

Abstract
Purpose
An association between low-dose hyper-radiosensitivity (HRS) and the “early” G2/M checkpoint has been established. An improved molecular understanding of the temporal dynamics of this relationship is needed before clinical translation can be considered. This study was conducted to characterize the dose response of the early G2/M checkpoint and then determine whether low-dose radiation sensitivity could be increased by synchronization or chemical inhibition of the cell cycle.
Methods and Materials
Two related cell lines with disparate HRS status were used (MR4 and 3.7 cells). A double–thymidine block technique was developed to enrich the G2-phase population. Clonogenic cell survival, radiation-induced G2-phase cell cycle arrest, and deoxyribonucleic acid double-strand break repair were measured in the presence and absence of inhibitors to G2-phase checkpoint proteins.
Results
For MR4 cells, the dose required to overcome the HRS response (approximately 0.2 Gy) corresponded with that needed for the activation of the early G2/M checkpoint. As hypothesized, enriching the number of G2-phase cells in the population resulted in an enhanced HRS response, because a greater proportion of radiation-damaged cells evaded the early G2/M checkpoint and entered mitosis with unrepaired deoxyribonucleic acid double-strand breaks. Likewise, abrogation of the checkpoint by inhibition of Chk1 and Chk2 also increased low-dose radiosensitivity. These effects were not evident in 3.7 cells.
Conclusions
The data confirm that HRS is linked to the early G2/M checkpoint through the damage response of G2-phase cells. Low-dose radiosensitivity could be increased by manipulating the transition of radiation-damaged G2-phase cells into mitosis. This provides a rationale for combining low-dose radiation therapy with chemical synchronization techniques to improve increased radiosensitivity.
Keywords: Ionizing radiation, Low dose, Cell cycle, G2 checkpoint arrest, Chk1, Chk2
INTRODUCTION
Hyper-radiosensitivity (HRS) defines the extreme sensitivity of cells to low doses of low–linear energy transfer radiation (1). Hyper-radiosensitivity cell killing has been attributed to apoptosis after the evasion of dose-dependent damage detection processes (2–5). The involvement of deoxyribonucleic acid (DNA) repair pathways and processes has previously been associated with overcoming HRS (1, 6), and the low-dose radiosensitivity transition is thought to be regulated by the “early” G2/M checkpoint (3, 7). Proof of this concept has already been shown in a preclinical study designed to exploit HRS, which showed some success combining a low-dose radiation scheme with cell cycle modulation (8). Because HRS is more prominent in proliferating malignant tissues than in quiescent normal tissues and remains active after fractionated irradiation, the potential exists for further therapeutic exploitation in a primary or recurrent setting (9–13). However, the molecular basis of HRS needs to be fully defined before widespread clinical translation can be considered.
Because failure to repair DNA damage in G2-phase cells can lead to cell death or mutations that threaten genomic stability, defects in the G2 checkpoint are linked to reduced viability (14, 15). Indeed, two distinct checkpoints exist to arrest radiation-damaged cells in the G2 phase of the cell cycle (16). The “classic” Sinclair checkpoint (17) is ataxia telangiectasia mutated (ATM) independent and functions to arrest cells damaged in the G1 or S phases after higher dose exposures. The second, or early, checkpoint is ATM dependent and critical for cells in the G2 phase at the time of low-dose X-irradiation (18). Ataxia telangiectasia mutated protein appears to be the main regulator in the early checkpoint (18), phosphorylating Chk2 and halting cell progression into mitosis. Significantly for our low-dose HRS studies, the dose response of the early G2/M checkpoint overlaps that of overcoming HRS, which initially suggested a role for G2 cellular damage responses in the HRS/increased radioresistance (IRR) transition (4).
Our previous studies have established the role of G2-phase cells in HRS and linked the HRS/IRR transition to the early G2/M checkpoint (3, 4, 7, 19). In this article the temporal dynamics of the early G2-phase checkpoint were further defined to determine whether HRS could be used to radiosensitize cells by combining low-dose X-irradiation and inhibition of specific proteins involved in the G2/M transition. The data show that modulation of proteins integral to the G2/M checkpoint can exert influence over the cell cycle response to low-dose radiation, which may have clinical merit for further exploiting HRS.
