

Blood groups, finger prints, iris scans and DNA bar codes are several ways to distinguish or identify individual humans. According to Arumugam and colleagues (2011) we can now also be distinguished by the microbial communities in our faeces, our ‘enterotypes’.
The human gut is one of the most densely populated ecosystems known, and although this ecosystem contains members of the three domains of life – bacteria, archaea and eucarya (Finegold et al., 1983) – it is dominated by bacteria. They are essential in digesting the food we eat and in keeping us healthy by stimulating our immune system and fighting off pathogenic bacteria. It is estimated that 1013–1014 microbes inhabit our gastrointestinal tract (GIT), with the greatest number residing in the distal gut, where they synthesize essential vitamins and process otherwise indigestible components of our diet such as plant polysaccharides (Backhed et al., 2005). Early studies used 16S ribosomal DNA (rDNA) analysis to type and enumerate the distal gut and faecal microbiota. More than 90% of the bacterial phylotypes present in the intestinal microbiota in healthy humans are members of only three bacterial divisions: the Bacteroidetes, the Firmicutes and the Actinobacteria (Zoetendal et al., 2006).
Nevertheless, each healthy adult's gut appears to have a unique and relatively stable microbiota (Zoetendal et al., 1998; Turnbaugh et al., 2007), which is a reflection of the numerous different phylogenetic clusters among the Firmicutes, Clostridium clusters IV, IX and XIVa, including the predominant genera Clostridium, Eubacterium, Roseburia and Ruminococcus. Furthermore, the Actinobacteria that encompass mainly the genera Bifidobacterium and Atopobium also represent important members of the gut microbial community (Harmsen et al., 2002; Turroni et al., 2008). Notably, recent estimates of the diversity of the human gut microbial ecosystem indicate it may encompass more than 1000 species and a multitude of strains (Backhed et al., 2005; Blaut and Clavel, 2007; Rajilic‐Stojanovic et al., 2007). Microbiota composition studies in humans have discovered that aberrations in the microbiome composition is present in obese individuals (Ley et al., 2006; Turnbaugh et al., 2009) as well as in individuals with a variety of other diseases (Zoetendal et al., 2008).
Since the largest part (∼80%) of gut microbes remains uncultured, metagenomics analysis has become fashionable in the past 5 years to estimate the types, relative abundance and genome content of microbes in various parts of the GIT. The first metagenomics analysis from just two faecal samples (Gill et al., 2006) led to early insight into enrichment of genes encoding specific metabolic pathways, including metabolism associated with glycans, amino acids and xenobiotics, but also methanogenesis and biosynthesis of vitamins and isoprenoids. However, this early study provided only a very fragmented view due to limitations in sample size, sequencing technology, number of bases sequenced and availability of suitable reference genomes of gut inhabitants.
Gut microbial reference genomes
Large efforts have now been initiated to make catalogues of reference genomes from the human gut microbiome. Research groups from around the world have launched an International Human Microbiome Consortium (IHMC) which together aims to sequence more than 1000 human bacterial reference genomes, including 900 genomes from many body sites (but mainly the GIT) by the Human Microbiome Jumpstart Reference Strains Consortium (Nelson et al., 2010) (http://www.hmpdacc.org/project_catalog.html) and 100 gut genomes by the MetaHIT Consortium (http://www.sanger.ac.uk/resources/downloads/bacteria/metahit/). The MetaHIT sequencing project, coordinated by the Sanger Institute in the UK and funded by the European Commission, aims to sequence 30 strains of cultured gut bacteria, and 70 individual isolated cells of uncultured gut bacteria by single‐cell genome sequencing (de Jager and Siezen, 2011).
Human gut microbial gene catalogues
Later metagenomics studies generated some 3 Gb of microbial sequence from faecal samples of 33 individuals from the USA and Japan (Kurokawa et al., 2007; Turnbaugh and Gordon, 2009; Turnbaugh et al., 2009). More recently, Illumina sequencing of DNA from faecal samples from 144 European adults generated 577 Gb of DNA sequence, almost 200 times more sequence than all previous studies (Qin et al., 2010). After assembly, a non‐redundant set of 3.3 million gut ORFs was obtained which mapped well to 89 reference gut genomes. The cohort harboured a total of 1000–1150 prevalent bacterial species, and each individual had at least 160 of these species. This large data set allowed an in‐depth assessment of the bacterial functions important for life in the gut, and gut‐specific functions for adhesion to the host epithelial proteins and for harvesting sugars of the globoseries glycolipids (Qin et al., 2010).
