

Abstract
Genome-wide association studies (GWAS) of responses to drugs, including clopidogrel, pegylated-interferon and carbamazepine, have led to the identification of specific patient subgroups that benefit from therapy. However, the identification and replication of common sequence variants that are associated with either efficacy or safety for most prescription medications at odds ratios (ORs) >3.0 (equivalent to >300% increased efficacy or safety) has yet to be translated to clinical practice. Although some of the studies have been completed, the results have not been incorporated into therapy, and a large number of commonly used medications have not been subject to proper pharmacogenomic analysis. Adoption of GWAS, exome or whole genome sequencing by drug development and treatment programs is the most striking near-term opportunity for improving the drug candidate pipeline and boosting the efficacy of medications already in use.
At least a third of the money that is spent on prescription drugs is wasted, amounting to more than $100 billion per year thrown away in the United States alone. This is because a substantial proportion of patients are prescribed medications that are, at an individual level, either ineffective or dangerous. Surprisingly similar challenges exist, to varying degrees, for both drugs in development and commonly prescribed drugs, in that the drugs either lack efficacy (Box 1) or produce serious adverse reactions in some patient subgroups. To remedy both hypo-innovation and a lineage of failures in drug development, some pharmaceutical companies have begun to embrace open-innovation models and multiple precompetitive partnerships, encompassing not only academic institutions but also other pharmaceutical companies1–4. Will these approaches suffice? Or should companies and clinicians pay more attention to the genetic basis of drug response?
Box 1. Missed opportunities in top grossing drugs.
Perhaps the best example of both waste and missed opportunity can be found with the three top-grossing prescription drugs worldwide; TNF α-receptor inhibitors (etanercept (Enbrel), infliximab (Remicade) and adalimumab (Humira)), are used in the treatment of rheumatoid arthritis with aggregate sales of nearly $30 billion. However, these three specific biological agents cost more than $15,000 per patient annually and only 40% of individuals respond to treatment (Fig. 2)81,82. Despite these agents having been available for over a decade, little work has been undertaken or published to define the biological signal that partitions the patients who respond from those who do not respond. Indeed, it is entirely possible that thousands of frozen samples still exist from previous clinical trials. Access to some of these samples may be difficult in some countries due to regulatory or ethical obstacles, but that should not be a limiting factor to accomplish systematic genomic assessment in an adequately sized cohort of patients. The evidence from our review of all pharmacogenomic GwAS published so far (Table 1) suggests it would be quite possible to identify common DNA sequence variant risk alleles that would be predictive of the therapeutic response to TNF-α blockers, potentially exploiting available residual samples from original clinical trials. Furthermore, GwAS represent only one of the approaches that can be employed to discover the determinants of drug responsiveness or major side effects. It remains possible that some of the lack of responsiveness is tied to antibody production to the drug, but that has not been adequately studied83. A few, limited GwAS attempts have been negative to date. Now that the technical accuracy, speed and cost of exome and whole genome sequencing have become remarkably conducive for research, there are even more systematic means of determining genomic markers of drug response. And that does not even take into account the full range of biological assays to understand variability between individuals including their proteins, RNA transcripts, metabolites, microbiome and epigenomics. Efforts to understand responsiveness to TNF-α blockers would not only facilitate identification of patients for whom these agents are appropriate but would crucially also set up an enormous opportunity to develop drugs for the majority of patients who are not responsive to this class of drugs. This win-win perspective for society and for the biotech-pharmaceutical industry has been overridden by the short-term, exceptional success of the drugs from a revenue-generation point-of-view. The incentives to pursue precision are sorely lacking. But the evidence that signals delineating those who will repond likely exist, left undiscovered so far, is overwhelming.
Until recently, the means for identifying the genomic variants that underlie differential drug responses in individual patients were limited. Although we know that genetics has an impact on drug absorption, metabolism, pharmacodynamics and excretion, candidate gene studies of the prior generation were intrinsically biased and were frequently not replicated in either the same or different populations. Performed without the advantage of hypothesis-free, genome-wide probing as pharmacogenomic studies are, the bias of those pharmacogenetic studies was attributable to testing only a limited number or the investigator’s favorite candidate gene variants. Even though Roche (Basel) received US Food and Drug Administration (FDA) approval for its AmpliChip Cytochrome P450 genotyping test in 2004, it has hardly been used to screen drug conversion or metabolic pathways in research or clinical practice. The field of pharmacogenomics seems to have stagnated with few recent clinically meaningful discoveries. For example, knowledge of the candidate genes found to influence the efficacy of warfarin (one of the most widely used medications), could have been used to guide more appropriate dosing, but has not been applied in the clinic.
In GWAS, which have assessed 500,000 to 1 million common DNA sequence variants in small patient cohorts, a substantial number of unanticipated and often striking signals have been associated with either efficacy or safety. This does not take into account the more comprehensive assessment of the genome through either exome or whole genome sequencing that can readily be performed today.
We argue here that genomic guidance should be embedded in all drug development and treatment programs. Furthermore, to improve efficacy, avoid serious side-effects and promote cost effectiveness, genomically guided research should now be undertaken for all commonly used medications in clinical practice.
The success of pharmacogenomics in GWAS
GWAS have been used to identify common DNA sequence variants that are associated with susceptibility to >250 complex disease traits (including heart attack, diabetes, most cancers, Alzheimer’s disease and most autoimmune diseases, such as Crohn’s, rheumatoid arthritis and multiple sclerosis) in hundreds of thousands of patients, resulting in >1,700 publications5. With few exceptions, the ORs for the effect are typically in the range of 1.05 to 1.15, indicating a remarkably small effect, and leaving the explanation for heritability of any of the traits studied largely unresolved. However, using GWAS to investigate drug responses has resulted in strikingly positive results. When considering all of the GWAS catalogued in the US National Human Genome Research Institute, pharmacogenomics studies are sevenfold more likely to achieve ORs >3.0 compared with common disease GWAS6.
Typically only a small number of cases and controls have been required to demonstrate extremely large ORs (Table 1), with some ranging from 20 to 80—nearly two log orders more than susceptibility variants for diseases. Unfortunately, this pharmacogenomic approach is not routinely applied to either experimental or currently marketed drugs. With clear evidence that some subpopulations are at risk from certain therapies, neglecting the usage of genotyping for known sequence variant markers, which can be performed by rapid, point-of-care platforms, compromises patient care and could be considered ethically unacceptable. Before delving into the specifics of the ability to link common sequence variations with drug efficacy or side effects, let us consider why GWAS for pharmacogenomics seem to provide such robust results.