METHODS AND MATERIALS
Cell lines
MR4 and 3.7 isogenic rat fibroblast cells (20) with disparate HRS status (19) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 2-mmol/L L-glutamine, and 1-mmol/L sodium pyruvate (Cellgro; Mediatech, Manassas, VA). For routine maintenance, each cell line was grown as a monolayer at 37°C under 5% O2/5% CO2 (balance N2) and subcultured once or twice weekly to maintain exponential growth. All cell lines were confirmed to be mycoplasma free before use. Plating efficiencies of MR4 and 3.7 cells were 0.46 ± 0.07 and 0.26 ± 0.02, respectively.
Irradiation
For the cell survival assays, the cells were irradiated as suspension cultures at 37°C in complete culture medium with single doses at a dose rate of 0.75 Gy/min by use of a Pantak HF320 X-ray unit (Pantak, East Haven, CT) (half-value layer of 3.6 mm Cu equivalent). For flow cytometry or slide immunofluorescence studies, the cells were irradiated as monolayers at a dose rate of 0.82 Gy/min.
G2-phase cell synchrony by double thymidine block
The production of highly enriched G2 cell populations was required for a number of assays in which sorting G2-phase cell populations by flow cytometry was not practical. Therefore S-phase arrest was achieved with a double thymidine block, a technique previously reported to produce high levels of S-phase synchronization (21), and then the cells were allowed to progress into the G2 phase. The 3.7 cells deviated from this general protocol somewhat because they did not tolerate a second treatment with thymidine. Consequently, a single thymidine block was used and proved effective at S-phase enrichment for this cell line.
Clonogenic survival assay
A fluorescence-activated cell sorter (FACSVantage; BD Biosciences, San Jose, CA) was used to accurately dispense 300 to 5,000 cells into individual 5-mL Falcon polystyrene round-bottom tubes (BD Biosciences) depending on the expected surviving fraction to ensure greater than 100 colonies after plating (4). This method was a modification of the original as first described by Durand (22).
After irradiation, both the asynchronous and cell cycle phase–enriched populations were plated into 100-mm culture dishes (Corning, Corning, NY) containing pre-warmed complete culture medium. After incubation, the resultant colonies were stained with 2% crystal violet in 95% ethanol, and those colonies consisting of greater than 50 cells were scored as representing surviving cells by use of ColCount (Oxford Optronix Ltd., Oxford, England). Surviving fractions were calculated from these data by reference to the mean plating efficiency of sorted and sham-irradiated controls. Conventional clonogenic assays were used to measure cell survival after thymidine synchronization.
Data analysis for survival assays
The clonogenic survival data were fitted to a modification of the linear–quadratic model called the induced-repair (IR) model (23). Surviving fractions were fitted with the linear–quadratic or IR equation by use of nonlinear least squares regression through the iterative method of Gauss-Newton with step halving (SAS JMP software; SAS Institute, Cary, NC) to produce the best-fit parameters for each model. All parameters were fitted simultaneously, and estimates of uncertainty were expressed as likelihood confidence intervals. The presence of low-dose HRS is deduced by values of αs (representing the enhanced response at low doses) and αr (representing the high-dose radioresponse) whose confidence limits do not overlap and a value of dc (dose transition between hyper-radiosensitivity and increased resistance) significantly greater than zero.
Assessment of H3 and H2AX by flow cytometry
The method for assessment of anti–phosphohistone H3 (Ser28) staining has been described previously (4). In brief, after alcohol fixation, cells were stained with anti-H3 primary (catalog No. 07-145; Millipore, Billerica, MA) and FITC (fluorescein isothiocyanate)-conjugated secondary antibodies (Molecular Probes, Eugene, OR) with propidium iodide (Calbiochem, San Diego, CA). A modified method based on that of MacPhail et al. (24) was developed to measure phosphorylated histone H3 and γH2AX concurrently. Ethanol-fixed samples were stained with anti-H3 primary antibody (catalog No. 06-570; Millipore), anti-rabbit Alexa Fluor 680-R-phycoerythrin antibody (catalog No. A-20984; Molecular Probes) and H2AX-FITC–conjugated antibody (catalog No. 17-344; Millipore). This technique measured the level of DNA double-strand breaks (DSBs) (γH2AX) in mitotic cells (H3-P).
