

Abstract
Pazopanib (Votrient™, GlaxoSmithKline), a multi-kinase inhibitor with activity against VEGFR and other receptors, was recently approved by the FDA for the treatment of advanced renal cell carcinoma (RCC). Here, we review the history of its development, together with an overview of VEGF and its receptors and co-receptors. Results from selected clinical trial data in RCC and other malignant diseases are presented. Based on available evidence, pazopanib is an effective VEGFR inhibitor with demonstrable clinical activity in metastatic RCC and promising activity in other diseases. Like most kinase inhibitors, its activity is not restricted to VEGF receptors, which is reflected in its side-effect profile.
Keywords: pazopanib, VEGFR, renal cell carcinoma
Introduction to VEGF biology and anti-VEGF therapy
Following the pioneering work of the late Judah Folkman demonstrating the role of angiogenesis in tumor development, the treatment of metastatic cancer has undergone a major revolution, especially in diseases such as renal cell carcinoma (RCC). Folkman’s initial work, now almost 40 years ago,1 led to the realization that small tumors (ie, 3–4 mm) could lie dormant in the absence of a dedicated vascular supply. This work also led to the partial purification an angiogenic activity, termed tumor angiogenesis factor. Vascular permeability factor, now known as VEGF-A, is produced by a wide variety of cancers of which RCC is one of the best examples. Moreover, in a small subset of RCCs, VEGF-A is probably the predominant or sole angiogenic factor, as evidenced by the ability of an anti-VEGF antibody to control the disease for a period of several years.2 However, multiple lines of evidence have now demonstrated that other angiogenic growth factors can maintain and expand the tumor vasculature in the face of VEGF blockade.3
Multiple isoforms of VEGF-A exist (ie, in humans VEGF121, 148, 165, 183 and 206), including an inhibitory variant (165b),4 which may constitute the majority of VEGF-A produced by some normal tissues, including kidney. Each of these VEGF isoforms results from alternative mRNA splicing of a single gene encoded on chromosome 6 (6p12). Some isoforms are cell-associated, while others are soluble and act at a distance. In addition to VEGF-A, there are other VEGFs and non-VEGF angiogenic factors that bind VEGF receptors, including VEGF-B (11q13), VEGF-C (4q34), VEGF-D (Xp22) and placental-like growth factor (PLGF, 14q24-31) (reviewed in5). Thus, VEGF receptor inhibitors affect multiple ligands whereas ligand-specific blocking antibodies are considerably more restricted in their biologic effects.
There are three primary VEGF receptors. For tumor angiogenesis, VEGFR2 activation has the greatest effect on endothelial cell motility and proliferation. Interestingly, VEGF-A binds VEGFR1 with higher affinity than VEGFR2, although activation of VEGFR2 results in greater phosphorylation and downstream signaling. This suggests that VEGFR1 might have an inhibitory function in some settings. On the other hand, VEGFR1 expression on macrophages has been shown to affect their ability to infiltrate and release metalloproteinases, which have been shown to play an important role in the angiogenic switch during progression of low-grade to high-grade lesions.6 VEGFR3 and its ligand, VEGF-C, are the major stimulatory components of lymphatic vessels, and elevated levels of VEGF-C correlate with higher rates of lymphatic metastases (eg,7,8 and references therein). The kinase domain of the VEGF receptors shares significant similarity with platelet-derived growth factor receptors (PDGFRs), colony-stimulating factor 1 receptor (CSF-1R/c-Fms), and the receptors for stem-cell factor (ie, KIT) and FLT ligand (ie, FLT3). This similarity explains the observed cross-reactivity of many current small molecule VEGFR inhibitors.