Enterotypes
Now, in a May 2011 issue of Nature, gut metagenomics data sets from 39 samples from six nationalities, including newly sequenced 22 European faecal samples, have been combined and the DNA sequence reads compared with 1511 reference genomes including 389 publicly available human microbiome genomes generated through the IHMC and MetaHIT consortia (Arumugam et al., 2011). Nearly 53% of the sequence reads could be robustly assigned to a genus in the reference genome set, and 80% could be assigned to a phylum. The phylogenetic composition confirmed that the Firmicutes and Bacteriodetes phyla are the most dominant in the human faecal samples. Multidimensional cluster analysis and principal component analysis (PCA) revealed that the gut samples separate into three robust clusters, designated as ‘enterotypes’. Each of the three enterotypes is identifiable by variation in the levels of one of three main genera: Bacteriodetes (enterotype 1), Prevotella (enterotype 2) and Ruminococcus (enterotype 3) (Fig. 1a). The same analysis performed on two larger published human microbiome data sets generated the same three enterotype clusters (Fig. 1b and c).
Figure 1.
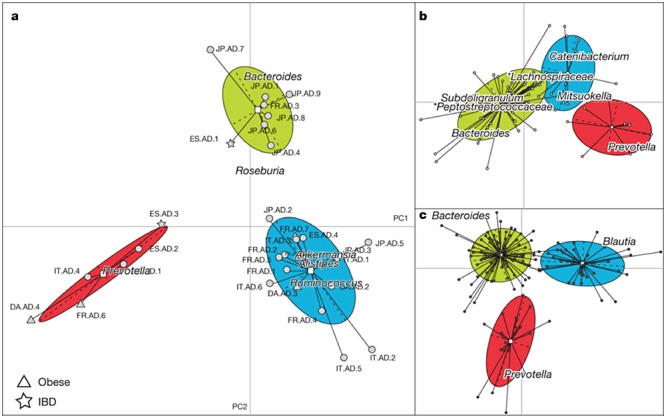
Phylogenetic differences between enterotypes. Between‐class analysis, which visualizes results from PCA and clustering, of the genus compositions of 33 Sanger metagenomes estimated by mapping the metagenome reads to 1511 reference genome sequences using an 85% similarity threshold (a), Danish subset containing 85 metagenomes from a published Illumina data set (b), and 154 pyrosequencing‐based 16S sequences (c) reveals three robust clusters that we call enterotypes. Two principal components are plotted using the ade4 package in R with each sample represented by a filled circle. The centre of gravity for each cluster is marked by a rectangle and the coloured ellipse covers 67% of the samples belonging to the cluster. IBD, inflammatory bowel disease. Adapted by permission from Macmillan Publishers: Nature (Arumugam et al., 2011), copyright 2011.
Phylogenetic and functional differences among enterotypes seem to reflect different combinations of microbial trophic chains. The abundances and co‐occurrence networks of the three enterotypes from the Sanger metagenomes are shown in Fig. 2. The drivers of enterotype 1 (mainly Bacteriodetes, co‐occurring with, e.g. Parabacteriodetes) appear to derive energy mainly from fermentation of carbohydrates as these genera have a very broad saccharolytic potential. In enterotype 2, the Prevotella and co‐occurring Desulfovibrio can act in synergy to degrade mucin glycoproteins of the gut mucosal layer, while in enterotype 3 the Ruminococcus and Akkermansia can bind mucins and transport and degrade the constituent sugars. Enterotypes 1 and 2 are also enriched in biosynthesis of different vitamins (Arumugam et al., 2011). Surprisingly, the enterotypes do not correlate with host properties such as nationality, age, gender or body mass index. However, data‐driven marker genes or functional modules were identified for most of these host properties.
Figure 2.
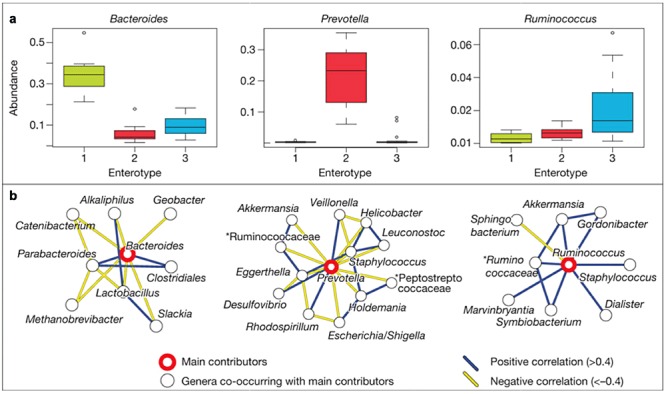
Main contributors to enterotypes. a. Phylum abundance box plots of the main contributors of each enterotype from the Sanger metagenomes as determined by read abundance. b. Co‐occurrence networks of the three enterotypes from the Sanger metagenomes. Unclassified genera under a higher rank are marked by asterisks. Adapted by permission from Macmillan Publishers: Nature (Arumugam et al., 2011), copyright 2011.
Future perspectives
This study indicates the existence of a limited number of well‐balanced host–microbial symbiotic states in human faeces that presumably reflect the microbial composition in the large intestine. Perhaps it would have been wiser to call these ‘faecotypes’ than ‘enterotypes’, since it is well known that the microbial abundance and composition changes dramatically throughout the GIT. It remains to be seen whether such stable and universal microbial consortia are also found in the small intestine where the genera Streptococcus, Clostridium and Veillonella are dominant (Booijink et al., 2007; 2010a,b; van den Bogert et al., 2011).
