

Abstract
Ginseng is known to have antistress effects. Previously, red ginseng (RG) was shown to repress stress-induced peptidyl arginine deiminase type IV (PADI4) via estrogen receptor β (ERβ) in the brain, thus inhibiting brain cell apoptosis. Moreover, tumor necrosis factor (TNF)-α plays a critical role in immobilization (IMO) stress. However, the signaling pathway of RG-mediated repressesion of inflammation is not completely understood. In this study, we determined how RG modulated gene expression in stressed brain cells. Since secretion of TNF-α is modulated via TNF-α converting enzyme (TACE) and nuclear factor (NF)-κB, we examined the inflammatory pathway in stressed brain cells. Immunohistochemistry revealed that TACE was induced by IMO stress, but RG repressed TACE induction. Moreover, PADI4 siRNA repressed TACE expression compared to the mock transfected control suggesting that PADI4 was required for TACE expression. A reporter assay also revealed that H2O2 oxidative stress induced NF-κB in neuroblastoma SK-N-SH cells, however, RG pretreatment repressed NF-κB induction. These findings were supported by significant induction of nitric oxide and reactive oxygen species (ROS) by oxidative stress, which could be repressed by RG administration. Taken together, RG appeared to repress stress-induced PADI4 via TACE and NF-κB in brain cells thus preventing production of ROS and subsequently protecting brain cells from apoptosis.
Keywords: Panax ginseng, Oxidative stress, Peptidyl arginine deiminase type IV, Tumor necrosis factor-α converting enzyme, Nuclear factor-κB
INTRODUCTION
The antistress effects of ginseng are well known, and its active ingredients have been identified as ginsenosides [1,2]. Immobilization (IMO) stress-induced polyamine levels were decreased by ginsenosides Rb1 and Rg3, and by ginseng total saponin in mice brains [3]. Rb1, the main ingredient of ginseng, has a neuroprotective effect against glutamate-induced neurotoxicity [4,5], ischemia [6], neurodegeneration [7], and seizures [8]. Rg3 in fermented red-ginseng has anti-stress [4] and neuroprotective effects [9].
The structure of ginsenoside is similar to that of steroids [10]. Ginsenoside Rh1 and Rb1 showed estrogen receptor (ER) activation and ER-dependent action, respectively, in the mammary gland and uterus [10]. Ginseng also activates ER in breast cancer cells in vitro but not in vivo [11]. Although ER was activated in endothelial cells by protopanaxadiol and protopanaxatriol ginsenosides in vitro [12], ER is not regulated by Rh2 in brain astrocytes [13]. These results indicate that the effect of ginseng on the brain should be further investigated.
Membrane lipids in the brain can undergo oxidative damage upon physical stress such as a cold swim, electric foot shock, and IMO [14]. However, investigation of the anti-stress effects of ginseng and ginsenosides has been focused on biochemical parameters such as plasma interleukin (IL)-6 [15], plasma cholesterol, glucose, serum corticosterone [16], in vitro free radical scavenging activity [17], proinflammatory cytokines (tumor necrosis factor [TNF]-α, IL-1β, and IL-6) [18], and malondialdehyde levels [19]. Rg1 also prevented glutathione reduction and superoxide dismutase activation induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in the brains of mice [20]. Moreover, ginseng increases the release of cytokines and the expression of toll-like receptor 4 [21]. Our previous study showed that IMO as well as in vitro oxidative stressors such as H2O2, acrylamide, and an endoplasmic reticulum stressor tunicamycin significantly induced peptidyl arginine deiminase type IV (PADI4) gene. Moreover, pretreatment with red ginseng (RG, steamed and dried ginseng) derepressed ERβ, which subsequently represses induction of PADI4. Consistently, RG pretreatment inhibits the production of cyclooxygenase 2 and malondialdehyde demonstrating that RG protects the brain from cell death by repressing PADI4 via ERβ stimulation [22]. However, the mechanism whereby RG affects gene expression of TNF-α and downstream nuclear factor (NF)-κB remains unknown. Here we found that RG repressed PADI4 via TNF-α convertase (TACE) and NF-κB in brain cells, thus preventing production of reactive oxygen species (ROS) and subsequently protecting brain cells from apoptosis.