For both staining methods, data were expressed as the “mitotic ratio”—a function of radiation dose, determined by calculating the ratio of irradiated vs. unirradiated cells that stained positive for phosphorylated histone H3 in two populations of matched asynchronously or blocked cell cultures. Presenting the data in this manner allows the effect of irradiation on the entry of cells into mitosis and the activity of the early G2/M checkpoint to be determined. Further analysis of these data was undertaken with the Student t test and determination of the area under the curve (AUC) as a measure of total time course kinetics. The AUC was calculated by taking the integral under each dose response curve and then comparing the resultant areas for each cell line and dose point.
Analysis of H2AX and H3 by immunofluorescence
MR4 and 3.7 cells were grown directly on chamber slides (BD Biosciences) in complete media or grown in flasks, fixed, and cytospun on to slides for staining via the protocol outlined by Wykes et al. (19).
Chk1 and Chk2 inhibitors
Two commercially available Chk1 and Chk2 inhibitors (SB-210787 and Gö6976) were used (EMD Chemicals, San Diego, CA). SB-218078 is a staurosporine-like inhibitor of the ATP (adenosine triphosphate)-binding pocket of Chk1, and Gö6976 is an indolocarbazole with a structure similar to UCN-01 that inhibits Chk1 and Chk2; cells were subjected to varying concentrations based on previously published studies (21, 25–28). All stock solutions of the compounds were dissolved in dimethyl sulfoxide at a concentration of 10 mmol/L and stored at −20°C in lightproof boxes (Sigma-Aldrich, St. Louis, MO).
For the inhibitor experiments, the cells were grown to 50% to 60% confluency in complete media and treated for 24 hours with complete media plus inhibitor. For the analysis of inhibitor effects on cell cycle, standard p-H3/propidium iodide flow assays were used as described previously. For cell survival assays, small adjustments were made in the procedure, because the cells were not sorted into flasks but rather were counted by hand and diluted to appropriate numbers for each radiation dose. This change was made because the inhibitor-treated cells were not able to tolerate the sorting process.
RESULTS
Thymidine double-block enrichment of MR4 G2/M cell increases HRS response
Previous studies indicated that HRS was a specific G2-phase response (1) and enhanced in G2 phase–enriched populations (4). However, defining the temporal response of overcoming HRS with increasing radiation dose was hindered by the limitation of obtaining large numbers of unperturbed G2-phase cells. To overcome this, we adapted a chemical synchronization technique using thymidine to obtain G2-phase MR4 and 3.7 cells in larger numbers than can be obtained by flow cytometry. The technique produces a good degree of S-phase synchronization (Fig. 1), which results in G2-phase enrichment a few hours after the block is released. Typically, a G2/M enrichment of approximately 40% was achieved for MR4 cells 3 to 4 hours after thymidine release and for 3.7 cells 5 to 6 hours after treatment (Fig. 1). Asynchronous populations of MR4 cells were confirmed to exhibit HRS, unlike the isogenic cell line 3.7, as defined by the IR model (23) (Fig. 2, Table 1). G2/M-enriched populations of MR4 cells were confirmed to exhibit an exaggerated HRS response as reported previously (4). G2/M-enriched populations of 3.7 cells were more radiosensitive than asynchronous populations (Fig. 2).
Fig. 1.

Thymidine double block. Left, Schematic diagram representing the enrichment strategy. Right, Cell cycle distributions as a function of time after the release of the thymidine block. Samples were fixed and stained with propidium iodide, and deoxyribonucleic acid content was determined by flow cytometry (n = 3). UB = unblocked IR = ionizing radiation.
Fig. 2.

Enrichment of MR4 G2/M cell modulates the hyper-radiosensitivity (HRS) response. Clonogenic cell survival data for MR4 (left) and 3.7 (right) cells. The insets show a low-dose expansion. The lines are fit to the raw data via the induced-repair model. MR4 cells showed evidence of HRS in asynchronous (squares) and G2/M-enriched (circles) cell populations (mean ± SD of n = 3, each with 4 replicates). Hyper-radiosensitivity was not evident in the asynchronous 3.7 cells (squares).
Table 1.
Parameters obtained from mathematical modeling via induced-repair model
| αs (mean ± SE) | αr (mean ± SE) | β (mean ± SE) | dc
|
||
|---|---|---|---|---|---|
| mean ± SE | CL | ||||
| MR4 control | 1.93 ± 1.06 | 0.13 ± 0.05 | 0.14 ± 0.03 | 0.08 ± 0.04 | 0.01–0.203 |
| MR4 blocked | 5.33 ± 0.91 | 0.57 ± 0.09 | 0.03 ± 0.04 | 0.21 ± 0.03 | 0.09–0.59 |
| 3.7 control | WNC | 0.35 ± 0.13 | 0.04 ± 0.05 | 0.001 | WNC |
| 3.7 blocked | 5.45 ± 1.35 | 0.79 ± 0.05 | 0.13 ± 0.02 | 0.15 ± 0.03 | 0.09–0.23 |
Abbreviations: CL = confidence limits; WNC = would not converge.