Table 1.
GWAS pharmacogenomics responses
| Drug | Response | Cases | Gene | Genotype OR (95% CI) |
Genome-wide significance/ replication |
Reference | |
|---|---|---|---|---|---|---|---|
| Efficacy | Metformin | Glycemic response | 3,960 | ATM | 1.35 (1.22–1.49) | Yes/Yes | 30 |
| Interferon a (Pegasys) | Sustained hepatitis C genotype 1 viral response |
1,137; 142; 293; 1,015 |
IL28B | 37.7 (16.7–83.9) | Yes/Yes | 13 – 16 | |
| Clopidogrel (Plavix) | Anti-platelet responsiveness | 429 | CYP2C19 | 2.42 (1.18–4.99) | Yes/Yes | 21 | |
| Warfarin (Coumadin) | Maintenance dose | 181; 1,053 |
VKORC1CYP2C9CYP4F2 | 1.11 (1.00–1.22) | Yes/Yes | 65,66 | |
| Glucocorticoids | Response to glucocorticoid therapy in asthma |
935 418 |
GLCC11T | 2.36 (1.27–4.41) | No | 31,32 | |
| Thiazide | Diastolic blood pressure change |
389 | 12q15 region | – | Yes/No | 67 | |
| Candesartan (Atacand) | Diastolic blood pressure change |
198 | FUT4 | – | No | 68 | |
| Dabigatran (Pradaxa) | Bleeding | 1,490 | CES1 | 0.67 (0.55–0.82) | No | 69 | |
| ADR | Simvastatin (Zocor) | Skeletal myopathy | 85 | SLCO1B1 | 17.4 (4.8–62.9) | Yes/Yes | 45 |
| Ximelagatran | Hepatotoxicity | 74; 10 |
HLA-DRB1*0701
HLA-DQA1*0201 |
4.4 (2.2–8.9) 4.4 (2.2–8.1) |
No | 70 | |
| Flucloxacillin (Floxapen) | Hepatotoxicity | 51 | HLA-B*5701 | 80.6 (22.8–284.9) | Yes/Yes | 40 | |
| Lumiracoxib (Prexige) | Hepatotoxicity | 41 51 |
HLA-DRB1*1501
HLA-DQB1*0602 |
7.5 (5.0–11.3) | Yes/Yes | ||
| Amoxicillin-clavulanate (Augmentin) |
Hepatotoxicity | 201 |
HLA-DRB1*1501
HLA-DQB1*0602 |
2.8 (2.1–3.8) | Yes/Yes | 41 | |
| Carbamazepine (Tegretol) |
SJS | 12; 53 | HLA-A*3103 | 25.93 (4.93– 116.18) |
Yes/Yes | 71,72 | |
| Interferon α2b/ribavirin | Hemolytic anemia | 988 | ITPA | – | Yes/Yes | 73 | |
| Methotrexate (Trexall) | Drug clearance and toxicity in pediatric leukemia patients |
434 | SLCO1B1 | 16.4 (8.7–26.7) | Yes/Yes | 74 |
SJS, Steven Johnson syndrome.
The large ORs associated with pharmacogenomics studies are likely due to the lack of an evolutionary response to modern exogenous agents. Unlike common disease gene variant hunting, where disease loci are located on haplotype blocks that have been selected to overcome environmental influences and selection pressure over thousands if not millions of years, the genome has had very little time to adapt to exposure to prescription medications.
Another possible explanation is that there are a relatively small number of genes, such as those involving drug absorption, metabolism, effect at the cell or tissue level, or excretion, involved in the mode of action of any given drug. Owing to the limited number of genes and pathways a drug interacts with, which is in contrast to the large number of genes involved in regulating physiological processes that underpin common diseases, small differences between individuals produce an amplified response that results in the large ORs observed.
Pharmacogenomics of drug efficacy
Before the GWAS era, many candidate gene studies suggested associations between sequence variation and responses to drugs such as codeine, abacavir (Ziagen) and nortiptyline (Pamelor, Aventyl)7. Many of these associations have either not been assessed or validated with hypothesis-free, more systematic genome-wide studies. Also, no study investigating the contribution of low frequency or rare variants in a population on the efficacy of a drug has yet been published, but these data could be easily obtained by exome or whole genome sequencing. With rare variants estimated to frequently occur in drug target genes (1 every 17 bases), it is highly probable sequencing will yield further insights into drug responses8,9. Even worse, little effort has so far been applied to identify the common variants linked with drug responses. However, for the commonly prescribed drugs interferon-α, clopidogrel (Plavix), warfarin and steroid inhalers for asthma, GWAS have shown particular sequence variants to have a marked relationship with drug efficacy (Table 1).
The prototype for the genome basis of drug efficacy thus far is polyethylene glycol–modified (pegylated) interferon-α, the standard drug used in the treatment of chronic infections of hepatitis C virus (HCV). Chronic HCV infection affects >200 million people globally and is among the leading causes of cirrhosis and liver cancer10. Both host and viral genetic variation contribute toward disease progression and treatment response among individuals. Although 11 different HCV genotypes exist, genotype 1 represents 80% of all HCV circulating in the United States and 60% worldwide11. Current treatment strategies consist of injectable pegylated interferon-α and oral ribavirin (Rebetol, Copegus), costing ~$50,000 per patient per year but the treatment is effective in <50% of patients12. Interestingly, interferon is an outlier to the proposed evolutionary hypothesis of drug response, as the molecule, although prescribed, is a naturally occurring protein produced in response to pathogens. Four GWAS have been carried out in various HCV-infected populations, and the results indicate that three single nucleotide polymorphisms (SNPs) in IL28B are mainly responsible for an individual’s responsiveness to pegylated interferon-α/ribavirin13–16. Of these three SNPs, rs12979860 is the most important, with a CC genotype at this SNP thought to be the best pretreatment predictor of therapy responsiveness and spontaneous viral clearance, whereas carriers of a T allele at the same SNP have poorer outcomes including a higher rate of diabetes mellitus after liver transplantation17–19. But currently, SNP genotyping as part of HCV therapy is not done, except in a very limited number of clinical centers.