VEGF receptors do not act alone. Neuropilins (NRP-1/2), originally identified as high-affinity receptors for the secreted class 3 semaphorins (reviewed in9), were subsequently identified as VEGF co-receptors. In mice, a deficiency in NRP-1 is embryonic-lethal with marked developmental defects in the cardiovascular system.10 NRP-2 defects are milder with abnormalities mainly affecting lymphatics.11 Although the class 3 semaphorins were initially identified as neural guidance molecules, SEMA3F and SEMA3B were subsequently found to be deleted or substantially suppressed in tumors.12–14 On a functional level, the secreted Sema3s were shown to inhibit endothelial cells by downregulating activated integrins,15 and also to inhibit tumor cells in vitro and in vivo.16–18 Mechanistically, SEMA3F was also shown to inhibit activated integrins on tumor cells with marked downregulation of various signaling pathways, with effects on HIF-1/2α and VEGF.19
Upregulation of NRPs occurs in various tumor types.20–24 Not only do NRPs bind VEGF, either alone or in combination with VEGF receptors, but they bind additional growth factors, such as hepatocyte growth factor (HGF), with enhanced effects on signaling.25,26 New therapeutic agents are being developed against the neuropilins, and the combination of blocking antibodies against Nrp-1 and Vegf-A was shown to be more potent than either agent alone.9 A schematic of these various interactions and effects is shown in Figure 1.
Figure 1.
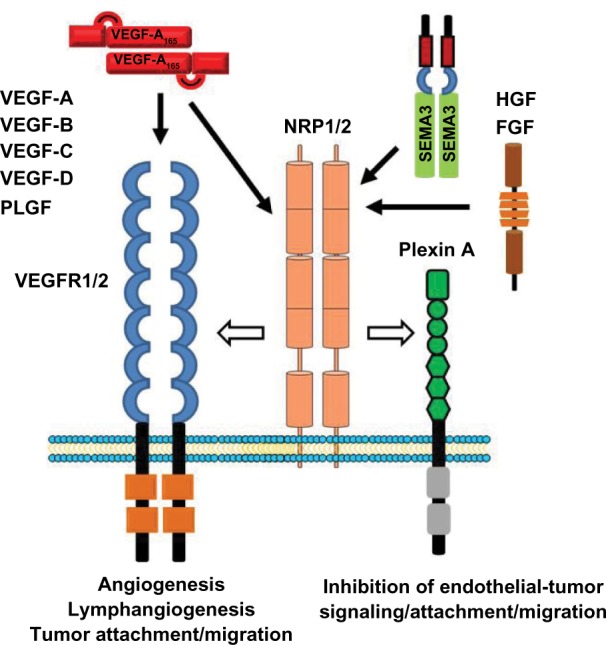
Schematic showing neuropilin1/2, VEGFR1/2 and plexin A receptors/co-receptors and their various ligands. The neuropilins function as co-receptors for various VEGF ligands, whereas the plexins are co-receptors for Sema3s.
Abbreviations: FGR, fibroblast growth factor; HGF, hepatocyte growth factor.
Development of pazopanib (GW786034)
The development of pazopanib [N4-(2,3-dimethyl-2H-indazol-6-yl)-N4-methyl-N2-(4-methyl-3-sulfonamidophenyl)-2,4-pyrimidinediamine] as an oral inhibitor of VEGFR2 followed a medicinal chemistry approach based on two initial inhibitory compounds and knowledge of the related FGFR, since the crystal structure for VEGFR2/KDR was unavailable at that time.27 An optimization process ensued, which consisted of systematically testing various chemical modifications and substitutions to a core molecule in order to identify a final compound that had the following properties: high oral bioavailability and low clearance to permit once a day dosing, inhibition of in vitro VEGFR kinase activity at nanomolar levels with the least effect on inhibiting P450 isozymes. A lead compound, indazolylpyrimidine 13, emerged (Figure 2) and its mono-HCL salt became known as GW786034 or pazopanib (Votrient™; GlaxoSmithKline).
Figure 2.
