

Abstract
Profiling phylogenetic marker genes, such as the 16S rRNA gene, is a key tool for studies of microbial communities but does not provide direct evidence of a community’s functional capabilities. Here we describe PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States), a computational approach to predict the functional composition of a metagenome using marker gene data and a database of reference genomes. PICRUSt uses an extended ancestral-state reconstruction algorithm to predict which gene families are present and then combines gene families to estimate the composite metagenome. Using 16S information, PICRUSt recaptures key findings from the Human Microbiome Project and accurately predicts the abundance of gene families in host-associated and environmental communities, with quantifiable uncertainty. Our results demonstrate that phylogeny and function are sufficiently linked that this ‘predictive metagenomic’ approach should provide useful insights into the thousands of uncultivated microbial communities for which only marker gene surveys are currently available.
Introduction
High-throughput sequencing has facilitated major advances in our understanding of microbial ecology and is now widespread in biotechnological applications from personalized medicine1 to bioenergy2. Markers such as the 16S ribosomal RNA gene (16S) of bacteria and archaea are frequently used to characterize the taxonomic composition and phylogenetic diversity of environmental samples. Because marker gene studies focus on one or a few universal genes, they cannot directly identify metabolic or other functional capabilities of the microorganisms under study. Conversely, metagenomic sequencing aims to sample all genes from a community and can produce detailed metabolic and functional profiles. Although relatively little sequencing is needed to characterize the diversity of a sample3, 4, deep, and therefore costly, metagenomic sequencing is required to access rare organisms and genes5. Thus, marker gene profiling of large sample collections is now routine, but deep metagenomic sequencing across many samples is prohibitively expensive.
Although marker gene and shotgun sequencing strategies differ in the type of information produced, phylogeny and biomolecular function are strongly, if imperfectly, correlated. Phylogenetic trees based on 16S closely resemble clusters obtained based on shared gene content6-9, and researchers often infer properties of uncultured organisms from cultured relatives. For example, the genome of a Bacteroides spp. might reasonably be inferred to contain many genes encoding glycoside hydrolase activity, based on the commonality of these activities in sequenced Bacteroides isolates10. This association is in turn closely related to the pan- and core-genomes of each phylogenetic subtree11, in that larger and more strongly conserved core genomes result in more confident linkages of genes with clades. Conversely, a clade’s core genome consists of genes its genomes can be expected to carry with high probability. The correlation between phylogeny and functional attributes depends on factors including the complexity of the trait12, but the overall degree of correlation suggests that it may be fruitful to predict the functions encoded in an organism’s genome on the basis of functions encoded in closely related genomes.
Recently, some 16S studies have extended these intuitions to infer the functional contribution of particular community members by mapping a subset of abundant 16S sequences to their nearest sequenced reference genome13-15. The accuracy of such approaches has not been characterized, but the correlation between gene content and phylogeny8, 9, 16 (excepting special cases such as laterally transferred elements and intracellular endosymbionts with reduced genomes) suggests that it may be possible to approximately predict the functional potential of microbial communities from phylogeny. Widespread and reproducible application of such a strategy requires an automated method that formalizes the relationship between evolutionary distance and functional potential across the entire metagenome, accounts for variation in marker gene copy number17, and accurately recaptures insights from shotgun metagenomic sequencing.
Here we describe PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States), a technique that uses evolutionary modeling to predict metagenomes from 16S data and a reference genome database. We investigated the accuracy of this approach as a function of the phylogenetic proximity of reference genomes to sampled environmental strains and the rate of decay of the phylogeny-function correlation owing to a variety of factors including gene duplication, loss, and lateral gene transfer. Lateral gene transfer is particularly relevant because it allows distantly related genomes to share functions that are missing from closer relatives and appears to be particularly widespread in microbes sharing a common environment, including constituents of the human microbiome18, 19 as well as extreme and contaminated environments20, 21 Quantitative predictions also depend on accurate modeling of community member abundance, which can be affected by 16S copy-number variation17 (Supplementary Results). Although these caveats could theoretically limit the accuracy of any inference of microbial function from 16S sequence data, their quantitative effects on this relationship have not previously been explored in detail.
Our results using published data show that PICRUSt recaptures key findings from the Human Microbiome Project and predicts metagenomes across a broad range of host-associated and environmental samples. We applied PICRUSt to a range of datasets from humans22, soils23, other mammalian guts24 and the hyper-diverse and underexplored Guerrero Negro microbial mat23, 24, which allowed us to model how the accuracy of PICRUSt varies based on the availability of reference genomes for organisms in each environment. In the best cases, correlations between inferred and metagenomically measured gene content approached 0.9 and averaged approximately 0.8. PICRUSt recaptured most of the variation in gene content obtained by metagenomic sequencing using only a few hundred 16S sequences and in some cases outperforms the metagenomes measured at particularly shallow sampling depths Additionally, we quantified the effects of several other factors on PICRUSt’s accuracy, including reference database coverage, phylogenetic error, gene functional category (a potential surrogate for the effects of lateral gene transfer), ancestral state reconstruction method, microbial taxonomy and 16S sequencing depth. Finally, we applied PICRUSt to several 16S-only datasets to identify previously undescribed patterns in gene content in oral, vaginal and coral mucus samples. Our implementation of these techniques, associated documentation and example datasets are made freely available via the PICRUSt software package at http://picrust.github.com.
