

Summary
The genus Lactobacillus includes a diverse group of bacteria consisting of many species that are associated with fermentations of plants, meat or milk. In addition, various lactobacilli are natural inhabitants of the intestinal tract of humans and other animals. Finally, several Lactobacillus strains are marketed as probiotics as their consumption can confer a health benefit to host. Presently, 154 Lactobacillus species are known and a growing fraction of these are subject to draft genome sequencing. However, complete genome sequences are needed to provide a platform for detailed genomic comparisons. Therefore, we selected a total of 20 genomes of various Lactobacillus strains for which complete genomic sequences have been reported. These genomes had sizes varying from 1.8 to 3.3 Mb and other characteristic features, such as G+C content that ranged from 33% to 51%. The Lactobacillus pan genome was found to consist of approximately 14 000 protein‐encoding genes while all 20 genomes shared a total of 383 sets of orthologous genes that defined the Lactobacillus core genome (LCG). Based on advanced phylogeny of the proteins encoded by this LCG, we grouped the 20 strains into three main groups and defined core group genes present in all genomes of a single group, signature group genes shared in all genomes of one group but absent in all other Lactobacillus genomes, and Group‐specific ORFans present in core group genes of one group and absent in all other complete genomes. The latter are of specific value in defining the different groups of genomes. The study provides a platform for present individual comparisons as well as future analysis of new Lactobacillus genomes.
Introduction
Lactobacilli are Gram‐positive, low G+C content and acid‐tolerant bacteria (Hugenholtz, 1998). They are lactic acid bacteria, belonging to the family of Lactobacillaceae, and include one of the most numerous groups of bacteria linked to humans with many species that are used for the industrial fermentation of dairy and other food products. Lactobacilli are naturally associated with mucosal surfaces, particularly the gastrointestinal tract, the vagina and the oral cavity (Tannock, 2004). Moreover, they are also indigenous to food‐related habitats, including wine, milk and meat environments, as well as plants, such as fruits, vegetables and cereal grains (Wood and Holzapfel, 1995; Wood and Warner, 2003). Finally, several strains of Lactobacillus spp. are marketed as probiotics as their consumption results in a health benefit to the host (Saxelin et al., 2005). Like other lactic acid bacteria, lactobacilli share the capacity to grow in nutritionally rich environments and rapidly convert sugars into lactic acid via simple metabolic pathways (de Vos and Hugenholtz, 2004). In general, lactobacilli are anaerobic and strictly fermentative, although some have rudimentary electron transport chains that, when grown in the presence of exogenously added cofactors such as haem, allows them to respire molecular oxygen and possibly nitrate (Brooijmans et al., 2009). When subject to standard fermentation conditions, the lactobacilli can be divided into three groups based on the characteristics of their metabolic products: obligately homofermentative, facultatively heterofermentative and obligately heterofermentative lactobacilli (Pot et al., 1994; Hammes and Vogel, 1995).
The ecological and phenotypic diversity of lactobacilli is reflected by their taxonomic diversity and currently 154 Lactobacillus species are known (http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=1578 as on 19 May 2010). It has been proposed earlier that the genus Lactobacillus could be subdivided into three groups based on type of fermentation (Dellaglio and Felis, 2005): the L. casei‐Pediococcus group, the Leuconostoc group and the L. delbrueckii group. The L. delbrueckii group was later renamed the L. acidophilus group, and the Lactobacillus casei group was split into further subgroups and a new genus. However, the main discrepancy in the taxonomy of the genus Lactobacillus is the non‐correlation between phylogeny and metabolic properties (Canchaya et al., 2006).
Next‐generation sequencing technology is revolutionizing the way that we practise research in microbial sciences and provides an unprecedented view on microbial diversity. The first Lactobacillus genome to be sequenced was the 3.3‐Mb L. plantarum WCFS1 genome (Kleerebezem et al., 2003), followed by the 2.0 Mb genomes of the probiotic L. johnsonii LA1 (Pridmore et al., 2004) and L. acidophilus NCFM (Altermann et al., 2005). In a comparative study that included 10 new and several known complete genomes of Lactobacillus, Pediococcus, Streptococcus, Lactococcus, Leuconostoc and Oenococcus, it was established that these lactic acid bacteria share a common ancestor with the bacilli and their gene complement is the result of a combination of extensive gene loss and horizontal gene transfer during evolution (Makarova et al., 2006). This analysis also defined for the first time a set of core genes shared by all LAB, the LaCOG, consisting of 567 genes. Using much more stringent criteria, a set of 141 core proteins was defined based on the comparison of 12 complete genomes of lactobacilli that provided further insight in the classification of Lactobacillus via phylogenomics (Claesson et al., 2008). Another comparative analysis, based on six Lactobacillus and several other genomes of lactic acid bacteria, aimed to link strain origin and genome but only identified a set of nine niche‐specific genes (O'Sullivan et al., 2009).
In less than 7 years, 20 complete Lactobacillus genomes have now become available within the NCBI database, with more than 100 incomplete or ongoing projects reported according to the GOLD database (as on 17 May 2010) (Nelson et al., 2010). In this study we divided 20 complete Lactobacillus genomes into various groups based on the phylogeny of their core genome, and subsequently examined the pan genome, niche specific genes and specific features of conserved genes. This study aims to capitalize on the emerging advances in Lactobacillus genome sequence analysis and provides a platform for present individual comparisons as well as future analysis of new Lactobacillus genomes.