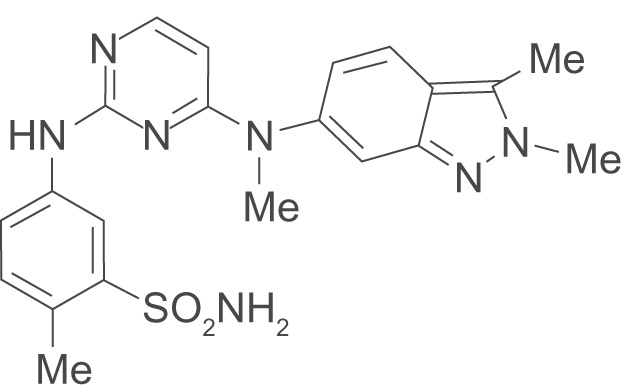
The structure of pazopanib (cmpd 13).
Subsequent in vitro studies demonstrated that GW786034 inhibited each of the VEGF receptors (VEGFR1-3) with IC50 values of 10 to 47 nM and similar levels inhibited PDGFRα/β and c-Kit (71–84 nM).28 Interestingly, Flt-3, which is closely related to c-Kit, was reportedly not affected. Also, closely related to the VEGFRs are the receptors for fibroblast growth factors (FGFRs) and CSF-1 (c-fms). These were inhibited by pazopanib at concentrations of 80 to 146 nM. In a panel of 225 kinases, 13 additional targets were inhibited at 300 nM, with Aurora-A, c-Raf, MLK1, PTK5 and TAO3 being the most sensitive. At higher concentrations, a number of additional kinases were inhibited, including p38a, FAK, LCK and ITK (IC50 ≤ 1 μmol), and at higher levels (IC50 2–10 μmol) members of the JNK and Src families, GSK3, Tie-2, Alk6, Met and IGF-1R were affected.
Despite these various determinations of in vitro activity, when a close analogue of pazopanib, GW771806, was given by continuous IV to tumor-bearing mice, micromolar concentrations were required to affect disease control. Using oral pazopanib in mice, concentrations greater than 40 micromolar were required to inhibit VEGFR2. The difference between the in vitro and in vivo levels required to inhibit VEGFR2 has been attributed to the high degree of protein binding of pazopanib, which is greater than 99.9%.28 Therefore, a steady-state level of ≥40 μmol was chosen as the target in phase I studies, which could be achieved in the majority of patients receiving ≥800 mg daily given once a day, or 300 mg given twice daily (see29 and references therein). Achieving these levels resulted in blood pressure increases in about half the patients, whereas lower levels resulted in fewer patients experiencing increased blood pressure. Thus, while the development of hypertension may be indicative of adequate therapeutic levels, the converse is not true. Doses up to 2000 mg daily were investigated in a limited number of patients,29 but not further pursued since a plateau in the steady-state exposure was observed at ≥800 mg per day. Hypertension, diarrhea, hair depigmentation and nausea were the most frequent adverse effects. Among a spectrum of 63 patients with advanced cancer, partial responses were noted in three patients — two with renal cell carcinoma and one with a neuroendocrine tumor.