MATERIALS AND METHODS
Cells, animals, immobilization stress, red ginseng, and treatment
Human neuroblastoma SK-N-SH cells (ATCC HTB-11; American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI 1640 (Lonza, Walkersville, MD, USA) media containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (10,000 U penicillin/mL, 10,000 mg streptomycin/mL), 1 mM HEPES (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid), 1 mM sodium pyruvate, 4.5 g/L glucose, 1.5 g/L bicarbonate, 2 mM L-glutamine at 37℃, and 5% CO2. Rat neuroglioma C6 (ATCC CCL-107, American Type Culture Collection) was cultured in Dulbecco’s modified Eagle medium media containing 10% FBS and 2% penicillin-streptomycin. RG stock was prepared at 10 mg/mL in phosphate buffer saline (PBS, pH 7.4), diluted with RPMI 1640 media containing 10% FBS and 2% penicillin-streptomycin to 1 mg/mL just prior to use and sterilized by filtration with a 0.22 mm bottle top filter (Nalgene, Rochester, NY, USA). Male ICR mice (20 to 25 g) were housed in a temperature-controlled environment (temperature 21±2℃, humidity 60±10%) and 12-hour dark: 12-hour light cycle under conditions where food and water were freely available. All experiments conformed to the animal care guidelines of the Korean Academy of Medical Sciences, and all efforts were made to minimize animal suffering. Stress procedures were approved and monitored by the ethical committee of Sungkyunkwan University. For stress experiments, mice were immobilized for 30 or 45 min in a tightly fitted, 50 mL conical tube. At the end of the stress period, the mice were sacrificed by cervical dislocation, and the brain was rapidly removed and frozen. Animals that were set free in their home cage in the absence of any stressors served as controls (the normal control group). Mice were divided into three groups: non-treated (no stress), stress + no treat, and stress + RG. The non-treated group was used as a control. The stress + no treat group was administrated only PBS. Lastly, the stress + RG group was administered RG orally (RG extract; Korea Ginseng Corporation, Daejeon, Korea) twice a day for one week. The mice began fasting on the evening of the 7th day and were sacrificed 3 h after the last administration of RG on the morning of the 8th day.
For in vitro experiments, a filtered RG solution was further diluted with RPMI 1640 media to 1 mg/mL prior to RG treatment. In addition, cells were treated with RG for 48 h.
Determination of nitric oxide and reactive oxygen species
For ROS determination, 2×105 SK-N-SH cells were cultured overnight, and treated with 1 mg/mL of RG for 48 h or RPMI 1640 media alone (control). After treatment with 0.5 mM H2O2 for 20 min, cells were washed with PBS, and the intracellular accumulation of ROS was measured using the fluorescent probe H2DCFDA. At the end of the treatments, cells were loaded with 20 μM H2DCFDA and incubated at 37℃ for 30 min in the dark. Cells were then collected and resuspended in PBS. The fluorescence was measured immediately by flow cytometry (Biorad, Hercules, CA, USA).
For nitric oxide (NO) determination, C6 cells were pretreated with 0.5 mg/mL of RG for 24 h, and then 1 μg/mL of lipopolysaccharides (LPS; Sigma, St. Louis, MO, USA) was added. The amount of NO2- in cell supernatants was measured spectrophotometrically by the Griess reaction. Samples were supplemented with 276 mU of nitrate reductase and 40 μM NADPH and then allowed to react with the Griess reagent (aqueous solution of 1% sulfanylamide and 0.1% naphthylethylenediamine dihydrochloride in 2.5% H3PO4) in order to form a stable chromophore absorbing at a wavelength of 546 nm.
Purification of total RNA
Total RNA from mouse brain or tissue cultured cells was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and purified using an RNeasy mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RNA quality was measured spectrophotometrically.
Reverse transcriptase polymerase chain reaction
Total RNA was reversely transcribed to complementary DNA by M-MLV reverse transcriptase (RT) (RexGene Biotech, Ochang, Korea). All polymerase chain reaction (PCR) primer pairs were designed for mRNA sequencing within 200 bp; p53, 5’-CTGAGG TTGGC-TCTGACTGTACCAC-CATCC3’ (forward) and 5’CTCATTCAGCTCTCGGAACATCTCGA-AGCG3’ (reverse); β-actin, 5’-TGG AAT CCT GTG GCA TCC ATG AAA-3’ (forward) and 5’-TAA AAC GCA GCT CAG TAA CAG TCC G-3’ (reverse). Relative quantification of select mRNA was performed on 20 μl of cDNA using StepOne (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. The RT PCR conditions were optimized to comprise an initial denaturation step of 10 min at 95℃, followed by 35 cycles of 95℃ for 10 s, 55℃ for 15 s and 72℃ for 20 s. Statistical analysis was by analysis of variance between groups (ANOVA).