Early G2/M checkpoint response of G2-enriched cell populations
Next, the temporal function of the early G2 checkpoint was determined in both cell lines by assessment of the progression of irradiated G2 cells into mitosis by use of a mitosis specific marker, phosphorylated histone H3 (Fig. 3) (4). As expected, 3.7 cells exhibited an immediate post-irradiation decrease in the mitotic ratio even at the lowest doses, indicating fewer radiation-damaged G2-phase cells progressing into mitosis. The extent of the block was dose dependent and occurred up to 60 minutes after exposure. Mitotic ratio values were significantly different from 0 Gy for all dose points above 0.05 Gy (p values < 0.05). These data show an active early G2 checkpoint in 3.7 cells, whereas the increase in the mitotic ratio that occurs 2 hours after exposure indicates a return to normal cell cycle progression.
Fig. 3.

Induced-repair dose and time for mitotic index response of G2-enriched cell populations. Mitotic ratio (H3-P cells) is assessed as a function of time after irradiation. MR4 cells continue to enter mitosis at a constant rate after doses lower than 0.3 Gy, indicating initiation of an early G2/M block (right). Conversely, 3.7 cells exhibit an active G2/M block at all doses. Data points represent mean ± SD (n = 3). AUC = area under the curve.
In contrast, no significant reduction in mitotic ratio was evident for MR4 cells until the radiation dose exceeded 0.2 Gy (mitotic ratios for doses in the range 0.05–0.3 Gy were not significantly different from 0 Gy [p > 0.05], unlike at higher doses [p = 0.02 for 0.6 Gy and p = 0.007 for 1.0 Gy]). We hypothesize that these data indicate that the early G2 checkpoint does not function effectively in MR4 cells at very low dose levels. In addition, once activated, cell cycle progression remained inhibited for approximately 60 minutes after treatment. Measurable differences between the early G2 checkpoint function of each cell line at doses above 0.2 Gy can be shown by AUC determination and analysis by the Student t test (p = 0.02 for 0.2 Gy, p = 0.006 for 0.3 Gy, p = 0.004 for 0.6 Gy, and p = 0.001 for 1.0 Gy) (Fig. 3). Overall, these data provide support to the relationship between the HRS response and failure of the early G2/M checkpoint (4).
Resolution of IR-induced DNA DSBs after G2 enrichment
The cellular response to radiation-induced DNA DSBs includes phosphorylation of ATM, followed by activation of downstream targets with γH2AX operating as a damage sensor (29). Measuring the reduction in foci number with time after irradiation is an established approach to evaluating the extent of DNA repair. To determine the extent of DNA DSB repair after irradiation, the disappearance of γH2AX foci was measured either in individual cells by counting distinct foci to give a quantitative measure of DNA DSBs or by flow cytometry to give a qualitative measure of repair simultaneously from the entire cell population.
In the thymidine-enriched population of MR4 cells, a reduction in foci number was seen with increasing time (0–60 minutes) after 2-Gy treatment, indicating active DNA repair (Fig. 4A). After 0.2 Gy, however, the number of foci remained stable for 60 minutes after treatment, indicating a lack of DNA DSB repair, as well as a response that is coincident with a failure to induce the early G2 checkpoint. The lack of DNA repair evident in Fig. 4A for G2/M-phase MR4 cells could also be explained if the recognition of damage occurred more slowly in MR4 cells. The data in Fig. 4B show that the maximum detection of γH2AX foci occurred at 1 hour for 3.7 cells but more slowly (2 hours) for MR4 cells, which is consistent with this hypothesis.
Fig. 4.

Resolution of repair–induced deoxyribonucleic acid (DNA) double-strand breaks (DSBs) after G2 enrichment. (A) Repair of DNA DSBs as a function of time for G2/M-enriched MR4 cells after irradiation with 0.2 and 2 Gy; for reference, the black bar represents the foci number immediately after sham irradiation (0 Gy). Repair is only seen after higher dose exposure. (B) Repair of DNA DSBs as a function of time after irradiation for 3.7 and MR4 cells as determined by flow cytometry. The 3.7 cells show maximum H2AX phosphorylation earlier after irradiation than the MR4 cells. Data points represent mean values (n = 3).
