K, Triller A. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron. 2009;62:670–682. doi: 10.1016/j.neuron.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral L, Cardullo F. Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J Pharmacol Exp Ther. 2010;335:636–644. doi: 10.1124/jpet.110.172544. [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001;288:301–308. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- Brailoiu GC, Brailoiu E, Chang JK, Dun NJ. Excitatory effects of human immunodeficiency virus 1 Tat on cultured rat cerebral cortical neurons. Neuroscience. 2008;151:701–710. doi: 10.1016/j.neuroscience.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G. The roles of receptor-associated protein (RAP) as a molecular chaperone for members of the LDL receptor family. Int Rev Cytol. 2001;209:79–116. doi: 10.1016/S0074-7696(01)09011-8. [DOI] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin I, Rostaing P, Triller A. Gephyrin accumulates at specific plasmalemma loci during neuronal maturation in vitro. J Comp Neurol. 1996;374:467–479. doi: 10.1002/(SICI)1096-9861(19961021)374:3<467::AID-CNE10>3.0.CO%3B2-S. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K. Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6:221–228. doi: 10.3109/13550280009015824. [DOI] [PubMed] [Google Scholar]
- Ding JD, Kennedy MB, Weinberg RJ. Subcellular organization of camkii in rat hippocampal pyramidal neurons. J Comp Neurol. 2013;521:3570–3583. doi: 10.1002/cne.23372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW. HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Xu R, Bull C, Buch SK, El-Hage N, Nath A, Knapp PE, Hauser KF. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol. 2010;177:1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 tat transgenic mice. Biol Psychiatry. 2013;73:443–453. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann JC, Kins S, Rostaing P, El Far O, Kirsch J, Sheng M, Triller A, Betz H, Kneussel M. Gephyrin interacts with dynein light chains 1 and 2, components of motor protein complexes. J Neurosci. 2002;22:5393–5402. doi: 10.1523/JNEUROSCI.22-13-05393.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, Keane RW, Resnick L, Mizrachi Y, Volsky DJ, Epstein LG, Gendelman HE. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LJ, Sharma A, Monaco MC, Major EO, Al-Harthi L. Human immunodeficiency virus type 1 (HIV-1) transactivator of transcription through its intact core and cysteine-rich domains inhibits Wnt/beta-catenin signaling in astrocytes: relevance to HIV neuropathogenesis. J Neurosci. 2012;32:16306–16313. doi: 10.1523/JNEUROSCI.3145-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- Kawaguchi SY, Hirano T. Sustained structural change of GABAA receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. J Neurosci. 2007;27:6788–6799. doi: 10.1523/JNEUROSCI.1981-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008a;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Waataja JJ, Thayer SA. Cannabinoids inhibit network-driven synapse loss between hippocampal neurons in culture. J Pharmacol Exp Ther. 2008b;325:850–858. doi: 10.1124/jpet.107.131607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Shin AH, Thayer SA. Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol Pharmacol. 2011;80:357–366. doi: 10.1124/mol.111.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kneussel M, Haverkamp S, Fuhrmann JC, Wang H, Wässle H, Olsen RW, Betz H. The gamma-aminobutyric acid type A receptor (GABAAR)-associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc Natl Acad Sci U S A. 2000;97:8594–8599. doi: 10.1073/pnas.97.15.8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Leil TA, Chen ZW, Chang CS, Olsen RW. GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci. 2004;24:11429–11438. doi: 10.1523/JNEUROSCI.3355-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ST, Matsushita M, Moriwaki A, Saheki Y, Lu YF, Tomizawa K, Wu HY, Terada H, Matsui H. HIV-1 Tat inhibits long-term potentiation and attenuates spatial learning. Ann Neurol. 2004;55:362–371. doi: 10.1002/ana.10844. [DOI] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med. 2000;6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE, Nath A. Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neurosci. 2003;117:43–53. doi: 10.1016/S0306-4522(02)00713-3. [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABAA receptors. J Neurosci. 2007;27:14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective translocation of Ca2+/calmodulin protein kinase IIα (CaMKIIα) to inhibitory synapses. Proc Natl Acad Sci U S A. 2010;107:20559–20564. doi: 10.1073/pnas.1010346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel MA, Ryan TJ, Bell KF, Fowler JH, McMahon A, Al-Mubarak B, Komiyama NH, Horsburgh K, Kind PC, Grant SG, Wyllie DJ, Hardingham GE. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron. 2012;74:543–556. doi: 10.1016/j.neuron.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Chang Q. Inhibitory synaptic regulation of motoneurons: a new target of disease mechanisms in amyotrophic lateral sclerosis. Mol Neurobiol. 2012;45:30–42. doi: 10.1007/s12035-011-8217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels G, Moss SJ. GABAA receptors: properties and trafficking. Crit Rev Biochem Mol Biol. 2007;42:3–14. doi: 10.1080/10409230601146219. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Constructing inhibitory synapses. Nat Rev Neurosci. 2001;2:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Ninan I, Arancio O. Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron. 2004;42:129–141. doi: 10.1016/S0896-6273(04)00143-6. [DOI] [PubMed] [Google Scholar]
- Nymann-Andersen J, Wang H, Chen L, Kittler JT, Moss SJ, Olsen RW. Subunit specificity and interaction domain between GABA(A) receptor-associated protein (GABARAP) and GABA(A) receptors. J Neurochem. 2002;80:815–823. doi: 10.1046/j.0022-3042.2002.00762.x. [DOI] [PubMed] [Google Scholar]
- Rannals MD, Kapur J. Homeostatic strengthening of inhibitory synapses is mediated by the accumulation of GABAA receptors. J Neurosci. 2011;31:17701–17712. doi: 10.1523/JNEUROSCI.4476-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- Rissman RA, De Blas AL, Armstrong DM. GABA(A) receptors in aging and Alzheimer's disease. J Neurochem. 2007;103:1285–1292. doi: 10.1111/j.1471-4159.2007.04832.x. [DOI] [PubMed] [Google Scholar]
- Sá MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM. Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol (Berl) 2004;107:97–110. doi: 10.1007/s00401-003-0781-3. [DOI] [PubMed] [Google Scholar]
- Schellenberg GD, D'Souza I, Poorkaj P. The genetics of Alzheimer's disease. Curr Psychiatry Rep. 2000;2:158–164. doi: 10.1007/s11920-000-0061-z. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shen M, Thayer SA. The cannabinoid agonist Win55,212–2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998;783:77–84. doi: 10.1016/S0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- Shin AH, Kim HJ, Thayer SA. Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 Tat. Br J Pharmacol. 2012;166:1002–1017. doi: 10.1111/j.1476-5381.2011.01805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speth C, Stöckl G, Mohsenipour I, Würzner R, Stoiber H, Lass-Flörl C, Dierich MP. Human immunodeficiency virus type 1 induces expression of complement factors in human astrocytes. J Virol. 2001;75:2604–2615. doi: 10.1128/JVI.75.6.2604-2516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/S0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Properties of neuroprotective cell-permeant Ca2+ chelators: effects on [Ca2+]i and glutamate neurotoxicity in vitro. J Neurophysiol. 1994;72:1973–1992. doi: 10.1152/jn.1994.72.4.1973. [DOI] [PubMed] [Google Scholar]
- Waataja JJ, Kim HJ, Roloff AM, Thayer SA. Excitotoxic loss of post-synaptic sites is distinct temporally and mechanistically from neuronal death. J Neurochem. 2008;104:364–375. doi: 10.1111/j.1471-4159.2007.04973.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature. 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- Wang ZW. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol Neurobiol. 2008;38:153–166. doi: 10.1007/s12035-008-8039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Zhang M, Zhu Y, Wang JH. Ca(2+)-calmodulin signalling pathway up-regulates GABA synaptic transmission through cytoskeleton-mediated mechanisms. Neuroscience. 2004;127:637–647. doi: 10.1016/j.neuroscience.2004.05.056. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Wu J, Wang Y, Rowan MJ, Anwyl R. Evidence for involvement of the neuronal isoform of nitric oxide synthase during induction of long-term potentiation and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 1997;78:393–398. doi: 10.1016/S0306-4522(97)84911-1. [DOI] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Dailey ME, Green SH. Role of Ca2+/calmodulin-dependent protein kinase II in dendritic spine remodeling during epileptiform activity in vitro. J Neurosci Res. 2009;87:1969–1979. doi: 10.1002/jnr.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Navia BA, Marra CM, Singer EJ, Chang L, Berger J, Ellis RJ, Kolson DL, Simpson D, Miller EN, Lipton SA, Evans SR, Schifitto G, Adult Aids Clinical Trial Group (ACTG) 301 Team Memantine for AIDS dementia complex: open-label report of ACTG 301. HIV Clin Trials. 2010;11:59–67. doi: 10.1310/hct1101-59. [DOI] [PMC free article] [PubMed] [Google Scholar]