Western blot analysis
Cells were collected by centrifugation after washing with PBS, and resuspended in 50 mM Tris-Cl pH 7.4, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulphonyl fluoride and 1X protease cocktail inhibitor. Cells were lysed by sonication and cell lysates were harvested after centrifugation at 12,000 rpm, 4℃ for 15 min. The amount of protein was determined by Bradford assay and 30 to 40 μg of protein was used for Western blot. Proteins separated by 10% or 15% SDS-polyacrylamide gel electrophoresis were electroblotted onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). The membrane was then incubated in blocking buffer (7% skim milk, in TPBS [PBS, 0.1% Tween 20]) at room temperature on a shaking incubator for more than 1 h. After washing 3 times with TPBS, the membrane was incubated in a primary antibody (1:1,000) for 1 h at room temperature with primary antibodies PADI4, TACE (Abcam, Cambridge, England), Bcl-2, caspase-3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), β-actin (Sigma), or p65 NF-κB (Cell Signaling, Beverly, MA, USA). After extensive washing, the membrane was incubated with a secondary antibody (horseradish peroxidase-conjugated anti-IgG antibody, 1:5,000 or 1:10,000 anti-rabbit, anti-mouse; Sigma) for 1 h. Bands were detected with Power Optic-ECL western blotting detection reagent (iNtRON Biotech, Seongnam, Korea).
Immunohistochemistry
In order to determine the level of TACE, immunohistochemistry was performed on mouse formalin-fixed, paraffin-embedded 3-μm-thick sections with Discovery XT (Ventana Medical Systems, Tucson, AZ, USA). TACE antibody (Abcam) was used at a 1:100 dilution. For immunofluorescence, anti-red fluorescent protein antibody (Rockland, Gilbertsville, PA, USA) was used at 1:200 followed by a Alexa Fluor 488 anti-rabbit secondary antibody (Invitrogen) according to the manufacturer’s protocol.
Transfection
To knock down PADI4 expression by siRNA, 100 nM of siRNA (Genolution Phamaceuticals, Seoul, Korea) was added to 250 μL of serum-free RPMI 1640 media without antibiotics, to which 5 μL of transfection reagent (TransIT-TKO; Mirus, Madison, WI, USA) had been added and incubated for 20 min at room temperature. SK-N-SH cells (3×105 cells/well) were cultured in 6-well plates overnight, washed once with serum-free media without antibiotics, and replaced with 1 mL of serum-free media containing antibiotics. Subsequently, the transfection reagent containing siPADI4 was added to the cell culture and incubated for 24 h. As a control, cells were transfected with mock siRNA. After 24 h incubation, siRNA was removed and replaced with new complete media containing 1 mg/mL RG (cells in RPMI 1640 media alone were used as a control) for 48 h. Subsequently, cells were washed with PBS twice, and exposed to 0.5 mM H2O2 for 2 h followed by washing and lysis.
Luciferase reporter gene assay
NF-κB was determined by the luciferase reporter gene assay according to the manufacturer’s protocol. Briefly, SK-N-SH cells were seeded in a 12-well plate with 1×105 cells per well in 1 mL of RPMI 640 media. Then, cells were transfected with 1 μg of NF-κB plasmid (pGL3-NFκB) with the Renilla luciferase reporter mRNA. After 24 h, cells were treated with RG for another 24 h followed by treatment with 0.1 mM H2O2 for 4 h. Then the cells were collected in a passive lysis buffer (Promega, Madison, WI, USA), and the supernatant was used for determination of luciferase activity with the reporter assay system (Promega).
Statistical analysis
Data were analyzed by ANOVA followed by Dunett’s t-test for comparisons between groups. Significance was accepted when p<0.05 (*p<0.05, **p<0.01, and ***p<0.001). Data are expressed as median±standard deviation for three to five independent experiments.
RESULTS
Reversion of cell death-associated gene expression by red ginseng
Previously, we demonstrated that RG represses PADI4 expression as well as cell death, and induced Bcl-2, an anti-apoptotic factor, in vivo. Moreover, RG repressed active p53 expression after oxidative stress in vitro [22]. However, other apoptosis-related factors such as caspase-3 and p53 were not examined in vivo. To confirm the anti-apoptotic nature of RG, we investigated these two markers after IMO stress. When mice were subjected to IMO stress, the level of p53 mRNA was significantly repressed. However, RG pretreatment reversed this repression back to normal (Fig. 1A). To corroborate anti-apoptosis at the protein level, we determined the expression levels of representative apoptotic/anti-apoptotic markers caspase-3 and Bcl-2 by Western blot. Consistently, RG pretreatment increased Bcl-2 and repressed caspase-3 (Fig. 1B) indicating that RG could reverse IMO-induced apoptosis.