Results and discussion
General features of 20 Lactobacillus genomes
The 20 completed genomes of Lactobacillus, representing 14 different species, vary in size from approximately 1.8 to 3.3 Mb and show a number of discriminating features (Table 1). The number of predicted protein‐coding sequences (CDS) in these Lactobacillus genomes ranges from 1721 to 3100 and such variation points at substantial gene loss/gain in their evolution, as has been presented previously for a smaller set of Lactobacillus genomes (Makarova et al., 2006). The pangenome, defined as the full complement of genes of these Lactobacillus genomes, consists of nearly 14 000 proteins (Table S1).
Table 1.
A general overview of the origin and genome statistics of the 20 Lactobacillus genomes.
| Genome | Length (bp) | G+C content (%) | Predicted ORFs | Isolated from | Genes assigned to COG | LPXTG genes | Genes encoding signal peptides predicted by SignalP (%) | Genes encoding signal peptides predicted by LocateP (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Lactobacillus acidophilus NCFM | 1 993 564 | 34.71 | 1864 | Infant faeces | 1433 | 13 | 21.03 | 9.40 | Altermann et al. (2005) |
| Lactobacillus helveticus DPC 4571 | 2 080 931 | 37.08 | 1757 | Cheese | 1396 | 2 | 17.07 | 6.96 | Callanan et al. (2008) |
| Lactobacillus gasseri ATCC 33323 | 1 894 360 | 35.26 | 1755 | Human Gut | 1316 | 14 | 17.89 | 6.38 | Azcarate‐Peril et al. (2008) |
| Lactobacillus crispatus ST1 | 2 043 161 | 36 | 2024 | Chicken faeces | 1499 | 8 | 13.58 | 9.39 | Ojala et al. (2010) |
| Lactobacillus johnsonii FI9785 | 1 781 645 | 34.43 | 1733 | Human faeces | 1320 | 12 | 24.58 | 6.49 | Wegmann et al. (2009) |
| Lactobacillus johnsonii NCC 533 | 1 992 676 | 34.61 | 1821 | Human faeces | 1403 | 18 | 19.17 | 7.41 | Pridmore et al. (2004) |
| Lactobacillus delbrueckii ssp bulgaricus ATCC BAA 365 | 1 856 951 | 49.69 | 1721 | Yoghurt | 1196 | 3 | 17.49 | 8.28 | Makarova et al. (2006) |
| Lactobacillus delbrueckii ssp bulgaricus ATCC 11842 | 1 864 998 | 49.72 | 2094 | Yoghurt | 1153 | 2 | 16.19 | 8.69 | van de Guchte et al. (2006) |
| Lactobacillus casei ATCC 334 | 2 924 325 | 46.58 | 2771 | Cheese | 1959 | 18 | 20.39 | 8.55 | Makarova et al. (2006) |
| Lactobacillus casei BL23 | 3 079 196 | 46.34 | 3044 | Cheese | 2152 | 20 | 20.47 | 8.40 | Mazéet al. (2010) |
| Lactobacillus rhamnosus GG | 3 010 111 | 46.69 | 2944 | Human Gut | 2032 | 18 | 30.16 | 7.83 | Kankainen et al. (2009) |
| Lactobacillus rhamnosus Lc 705 | 3 033 106 | 46.68 | 2992 | Cheese | 2099 | 15 | 31.45 | 8.34 | Kankainen et al. (2009) |
| Lactobacillus sakei 23k | 1 884 661 | 41.26 | 1879 | Meat | 1462 | 7 | 19.96 | 8.46 | Chaillou et al. (2005) |
| Lactobacillus brevis ATCC 367 | 2 340 228 | 46.06 | 2218 | Human | 1678 | 12 | 21.06 | 9.38 | Makarova et al. (2006) |
| Lactobacillus plantarum JDM1 | 3 197 759 | 44.66 | 2948 | Human saliva | 2248 | 34 | 28.05 | 7.94 | Zhang et al. (2009) |
| Lactobacillus plantarum WCFS1 | 3 348 625 | 44.42 | 3100 | Adult Intestine | 2305 | 35 | 19.87 | 7.88 | Kleerebezem et al. (2003) |
| Lactobacillus fermentum IFO 3956 | 2 098 684 | 51.47 | 1843 | Adult Intestine | 1519 | 5 | 14.81 | 5.48 | Morita et al. (2008) |
| Lactobacillus reuteri DSM 20016 | 1 999 618 | 38.87 | 1935 | Silage | 1529 | 4 | 15.76 | 4.84 | A. Copeland, S. Lucas, A. Lapidus, K. Barry, J.C. Detter, T. Glavina del Rio, N. Hammon et al. (unpublished) |
| Lactobacillus reuteri JCM 1112 | 2 039 414 | 38.88 | 1820 | Fermented plant material | 1495 | 5 | 16.65 | 5.22 | Morita et al. (2008) |
| Lactobacillus salivarius | 2 133 977 | 33.04 | 2073 | Terminal ileum of human | 1476 | 5 | 14.52 | 6.58 | Claesson et al. (2007) |
The Lactobacillus secretome has received considerable attention as it includes proteins that may interact with the environment (Kleerebezem et al., 2010). Both SignalP (Emanuelsson et al., 2007) and LocateP (Zhou et al., 2008) were used to predict the secretome of the lactobacilli (Table 1). While secretome predictions via SignalP suffer from some inaccuracy not present in LocateP, it predicted the largest secretome. It is of interest to note that the fraction of genes that were predicted to encode signal sequences is highly variable. The largest set (over 30% of the predicted proteome) was found to be encoded by the genomes of the L. rhamnosus GG and Lc705 that are marketed as probiotics (Kankainen et al., 2009). However, several other probiotic strains, including L. johnsonii NCC533 and L. acidophilus NCFM, were predicted to encode a higher fraction of secreted proteins than L. helveticus or L. delbrueckii that contain an equally sized genome but derive from a well‐known dairy background. Similarly, the latter starter strains were predicted to have the lowest number of proteins that are cell wall anchored via sortases that recognize the LPXTG‐like motif (termed here LPXTG genes) located at the C‐terminal end (Boekhorst et al., 2005). The 20 Lactobacillus genomes showed a highly diverse G+C content varying from 33% to 51%. This represents a span of G+C values that is about twice as large as that normally observed in well‐defined bacterial genera, raising the question whether the Lactobacillus species analysed here belong to a single genus (Fujisawa et al., 1992).