This is an exciting field of research, where much is still to be learned about how stable enterotypes are formed and maintained in humans and other animals, and whether they can be influenced by diets, probiotics, drugs or diseases (Kau et al., 2011; Walter and Ley, 2011).
Acknowledgments
The research that led to the discovery of the enterotypes was in part funded by the European Community's Seventh Framework Programme (FP7/2007‐2013) in the project MetaHIT; grant agreement HEALTH‐F4‐2007‐201052.
References
- Arumugam M., Raes J., Pelletier E., Le Paslier D., Yamada T., Mende D.R. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. , and et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F., Ley R.E., Sonnenburg J.L., Peterson D.A., Gordon J.I. Host–bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Blaut M., Clavel T. Metabolic diversity of the intestinal microbiota: implications for health and disease. J Nutr. 2007;137:751S–755S. doi: 10.1093/jn/137.3.751S. [DOI] [PubMed] [Google Scholar]
- van den Bogert B., de Vos W.M., Zoetendal E.G., Kleerebezem M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl Environ Microbiol. 2011;77:2071–2080. doi: 10.1128/AEM.02477-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booijink C.C., Zoetendal E.G., Kleerebezem M., de Vos W.M. Microbial communities in the human small intestine: coupling diversity to metagenomics. Future Microbiol. 2007;2:285–295. doi: 10.2217/17460913.2.3.285. [DOI] [PubMed] [Google Scholar]
- Booijink C.C., Boekhorst J., Zoetendal E.G., Smidt H., Kleerebezem M., de Vos W.M. Metatranscriptome analysis of the human fecal microbiota reveals subject‐specific expression profiles, with genes encoding proteins involved in carbohydrate metabolism being dominantly expressed. Appl Environ Microbiol. 2010a;76:5533–5540. doi: 10.1128/AEM.00502-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booijink C.C., El‐Aidy S., Rajilic‐Stojanovic M., Heilig H.G., Troost F.J., Smidt H. High temporal and inter‐individual variation detected in the human ileal microbiota. Environ Microbiol. 2010b;12:3213–3227. doi: 10.1111/j.1462-2920.2010.02294.x. et al. [DOI] [PubMed] [Google Scholar]
- Finegold S.M., Sutter V.L., Mathiesen G.E. Microflora composition and development. In: Hentges D.J., editor. Academic Press; 1983. pp. 3–119. [Google Scholar]
- Gill S.R., Pop M., Deboy R.T., Eckburg P.B., Turnbaugh P.J., Samuel B.S. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen H.J., Raangs G.C., He T., Degener J.E., Welling G.W. Extensive set of 16S rRNA‐based probes for detection of bacteria in human feces. Appl Environ Microbiol. 2002;68:2982–2990. doi: 10.1128/AEM.68.6.2982-2990.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager V., Siezen R.J. Single‐cell genomics: unravelling the genomes of unculturable microorganisms. Microb Biotechnol. 2011;4:431–437. doi: 10.1111/j.1751-7915.2011.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau A.L., Ahern P.P., Griffin N.W., Goodman A.L., Gordon J.I. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K., Itoh T., Kuwahara T., Oshima K., Toh H., Toyoda A. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R.E., Turnbaugh P.J., Klein S., Gordon J.I. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Nelson K.E., Weinstock G.M., Highlander S.K., Worley K.C., Creasy H.H., Wortman J.R. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J., Li R., Raes J., Arumugam M., Burgdorf K.S., Manichanh C. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajilic‐Stojanovic M., Smidt H., de Vos W.M. Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol. 2007;9:2125–2136. doi: 10.1111/j.1462-2920.2007.01369.x. [DOI] [PubMed] [Google Scholar]
- Turnbaugh P.J., Gordon J.I. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., Ley R.E., Hamady M., Fraser‐Liggett C.M., Knight R., Gordon J.I. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., Hamady M., Yatsunenko T., Cantarel B.L., Duncan A., Ley R.E. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F., Ribbera A., Foroni E., van Sinderen D., Ventura M. Human gut microbiota and bifidobacteria: from composition to functionality. Antonie Van Leeuwenhoek. 2008;94:35–50. doi: 10.1007/s10482-008-9232-4. [DOI] [PubMed] [Google Scholar]
- Walter J., Ley R.E. The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol. 2011;65 doi: 10.1146/annurev-micro-090110-102830. [DOI] [PubMed] [Google Scholar]
- Zoetendal E.G., Akkermans A.D., De Vos W.M. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host‐specific communities of active bacteria. Appl Environ Microbiol. 1998;64:3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoetendal E.G., Vaughan E.E., de Vos W.M. A microbial world within us. Mol Microbiol. 2006;59:1639–1650. doi: 10.1111/j.1365-2958.2006.05056.x. [DOI] [PubMed] [Google Scholar]
- Zoetendal E.G., Rajilic‐Stojanovic M., de Vos W.M. High‐throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut. 2008;57:1605–1615. doi: 10.1136/gut.2007.133603. [DOI] [PubMed] [Google Scholar]