Fig. 1. Decrease of cell death-associated gene expression by red ginseng (RG) in vivo. (A) Mice (3 mice/group) were administered RG for 1 wk followed by immobilization (IMO) stress for 45 min. RNA was purified from brain, and two to three samples from each group were used for reverse transcriptase polymerase chain reaction analyses. (B) Mice (5 mice/group) were administered RG for 1 wk followed by IMO stress for 45 min. Five samples from each group were mixed and used for Western blot analyses. Experiments were performed twice independently, and representative data from two experiments are shown. PBS, phosphate buffer saline. *p<0.05 and **p<0.01.

Repression of tumor necrosis factor-α convertase by red ginseng
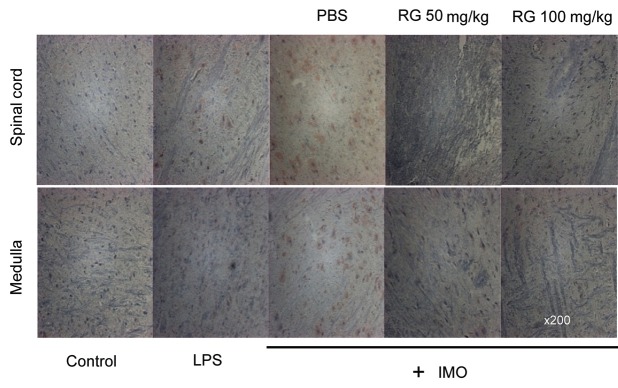
System biology analyses previously showed that IMO stress up-regulated immuneresponse-induced genes, and TNF-α was found at the center of the network [22]. Moreover, TNF-α was significantly induced by IMO stress but RG administration reversed TNF-α induction [22]. Normally, TNF-α is bound on the cell membrane in an inactive form, and is cleaved by a TACE (synonyms: ADAM-17 [a disintegrinandmetalloproteinase 17], CD156b, cSVP and MGC71942) to be released in an active soluble form [23]. To further elucidate the function of RG in TNF-α induction, the TACE level in the brains of IMO-stressed mice was determined by immunohistochemistry. TACE was induced by IMO specifically in the medulla and the spinal cord (Fig. 2). In addition, LPS, a well-known inducer of TNF-α, also induced TACE. However, pre-administration of RG repressed induction of TNF-α (Fig. 2) indicating that TNF-α induction in the IMO-stressed brain could be ascribed to TACE activation.
Fig. 2. Repression of immobilization (IMO)-induced tumor necrosis factor-α converting enzyme (TACE) by red ginseng (RG) in vivo. Mice were stressed by IMO for 30 min and immunohistochemistry analysis using a TACE antibody. PBS, phosphate buffer saline; Control, normal non-stressed control; LPS, lipopolysaccharides-injected group.
TNF-α and ROS activate c-Jun N-terminal kinases (JNK) [24], and JNK further activates p53 as well as other apoptotic factors [25]. Therefore, to further investigate how RG affected cell death after PADI4 induction, SK-N-SH cells were transfected with siPADI4 prior to H2O2 stress, and expression of TACE was determined by Western blot. As a control, siPADI4 was shown to repress PADI4 expression (Fig. 3).
Fig. 3. Reversal of peptidyl arginine deiminase type IV (PADI4)-induced tumor necrosis factor-α convertase (TACE) expression by red ginseng (RG) in vitro. SK-N-SH cells transfected with siPADI4 were treated with 1 mg/mL RG for 48 h, and exposed to 0.5 mM H2O2 for 2 h. Cells were then lysed, and cell lysates were used for Western blot analysis. Representative data from two experiments are shown.
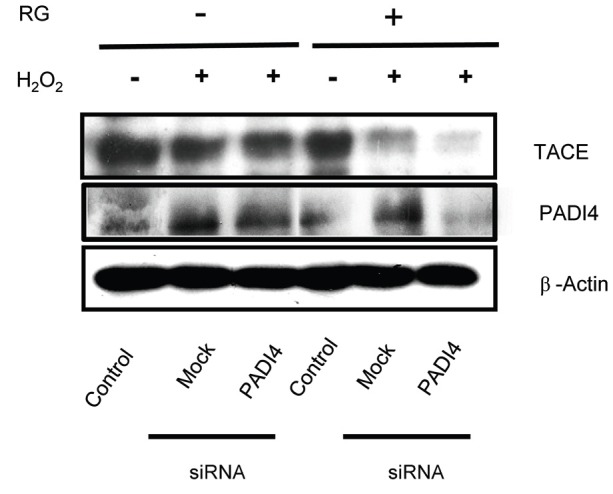
Interestingly, expression of TACE was not affected by siPADI4 in the non-RG-treated stressed condition. However, RG pretreatment repressed expression of TACE compared to the mock transfected control under oxidative stress conditions (Fig. 3). These results indicated that oxidative stress induced PADI4, which subsequently activated TACE. However, RG repressed PADI4 induction, suggesting that PADI4 may be an upstream regulator of TACE.