The Lactobacillus core genome
To study the relation between the genes in the 20 genomes, we determined the set of shared orthologous genes, termed the Lactobacillus core genome (LCG). A total of 383 sets of orthologous genes were calculated to constitute this LCG (Table S2). This LCG is larger than the gene set for 141 core proteins defined based on the comparison of 12 Lactobacillus genomes (Claesson et al., 2008). This can be mainly ascribed to the more stringent criteria and the classification of genes into COGs that was used to select the core genes in this previous study.
Close inspection of the order of the genes in the LCG revealed that over 100 genes were organized in operon‐like clusters that were conserved in all 20 genomes. This indicated that apart from a shared function, these genes also had a conserved organization and control. This reflects a common ancestry that likely extends beyond the Lactobacillus group as many of the genes in the LCG are also conserved in other related Gram‐positive bacteria. Among those genes, we found the canonical large gene clusters for the ribosomal proteins, the major proton‐translocating ATPase and many house‐keeping functions. Moreover, the LCG contained all genes of the dlt operon coding for the d‐alanylation of lipotechoic acids that are involved in specific signalling to the host (de Vos, 2005). In addition, three conserved two‐component regulatory systems were found to be present in all 20 Lactobacillus genomes that could form a basic network of responses to the environment although it is not known yet what they control. Moreover, the ccpA gene for the carbon catabolite control protein was always located adjacent to that of the pepQ gene for a prolidase. This was earlier observed in L. delbrueckii where the specific CcpA‐mediated control of the prolidase gene expression was experimentally verified (Morel et al., 1990; Schick et al., 1999). This common organization indicates a link between control of sugar and nitrogen metabolism that is conserved in all lactobacilli. While the LCG contains over 80 genes for hypothetical proteins, one gene with an assigned function stands out – this is the gene annotated to encode FbpA that is present in all sequenced Lactobacillus genomes, including those not yet completed, such as that of the intestinal L. buchneri and L. coleohominis (Nelson et al., 2010). This over‐500‐residue FbpA protein has first been described in S. pyogenes as a fibronectin‐binding protein (Courtney et al., 1994). It is highly conserved in many lactic acid bacteria as well as some bacilli, and belongs to the PF05833 family of proteins. Given its widespread occurrence in Gram‐positive bacteria and absence of signal and other cognate topogenic sequences, it is doubtful whether binding to fibronectin is the natural function of this protein in lactobacilli. It is tempting to speculate that the FbpA‐like proteins share a common function relating to environmental interactions such as biofilm formation.
In order to further characterize the Lactobacillus gene pool, we classified it using the COG classification that annotated the vast majority of the LCG genes (Fig. 1). This functional prediction of the LCG showed 26% of genes belonging to ‘Translation, ribosomal structure and biogenesis’, most likely acting as house‐keeping genes, while 10% of the genes belonged to ‘Replication, recombination and repair’, 14% to ‘unknown function or general function prediction only’, 7% to ‘Transciption’, and 6% to ‘Carbohydrate transport and metabolism’ (Fig. 1). Remarkably in view of the large predicted secretome of the lactobacilli (Table 1) is that only a small fraction (5%) of the proteins encoded by the LCG were predicted to be secreted, indicating that many secreted proteins are encoded by strain‐specific genes.
Figure 1.
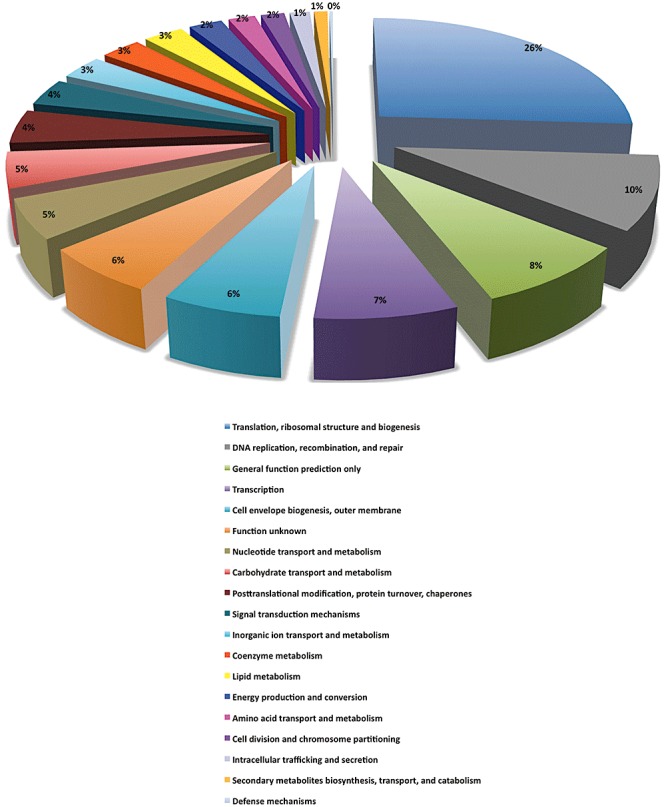
COG distribution of the predicted function of the LCG genes.