With >2 million coronary stenting procedures performed annually, variants that determine the differential responsiveness to clopidogrel, the standard adjunctive anti-platelet agent, are of huge public health importance20. A GWAS validated previous candidate gene studies and implicated CYP2C19 loss-of-function (LOF) polymorphisms with reduced anti-platelet effects and a threefold increased risk of stent thrombosis21,22. The LOF variants are remarkably common, such that the inert pro-drug clopidogrel is not normally metabolized into an active drug. At least a third of individuals carry one or more LOF alleles, and the frequency is higher among those with Asian and African ancestries than with European ancestry. Although the absolute risk of stent thrombosis after stenting is relatively low, in the range of 2–3% of patients, there is an extremely large population of more than 2 million patients at risk. When stent thrombosis occurs it usually results in heart attack or death. Given the exceedingly large number of patients undergoing stenting, the high proportion of individuals who carry LOF alleles and the alternative medications that are available to avoid this interaction, the evidence and necessity to use this genomic information appears overwhelming. Furthermore, rapid point-of-care genotyping has enabled CYP2C19 LOF carriers to be readily identified and receive more potent anti-platelet agents, which successfully achieve full platelet inhibition and would be expected to protect against stent thrombosis23.
Widespread uptake of genotyping before therapy starts, or during therapy, has been hindered by an education gap in physicians with >90% of physicians feeling they are not proficient in delivering genomically guided care24. An accompanying problem is a lack of continuity of care; put simply, the physician inserting the stent is unlikely to manage the patient long-term. With genotyping data currently taking several days to be processed, the patient has often been discharged and already initiated on an anti-platelet agent, leaving genotype data largely redundant. There are also the issues of the need to carry out the genotyping in a Clinical Laboratory Improvement Amendments–certified lab and the diversity of genotyping and sequencing platforms. To further complicate matters, reimbursement related to pharmacogenomic testing remains uncertain in the United States. It is hoped that improved point-of-care genotyping platforms will reduce the time lag and facilitate the prescription of anti-platelet agents once the patients genotype is known. The clear-cut relationship between cytochrome gene variants and the vitamin K receptor in the response to warfarin has been established in multiple GWAS. Adverse reaction to warfarin is one of the most important causes of drug-related hospitalizations25 and it has been shown that taking GWAS-derived SNPs into consideration in dosage calculations delivers superior dosage predictions and reduces bleeding events by a third compared with traditional dosing algorithms26–28.
The GWAS investigating metformin (Glucophage) nonresponsiveness, an effect observed in 25% of individuals receiving the top-selling drug for glycemic control, contradicted previous pharmacogenetic evidence. Instead of validating the organic cation transporter 1 (OAT1) gene29, the GWAS suggested that a SNP close to the ataxia telangiectasia gene was involved in drug response, which disappointingly accounted for only 2.5% of the total glycemic variability observed30. Perhaps this unexpected observation was due to poor phenotyping of participants, which was compounded by retrospective recruitment of participants from previously conducted clinical trials30. This may have introduced undesirable confounding factors.
Although SNPs with genome-wide significance have not been linked to the management of asthma patients, several SNPs have shown functional relevance31–33. For example, homozygotes for the rs37973 allele within the GLCCI1 gene have a threefold reduction in forced expiratory volume in 1 s, a standard test measuring lung function, with a significantly higher risk of poor response (OR, 2.36; 95% confidence interval (CI), 1.27 to 4.41)32. Recently, whole genome sequencing revealed a rare variant allele in the amyloid precursor protein (APP) gene, which was found to be protective for Alzheimer’s disease and cognitive decline34. Although not achieving genome-wide significance—that is, the statistical correction for a variant after adjustment for more than a million comparisons—the APP protective allele was nevertheless replicated in multiple large cohorts and exhibited salutary functional biologic effects. This finding suggests that the lack of genome-wide significance itself may not disprove the clinical significance of a sequence variant.
GWAS that investigated responses to the diabetes medication metformin and glucocorticoids were conducted post hoc on poorly phenotyped cohorts that had been established to answer different research questions. Such poor experimental set-ups reduce the overall discovery power of GWAS and can potentially result in false-positive associations. Similarly, GWAS that investigate human-measured subjective score phenotypes generally do not detect significant levels of association (Table 2). It is therefore imperative that cohorts are assembled appropriately (Fig. 1). With large effect sizes expected, it is likely that few ethnically matched cases, phenotypically defined through prospective, objective assessment, would be required. Importantly, simple population controls are inappropriate, as all individuals recruited should receive the same drug without objectively demonstrating the same phenotype. Here the cases are the individuals with the most extreme response and controls are those lacking signs of efficacy to the drug in question. Controls should not receive any additional medications as this would confound interpretation of results. A prime example for the lack of efficacy in drugs today is for tumor necrosis factor (TNF) α-receptor blockers, the leading group of prescription drugs worldwide by gross sales (Box 1 and Fig. 2).
Table 2.
GWAS investigating subjective phenotypes
| Drug | Response | Cases | Lowest p-value |
Reference |
|---|---|---|---|---|
| Interferon β | Response to multiple sclerosis clinical scoring |
206 | 0.004 | 75 |
| Anti-tumor necrosis factor |
Response to rheumatoid arthritis by clinical scoring |
89 | 0.009 | 76 |
| Methylphenidate (Ritalin) |
Response to ADHD by clinical scoring |
309 | 3 × 10−6 | 77 |
| Iloperoidone (Fanapt) |
Response to schizophrenia by clinical scoring |
210 | 1 × 10−7 | 78 |
| Antidepressants | Response to depression by clinical scoring |
339 | 8 × 10−7 | 79 |
| Citalopram (Celexa) | Response to depression by clinical scoring |
1,948 | 5 × 10−7 | 80 |
In contrast to Table 1, none of these GWAS studies used a clinical phenotype or even a laboratory surrogate. Instead, each study assessed drug responsiveness on the basis of a semiquantitative, subjective scoring by a healthcare professional. The negative results may thus reflect an artificial, nonorganic, subjective phenotype. ADHD, attention deficit hyperactivity disorder.
Figure 1.