Pazopanib in RCC
A phase II study of pazopanib in metastatic RCC has now been reported.30 Among 225 patients, there was an overall response rate of 35% with a median duration of response of 68 weeks (15.7 months). Stable disease, lasting a minimum of 8 weeks, was observed in 45%. In a large majority of patients (n = 195), some degree of tumor reduction was noted. A phase III trial of pazopanib in RCC was presented at the 2009 ASCO meeting.31 This study involved 435 patients randomized to receive 800 mg/d of pazopanib (n = 290) or placebo (n = 145). Patients were either treatment-naïve or had received one prior cytokine-based therapy. The primary endpoint was progression-free survival (PFS); secondary end-points were overall survival, response rate and safety. A companion study allowed patients receiving placebo to cross-over at the time of disease progression. The PFS was significantly prolonged with pazopanib (overall survival 9.2 vs 4.2 months; hazard ratio: 0.46; 95% confidence interval [CI]: 0.34, 0.62; P < 0.0000001). The response rate was 30% with pazopanib vs 3% with placebo and the median duration of response was 58.7 weeks (13.5 months). The most common adverse effects with pazopanib were diarrhea (52%), hypertension (40%), hair color change (38%), nausea (26%), anorexia (22%) and vomiting (21 %) with grade 3 and 4 toxicities occurring in 4% or less of patients. Elevation of alanine-leucine transaminase (ALT) was the most common laboratory abnormality occurring in 53% (10% grade 3; 2% grade 4). Similar side-effect frequencies were reported in the phase II study.31
Pazopanib compared to other VEGFR inhibitors in RCC
The reported objective response rates (discussed above) with pazopanib are 30% to 35%. As noted by Hutson et al,30 this is comparable to the response rate observed with sunitinib (31 %) in a large randomized phase III trial.32 Furthermore, the PFS with both agents was similar. In contrast, both pazopanib and sunitinib appear more active than sorafenib, which had only a 10% objective response rate and PFS of 5.5 months in patients who had progressed after previous treatment.33
Making therapeutic efficacy judgments between therapeutic agents based on separate studies is notoriously error-prone, but commonly done. For example, sunitinib has not been directly compared to sorafenib, although it is often considered to be more active based on the above-mentioned reports. Of note, Heng et al34 recently reported on prognostic factors in 645 RCC patients treated with sunitinib (n = 396), sorafenib (n = 200), or bevacizumab (n = 49). Interestingly, there was no apparent difference in overall survival among the groups. Fortunately, pazopanib is being directly compared to sunitinib in an ongoing study, VEG108844 (NCT00720941 – www.clinicaltrials.gov).
Sorafenib, sunitinib and pazopanib have been directly compared for their effects on in vitro and in vivo kinase activity, with an intent to understand the mechanism of myelosuppression.35 Among 242 kinases, and using each of the agents at 0.3 and 10 μmol concentrations, sunitinib was the most promiscuous agent. At the lower concentration, sunitinib inhibited 49 kinases by >50%, whereas pazopanib and sorafenib inhibited 29 and 26, respectively. At 10 μmol, substantially more kinases were inhibited (eg, sunitinib inhibited 149). Of note, the ability of pazopanib, sunitinib and sorafenib to inhibit VEGFR2, either using purified kinases or cell-based assays, was nearly identical. In the cell-based assays, sorafenib had less activity against c-KIT than either pazopanib or sunitinib, whereas pazopanib was inactive against FLT3, confirming the previously reported results.28
The mechanism of myelosuppression was studied using bone marrow-derived colony forming assays in the presence of GM-CSF alone, or together with stem-cell factor (SCF) and/or FLT-3 ligand, which significantly increased the number of colonies. Of note, the addition of either SCF or FLT-3 ligand sensitized the CFU-GM colony growth to inhibitors. However, FLT-3 ligand plus GM-CSF did not sensitize colony growth to pazopanib, which is consistent with its lack of activity against FLT-3. For each growth factor combination, sunitinib was more myelosuppressive than pazopanib or sorafenib. Thus, it is likely that hematologic toxicity in vivo reflects the effects of these agents on multiple kinases including KIT, FLT-3 and possibly others.
How pazopanib will fare with regard to other toxicities, such as cardiovascular damage, will require longer follow-up. Hepatic toxicity has emerged as more common with pazopanib than the other VEGFR inhibitors. Thus, it would be prudent for patients to avoid concomitant potential hepatotoxins, including those sold as nutritional supplements, and to routinely monitor liver function.
The use of pazopanib in other cancers – selected examples
Pazopanib appears to have activity in other malignant diseases, although few results have been reported in other than abstract form. In a phase II study of soft tissue sarcoma,36 pazopanib appeared to modestly prolong the overall time to progression compared to historical controls. However, partial responses were observed in 9/142 patients with 2 individuals still in remission at 415 and 812 days.