Measurement of early G2/M checkpoint after treatment with Chk1 and Chk2 inhibitors
Our previous work showed that overcoming HRS was associated with the ATM-dependent early G2/M checkpoint but not ATM activation per se (4), and although the activation of a cell cycle checkpoint was regarded as the most important factor, we have yet to identify specific downstream proteins tied into this response. Consequently, we investigated whether inhibition of Chk1 and Chk2 would allow cells to overcome HRS because these proteins are downstream regulators of G2/M progression. Working concentrations of two specific inhibitors, Gö6976 and SB-218078, were determined by use of an MTT proliferation assay (data not shown). A concentration of 5 μmol/L did not inhibit cell proliferation or produce notable toxicity 24 hours after exposure and so was used. No G2/M arrest was seen for MR4 cells over the dose range 0 to 1 Gy as signified by mitotic index values greater than 1, whereas a complete cell cycle arrest was evident for 3.7 cells regardless of radiation dose (Fig. 5). Treatment with the inhibitors did produce greater variability in p-H3 levels than usually seen in this assay; however, the mean mitotic index was found to be significantly different between the two cell lines across all doses for both inhibitors (p < 0.05 [p = 0.002–0.04]) for SB-218078 over 0- to 1-Gy dose range and p < 0.05 [p = 0.0009–0.02] for Gö6976 over 0- to 1-Gy dose range according to Student t test). The distinct responses after inhibitor treatment suggested that these cell lines were exhibiting different early G2/M checkpoint activity, likely because of their disparate HRS status.
Fig. 5.
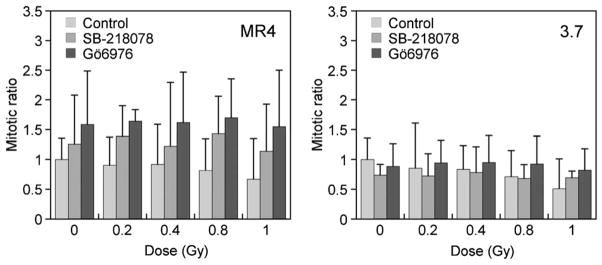
Measurement of early G2/M checkpoint after inhibitor treatment, showing mitotic ratio as a function of dose. The inhibitors changed the typical radiation-induced checkpoint responses as seen in Fig. 3, signifying modulation of the early G2 checkpoint. Data points represent mean ± SD (n = 3).
Measurement of cell survival and low-dose HRS after treatment with Chk1 and Chk2 inhibitors
Clonogenic cell survival assays were performed in the presence of the two inhibitors to determine the effects on the HRS/IRR transition. We hypothesized that Chk1/Chk2 inhibition would prevent the activation of the early G2 checkpoint in HRS-positive cells (MR4) and inhibit the development of the IRR response by extending the HRS response to higher doses. Treatment of asynchronous populations of MR4 cells with Gö6976 resulted in an enhanced HRS response as predicted and also unexpectedly amplified the IRR response (Fig. 6). This pattern of survival response is similar to that reported for flow cytometry–synchronized G2/M MR4 cells (4). No changes in survival were seen for 3.7 cells treated with Gö6976. Treatment with SB-218078 did not significantly change the low-dose survival response of either cell line and did not affect the HRS response of MR4 cells (data not shown).
Fig. 6.
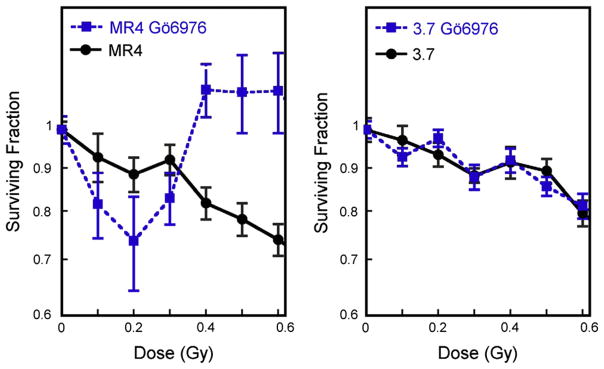
Inhibitors used to modulate hyper-radiosensitivity (HRS). Clonogenic survival of MR4 and 3.7 cells occurred in the presence and absence of Gö6976, a specific inhibitor of Chk1 and Chk2. Treatment with Gö6976 produced an enhanced HRS and amplified increased radioresistance response in MR4 cells but not 3.7 cells. Data points represent mean ± SD (n = 5).