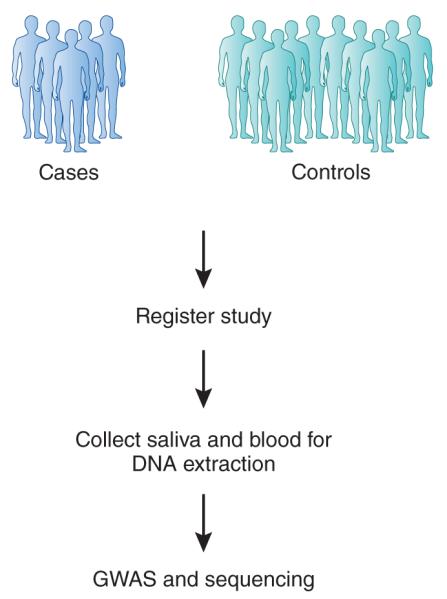
Assembling cohorts for drug-response GwAS. Ethnically matched cases and controls should be chosen, ideally with collaborative academic and community medical center networks, to recruit participants. All cases and controls should ideally receive only a single medication to avoid inducing or inhibiting medication interactions. Cases must have an objectively measured phenotype, and the same phenotype must be absent from controls.
Figure 2.
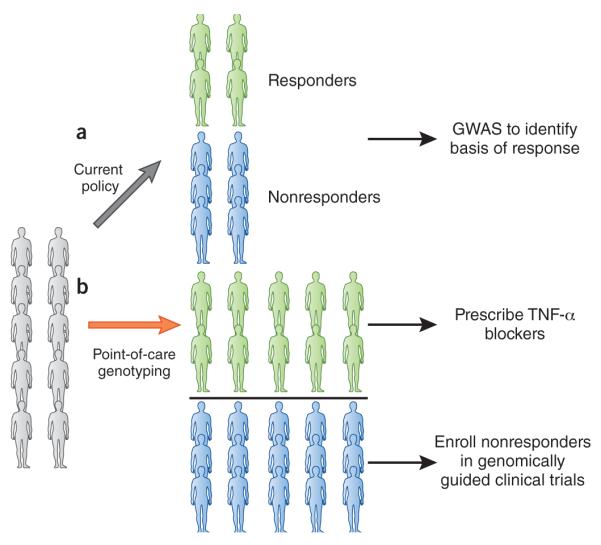
Current and future strategies for treatment with TNF-α blockers. (a) From a total of ten people receiving TNF-α blockers with rheumatoid arthritis only four individuals benefit. For ten patients, TNF-α blockers cost on average $150,000 per year, meaning $90,000 dollars is wasted. To identify variants underlying response, a GwAS needs to be done. (b) Point-of-care genotyping, following results of genome-wide genotyping and/or sequencing would facilitate TNF-α prescription only to individuals likely to benefit. Those unlikely to respond could be recruited to genomically guided drug trials.
Pharmacogenomics of adverse drug reactions
Multiple adverse drug reactions (ADRs) exist and the international Serious Adverse Events Consortium (http://www.saeconsortium.org/), a collection of academic institutes and pharmaceutical companies, are attempting to determine genetic (DNA) variants associated with serious ADRs, including drug-induced liver injury, drug-induced renal injury and serious skin rashes.
Hepatotoxicity is the most common cause of clinical trial termination and drug withdrawal from the market35. Traditionally, the highly polymorphic cytochrome P450 system was investigated as a cause of hepatotoxicity because six cytochrome P450 enzymes are responsible for metabolizing over 90% of drugs36. However, it is now known that drug-induced hepatotoxicity (DIHT) is largely attributed to variation within alleles for human leukocyte antigens (HLA), triggered by a wide variety of structurally different therapeutic agents37. Although rare, this ADR accounts for half of all liver failure admissions with 75% of patients requiring liver transplantation38.
Multiple candidate gene studies have previously described an association between HLA complex alleles and DIHT. GWAS have validated several of these associations and delineated additional loci. The earliest GWAS investigating DIHT studied only 74 patients with abnormal liver function tests in response to the direct thrombin inhibitor ximelagatran. Only a moderate association within the HLA DRB1 gene was detected (P = 6.0 × 10−6), and further direct genotyping was required to delineate HLA-DRB1*0701 (P = 4 × 10−5)39. Huge ORs were derived from a GWAS performed on 51 patients of northern European ancestry with flucloxacillin (Floxapen)-induced DIHT, which identified class 1 HLA-B*5701 as the risk factor (OR, 80.6; 95% CI, 23–285)40. With flucloxaxicillin-induced hepatotoxicity occurring in fewer than 1 in 10,000 patients taking the drug, it is estimated that screening for HLA B*5701 would benefit only 1 in 500–1,000 individuals positive for HLA B*5701 (ref. 37). The problem of a poor positive predictive value also emerged from the largest DIHT GWAS, which associated haplotype DRB1*1501-DQB1*0602-DQA1*0102 with amoxicillin-clavulanate related hepatoxicity (DRB1*1501-DQB1*0602 OR, 3.3; 95% CI, 2.0–5.7; DQA1*0102 OR, 2.2; 95% CI, 1.6–3.2)41.
Carbamazepine (Tegretol), a commonly used drug for a variety of neurologic and psychiatric indications, is associated with a skin rash in 10% of individuals, an ADR that can manifest and cause fatal hypersensitivity reactions either through Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN) in ~1 in 1,000 individuals exposed42. A recent GWAS in individuals of European ancestry has shown that HLA-A*3103 is strongly associated with carbamazepine-induced hypersensitivity reactions and SJS/TEN (OR, 25.9)43. The well established ‘Asian’ risk variant HLA-B*1502 is screened for in all patients in Taiwan who are prescribed carbamazepine and such screening has been shown to markedly lower the risk of SJS44. However, in the United States no screening of individuals of European ancestry who are prescribed carbamazepine for the relevant variant allele has yet to be initiated.
Another example of actionable data derived from GWAS involves statins and skeletal muscle toxicity. Although usually well tolerated, treatment with statins occasionally results in severe rhabdomyolysis or death. GWAS delineated a common variant within the SLCO1B1 gene that was associated with statin-induced myopathy (OR, 17.4; 95% CI, 4.8–62.9)45. This finding led the FDA and the Clinical Pharmacogenetics Implementation Consortium to suggest that genotyping should be carried out before high-dose simvastatin dosing46. However, despite attempts, genotyping has not been incorporated into clinical practice. Instead, statins are administered using a trial-and-error model, whereby serum creatine kinase levels are only monitored in patients who develop signs of severe muscle inflammation.