Grouping of Lactobacillus genomes
The 383 genes of the LCG were used for the construction of a phylogenetic tree of the lactobacilli (method described in detail in the Experimental procedures section). The obtained tree differs slightly from the well‐known 16S rRNA‐based grouping but adds a higher level of confidence as it is based on comparisons of the complete LCG with ∼130 kb per genome.
The generated whole‐genome‐based phylogeny revealed the presence of three distinct and large clusters of lactobacilli (Fig. 2). These clusters were named after the strain designation of the largest or most well‐known genome they contained. In this way the NCFM, WCFS and GG clusters were defined that consisted of 8, 7 and 5 genomes respectively. The NCFM cluster is not only the largest but also the most coherent. In contrast, the WCFS and GG clusters contain each an outgroup genome, that of L. salivarius and L. sakei respectively.
Figure 2.
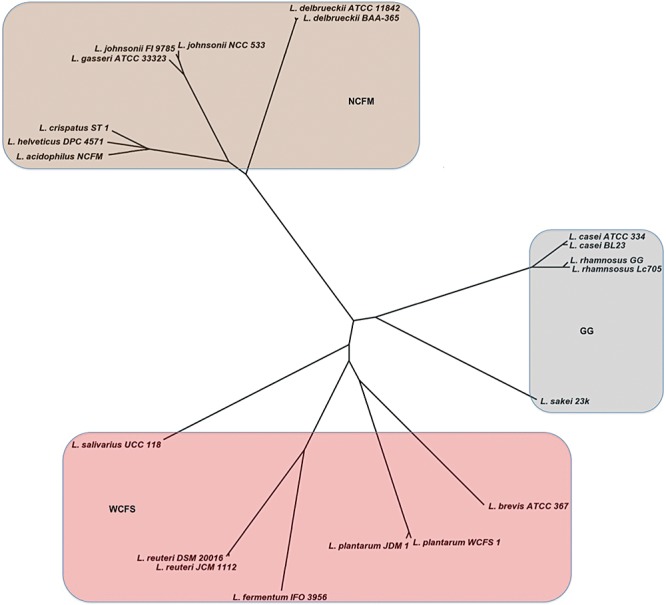
Phylogenetic grouping of the Lactobacillus spp. with known genomes based on the features of their LCG. Three groups are shaded with different colours and termed NCFM, WCFS and GG groups (for further explanation see text).
The COG distribution of all 20 genomes was compared to reveal specific features (Table S3). The Lactobacillus genomes were dominated by COG categories including ‘Amino acid transport and metabolism’, ‘Carbohydrate transport and metabolism’, ‘Replication, recombination and repair’, ‘Transcription’ and ‘Translation, ribosomal structure and biogenesis’. Remarkably, the first two categories were only moderately represented in the LCG as they included only 8 and 19 genes of the total of 383 genes respectively. The NCFM group was characterized by more than average number of genes in the ‘Translation, ribosomal structure and biogenesis’, while the GG group had the smallest number of genes in this category. The categories ‘Transcription’ and ‘Replication, recombination and repair’ also showed variation among different Lactobacillus groups with some exceptions. It was also interesting to notice that the largest genomes (L. casei BL23, L. rhamnosus GG and Lc705, and L. plantarum WCSF1) are having most carbohydrate utilization proteins as reported earlier for L. plantarum WCFS1 genome (Kleerebezem et al., 2003). Apart from this no clear trends could be observed when the COG distribution was analysed.
Specific signatures in the Lactobacillus genomes
Subsequently, we defined additional groups of core genes, next to the LCG, including the set of genes that are present in all the genomes of one group (termed the core group genes) and the set of genes that are present in all genomes of one group and absent in all other Lactobacillus genomes (termed the signature group genes). The core group gene numbers are similar and vary from 771, 636 to 991 (Table 2, Tables S4–S6), but the signature group genes vary from 119, 14 to 88 in the NCFM, WCFS and GG groups respectively (Table 2, Tables S7–9). The low level of signature group genes in the WCFS group can be explained by the fact that this group is the least coherent as indicated above (Fig. 2).
Table 2.
Proteins found in core group and signature group genes of Lactobacillus genomes.
| NCFM | WCFS | GG | |
|---|---|---|---|
| Core group genes | 771 | 636 | 991 |
| Signature group genes | 119 | 14 | 88 |
The core group genes were further used to define the LCG‐specific ORFans and the Group‐specific ORFans. ORFans are the genes present in genome of one species and absent in all other. LCG‐specific ORFans are the genes present in LCG and absent in all other genomes while Group‐specific ORFans are the genes present in core group genes of one group and absent in all other genomes. As can be expected from the different level of coherence of the three groups (see above), there were large differences between the number of Group‐specific ORFans, including 56, 4 and 30 for the NCFM, WCFS and GG group respectively (Table 3, Tables S10–12) while LCG‐specific ORFans consisted of 41 genes (Table 3, Table S13). Here we describe the salient features of these LCG‐specific ORFans and the Group‐specific ORFans that are characteristic of the lifestyle of the members of these groups.
Table 3.