DISCUSSION
Over the last decade, several groups have shown that a variety of mammalian cells exhibit low-dose HRS [as previously reviewed (1)]. In vitro studies have established a relationship between HRS/IRR and evasion of the early G2/M checkpoint (3, 4), with HRS-associated cell death linked to apoptosis (2, 5). In vivo, HRS has been shown in normal tissues (11), metastatic tumor nodules (30), and some tumor xenograft experiments (8, 31). For this reason, we undertook this study to define the temporal response of the cell cycle checkpoint in two closely related cell lines that inhibit different HRS statuses.
To accurately define the dose response of the early G2/M checkpoint, cells were enriched into the G2 phase by use of a modified thymidine block technique (Fig. 1). As expected, asynchronous MR4 cells exhibited a distinct HRS response that was amplified in the G2 phase–enriched populations (Fig. 2). However, no distinct evidence for an HRS response was seen for asynchronous 3.7 cells, which is consistent with previous studies (Fig. 2, Table 1) (3, 4). Interestingly, G2 phase–enriched populations of 3.7 cells showed some evidence of an HRS response (Fig. 2). This was unexpected given the robust early G2-phase checkpoint (Fig. 3). We speculate that the HRS response of the enriched population could be attributed to cells that were synchronized at a point in the G2 phase of the cell cycle that is beyond the checkpoint, and these cells are therefore incapable of G2 arrest and proceed into mitosis with unrepaired damage producing HRS.
The ATM-dependent early G2/M checkpoint was previously suggested to have an activation threshold of approximately 0.4 Gy, but lower radiation doses were not examined (18). The data in Fig. 3 provide a more complete dose response of the early G2/M checkpoint activation. Activation was evident only after exposure to doses in excess of 0.2 Gy, a dose that approximates the activation profile of ATM in this cell line (4) and the dose required to overcome HRS (Fig. 1). In marked contrast, a dose-independent activation of the early G2/M checkpoint was shown for 3.7 cells (Fig. 3). In addition, using γH2AX as a marker of DNA DSBs, we showed that damage was sustained after irradiation in mitotic MR4 cells, suggesting a reduced rate or lack of DNA break repair with time after exposure (Fig. 4). A comparison of the data for MR4 and 3.7 cells in Figs. 2, 3, and 4 verifies the link between overcoming HRS and dose dependency of the early G2/M checkpoint.
To take advantage of HRS as a clinical modality, multiple small dose fractions need to be given. This presents a logistic challenge for busy radiotherapy clinics. To overcome this limitation, pulsed dosing techniques have been developed that separate 0.2-Gy pulses by 3-minute time intervals (10). This approach appears to be an effective strategy for controlling locoregional recurrence in patients who have previously undergone irradiation (9). Combined pulsed treatments with cell cycle modulation may prove more effective for tumor control. The connection between low-dose HRS and the G2/M checkpoint is important because modulation of this checkpoint has long been perceived as a possible important target in cancer therapy. Modest clinical success has been achieved using UCN-01 as a chemical synchronization agent (32, 33), although its avid binding to human plasma protein α1-acid glycoprotein resulted in an extremely long half-life and decreased bioavailability (34). In our current study using two similar compounds (Gö6976 and SB-218078) that affect cell cycle regulation, we have been able to show differential effects of G2-phase checkpoint function in cell lines with disparate HRS responses and the modulation of HRS in MR4 cells by Gö6976, implicating Chk1 and Chk2 as potential regulators of the HRS/IRR transition. The variability in survival response produced by Gö6976 treatment, combined with the inherent complexity of measuring cell survival after low-dose exposures, prevents firmer conclusions being drawn from these data. Continued studies will be necessary to determine the importance of these proteins in the HRS/IRR response, and we anticipate that further work in this area could lead to the mechanism that links detection and repair of radiation-induced DNA damage to the HRS response through a specific cell cycle protein.
Acknowledgments
Supported by the Radiation Oncology Research Development Corporation, Detroit, Michigan, and the Departments of Radiation Oncology, Wayne State University and William Beaumont Hospital. Dr. Krueger was generously supported by the National Institutes of Health Training Program in the Biology of Cancer (T32 CA 09531-19).