Knowing associations exist between statins and common variants, it could be hypothesized that underlying genetic variants contributed toward 52 deaths relating to drug-induced rhabdomyolysis, witnessed within patients receiving cerivastatin between 1998 and 2001, before the drug was withdrawn from the market47. With no study ever carried out, we will unfortunately never know why the patients died, leaving cervistatin’s mechanism of action poorly defined and future statins susceptible to similar difficulties. Many of the 150 drug withdrawals from the market since 1960 due to ADRs have been attributed to genetic variants48. Indeed, the widely used medications involved in two of the largest drug withdrawals in history—rofecoxib and rosiglitazone—were never subjected to state-of-the-art pharmacogenetic investigations. Prime examples rofecoxib and rosiglitazone, while highlighting pharmacogenetic negligence, also exemplify a larger problem plaguing drug development, where some pharmaceutical companies have in the past taken extraordinary measures to bury evidence associated with nonfavorable outcomes, especially ADRs49.
One drug withdrawn from the market due to rare but fulminant hepatitis was however studied through GWAS. Lumiracoxib, a selective cyclooxygenase-2 inhibitor, was developed to control osteoarthritic symptoms and was shown to be superior to existing nonselective, nonsteroidal anti-inflammatory drugs at reducing ulcer complications50. However, data from the phase 3 clinical trial showed hepatotoxicity in 2.6% of individuals receiving lumiracoxib50. A GWAS was carried out in a subgroup of the large pivotal efficacy trial that identified a significant association between a HLA haplotype (HLA-DRB1*1501-DQB1*0602-DRB5*0101-DQA1*0102) and lumiracoxib-induced hepatotoxicity (P = 2.8 × 10−10)51. This is the same haplotype associated with amoxicillin-clavulanate hepatotoxicity, despite no obvious structural similarity between the two therapeutic agents. Although DRB1*1501 produced the largest signal (OR, 5.0; 95% CI, 3.6–7.0), the HLA-DQA1*0102 allele represents the most robust marker for screening (sensitivity of 74% and negative predictive value of 99%). Aithal and Daly52 predicted that using HLA-DQA1*0102 as a screening tool the incidence of hepatotoxicity would be reduced to 1.0% for those not carrying an HLA-DQA1*0102 allele and therefore facilitate the reintroduction of the drug to market52.
In summary, there are multiple precedents for identifying sequence variants that are associated with serious, yet infrequent side effects. Some of these variants are ancestry specific, reinforcing the need to study such effects across all ancestries. It is striking that even though the incidence of such side effects is quite low, the studies used to discover the variants have required as few as 12 cases and for many less than 100 individuals affected. Clearly, GWAS have been shown to be an extraordinarily powerful tool for finding sequence variants tied to key drug side effects.
Genomically guided clinical trials
The track record of randomized trials that incorporate clinical phenotypes is fraught, with very large trials that cost hundreds of millions of dollars at best demonstrating small relative benefit of a 15–20% relative reduction in a composite clinical endpoint. This very inefficient and expensive model for the conduct of clinical trials and drug development is untenable. The small magnitude of efficacy can be easily overridden by a low incidence of major side effects, making a positive impact for a drug all the less likely. All of these problems could potentially be addressed by adopting genomically guided clinical trials. We are equipped with actionable knowledge that can be used to prevent patients from experiencing harm when receiving prescribed medications. Elimination of major side effects is a priority and a realistic goal. Subsequent efforts focussing on establishing overwhelming efficacy for all therapeutic agents may be more complex, but would dramatically reduce wastage within society. Both goals are attainable and it is foreseeable that, eventually, this will become the routine standard of care practiced.
Although individuals enrolled to randomized clinical trials may appear phenotypically similar and balanced between cases and controls, they are profoundly heterogeneous at the molecular level. Hypothesis-free genomic screening of potential trial participants and enrollment of individuals with the same key genomic biomarkers of interest would further reduce heterogeneity and enable testing of very specific biologically based hypotheses. Once a key genomic variant marker was identified, it would be appropriate to perform the randomized trial of the new drug comparing placebo or standard treatment only in those patients carrying the sequence variant of interest. Ideally, the critical sequence variants could be established for all drugs in development before phase 3 clinical trials were initiated.
We have directed our attention to germline DNA sequence variants because the use of tumor somatic mutations in both clinical practice and for new drug development programs has been more widely accepted, such as for specific mutations in the genes KRAS, BRAF and anaplastic lymphoma kinase (ALK) for recently approved cancer therapies. This is highlighted through somatic genotyping of specific tumors assessing epidermal growth factor receptor (EGFR) mutations or overexpression of human epidermal growth factor receptor 2 (HER2) to guide appropriate first line therapy53,54. This concept has led to the creation of adaptive trial designs, capable of simultaneously identifying appropriate individuals, dosage and therapeutic combinations faster than traditional designs55. Furthermore, the landmark I-SPY2 trial (NCT01042379) demonstrates how an adaptive trial can incorporate multiple developmental drugs from numerous pharmaceutical companies56.
But now we are beginning to see successful drug development programs anchored to germline sequence variants, such as a new treatment for a rare form of cystic fibrosis57 and the use of an exon-skipping antisense compound, eteplirsen, in Duchenne muscular dystrophy (DMD)58. In each of these examples, a particular mutation is the target for the specific biologically matched, and thus genomically guided, drug intervention. Of note, the discovery of the mutated genes for these two diseases, the cystic fibrosis transconductance regulator (CFTR) and dystrophin in DMD, were made as far back as 1989 and 1987, respectively. Accordingly, the recent genomic variant driven drug development process has propelled successful efforts after more than two decades without any progress.
A similar approach is to use surrogate markers for disease phenotypes within well-defined populations, as is currently being done in an ongoing trial investigating an amyloid binding monoclonal antibody for use in the prevention of Alzheimer’s disease59. The trial, which involves the US National Institutes of Health, Banner Alzheimer’s Institute (Phoenix), the University of Antioquia in Medellín, Colombia and Genentech (S. San Francisco, CA, USA) is investigating a presenilin gene mutation, commonly known as the Paisa mutation, in 300 family members that are all prone to develop premature Alzheimer’s disease in their forties. Rather than measuring dementia as an end-point, surrogate markers are being used to inform investigators of the drug’s efficacy. If successful within this well demarcated subgroup, the therapeutic agent could then be investigated in a broader genomically defined population.