General statistics of proteins predicted to be ORFans from the three specific core groups of Lactobacillus genomes.
| Data set | Genes blasted | ORFans found | Hypothetical | Annotated |
|---|---|---|---|---|
| Complete core (LCG) | 383 | 41 | 13 | 28 |
| NCFM | 119 | 56 | 34 | 22 |
| WCFS | 14 | 4 | 3 | 1 |
| GG | 88 | 30 | 15 | 15 |
The LCG‐specific ORFans are the genes that are only found in the genomes of 20 complete lactobacilli. Remarkably, all ORFans were predicted to encode small proteins with an average size of 75 residues and this may be due to the method of calculating the ORFans. As these ORFans are unique for lactobacilli it is not a surprise that 13 out of 41 ORFans were predicted to encode hypothetical proteins. Several of these were found in operon structures but their function remains to be elucidated. Many of the annotated ORFans (a total 13) were predicted to code for ribosomal proteins and some of them also existed in conserved operon‐like clusters.
The NCFM Group‐specific ORFans are found in the genomes of L. acidophilus, L. helveticus, L. crispatus, L. gasseri, L. johnsonii and L. delbrueckii and the 56 representatives include a majority (34) of genes coding for not‐yet‐annotated proteins and many that have been annotated inconsistently, such as LBA1852 that is annotated as a potential d‐ala,d‐ala ligase in L. acidophilus but a TAT‐pathway signal in L. gasseri and a conserved hypothetical protein in all other representatives of the NCFM group. Evidently, this hampers the possibility to speculate about the function of these genes. Other NCFM Group‐specific ORFans include LBA0044 for a GDSL‐like lipase/acylhydrolase, LBA0342 for a 2′,3′‐cyclic nucleotide 3′‐phosphodiesterase with a polynucleotide kinase domain conserved in all NCFM group members, and LBA0189 predicted to code for the glycerol‐3‐phosphate acyltransferase PlsY involved in the early stages of glycerolipid biosynthesis.
The WCFS Group‐specific ORFans are found in the genomes of L. plantarum, L. brevis, L. fermentum, L. reuteri and L. salivarius. There are only four of these detected and three of these encode hypothetical proteins. The remaining one is represented by Lp_2528 in L. plantarum and is annotated in various ways, including a dioxygenase, a bleomycin resistance protein and a lactoylglutathione lyase glyoxalase. The latter is likely to be the correct annotation based on extensive blast analysis and lactoylglutathione lyase is involved in the detoxification of methylglyoxal, a highly toxic byproduct of triosephosphates that are abundant glycolytic intermediates in lactobacilli. Recently, it has been observed that in S. mutans the lactoylglutathione lyase glyoxalase gene was upregulated during acid stress while its inactivation results in loss of acid resistance (Korithoski et al., 2007). It is tempting to speculate that members of the WCFS group that include species that are known to tolerate acidity below pH 3 have adapted a specific form of acid resistance effected by a highly related lactoylglutathione lyase glyoxalase.
The GG group includes L. rhamnosus, L. casei and L. sakei, and the GG group‐specific Orfans include 30 genes from which 15 code for hypothetical proteins. Many of the annotated ones have discrete features in spite of their small size. These include LGG02390, a small hydrophobic protein coding for bacteriocin immunity. In L. sakei this is likely to be Sakacin P but its function in L. rhamnosus GG is not clear – while a potential bacteriocin operon was predicted from the genome (Kankainen et al., 2009) as experimental analysis suggested that this strain does not seem to produce bacteriocins (De Keersmaecker et al., 2006). However, this may be due to the laboratory growth conditions employed in this study that are known to induce different gene expression than the intestinal environment (Marco et al., 2010). Another small protein is that encoded by LGG01384 in L. rhamnosus GG which has all features of a 4Fe‐4S ferredoxin, found in many anaerobic bacteria and archaea. The question remains in what redox reaction this ferredoxin is involved, as the members of the GG group are considered to grow only by fermentation and do not respire.
In our analysis we could not identify any niche‐specific genes when considering the source of the isolated strains. Such genes were previously reported for the analysis of a smaller set of genomes (O'Sullivan et al., 2009). All the nine niche‐specific genes identified in that study were found to be present in other niches as well based on the present set of Lactobacillus genomes. However, it remains to be seen whether the source of isolation is really the natural niche, the more so as some species, such as L. plantarum, are found in plant fermentations, dairy products and the intestinal tract (De‐Vries et al., 2006). Within this cosmopolitan species, a set of characteristic genes can be detected, as was already indicated by complete genome hybridization (Molenaar et al., 2005). The observation that many L. plantarum genes are expressed in the intestine of humans and mice but are transcriptionally silent in laboratory media indicate the presence of a core of genes specific for the intestinal niche (Bron et al., 2004; Marco et al., 2010). Further comparative and functional genome sequencing will show whether more of these niche‐specific genes can be detected and how widely these are distributed.
Conclusions
Detailed comparative analysis of the 20 Lactobacillus genomes revealed a platform for present individual comparisons as well as future analysis of new Lactobacillus genomes. A set of features were defined that included the total of 383 sets of orthologous genes defining the LCG that allowed the classification of all 20 genomes into the NCFM, WCFS and GG groups. Notably the Group‐specific ORFans appeared to be of specific value in defining the different genomic groups and providing insight in the origin and function of the species they include.