Inhibition of nuclear factor-κB by red ginseng
To further examine whether RG could inhibit inflammation, we investigated induction of NF-κB by oxidative stress [24] using a reporter gene assay. We found that exposure to H2O2 significantly increased the NF-κB level compared to non-oxidative conditions. However, RG pretreatment significantly repressed NF-κB induction (Fig. 4A). To confirm repression of NF-κB induction by RG, translocation of NF-κB from the cytosol to the nucleus was determined. After exposure to H2O2, the NF-κB level in the nucleus was significantly increased whereas the NF-κB level was significantly decreased by RG pretreatment (Fig. 4B, C). These results indicated that translocation of NF-κB to the nucleus as well as NF-κB induction were significantly repressed by RG treatment.
Fig. 4. Repression of nuclear factor (NF)-κB induction and translocation by red ginseng (RG) in vitro. (A) SK-N-SH cells were treated with 1 mg/mL of RG and then exposed to 0.1 mM of H2O2 followed by determination of NF-κB activity using a reporter gene assay. (B,C) Increase of NF-κB translocation from cytosol to nucleus by RG. SK-N-SH cells (8×105 cells) were pretreated with 1 mg/mL of RG extract for 48 h, and then washed with phosphate buffer saline (PBS) and exposed to 0.5 mM H2O2 for 0 or 1 h. Cells were then washed with PBS, and nuclear lysates were prepared followed by Western blot with p65 antibody. Black and gray bars are non-RG-treated and RG-treated cells, respectively. Control, normal non-stressed control. ***p<0.001.

Anti-inflammatory effect of red ginseng
Since NF-κB induction could produce ROS [24], we examined effect of RG on ROS production and NO release to further corroborate repression of NF-κB. When SK-N-SH cells were exposed to H2O2, the level of ROS increased significantly. However, RG pretreatment significantly repressed ROS induction (Fig. 5A). To confirm repression of ROS production by RG, LPS-induced NO release was examined. When C6 glioma cells were treated with LPS, the NO level increased in a time-dependent manner. Moreover, RG pretreatment significantly inhibited the level of release of LPS-induced NO (Fig. 5B) suggesting that RG repressed LPS-induced NO release.
Fig. 5. Repression of reactive oxygen species (ROS) and nitric oxide (NO) levels by red ginseng (RG) in vitro. (A) SK-N-SH cells were treated with 1 mg/mL of RG for 48 h or with media alone (control). After treatment with 0.5 mM H2O2 for 20 min, the intracellular accumulation of ROS was measured using the fluorescent probe H2DCFDA by flow cytometry. (B) C6 cells were pretreated with 0.5 mg/mL of RG extract for 24 h followed by the addition of 1 μg/mL of lipopolysaccharides (LPS). The amount of NO2- in cell supernatants was determined. Control, normal non-stressed control; LPS, LPS-treated group. *p<0.05, **p<0.01, and ***p<0.001 vs. without RG or with RG, by ANOVA.

These results demonstrated that RG inhibited the expression of genes involved in inflammation and oxidative stress thereby decreasing production of oxidative compounds and subsequently protecting the brains of mice from oxidative damage.
DISCUSSION
IMO stress was previously shown to upregulate the immune response-associated gene TNF-α in the brains of mice [22]. Moreover, restraint stress increases expression of the inducible isoform of NO synthase (iNOS) in rat brains, and iNOS inhibitor aminoguanidine protects from stress-induced pathophysiology [26]. However, how TNF-α expression could be controlled by RG remained unknown. In this study we demonstrated that RG inhibited PADI4 via TACE expression followed by inflammation and apoptosis.
Once immune-response associated genes are induced, they could disrupt homeostasis and eventually result in a disease state. Our previous result demonstrated that cell death-associated genes were induced by IMO stress [22]. Moreover, IMO stress increased apoptosis [27]. Apoptosis is induced by activation of caspases such as caspase-3 [28]. However, in this study we showed that RG repressed caspase-3 expression and increased anti-apoptotic Bcl-2 and p53, indicating that RG repressed apoptosis. RG also repressed TACE expression in vivo (Fig. 2). Moreover, siPADI4 repressed both TACE and PADI4 expression collaterally (Fig. 3), indicating that PADI4 induced TACE expression in the presence of RG. Consistently, RG could repress NF-κB induction as well as NF-κB translocation under oxidative stress conditions (Fig. 4), which subsequently repressed ROS and NO production (Fig. 5). Since restraint stress increases expression of the iNOS in rat brains, and iNOS inhibitor aminoguanidine protects from stress-induced pathophysiology [26], our results suggest that RG can modulate gene expression via various activities comprising PADI4, TACE, NF-κB, and ROS followed by a net reduction in cell death.