In a study of 19 evaluable patients with recurrent or metastatic breast cancer.37 there was one observed partial response (5%) and 11 patients with stable disease (58%). The median time to progression was 3.7 months (95% CI: 1.7 months – not reached). In half of the 18 patients with measurable target lesions, there was some reduction in size, as is typically seen with anti-angiogenic agents in RCC. Here, the estimated PFS was 55% at 3 months and 28% at 6 months, suggesting that a subset of patients would obtain some benefit. In advanced differentiated thyroid cancer, a majority of patients experienced over 50% reduction in thyroglobulin levels, with a few patients demonstrating partial response, after treatment with pazopanib.38
Two phase I trials have addressed dosing of pazopanib in gastrointestinal cancers. In locally unresectable and/or advanced hepatocellular carcinoma, the maximum tolerated dose was 600 mg daily, and there was preliminary evidence of anti-tumor activity with changes in tumor permeability, as measured by dynamic contrast enhanced MRI.39 A second study of patients with previously untreated advanced or metastatic colorectal cancer combined pazopanib with either FOLFOX6 or CapeOx (capecitabine/oxaliplatin combination) to determine the optimally tolerated regimen (OTR). The OTR for pazopanib with full-dose FOLFOX6 was 800 mg, while a dose-reduction of capecitabine to 850 mg/m2 twice daily was necessary to achieve an OTR using 800 mg of pazopanib with CapeOx.40
Dozens of other clinical trials involving pazopanib are ongoing, addressing a variety of diseases and drug combinations. Among the malignant diseases being examined are cervical cancer, neuroendocrine tumors, non-small cell and small-cell lung cancer, nasopharyngeal cancers and multiple myeloma. Many non-malignant conditions are also being studied, such as macular degeneration, lymphedema and plaque psoriasis. Trials with combinations of pazopanib with other multi-kinase inhibitors, as well as traditional chemotherapeutic agents are also ongoing.
Concluding remarks
Although it has taken nearly four decades from the pioneering studies of Folkman, anti-angiogenic therapy represents a new dimension in the treatment of metastatic cancer. However, like most new developments, the problem is more complex than initially envisaged. Moreover, new data continue to emerge that challenge preconceived notions. For example, the multiple isoforms of VEGF-A represent only one of many different angiogenic factors. Far more surprising has been recent evidence from tumor model systems demonstrating that VEGF inhibition can enhance tumor invasion/metastasis,41 possibly via increased hypoxia within the tumor.
Pazopanib represents a new VEGFR-targeted inhibitor, recently FDA approved for the treatment of advanced RCC. It has clear activity in this disease, but whether it will be more active or less toxic than other agents is under investigation. Like other small-molecule VEGFR inhibitors, pazopanib is not specific. However, this lack of specificity can be advantageous (eg, to target FGFRs).
In certain malignant diseases, most notably RCC, anti-VEGFR targeted therapy is initially successful in a majority of patients. However, in most individuals the disease becomes resistant to continuous VEGF blockade. Understanding this resistance is an immediate challenge to improving patient outcome. It is likely that non-VEGF angiogenic factors mediate at least part of this resistance, although additional mechanisms have also been implicated involved.3 Thus, it may be necessary to continue VEGF blockade while adding agents that target other factors. The availability of VEGFR inhibitors with different side-effect profiles, such as pazopanib, will facilitate that process.
Footnotes
Disclosure
The author discloses no conflicts of interest.