Experimental procedures
Orthology estimation and genome comparisons
To estimate orthologous genes an all‐against‐all comparison of the genes of all genomes was performed using blastp (Altschul et al., 1997) with the standard scoring matrix BLOSUM62 and an initial E‐value cut‐off of 1e−04. The score of every blast hit was set into proportion to the best score possible, the score of a hit of the query gene against itself. This resulted in a so‐called score ratio value (SRV) between 0 and 100 that reflected the quality of the hit much better than the raw blast bit score (Lerat et al., 2003).
Two genes were considered orthologous if there existed a reciprocal best blast hit between these genes, and both hits had an SRV > 35. Based on this orthology criterion the core genome was calculated as the set of genes that had orthologous genes in all other analysed strains. The group‐wise comparisons were also calculated based on this orthology threshold. A core genome was calculated for the five groups created based on phylogeny. Subsequently, all genes were filtered out of these five groups that had an orthologue in any strain outside the subset.
The Pan genome was calculated as the set of all unique genes of a set of genomes. All genes of one reference genome are taken as basic set for the calculation. Subsequently, the genes of a second genome were compared with this set, and all genes in the second genome that had no orthologous gene in the starting gene set were added to this set. This process was iteratively repeated for all genomes of the compared set, resulting in the pan genome.
For all orthology calculations we used the comparative genomics platform EDGAR (Blom et al., 2009). Signal sequences were predicted with SignalP v3.0 (Emanuelsson et al., 2007– see http://www.cbs.dtu.dk/services/SignalP‐3.0/) and LocateP (Zhou et al., 2008– see http://www.cmbi.ru.nl/locatep‐db/cgi‐bin/locatepdb.py). Additionally, various custom Perl scripts were used to support the analyses.
Search for LCG‐specific genes (ORFans)
To identify LCG‐specific genes we created a database of the protein sequences of all completely sequenced genomes present in the NCBI database. All Lactobacillus genomes were excluded from this set. The final database comprised of 3 505 217 proteins from 1047 genomes. We compared the core genes of all Lactobacillus strains to this database using blastp with an initial E‐value cut‐off of 1e−30. Genes that had no blast hit against any of the proteins in the database were considered to be Lactobacillus specific. The genes specific for the three genomic subsets NCFM, WCFS and GG were analysed using the same approach.
Phylogenetic tree
The phylogenetic tree was calculated using a slightly adapted version of the pipeline proposed by Zdobnov and Bork (2007). Every gene of the core genome was aligned together with all its orthologous genes using MUSCLE (Edgar, 2004). The numerous resulting multiple alignments were concatenated and poorly aligned positions were eliminated using GBLOCKS (Talavera and Castresana, 2007). The trimmed multiple alignment was used to create a phylogenetic tree using the neighbour‐joining implementation of PHYLIP (Felsenstein, 1995).
COG and LPXTG genes
Genes from all 20 Lactobacillus genomes were assigned to COGs using rps‐blast (Reverse Position Specific blast) and NCBI's Conserved Domain Database (CDD). Top hits were taken with an E‐value cut‐off of 10−2. HMMER software (http://hmmer.org/) package was used to scan a set of protein sequences for the generic sortase substrate HMM (Boekhorst et al., 2005).
Acknowledgments
This work was supported by Center of Excellence in Microbial Food Safety Research (MiFoSa), Academy of Finland. RK would like to thank Dr Janne Nikkila and Dr Anne Salonen for reading the text. JB acknowledges financial support by the BMBF (grant 0313805A ‘GenoMik‐Plus’). Authors would like to thank Miaomiao Zhou for providing the LocateP predictions of 20 Lactobacillus genomes.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Pangenome of twenty Lactobacillus.
Lactobacillus core genome.
COG distribution of the 20 Lactobacillus genomes.
Group Core NCFM.
Group Core WCFS.
Group Core GG.
Specific Core NCFM.
Specific Core WCFS.
Specific Core GG.
ORFans GG.
ORFans NCFM.
ORFans WCFS.
ORFans Lactobacillus Core Genome.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Altermann E., Russell W.M., Azcarate‐Peril M.A., Barrangou R., Buck B.L., McAuliffe O. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc Natl Acad Sci USA. 2005;102:3906–3912. doi: 10.1073/pnas.0409188102. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcarate‐Peril M.A., Altermann E., Goh Y.J., Tallon R., Sanozky‐Dawes R.B., Pfeiler E.A. Analysis of the genome sequence of Lactobacillus gasseri ATCC 33323 reveals the molecular basis of an autochthonous intestinal organism. Appl Environ Microbiol. 2008;15:4610–4625. doi: 10.1128/AEM.00054-08. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom J., Albaum S.P., Doppmeier D., Puhler A., Vorholter F.J., Zakrzewski M., Goesmann A. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhorst J., de Been M.W., Kleerebezem M., Siezen R.J. Genome‐wide detection and analysis of cell wall‐bound proteins with LPxTG‐like sorting motifs. J Bacteriol. 2005;14:4928–4934. doi: 10.1128/JB.187.14.4928-4934.