Physical, psychological or mixed stress in the brain triggers inflammatory responses including the release of several inflammatory cytokines, free radicals, prostanoids and transcription factors, activation of neutrophils, and the release of protein oxidation marker myeloperoxidase culminating in damaged brain cells [29]. In this study, we showed that IMO stress could induce inflammatory responses in the brain, leading to tissue damage. Interestingly, anti-inflammatory pathways are also activated in the brain in response to stress, to defend against inflammation-induced damage, thus resulting in dualizing pro- and anti-inflammatory responses [29]. RG may thus also induce an anti-inflammatory response to dampen the pro-inflammatory response incurred by oxidative stress. Further studies will be needed to elucidate the underlying mechanism of our observations.
Transcription in eukaryotic cells can be regulated at the post-translational level by histone modifications including methylation, phosphorylation, and ubiquitination, and histone arginine methylation is catalyzed by protein arginine methyltransferases [30]. PADI4 demethylates histone arginine at the p21 promoter region [31]. PADI4 thus represses the expression of genes induced by estrogen and retinoic acid receptors [32,33], serves as a p53 corepressor and represses the expression of p53 target genes p21/WAF1/CIP1 [31]. Although we demonstrated that siPADI4 attenuated cell death [22] and RG pretreatment increased p53 expression (Fig. 1A), we cannot exclude other mechanisms of anti-apoptosis. Our system biology analysis and network analyses showed that IMO stress shows significantly higher induction of cell death-associated genes such as HOXA5 (apoptosis), ITGB3BP (killing and adhesion), and LGALS3BP (cell death) than other genes. Moreover, RG pretreatment up-regulates apoptosis-associated genes including caspase, CASP9, and HOXA5, yet RG pretreatment did not result in increased cell death. Therefore, RG may also induce other as-yet-unknown inhibitors of apoptosis proteins such as Bruce, cIAP1, cIAP2, Survivin (BIRC5), or XIAP [34-36] alone or in combination, followed by inhibition of apoptosis. In this case, RG would show anti-apoptotic activity via caspase-independent pathways. Further studies are warranted to determine which mechanism is involved.
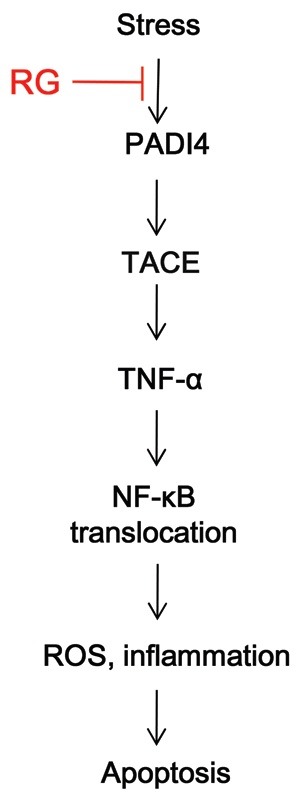
Taken together, we found that oxidative stress by IMO induced PADI4, which activated TACE and subsequently the release of TNF-α and NF-κB activation/translocation, which further induced inflammation and ROS secretion. In contrast, RG repressed PADI4 expression, thus reversing stress-induced gene expression and subsequently inhibiting apoptosis by repressing TACE and NF-κB activation/translocation, and by repressing ROS and NO production (Fig. 6).
Fig. 6. Red ginseng (RG) inhibits apoptosis in an oxidatively-stressed brain via tumor necrosis factor (TNF)-α convertase (TACE). Oxidative stress induces peptidyl arginine deiminase type IV (PADI4), which activates TACE and nuclear factor (NF)-κB translocation, triggering cues of inflammation as well as production of reactive oxygen species (ROS). Activation of PADI4 and TACE results in apoptosis. RG represses TACE via PADI4, thereby inhibiting apoptosis in the brain cells.
Acknowledgments
This work was supported by the 2011 grant from the Korean Society of Ginseng and a grant from the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (no. 111035-3).