Indeed, with the appropriate assumption of very high efficacy, genomically guided trials require fewer patients to be monitored over a shorter time frame but still with sufficient statistical power, thereby markedly reducing costs for biotech/pharmaceutical companies or other providers operating trials. Any concerns about a limited market resulting from drug efficacy only in patient subgroups could be assessed by exploration of a broader group of patients once the ideal proof-of-efficacy has been achieved. All of this is predicated upon systematic GWAS and sequencing studies to determine the pivotal genomic variants linked to the new drug’s efficacy and safety.
Despite the FDA recognizing the need to determine the pharmacogenomics response and promoting voluntary genomic data submission, we need to go far beyond the nudge that the European Medicines Agency and the FDA have recently made—suggesting integration of GWAS within drug development pipelines to reduce drug attrition and development costs60–62. Ideally, systematic assessment should be a requirement for all new drug development programs. Furthermore, just as there is a registry (http://www.clinicaltrials.gov/) for all clinical trials that are being conducted, thereby reducing the bias in the literature resulting from unpublished studies, there needs to be a pharmacogenomic GWAS and sequencing registry to prospectively record any pharmacogenomic investigation conducted on a drug. This would overcome the disadvantages with current generic GWAS databases, which only include results from studies deemed suitable for publication. Although it is essential for this approach to be conducted before drug approval, we should not neglect currently marketed drugs. Pharmaceutical companies have no business incentive to advocate precision medicine, as this strategy would potentially compromise the market share of their drugs. Therefore, direct involvement of industry is unlikely, with future studies investigating marketed drugs likely to stem from public-private initiatives.
Barriers to clinical pharmacogenetic testing
Currently, we have substantial evidence to support the incorporation of genomic data in patient’s management, but there are barriers that have limited adoption into clinical practice. Pharmacogenomic testing (genotyping or sequencing) is generally outsourced from hospitals to private companies, a time-consuming and costly process. Unfortunately, this is further compounded by a lack of understanding within the medical community regarding genomics, as highlighted by a recent survey indicating that only 10% of physicians felt adequately informed about pharmacogenomic testing63. A major problem going forwards is that many of the drugs that are commercially available and frequently used have lost their proprietary status and there is essentially no commercial interest in evaluating the pharmacogenomics of the drug’s efficacy and safety. Yet we know that every drug has substantial variability in both effectiveness and the occurrence of side effects. Although much interest has focused on comparative effectiveness trials sponsored by government agencies, it seems that the opportunity to define with precision who should receive a particular drug, and at what dose, should be a valuable field of research in the future and may be at least as important as comparing effectiveness of two or more drugs whose genomic basis of effects are unknown.
To date, the one group that has been particularly interested in the use of pharmacogenetic testing has been the pharmacy benefit managers (PBM), which in the United States are involved with authorizing of fulfilling most prescriptions. Here the underlying rationale is that if a PBM can save employers (their customers) the cost of a drug through genotyping, this would make the PBM more competitive64. For example, for generic clopidogrel, the retail price of a 90-day supply is ~$36 and the new drugs ticagrelor or prasugrel, which are not affected by LOF variants altering clopidogrel’s activation, cost >$700, approximately 20 times as much. If the PBM can understand who can be appropriately treated with a generic drug, there is a clear incentive for cost saving that is passed on to employers. Similarly, avoidance of a major sideeffect requiring hospitalization, such as fulminant hepatitis or SJS, would provide a major economic benefit. Of course, we would hope that the pursuit of discovery of the pivotal genomic variations that affect drug response would transcend pure financial considerations.
Conclusions
The time has come to embrace the untapped potential of genome sequencing and related omics technologies to transform both the future of drug development and the administration of commonly used prescription medications. Although no one can dispute the pivotal importance of the human genome for influencing how drugs achieve efficacy or compromise safety, far beyond that which was anticipated, relatively little has been done to incorporate such strategies into the routine medical practice of today or the new drugs of the future. Furthermore, profiling epigenomic changes in the context of drug response is also a field that has been relatively unexplored. Only when the life sciences industry and the medical community leverage the extraordinary leaps in technology and bioinformatics will we actualize the potential of precision, individualized medicine.
ACKNOWLEDGMENTS
E.J.T. was funded by the National Institutes of Health/National Center for Advancing Translational Sciences TR000109.
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Allison M. Reinventing clinical trials. Nat. Biotechnol. 2012;30:41–49. doi: 10.1038/nbt.2083. [DOI] [PubMed] [Google Scholar]
- 2.Mullard A. Partnering between pharma peers on the rise. Nat. Rev. Drug Discov. 2011;10:561–562. doi: 10.1038/nrd3526. [DOI] [PubMed] [Google Scholar]
- 3.Norman TC, Bountra C, Edwards AM, Yamamoto KR, Friend SH. Leveraging crowdsourcing to facilitate the discovery of new medicines. Sci. Transl. Med. 2011;3:88mr1. doi: 10.1126/scitranslmed.3002678. [DOI] [PubMed] [Google Scholar]
- 4.Pollack A. Drug makers join efforts in research. The New York Times. 2012:B3. [Google Scholar]
- 5.Hindorff LA, et al. [accessed 22 October 2012];A catalog of published genome-wide association studies. < http://www.genome.gov/gwastudies>.