References
- 1.Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med. 1971;133(2):275–288. doi: 10.1084/jem.133.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang JC. Bevacizumab for patients with metastatic renal cancer: an update. Clin Cancer Res. 2004;10(18 Pt 2):6367S–70S. doi: 10.1158/1078-0432.CCR-050006. [DOI] [PubMed] [Google Scholar]
- 3.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woolard J, Wang WY, Bevan HS, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64(21):7822–7835. doi: 10.1158/0008-5472.CAN-04-0934. [DOI] [PubMed] [Google Scholar]
- 5.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312(5):549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen G, Liu XY, Wang Z, Liu FY. Vascular endothelial growth factor C: the predicator of early recurrence in patients with N2 non-small-cell lung cancer. Eur J Cardiothorac Surg. 2009 Sep 14; doi: 10.1016/j.ejcts.2009.08.005. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 8.Ko YH, Jung CK, Lee MA, et al. Clinical Significance of Vascular Endothelial Growth Factors (VEGF)-C and -D in Resected Non-Small Cell Lung Cancer. Cancer Res Treat. 2008;40(3):133–140. doi: 10.4143/crt.2008.40.3.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bagri A, Tessier-Lavigne M, Watts RJ. Neuropilins in tumor biology. Clin Cancer Res. 2009;15(6):1860–1864. doi: 10.1158/1078-0432.CCR-08-0563. [DOI] [PubMed] [Google Scholar]
- 10.Kawasaki T, Kitsukawa T, Bekku Y, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126(21):4895–4902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- 11.Yuan L, Moyon D, Pardanaud L, et al. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002;129(20):4797–4806. doi: 10.1242/dev.129.20.4797. [DOI] [PubMed] [Google Scholar]
- 12.Sekido Y, Bader S, Latif F, et al. Human semaphorins A(V) and IV reside in the 3p21.3 small cell lung cancer deletion region and demonstrate distinct expression patterns. Proc Natl Acad Sci U S A. 1996;93(9):4120–4125. doi: 10.1073/pnas.93.9.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roche J, Boldog F, Robinson M, et al. Distinct 3p21.3 deletions in lung cancer and identification of a new human semaphorin. Oncogene. 1996;12(6):1289–1297. [PubMed] [Google Scholar]
- 14.Brambilla E, Constantin B, Drabkin H, Roche J. Semaphorin SEMA3F localization in malignant human lung and cell lines: A suggested role in cell adhesion and cell migration. Am J Pathol. 2000;156(3):939–950. doi: 10.1016/S0002-9440(10)64962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serini G, Valdembri D, Zanivan S, et al. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature. 2003;424(6947):391–397. doi: 10.1038/nature01784. [DOI] [PubMed] [Google Scholar]
- 16.Xiang R, Davalos AR, Hensel CH, Zhou XJ, Tse C, Naylor SL. Semaphorin 3F gene from human 3p21.3 suppresses tumor formation in nude mice. Cancer Res. 2002;62(9):2637–2643. [PubMed] [Google Scholar]
- 17.Kusy S, Nasarre P, Chan D, et al. Selective suppression of in vivo tumorigenicity by semaphorin SEMA3F in lung cancer cells. Neoplasia. 2005;7(5):457–465. doi: 10.1593/neo.04721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bielenberg DR, Hida Y, Shimizu A, et al. Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest. 2004;114(9):1260–12671. doi: 10.1172/JCI21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Potiron VA, Sharma G, Nasarre P, et al. Semaphorin SEMA3F affects multiple signaling pathways in lung cancer cells. Cancer Res. 2007;67(18):8708–8715. doi: 10.1158/0008-5472.CAN-06-3612. [DOI] [PubMed] [Google Scholar]
- 20.Klagsbrun M, Takashima S, Mamluk R. The role of neuropilin in vascular and tumor biology. Adv Exp Med Biol. 2002;515:33–48. doi: 10.1007/978-1-4615-0119-0_3. [DOI] [PubMed] [Google Scholar]