Results
The PICRUSt algorithm
We developed PICRUSt to predict the functional composition of a microbial community’s metagenome from its 16S profile. This is a two-step process. In the initial ‘gene content inference’ step, gene content is precomputed for each organism in a reference phylogenetic tree. This reconstructs a table of predicted gene family abundances for each organism (tip) in the 16S-based phylogeny. Because this step is independent of any particular microbial community sample, it is pre-calculated only once. The subsequent ‘metagenome inference’ step combines the resulting gene content predictions for all microbial taxa with the relative abundance of 16S rRNA genes in one or more microbial community samples, corrected for expected 16S rRNA gene copy number, to generate the expected abundances of gene families in the entire community (Fig. 1).
Figure 1.
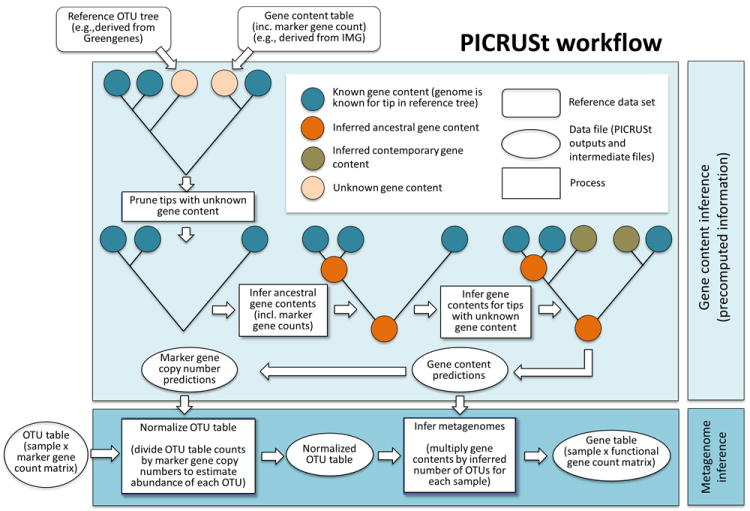
The PICRUSt workflow. PICRUSt is composed of two high-level workflows: gene content inference (top box) and metagenome inference (bottom box). Beginning with a reference OTU tree and a gene content table (i.e., counts of genes for reference OTUs with known gene content), the gene content inference workflow predicts gene content for each OTU with unknown gene content, including predictions of marker gene copy number. This information is precomputed for 16S based on Greengenes28 and IMG25, but all functionality is accessible in PICRUSt for use with other marker genes and reference genomes. The metagenome inference workflow takes an OTU table (i.e., counts of OTUs on a per sample basis), where OTU identifiers correspond to tips in the reference OTU tree, as well as the copy number of the marker gene in each OTU and the gene content of each OTU (as generated by the gene content inference workflow) and outputs a metagenome table (i.e. counts of gene families on a per-sample basis).
In the genome prediction step, PICRUSt predicts genes present in organisms that have not yet been sequenced based on the genes observed in their sequenced evolutionary relatives. To do this, PICRUSt uses existing annotations of gene content and 16S copy number from reference bacterial and archaeal genomes in the IMG database25. Any functional classification scheme can be used with PICRUSt; here, we demonstrate the use of the popular KEGG Orthology (KOs)26 and Clusters of Orthologs Groups (COGs)27 classification schemes. PICRUSt uses ancestral state reconstruction, along with a weighting method we developed for this work, to make predictions of gene content (with estimates of uncertainty)for all organisms represented in the Greengenes phylogenetic tree of 16S sequences28.
Prediction of a microbe’s gene content starts by inferring the content of the organism’s last common ancestor with one or more sequenced genomes. Inference of the genes in each ancestor (and uncertainty in that estimate) is handled by existing methods for ancestral state reconstruction (ASR). ASR algorithms infer the traits of ancestral organisms by fitting evolutionary models to the distribution of traits observed in living organisms using criteria such as maximum likelihood or Bayesian posterior probability. PICRUSt extends existing ASR methods to predict the traits of extant (in addition to ancestral) organisms. This allows the contents of the genomes of environmental strains to be inferred, with uncertainty in that inference quantified based on each gene family’s rate of change. This approach accounts both for gene families’ propensities for lateral transfer and for the degree to which each gene family is part of a ‘core’conserved within particular microbial clades. The gene contents of each reference genome and inferred ancestral genomes are then used to predict the gene contents of all microorganisms present in the reference phylogenetic tree. This initial genome prediction step is computationally intensive, but it is independent of any specific experiment and needs to be performed only once, allowing a single reference to be pre-computed ‘off-line’ and provided to users.