References
- 1.Bhattacharya SK, Muruganandam AV. Adaptogenic activity of Withania somnifera: an experimental study using a rat model of chronic stress. Pharmacol Biochem Behav. 2003;75:547–555. doi: 10.1016/S0091-3057(03)00110-2. [DOI] [PubMed] [Google Scholar]
- 2.Kaneko H, Nakanishi K. Proof of the mysterious efficacy of ginseng: basic and clinical trials: clinical effects of medical ginseng, Korean red ginseng: specifically, its anti-stress action for prevention of disease. J Pharmacol Sci. 2004;95:158–162. doi: 10.1254/jphs.FMJ04001X5. [DOI] [PubMed] [Google Scholar]
- 3.Lee SH, Jung BH, Kim SY, Lee EH, Chung BC. The antistress effect of ginseng total saponin and ginsenoside Rg3 and Rb1 evaluated by brain polyamine level under immobilization stress. Pharmacol Res. 2006;54:46–49. doi: 10.1016/j.phrs.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Kim YC, Kim SR, Markelonis GJ, Oh TH. Ginsenosides Rb1 and Rg3 protect cultured rat cortical cells from glutamate-induced neurodegeneration. J Neurosci Res. 1998;53:426–432. doi: 10.1002/(SICI)1097-4547(19980815)53:4<426::AID-JNR4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Radad K, Gille G, Moldzio R, Saito H, Rausch WD. Ginsenosides Rb1 and Rg1 effects on mesencephalic dopaminergic cells stressed with glutamate. Brain Res. 2004;1021:41–53. doi: 10.1016/j.brainres.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 6.Lim JH, Wen TC, Matsuda S, Tanaka J, Maeda N, Peng H, Aburaya J, Ishihara K, Sakanaka M. Protection of ischemic hippocampal neurons by ginsenoside Rb1, a main ingredient of ginseng root. Neurosci Res. 1997;28:191–200. doi: 10.1016/S0168-0102(97)00041-2. [DOI] [PubMed] [Google Scholar]
- 7.Lian XY, Zhang Z, Stringer JL. Protective effects of ginseng components in a rodent model of neurodegeneration. Ann Neurol. 2005;57:642–648. doi: 10.1002/ana.20450. [DOI] [PubMed] [Google Scholar]
- 8.Lian XY, Zhang ZZ, Stringer JL. Anticonvulsant activity of ginseng on seizures induced by chemical convulsants. Epilepsia. 2005;46:15–22. doi: 10.1111/j.0013-9580.2005.40904.x. [DOI] [PubMed] [Google Scholar]
- 9.Bae EA, Hyun YJ, Choo MK, Oh JK, Ryu JH, Kim DH. Protective effect of fermented red ginseng on a transient focal ischemic rats. Arch Pharm Res. 2004;27:1136–1140. doi: 10.1007/BF02975119. [DOI] [PubMed] [Google Scholar]
- 10.Hao K, Gong P, Sun SQ, Hao HP, Wang GJ, Dai Y, Liang Y, Xie L, Li FY. Beneficial estrogen-like effects of ginsenoside Rb1, an active component of Panax ginseng, on neural 5-HT disposition and behavioral tasks in ovariectomized mice. Eur J Pharmacol. 2011;659:15–25. doi: 10.1016/j.ejphar.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Shim MK, Lee YJ. Estrogen receptor is activated by Korean red ginseng in vitro but not in vivo. J Ginseng Res. 2012;36:69–175. doi: 10.5142/jgr.2012.36.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung KW, Leung FP, Mak NK, Tombran-Tink J, Huang Y, Wong RN. Protopanaxadiol and protopanaxatriol bind to glucocorticoid and oestrogen receptors in endothelial cells. Br J Pharmacol. 2009;156:626–637. doi: 10.1111/j.1476-5381.2008.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shieh PC, Tsao CW, Li JS, Wu HT, Wen YJ, Kou DH, Cheng JT. Role of pituitary adenylate cyclase-activating polypeptide (PACAP) in the action of ginsenoside Rh2 against beta-amyloid-induced inhibition of rat brain astrocytes. Neurosci Lett. 2008;434:1–5. doi: 10.1016/j.neulet.2007.12.032. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Wang X, Shigenaga MK, Yeo HC, Mori A, Ames BN. Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J. 1996;10:1532–1538. [PubMed] [Google Scholar]
- 15.Kim DH, Jung JS, Moon YS, Sung JH, Suh HW, Kim YH, Song DK. Inhibition of intracerebroventricular injection stress-induced plasma corticosterone levels by intracerebroventricularly administered compound K, a ginseng saponin metabolite, in mice. Biol Pharm Bull. 2003;26:1035–1038. doi: 10.1248/bpb.26.1035. [DOI] [PubMed] [Google Scholar]
- 16.Rai D, Bhatia G, Sen T, Palit G. Anti-stress effects of Ginkgo biloba and Panax ginseng: a comparative study. J Pharmacol Sci. 2003;93:458–464. doi: 10.1254/jphs.93.458. [DOI] [PubMed] [Google Scholar]