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron P.A., Grangette C., Mercenier A., de Vos W.M., Kleerebezem M. Identification of Lactobacillus plantarum genes that are induced in the gastrointestinal tract of mice. J Bacteriol. 2004;17:5721–5729. doi: 10.1128/JB.186.17.5721-5729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooijmans R.J., de Vos W.M., Hugenholtz J. Lactobacillus plantarum WCFS1 electron transport chains. Appl Environ Microbiol. 2009;11:3580–3585. doi: 10.1128/AEM.00147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callanan M., Kaleta P., O'Callaghan J., O'Sullivan O., Jordan K., McAuliffe O. Genome sequence of Lactobacillus helveticus, an organism distinguished by selective gene loss and insertion sequence element expansion. J Bacteriol. 2008;2:727–735. doi: 10.1128/JB.01295-07. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchaya C., Claesson M.J., Fitzgerald G.F., van Sinderen D., O'Toole P.W. Diversity of the genus Lactobacillus revealed by comparative genomics of five species. Microbiology. 2006;152:3185–3196. doi: 10.1099/mic.0.29140-0. [DOI] [PubMed] [Google Scholar]
- Chaillou S., Champomier‐Vergès M.C., Cornet M., Crutz‐Le Coq A.M., Dudez A.M., Martin V. The complete genome sequence of the meat‐borne lactic acid bacterium Lactobacillus sakei 23K. Nat Biotechnol. 2005;12:1527–1533. doi: 10.1038/nbt1160. et al. [DOI] [PubMed] [Google Scholar]
- Claesson M.J., van Sinderen D., O'Toole P.W. The genus Lactobacillus– a genomic basis for understanding its diversity. FEMS Microbiol Lett. 2007;1:22–28. doi: 10.1111/j.1574-6968.2006.00596.x. [DOI] [PubMed] [Google Scholar]
- Claesson M.J., van Sinderen D., O'Toole P.W. Lactobacillus phylogenomics–towards a reclassification of the genus. Int J Syst Evol Microbiol. 2008;58:2945–2954. doi: 10.1099/ijs.0.65848-0. [DOI] [PubMed] [Google Scholar]
- Courtney H.S., Li Y., Dale J.B., Hasty D.L. Cloning, sequencing, and expression of a fibronectin/fibrinogen‐binding protein from group A streptococci. Infect Immun. 1994;9:3937–3946. doi: 10.1128/iai.62.9.3937-3946.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker S.C., Verhoeven T.L., Desair J., Marchal K., Vanderleyden J., Nagy I. Strong antimicrobial activity of Lactobacillus rhamnosus GG against Salmonella typhimurium is due to accumulation of lactic acid. FEMS Microbiol Lett. 2006;259:89–96. doi: 10.1111/j.1574-6968.2006.00250.x. [DOI] [PubMed] [Google Scholar]
- De Vos W.M. Lipotechoic acid in lactobacilli: d‐alanine makes the difference. Proc Natl Acad Sci USA. 2005;102:10763–10764. doi: 10.1073/pnas.0504966102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellaglio F., Felis G.E. Taxonomy of Lactobacillus and bifidobacteria. In: Tannock G.W., editor. Caister Academic Press; 2005. pp. 25–49. [Google Scholar]
- De‐Vries M.C., Vaughan E.E., Kleerebezem M., de Vos W.M. Lactobacillus plantarum– survival, functional and potential probiotic properties in the human intestinal tract. Int Dairy J. 2006;16:1018–1028. [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O., Brunak S., von Heijne G., Nielsen H. Locating proteins in the cell using TargetP, SignalP, and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Department of Genetics, University of Washington; 1995. ) PHYLIP (Phylogeny Inference Package. Version 3.57 c. Seattle, WA, USA: [Google Scholar]
- Fujisawa T., Benno Y., Yaeshima T., Mitsuoka T. Taxonomic study of the Lactobacillus acidophilus group, with recognition of Lactobacillus gallinarum sp. nov. and Lactobacillus johnsonii sp. nov. and synonymy of Lactobacillus acidophilus group A3 (Johnson et al. 1980) with the type strain of Lactobacillus amylovorus (Nakamura 1981) Int J Syst Bacteriol. 1992;42:487–491. doi: 10.1099/00207713-42-3-487. [DOI] [PubMed] [Google Scholar]
- van de Guchte M., Penaud S., Grimaldi C., Barbe V., Bryson K., Nicolas P. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. Proc Natl Acad Sci USA. 2006;24:9274–9279. doi: 10.1073/pnas.0603024103. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes W.P., Vogel R.F. The genus Lactobacillus. In: Wood B.J.B., Holzapfel W.H., editors. Blackie Academic & Professional; 1995. pp. 19–54. [Google Scholar]
- Hugenholtz P. Blackie Academic & Professional; 1998. [Google Scholar]
- Kankainen M., Paulin L., Tynkkynen S., von Ossowski I., Reunanen J., Partanen P. Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human‐ mucus binding protein. Proc Natl Acad Sci USA. 2009;40:17193–17198. doi: 10.1073/pnas.0908876106. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleerebezem M., Boekhorst J., van Kranenburg R., Molenaar D., Kuipers O.P., Leer R. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci USA. 2003;100:1990–1995. doi: 10.1073/pnas.0337704100. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleerebezem M., Hols P., Bernard E., Rolain T., Zhou M., Siezen R.J., Bron P. The extracellular biology of the lactobacilli. FEMS Microbiol Rev. 2010;34:199–230. doi: 10.1111/j.1574-6976.2010.00208.x. [DOI] [PubMed] [Google Scholar]