Footnotes
Conflict of interest: none.
References
- 1.Marples B, Collis SJ. Low-dose hyper-radiosensitivity: Past, present, and future. Int J Radiat Oncol Biol Phys. 2008;70:1310–1318. doi: 10.1016/j.ijrobp.2007.11.071. [DOI] [PubMed] [Google Scholar]
- 2.Enns L, Bogen KT, Wizniak J, et al. Low-dose radiation hypersensitivity is associated with p53-dependent apoptosis. Mol Cancer Res. 2004;2:557–566. [PubMed] [Google Scholar]
- 3.Marples B, Wouters BG, Joiner MC. An association between the radiation-induced arrest of G2 phase cells and low-dose hyper-radiosensitivity: A plausible underlying mechanism? Radiat Res. 2003;160:38–45. doi: 10.1667/rr3013. [DOI] [PubMed] [Google Scholar]
- 4.Krueger SA, Collis SJ, Joiner MC, et al. Transition in survival from low-dose hyper-radiosensitivity to increased radioresistance is independent of activation of ATM Ser1981 activity. Int J Radiat Oncol Biol Phys. 2007;69:1262–1271. doi: 10.1016/j.ijrobp.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Krueger SA, Joiner MC, Weinfeld M, et al. Role of apoptosis in low-dose hyper-radiosensitivity. Radiat Res. 2007;167:260–267. doi: 10.1667/RR0776.1. [DOI] [PubMed] [Google Scholar]
- 6.Chalmers A, Johnston P, Woodcock M, et al. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int J Radiat Oncol Biol Phys. 2004;58:410–419. doi: 10.1016/j.ijrobp.2003.09.053. [DOI] [PubMed] [Google Scholar]
- 7.Short SC, Woodcock M, Marples B, et al. The effects of cell cycle phase on low dose hyper-radiosensitivity. Int J Radiat Biol. 2003;79:99–105. [PubMed] [Google Scholar]
- 8.Spring PM, Arnold SM, Shajahan S, et al. Low dose fractionated radiation potentiates the effects of taxotere in nude mice xenografts of squamous cell carcinoma of head and neck. Cell Cycle. 2004;3:479–485. [PubMed] [Google Scholar]
- 9.Richards GM, Tome WA, Robins HI, et al. Pulsed reduced dose-rate radiotherapy: A novel locoregional retreatment strategy for breast cancer recurrence in the previously irradiated chest wall, axilla, or supraclavicular region. Breast Cancer Res Treat. 2009;114:307–313. doi: 10.1007/s10549-008-9995-3. [DOI] [PubMed] [Google Scholar]
- 10.Tome WA, Howard SP. On the possible increase in local tumour control probability for gliomas exhibiting low dose hyper-radiosensitivity using a pulsed schedule. Br J Radiol. 2007;80:32–37. doi: 10.1259/bjr/15764945. [DOI] [PubMed] [Google Scholar]
- 11.Turesson I, Joiner MC. Clinical evidence of hypersensitivity to low doses in radiotherapy. Radiother Oncol. 1996;40:1–3. doi: 10.1016/0167-8140(96)01765-3. [DOI] [PubMed] [Google Scholar]
- 12.Short SC, Kelly J, Mayes CR, et al. Low-dose hypersensitivity after fractionated low-dose irradiation in vitro. Int J Radiat Biol. 2001;77:655–664. doi: 10.1080/09553000110041326. [DOI] [PubMed] [Google Scholar]
- 13.Joiner MC, Marples B, Lambin P, et al. Low-dose hypersensitivity: Current status and possible mechanisms. Int J Radiat Oncol Biol Phys. 2001;49:379–389. doi: 10.1016/s0360-3016(00)01471-1. [DOI] [PubMed] [Google Scholar]
- 14.Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer. 2007;7:861–869. doi: 10.1038/nrc2248. [DOI] [PubMed] [Google Scholar]
- 15.Krempler A, Deckbar D, Jeggo PA, et al. An imperfect G2M checkpoint contributes to chromosome instability following irradiation of S and G2 phase cells. Cell Cycle. 2007;6:1682–1686. doi: 10.4161/cc.6.14.4480. [DOI] [PubMed] [Google Scholar]
- 16.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 17.Sinclair WK. Cyclic x-ray responses in mammalian cells in vitro. Radiat Res. 1968;33:620–643. [PubMed] [Google Scholar]