The metagenome inference step relies on a user-provided table of operational taxonomic units (OTUs) for each sample with associated Greengenes identifiers. Such tables are typically produced as one of the main data products in a 16S rRNA gene sequencing assay by analysis systems such as QIIME29. Because 16S rRNA copy number varies greatly among different bacteria and archaea, the user’s table of OTUs is normalized by dividing the abundance of each organism by its predicted 16S copy number. The 16S rRNA copy numbers for each organism are themselves inferred as a quantitative trait by ASR during the genome prediction step. Normalized OTU abundances are then multiplied by the set of gene family abundances pre-calculated for each taxon during the gene content inference step The final output from metagenome prediction is thus an annotated table of predicted gene family counts for each sample, where gene families can be orthologous groups or other identifiers such as KOs, COGs, or Pfams. The resulting tables are directly comparable to those generated by metagenome annotation pipelines such as HUMAnN30 or MG-RAST31. As with metagenome sequence data, the table of gene family counts can optionally be further summarized as pathway-level categories. However, in addition to estimating the aggregate metagenome for a community, PICRUSt also estimates the contribution of each OTU to a given gene function, which is not as easily obtained from shotgun metagenome sequencing32.
PICRUSt recapitulates Human Microbiome Project metagenomes
The value of PICRUSt depends on the accuracy of its predicted metagenomes from marker gene samples and the corresponding ability to recapitulate findings from metagenomic studies. The performance of PICRUSt was first evaluated using the set of 530 HMP samples that were analyzed using both 16S rRNA gene and shotgun metagenome sequencing22. Although a shotgun metagenome is itself only a subset of the underlying biological metagenome, accurate prediction of its composition constitutes a critical test for PICRUSt. Human-associated microbes have been the subject of intensive research for decades, and the HMP alone produced >700 draft and finished reference genomes, suggesting that the human microbiome would be a worthwhile benchmark for testing the accuracy of PICRUSt’s metagenome predictions. We tested the accuracy of PICRUSt by treating HMP metagenomic samples as a reference and calculating the correlation of PICRUSt predictions from paired 16S samples across 6,885 resulting KO groups.
PICRUSt predictions had high agreement with metagenome sample abundances across all body sites (Spearman r=0.82, p<0.001, Fig. 2a, Supplemental Fig. 1). Using two synthetic communities from the HMP constructed from a set of known microorganisms33, we used PICRUSt to make predictions that were even more accurate for both communities (Spearman r=0.9, p<0.001, Supplemental Fig. 2). As a targeted example, we also tested PICRUSt’s accuracy in specifically predicting the abundance of glycosaminoglycan (GAG) degradation functions, which are more abundant in the gut than elsewhere in the body30. Using the same differential enrichment analysis on both PICRUSt and metagenomic data yielded identical rankings across body sites and very similar quantitative results (Fig. 2b), suggesting that PICRUSt predictions can be used to infer biologically meaningful differences in functional abundance from 16S surveys even in the absence of comprehensive metagenomic sequencing.
Figure 2.

PICRUSt recapitulates biological findings from the Human Microbiome Project. A) PCA plot comparing KEGG Module predictions using 16S data with PICRUSt (lighter colored triangles) and sequenced shotgun metagenome (darker colored circles) along with relative abundances for five specific KEGG Modules, B) M00061: Uronic acid metabolism, C) M00076: Dermatan sulfate degradation, D) M00077: Chondroitin sulfate degradation, E) M00078: Heparan sulfate degradation, and F) M00079: Keratan sulfate degradation, all involved in glycosaminosglycan degradation (KEGG pathway ko00531) using 16S with PICRUSt (P, lighter colored) and WGS (W, darker colored) across human body sites: nasal (blue), gastrointestinal tract (brown), oral (green), skin (red), and vaginal (yellow).
Inferring host-associated and environmental metagenomes
Next, we then evaluated the prediction accuracy of PICRUSt in metagenomic samples from a broader range of habitats including mammalian guts24, soils from diverse geographic locations34 and a phylogenetically complex hypersaline mat community23, 24. These habitats represent more challenging validations than the human microbiome, as they have not generally been targeted for intensive reference genome sequencing. Because PICRUSt benefits from reference genomes that are phylogenetically similar to those represented in a community, this evaluation allowed us to quantify the impact of increasing dissimilarity between reference genomes and the metagenome.