- Korithoski B., Lévesque C.M., Cvitkovitch D.G. Involvement of the detoxifying enzyme lactoylglutathione lyase in Streptococcus mutans aciduricity. J Bacteriol. 2007;189:7586–7592. doi: 10.1128/JB.00754-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat E., Daubin V., Moran N.A. From gene trees to organismal phylogeny in prokaryotes: the case of the gamma‐proteobacteria. PLoS Biol. 2003;1:E19. doi: 10.1371/journal.pbio.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K., Slesarev A., Wolf Y., Sorokin A., Mirkin B., Koonin E. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci USA. 2006;103:15611–15616. doi: 10.1073/pnas.0607117103. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco M.L., de Vries M.C., Wels M., Molenaar D., Mangell P., Ahrne S. Convergence in probiotic Lactobacillus gut‐adaptive responses in humans and mice. ISME J. 2010 doi: 10.1038/ismej.2010.61. et al (in press): doi:10.1038/ismej.2010.61. [DOI] [PubMed] [Google Scholar]
- Mazé A., Boël G., Zúñiga M., Bourand A., Loux V., Yebra M.J. Complete genome sequence of the probiotic Lactobacillus casei strain BL23. J Bacteriol. 2010;10:2647–2648. doi: 10.1128/JB.00076-10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar D., Bringel F., Schuren F.H., de Vos W.M., Siezen R.J., Kleerebezem M. Exploring Lactobacillus plantarum genome diversity by using microarrays. J Bacteriol. 2005;187:6119–6127. doi: 10.1128/JB.187.17.6119-6127.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel F., Frot‐Coutaz J., Aubel D., Portalier R., Atlan D. Characterization of a prolidase from Lactobacillus delbrueckii subsp. bulgaricus CNRZ 397 with an unusual regulation of biosynthesis. Microbiology. 1990;145:437–446. doi: 10.1099/13500872-145-2-437. [DOI] [PubMed] [Google Scholar]
- Morita H., Toh H., Fukuda S., Horikawa H., Oshima K., Suzuki T. Comparative genome analysis of Lactobacillus reuteri and Lactobacillus fermentum reveal a genomic island for reuterin and cobalamin production. DNA Res. 2008;3:151–161. doi: 10.1093/dnares/dsn009. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K.E., Weinstock G.M., Highlander S.K., Worley K.C., Creasy H.H., Wortman J.R. Human Microbiome Jumpstart Reference Strains Consortium. A catalog of reference genomes from the human microbiome. Science. 2010;5981:994–999. doi: 10.1126/science.1183605. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala T., Kuparinen V., Koskinen J.P., Alatalo E., Holm L., Auvinen P. Genome sequence of Lactobacillus crispatus ST1. J Bacteriol. 2010;13:3547–3548. doi: 10.1128/JB.00399-10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan O., O'Callaghan J., Sangrador‐Vegas A., McAuliffe O., Slattery L., Kaleta P. Comparative genomics of lactic acid bacteria reveals a niche‐specific gene set. BMC Microbiol. 2009;9:50. doi: 10.1186/1471-2180-9-50. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pot B., Ludwig W., Kersters K., Schleifer K.H. Taxonomy of lactic acid bacteria. In: de Vuyst L., Vandamme E.J., editors. Chapman & Hall; 1994. pp. 13–89. [Google Scholar]
- Pridmore R.D., Berger B., Desiere F., Vilanova D., Barretto C., Pittet A.C. The genome sequence of the probiotic intestinal bacterium Lactobacillus johnsonii NCC 533. Proc Natl Acad Sci USA. 2004;101:2512–2517. doi: 10.1073/pnas.0307327101. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxelin M., Tynkkynen S., Mattila‐Sandholm T., de Vos W.M. Probiotic and other functional microbes: from markets to mechanisms. Curr Opin Biotechnol. 2005;16:204–211. doi: 10.1016/j.copbio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Schick J., Weber B., Klein J.R., Henrich B. PepR1, a CcpA‐like transcription regulator of Lactobacillus delbrueckii subsp. lactis. Microbiology. 1999;145:3147–3154. doi: 10.1099/00221287-145-11-3147. [DOI] [PubMed] [Google Scholar]
- Talavera G., Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Tannock G.W. A special fondness for lactobacilli. Appl Environ Microbiol. 2004;70:3189–3194. doi: 10.1128/AEM.70.6.3189-3194.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos W.M., Hugenholtz J. Engineering metabolic highways in Lactococci and other lactic acid bacteria. Trends Biotechnol. 2004;22:72–79. doi: 10.1016/j.tibtech.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Wegmann U., Overweg K., Horn N., Goesmann A., Narbad A., Gasson M.J., Shearman C. Complete genome sequence of Lactobacillus johnsonii FI9785, a competitive exclusion agent against pathogens in poultry. J Bacteriol. 2009;22:7142–7143. doi: 10.1128/JB.01182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood B.J.B., Holzapfel W.H. 1st. Blackie Academic and Professional; 1995. [Google Scholar]
- Wood B.J.B., Warner P.J. Kluwer Academic/Plenum Publishers; 2003. [Google Scholar]
- Zdobnov E.M., Bork P. Quantification of insect genome divergence. Trends Genet. 2007;23:16–20. doi: 10.1016/j.tig.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Zhang Z.Y., Liu C., Zhu Y.Z., Zhong Y., Zhu Y.Q., Zheng H.J. Complete genome sequence of Lactobacillus plantarum JDM1. J Bacteriol. 2009;15:5020–5021. doi: 10.1128/JB.00587-09. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Boekhorst J., Francke C., Siezen R.J. LocateP: genome‐scale subcellular‐location predictor for bacterial proteins. BMC Bioinformatics. 2008;9:173. doi: 10.1186/1471-2105-9-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pangenome of twenty Lactobacillus.
Lactobacillus core genome.
COG distribution of the 20 Lactobacillus genomes.
Group Core NCFM.
Group Core WCFS.
Group Core GG.
Specific Core NCFM.
Specific Core WCFS.
Specific Core GG.
ORFans GG.
ORFans NCFM.
ORFans WCFS.
ORFans Lactobacillus Core Genome.