- 21.Roche J, Drabkin H, Brambilla E. Neuropilin and its ligands in normal lung and cancer. Adv Exp Med Biol. 2002;515:103–114. doi: 10.1007/978-1-4615-0119-0_9. [DOI] [PubMed] [Google Scholar]
- 22.Vanveldhuizen PJ, Zulfiqar M, Banerjee S, et al. Differential expression of neuropilin-1 in malignant and benign prostatic stromal tissue. Oncol Rep. 2003;10(5):1067–1071. [PubMed] [Google Scholar]
- 23.Fukahi K, Fukasawa M, Neufeld G, Itakura J, Korc M. Aberrant expression of neuropilin-1 and -2 in human pancreatic cancer cells. Clin Cancer Res. 2004;10(2):581–590. doi: 10.1158/1078-0432.ccr-0930-03. [DOI] [PubMed] [Google Scholar]
- 24.Hansel DE, Wilentz RE, Yeo CJ, Schulick RD, Montgomery E, Maitra A. Expression of neuropilin-1 in high-grade dysplasia, invasive cancer, and metastases of the human gastrointestinal tract. American J Surg Pathol. 2004;28(3):347–356. doi: 10.1097/00000478-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 25.Sulpice E, Plouet J, Berge M, Allanic D, Tobelem G, Merkulova-Rainon T. Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood. 2008;111(4):2036–2045. doi: 10.1182/blood-2007-04-084269. [DOI] [PubMed] [Google Scholar]
- 26.Hu B, Guo P, Bar-Joseph I, et al. Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene. 2007;26(38):5577–5586. doi: 10.1038/sj.onc.1210348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51(15):4632–4640. doi: 10.1021/jm800566m. [DOI] [PubMed] [Google Scholar]
- 28.Kumar R, Knick VB, Rudolph SK, et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther. 2007;6(7):2012–2021. doi: 10.1158/1535-7163.MCT-07-0193. [DOI] [PubMed] [Google Scholar]
- 29.Hurwitz HI, Dowlati A, Saini S, et al. Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res. 2009;15(12):4220–4227. doi: 10.1158/1078-0432.CCR-08-2740. [DOI] [PubMed] [Google Scholar]
- 30.Hutson TE, Davis ID, Machiels JP, et al. Efficacy and Safety of Pazopanib in Patients With Metastatic Renal Cell Carcinoma. J Clin Oncol. 2010;28(3):475–480. doi: 10.1200/JCO.2008.21.6994. [DOI] [PubMed] [Google Scholar]
- 31.Sternberg CN, Szczylik C, Lee E, et al. A randomized, double-blind phase III study of pazopanib in treatment-naive and cytokine-pretreated patients with advanced renal cell carcinoma (RCC) J Clin Oncol. 2009;27(15s(Suppl)) abstr 5021. [Google Scholar]
- 32.Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 33.Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 34.Heng DY, Xie W, Regan MM, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol. 2009;27(34):5794–5799. doi: 10.1200/JCO.2008.21.4809. [DOI] [PubMed] [Google Scholar]
- 35.Kumar R, Crouthamel MC, Rominger DH, et al. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer. 2009;101(10):1717–1723. doi: 10.1038/sj.bjc.6605366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043) J Clin Oncol. 2009;27(19):3126–3132. doi: 10.1200/JCO.2008.21.3223. [DOI] [PubMed] [Google Scholar]
- 37.Taylor SK, Chia S, Dent S, et al. A phase II study of GW786034 (pazopanib) in patients with recurrent or metastatic invasive breast carcinoma: Results after completion of stage I: A trial of the Princess Margaret Hospital Phase II Consortium. J Clin Oncol. 2009;27(15s(Suppl)) doi: 10.1634/theoncologist.2010-0081. abstr 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bible KC, Smallridge RC, Maples WJ, et al. Phase II trial of pazopanib in progressive, metastatic, iodine-insensitive differentiated thyroid cancers. J Clin Oncol. 2009;27(15s(Suppl)) abstr 3521. [Google Scholar]
- 39.Yau CC, Chen PJ, Curtis CM, et al. A phase I study of pazopanib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2009;27(15s(Suppl)) abstr 3561. [Google Scholar]
- 40.Brady J, Middleton M, Midgley RS, et al. A phase I study of pazopanib in combination with FOLFOX 6 or capeOx in subjects with colorectal cancer. J Clin Oncol. 2009;27(15s(Suppl)) abstr 4133. [Google Scholar]
- 41.Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]