- 6.Giacomini KM, et al. Pharmacogenomics and patient care: one size does not fit all. Sci Transl Med. 2012;4:153ps118. doi: 10.1126/scitranslmed.3003471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pirmohamed M. Pharmacogenetics: past, present and future. Drug Discov. Today. 2011;16:852–861. doi: 10.1016/j.drudis.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Nelson MR, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramirez AH, et al. Novel rare variants in congenital cardiac arrhythmia genes are frequent in drug-induced torsades de pointes. Pharmacogenomics J. 2012 May 15; doi: 10.1038/tpj.2012.14. advance online publication, doi:10.1038/tpj.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosen HR. Chronic hepatitis C infection. N. Engl. J. Med. 2011;364:2429–2438. doi: 10.1056/NEJMcp1006613. [DOI] [PubMed] [Google Scholar]
- 11.Soriano V, et al. Pharmacogenetics of hepatitis C. J. Antimicrob. Chemother. 2012;67:523–529. doi: 10.1093/jac/dkr506. [DOI] [PubMed] [Google Scholar]
- 12.McHutchison JG, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N. Engl. J. Med. 2009;361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 13.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka Y, et al. Genome-wide association of IL28B with response to pegylated inter-feron-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 15.Suppiah V, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 16.Rauch A, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345. 1345, e1331–1337. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 17.Veldt BJ, et al. Recipient IL28B polymorphism is an important independent predictor of posttransplant diabetes mellitus in liver transplant patients with chronic hepatitis C. Am. J. Transplant. 2012;12:737–744. doi: 10.1111/j.1600-6143.2011.03843.x. [DOI] [PubMed] [Google Scholar]
- 18.Clark PJ, Thompson AJ, McHutchison JG. IL28B genomic-based treatment paradigms for patients with chronic hepatitis C infection: the future of personalized HCv therapies. Am. J. Gastroenterol. 2011;106:38–45. doi: 10.1038/ajg.2010.370. [DOI] [PubMed] [Google Scholar]
- 19.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topol EJ, Teirstein PS. Textbook of Interventional Cardiology. 6th edn Elsevier Saunders; 2011. [Google Scholar]
- 21.Shuldiner AR, et al. Association of cytochrome P450 2C19 genotype with the anti-platelet effect and clinical efficacy of clopidogrel therapy. J. Am. Med. Assoc. 2009;302:849–857. doi: 10.1001/jama.2009.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mega JL, et al. Dosing clopidogrel based on cyp2c19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. J. Am. Med. Assoc. 2011;306:2221–2228. doi: 10.1001/jama.2011.1703. [DOI] [PubMed] [Google Scholar]
- 23.Roberts JD, et al. Point-of-care genetic testing for personalisation of antiplatelet treatment (RAPID GENE): a prospective, randomised, proof-of-concept trial. Lancet. 2012;379:1705–1711. doi: 10.1016/S0140-6736(12)60161-5. [DOI] [PubMed] [Google Scholar]
- 24.Patay BA, Topol EJ. The unmet need of education in genomic medicine. Am. J. Med. 2012;125:5–6. doi: 10.1016/j.amjmed.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Moore TJ, Furberg CD, Cohen MR. QuarterWatch, Monitoring FDA MedWatch Reports. Institute for Safe Medication Practices; [accessed 22 October 2012]. 2012. Anticoagulants the leading reported drug risk in 2011. < http://www.ismp.org/quarterwatch/pdfs/2011Q4.pdf>. [Google Scholar]
- 26.Klein TE, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N. Engl. J. Med. 2009;360:753–764. doi: 10.1056/NEJMoa0809329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epstein RS, et al. Warfarin genotyping reduces hospitalization rates results from the MM-wES (Medco-Mayo warfarin Effectiveness study) J. Am. Coll. Cardiol. 2010;55:2804–2812. doi: 10.1016/j.jacc.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Anderson JL, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II) Circulation. 2012;125:1997–2005. doi: 10.1161/CIRCULATIONAHA.111.070920. [DOI] [PubMed] [Google Scholar]
- 29.Shu Y, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 2008;83:273–280. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou K, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat. Genet. 2011;43:117–120. doi: 10.1038/ng.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tantisira KG, et al. Genome-wide association identifies the T gene as a novel asthma pharmacogenetic locus. Am. J. Respir. Crit. Care Med. 2012;185:1286–1291. doi: 10.1164/rccm.201111-2061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tantisira KG, et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N. Engl. J. Med. 2011;365:1173–1183. doi: 10.1056/NEJMoa0911353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Himes BE, et al. Genome-wide Association Analysis in Asthma Subjects Identifies SPATS2L as a Novel Bronchodilator Response Gene. PLoS Genet. 2012;8:e1002824. doi: 10.1371/journal.pgen.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonsson T, et al. A mutation in APP protects against Alzheimer/’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 35.Smith DA, Schmid EF. Drug withdrawals and the lessons within. Curr. Opin. Drug Discov. Devel. 2006;9:38–46. [PubMed] [Google Scholar]
- 36.Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician. 2007;76:391–396. [PubMed] [Google Scholar]
- 37.Daly AK. Using genome-wide association studies to identify genes important in serious adverse drug reactions. Annu. Rev. Pharmacol. Toxicol. 2012;52:21–35. doi: 10.1146/annurev-pharmtox-010611-134743. [DOI] [PubMed] [Google Scholar]
- 38.Lee WM. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003;349:474–485. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- 39.Kindmark A, et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008;8:186–195. doi: 10.1038/sj.tpj.6500458. [DOI] [PubMed] [Google Scholar]
- 40.Daly AK, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009;41:816–819. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 41.Lucena MI, et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology. 2011;141:338–347. doi: 10.1053/j.gastro.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roujeau JC. Clinical heterogeneity of drug hypersensitivity. Toxicology. 2005;209:123–129. doi: 10.1016/j.tox.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 43.McCormack M, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 2011;364:1134–1143. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen P, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N. Engl. J. Med. 2011;364:1126–1133. doi: 10.1056/NEJMoa1009717. [DOI] [PubMed] [Google Scholar]
- 45.Link E, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N. Engl. J. Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 46.Wilke RA, et al. The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin. Pharmacol. Ther. 2012;92:112–117. doi: 10.1038/clpt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Furberg CD, Pitt B. Withdrawal of cerivastatin from the world market. Curr. Control. Trials Cardiovasc. Med. 2001;2:205–207. doi: 10.1186/cvm-2-5-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang W, Roederer M.w., Chen WQ, Fan L, Zhou HH. Pharmacogenetics of drugs withdrawn from the market. Pharmacogenomics. 2012;13:223–231. doi: 10.2217/pgs.11.137. [DOI] [PubMed] [Google Scholar]
- 49.Doshi P, Jefferson T, Del Mar C. The imperative to share clinical study reports: recommendations from the Tamiflu experience. PLoS Med. 2012;9:e1001201. doi: 10.1371/journal.pmed.1001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schnitzer TJ, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364:665–674. doi: 10.1016/S0140-6736(04)16893-1. [DOI] [PubMed] [Google Scholar]
- 51.Singer JB, et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat. Genet. 2010;42:711–714. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 52.Aithal GP, Daly AK. Preempting and preventing drug-induced liver injury. Nat. Genet. 2010;42:650–651. doi: 10.1038/ng0810-650. [DOI] [PubMed] [Google Scholar]
- 53.Shih JY, Gow CH, Yang PC. EGFR mutation conferring primary resistance to gefitinib in non-small-cell lung cancer. N. Engl. J. Med. 2005;353:207–208. doi: 10.1056/NEJM200507143530217. [DOI] [PubMed] [Google Scholar]
- 54.Geyer CE, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 55.Berry DA. Adaptive clinical trials in oncology. Nat. Rev. Clin. Oncol. 2012;9:199–207. doi: 10.1038/nrclinonc.2011.165. [DOI] [PubMed] [Google Scholar]
- 56.Barker AD, et al. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin. Pharmacol. Ther. 2009;86:97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 57.Ramsey B.w., et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. [Accessed 16 October 2012];Sarepta Therapeutics announces eteplirsen meets primary endpoint of increased novel dystrophin. < http://finance.yahoo.com/news/sarepta-therapeutics-announces-eteplirsen-meets-110000135.html>.