- 18.Xu B, Kim ST, Lim DS, et al. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22:1049–1059. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wykes SM, Piasentin E, Joiner MC, et al. Low-dose hyper-radiosensitivity is not caused by a failure to recognize DNA double-strand breaks. Radiat Res. 2006;165:516–524. doi: 10.1667/RR3553.1. [DOI] [PubMed] [Google Scholar]
- 20.Gupta AK, Bakanauskas VJ, Cerniglia GJ, et al. The Ras radiation resistance pathway. Cancer Res. 2001;61:4278–4282. [PubMed] [Google Scholar]
- 21.Jiang X, Zhao B, Britton R, et al. Inhibition of chk1 by the G2 DNA damage checkpoint inhibitor isogranulatimide. Mol Cancer Ther. 2004;3:1221–1227. [PubMed] [Google Scholar]
- 22.Durand RE. Use of a cell sorter for assays of cell clonogenicity. Cancer Res. 1986;46:2775–2778. [PubMed] [Google Scholar]
- 23.Marples B, Joiner MC. The response of Chinese hamster V79 cells to low radiation doses: Evidence of enhanced sensitivity of the whole cell population. Radiat Res. 1993;133:41–51. [PubMed] [Google Scholar]
- 24.MacPhail SH, Banath JP, Yu Y, et al. Cell cycle-dependent expression of phosphorylated histone H2AX: Reduced expression in unirradiated but not X-irradiated G1-phase cells. Radiat Res. 2003;159:759–767. doi: 10.1667/rr3003. [DOI] [PubMed] [Google Scholar]
- 25.Jackson JJ, Gilmartin A, Imburgia C, et al. An indolocarbazole inhibitor of human checkpoint kinase (Chk1) abrogates cell cycle arrest caused by DNA damage. Cancer Res. 2000;60:566–572. [PubMed] [Google Scholar]
- 26.Curman D, Cinel B, Williams DE, et al. Inhibition of the G2 damage checkpoint and of protein kinases Chk1 and Chk2 by marine sponge alkaloid debromohymenialdisine. J Biol Chem. 2001;276:17914–17919. doi: 10.1074/jbc.M100728200. [DOI] [PubMed] [Google Scholar]
- 27.Kohn EA, Yoo CJ, Eastman A. The protein kinase C inhibitor Go6976 is a potent inhibitor of DNA damage-induced S and G2 cell cycle checkpoints. Cancer Res. 2003;63:31–35. [PubMed] [Google Scholar]
- 28.Zhao B, Bower MJ, McDevitt PJ, et al. Structural basis for Chk1 inhibition by UCN-01. J Biol Chem. 2002;277:46609–46615. doi: 10.1074/jbc.M201233200. [DOI] [PubMed] [Google Scholar]
- 29.Lobrich M, Jeggo PA. Harmonising the response to DSBs: A new string in the ATM bow. DNA Repair (Amst) 2005;4:749–759. doi: 10.1016/j.dnarep.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Harney J, Short SC, Shah N, et al. Low dose hyper-radiosensitivity in metastatic tumors. Int J Radiat Oncol Biol Phys. 2004;59:1190–1195. doi: 10.1016/j.ijrobp.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 31.Dey S, Spring PM, Arnold S, et al. Low-dose fractionated radiation potentiates the effects of Paclitaxel in wild-type and mutant p53 head and neck tumor cell lines. Clin Cancer Res. 2003;9:1557–1565. [PubMed] [Google Scholar]
- 32.Hotte SJ, Oza A, Winquist EW, et al. Phase I trial of UCN-01 in combination with topotecan in patients with advanced solid cancers: A Princess Margaret Hospital Phase II Consortium study. Ann Oncol. 2006;17:334–340. doi: 10.1093/annonc/mdj076. [DOI] [PubMed] [Google Scholar]
- 33.Dees EC, Baker SD, O’Reilly S, et al. A phase I and pharmacokinetic study of short infusions of UCN-01 in patients with refractory solid tumors. Clin Cancer Res. 2005;11:664–671. [PubMed] [Google Scholar]
- 34.Fuse E, Tanii H, Kurata N, et al. Unpredicted clinical pharmacology of UCN-01 caused by the specific binding to human alpha1-acid glycoprotein. Cancer Res. 1998;58:3248–3253. [PubMed] [Google Scholar]