- 17.Kang KS, Kim HY, Pyo JS, Yokozawa T. Increase in the free radical scavenging activity of ginseng by heat-processing. Biol Pharm Bull. 2006;29:750–754. doi: 10.1248/bpb.29.750. [DOI] [PubMed] [Google Scholar]
- 18.Joo SS, Won TJ, Lee DI. Reciprocal activity of ginsen-osides in the production of proinflammatory repertoire, and their potential roles in neuroprotection in vivo. Planta Med. 2005;71:476–481. doi: 10.1055/s-2005-864145. [DOI] [PubMed] [Google Scholar]
- 19.Voces J, Cabral de Oliveira AC, Prieto JG, Vila L, Perez AC, Duarte ID, Alvarez AI. Ginseng administration protects skeletal muscle from oxidative stress induced by acute exercise in rats. Braz J Med Biol Res. 2004;37:1863–1871. doi: 10.1590/S0100-879X2004001200012. [DOI] [PubMed] [Google Scholar]
- 20.Chen XC, Zhou YC, Chen Y, Zhu YG, Fang F, Chen LM. Ginsenoside Rg1 reduces MPTP-induced substantia nigra neuron loss by suppressing oxidative stress. Acta Pharmacol Sin. 2005;26:56–62. doi: 10.1111/j.1745-7254.2005.00019.x. [DOI] [PubMed] [Google Scholar]
- 21.Pannacci M, Lucini V, Colleoni F, Martucci C, Grosso S, Sacerdote P, Scaglione F. Panax ginseng C.A. Mayer G115 modulates pro-inflammatory cytokine production in mice throughout the increase of macrophage toll-like receptor 4 expression during physical stress. Brain Behav Immun. 2006;20:546–551. doi: 10.1016/j.bbi.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Kim EH, Kim IH, Lee MJ, Thach Nguyen C, Ha JA, Lee SC, Choi S, Choi KT, Pyo S, Rhee DK. Anti-oxidative stress effect of red ginseng in the brain is mediated by peptidyl arginine deiminase type IV (PADI4) repression via estrogen receptor (ER) β up-regulation. J Ethnopharmacol. 2013;148:474–485. doi: 10.1016/j.jep.2013.04.041. [DOI] [PubMed] [Google Scholar]
- 23.Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. FASEB J. 2006;20:1589–1598. doi: 10.1096/fj.05-5603rev. [DOI] [PubMed] [Google Scholar]
- 25.Fanger GR, Gerwins P, Widmann C, Jarpe MB, Johnson GL. MEKKs, GCKs, MLKs, PAKs, TAKs, and tpls: upstream regulators of the c-Jun amino-terminal kinases? Curr Opin Genet Dev. 1997;7:67–74. doi: 10.1016/S0959-437X(97)80111-6. [DOI] [PubMed] [Google Scholar]
- 26.Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C. Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proc Natl Acad Sci U S A. 1998;95:10966–10971. doi: 10.1073/pnas.95.18.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yun SJ, Lee DJ, Kim MO, Jung B, Kim SO, Sohn NW, Lee EH. Reduction but not cleavage of poly(ADP-ribose) polymerase during stress-mediated cell death in the rat hippocampus. Neuroreport. 2003;14:935–939. doi: 10.1097/01.wnr.0000074340.81633.f1. [DOI] [PubMed] [Google Scholar]
- 28.Mehmet H. Caspases find a new place to hide. Nature. 2000;403:29–30. doi: 10.1038/47377. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Bueno B, Caso JR, Leza JC. Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci Biobehav Rev. 2008;32:1136–1151. doi: 10.1016/j.neubiorev.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li P, Yao H, Zhang Z, Li M, Luo Y, Thompson PR, Gilmour DS, Wang Y. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol. 2008;28:4745–4758. doi: 10.1128/MCB.01747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306:279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 33.Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, et al. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 34.Wheatley SP, McNeish IA. Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol. 2005;247:35–88. doi: 10.1016/S0074-7696(05)47002-3. [DOI] [PubMed] [Google Scholar]
- 35.Hou YC, Chittaranjan S, Barbosa SG, McCall K, Gorski SM. Effector caspase Dcp-1 and IAP protein Bruce regulate starvation-induced autophagy during Drosophila melanogaster oogenesis. J Cell Biol. 2008;182:1127–1139. doi: 10.1083/jcb.200712091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanculescu A, Bembinster LA, Borgen K, Bergamaschi A, Wiley E, Frasor J. Estrogen promotes breast cancer cell survival in an inhibitor of apoptosis (IAP)-dependent manner. Horm Cancer. 2010;1:127–135. doi: 10.1007/s12672-010-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]