- 59.Belluck P. New drug trial seeks to stop Alzheimer’s before it starts. The New York Times. 2012 May 16; [Google Scholar]
- 60.Pharmacogenomics working Party (PGwP) [accessed 22 October 2012];European Medicines Agency. 2011 < http://bit.ly/OWZHTh>.
- 61.US Department of Health and Human Services. US Food and Drug Administration. Center for Drug Evaluation and Research & Center for Biologics Evaluation and Research [accessed 22 October 2012];E16 Biomarkers Related to Drug or Biotechnology Product Development: Context, Structure, and Format of Qualification Submissions. International Conference on Harmonisation. 2011 < http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM267449.pdf>.
- 62.US Department of Health and Human Services. US Food and Drug Administration [accessed 22 October 2012];Voluntary exploratory data submissions (vXDS) < http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083673.htm>.
- 63.Stanek EJ, et al. Adoption of pharmacogenomic testing by US physicians: results of a nationwide survey. Clin. Pharmacol. Ther. 2012;91:450–458. doi: 10.1038/clpt.2011.306. [DOI] [PubMed] [Google Scholar]
- 64.Topol EJ. Pharmacy benefit managers, pharmacies, and pharmacogenomic testing: prescription for progress? Sci. Transl. Med. 2010;2:44cm22. doi: 10.1126/scitranslmed.3001067. [DOI] [PubMed] [Google Scholar]
- 65.Takeuchi F, et al. A genome-wide association study confirms vKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009;5:e1000433. doi: 10.1371/journal.pgen.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cooper GM, et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008;112:1022–1027. doi: 10.1182/blood-2008-01-134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turner ST, et al. Genomic association analysis suggests chromosome 12 locus influencing antihypertensive response to thiazide diuretic. Hypertension. 2008;52:359–365. doi: 10.1161/HYPERTENSIONAHA.107.104273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Turner ST, et al. Genomic association analysis identifies multiple loci influencing antihypertensive response to an angiotensin II receptor blocker. Hypertension. 2012;59:1204–1211. doi: 10.1161/HYP.0b013e31825b30f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paré G, et al. [accessed October 22, 2012];RELY-Genetics: Genetic determinants of dabigatran plasma levels and their relation to clinical response at the European Society of Cardiology 2012 Congress, Munich. 2012 Aug 25-29; < http://spo.escardio.org/SessionDetails.aspx?id=398932#.UIwYgGl25GE>.
- 70.Kindmark A, et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008;8:186–195. doi: 10.1038/sj.tpj.6500458. [DOI] [PubMed] [Google Scholar]
- 71.McCormack M, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 2011;364:1134–1143. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ozeki T, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet. 2011;20:1034–1041. doi: 10.1093/hmg/ddq537. [DOI] [PubMed] [Google Scholar]
- 73.Fellay J, et al. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature. 2010;464:405–408. doi: 10.1038/nature08825. [DOI] [PubMed] [Google Scholar]
- 74.Trevino LR, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J. Clin. Oncol. 2009;27:5972–5978. doi: 10.1200/JCO.2008.20.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Byun E, et al. Genome-wide pharmacogenomic analysis of the response to inter-feron beta therapy in multiple sclerosis. Arch. Neurol. 2008;65:337–344. doi: 10.1001/archneurol.2008.47. [DOI] [PubMed] [Google Scholar]
- 76.Liu C, et al. Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Mol. Med. 2008;14:575–581. doi: 10.2119/2008-00056.Liu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mick E, Neale B, Middleton FA, McGough JJ, Faraone S.v. Genome-wide association study of response to methylphenidate in 187 children with attention-deficit/hyperactivity disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2008;147B:1412–1418. doi: 10.1002/ajmg.b.30865. [DOI] [PubMed] [Google Scholar]
- 78.Lavedan C, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol. Psychiatry. 2009;14:804–819. doi: 10.1038/mp.2008.56. [DOI] [PubMed] [Google Scholar]
- 79.Ising M, et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch. Gen. Psychiatry. 2009;66:966–975. doi: 10.1001/archgenpsychiatry.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Garriock HA, et al. A genomewide association study of citalopram response in major depressive disorder. Biol. Psychiatry. 2010;67:133–138. doi: 10.1016/j.biopsych.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Vollenhoven RF. Switching between anti-tumour necrosis factors: trying to get a handle on a complex issue. Ann. Rheum. Dis. 2007;66:849–851. doi: 10.1136/ard.2007.069872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bonafede MM, Gandra SR, Watson C, Princic N, Fox KM. Cost per treated patient for etanercept, adalimumab, and infliximab across adult indications: a claims analysis. Adv. Ther. 2012;29:234–248. doi: 10.1007/s12325-012-0007-y. [DOI] [PubMed] [Google Scholar]
- 83.Krintel SB, et al. Investigation of single nucleotide polymorphisms and biological pathways associated with response to TNFalpha inhibitors in patients with rheumatoid arthritis. Pharmacogenet. Genomics. 2012;22:577–589. doi: 10.1097/FPC.0b013e3283544043. [DOI] [PubMed] [Google Scholar]