To characterize this effect, we developed the Nearest Sequenced Taxon Index (NSTI) to quantify the availability of nearby genome representatives for each microbiome sample (Methods). NSTI is the sum of phylogenetic distances for each organism in the OTU table to its nearest sequenced reference genome, measured in terms of substitutions per site in the 16S rRNA gene and weighted by the frequency of that organism in the OTU table. As expected, NSTI values were greatest for the phylogenetically diverse hypersaline mat microbiome (mean NSTI=0.23 +/- 0.07 s.d.), least for the well-covered HMP samples (mean NSTI=0.03 +/- 0.02 s.d.), mid-range for the soils (mean NSTI=0.17 +/- 0.02 s.d.) and varied for the mammals (mean NSTI=0.14 +/- 0.06 s.d.) (Fig. 3). Also as expected, the accuracy of PICRUSt in general decreased with increasing NSTI across all samples (Spearman r=-0.4, p< 0.001) and within each microbiome type (Spearman r=-0.25 to -0.82, p<0.05). For a subset of mammal gut samples (NSTI<0.05) and all of the soil samples that we tested, PICRUSt produced accurate metagenome predictions (Spearman r=0.72 and 0.81, respectively, both p<0.001). It should be noted that both the mammal and hypersaline metagenomes were shallowly sequenced at a depth expected to be insufficient to fully sample the underlying community’s genomic composition, thus likely causing the accuracy of PICRUSt to appear artificially lower for these communities (see below). Although the lower accuracy on the hypersaline microbial mats community (Spearman r =0.25, p<0.001) confirms that PICRUSt must be applied with caution to the most novel and diverse communities, the ability to calculate NSTI values within PICRUSt from 16S data allows users to determine whether their samples are tractable for PICRUSt prediction prior to running an analysis. Moreover, the evaluation results verify that PICRUSt provides useful functional predictions for a broad range of environments beyond the well-studied human microbiome.
Figure 3.

PICRUSt accuracy across various environmental microbiomes. Prediction accuracy for paired 16S rRNA marker gene surveys and shotgun metagenomes (y-axis) are plotted against the availability of reference genomes as summarized by the Nearest Sequenced Taxon Index (NSTI; x-axis). Accuracy is summarized using the Spearman correlation between the relative abundance of gene copy number predicted from 16S data using PICRUSt versus the relative abundance observed in the sequenced shotgun metagenome. In the absence of large differences in metagenomic sequencing depth (see text), relatively well-characterized environments, such as the human gut, have low NSTI values and can be predicted accurately from 16S surveys. Conversely, environments containing much unexplored diversity (e.g. phyla with few or no sequenced genomes), such as the Guerrero Negro hypersaline microbial mats, tended to have high NSTI values.
PICRUSt outperforms shallow metagenomic sequencing
These validations showed that other factors in addition to NSTI also influence PICRUSt accuracy. Because sequenced metagenomes were used as a proxy for the true metagenome in our control experiments, metagenome sequencing depth was an additional contributing factor to the (apparent) accuracy of PICRUSt. This is because sequenced metagenomes themselves are incomplete surveys of total underlying functional diversity. Indeed, we found that metagenome sequencing depth for each sample correlated with PICRUSt accuracy (Spearman r=0.4, p<0.001), suggesting that samples with particularly low sequencing depth may be poor proxies for the community’s true metagenome and may lead to conservative estimates of PICRUSt accuracy (Supplemental Fig. 3). Similarly, we found a weak correlation between 16S rRNA gene sequencing depth and PICRUSt accuracy (Spearman r=0.2, p<0.001), also suggesting a statistically significant but numerically smaller impact on PICRUSt predictions (Supplemental Fig. 4). This is likely because proportionally more sequencing is needed to profile functional diversity than phylogenetic diversity.
To test the relationship between sequencing depth and accuracy, we used rarefaction analysis of the soil dataset to assess the effects of subsampling either the 16S rRNA genes (for PICRUSt predictions) or the shotgun metagenomic data (Fig. 4). We found that PICRUSt predictions converged rapidly with increasing sequencing depth and reached a maximum accuracy with only 105 16S sequences assigned to OTUs per sample (final Spearman r=0.82, p<0.001). This suggests that PICRUSt predictions could be performed on 16S data even from shallow sequencing (including many clone library/Sanger datasets) with little loss of accuracy. At this sequencing depth, subsamples from the full metagenome were very poor (though still significant) predictors of overall metagenome content (Spearman r=0.18, p < 0.001). Approximately 15,000 annotated metagenomic sequences per sample were required before being able to provide the same accuracy as PICRUSt with 105 assigned 16S reads. Accounting for the percent of genes surviving annotation (17.3% of metagenomic reads) or closed-reference OTU-picking (68.9% of post-QC 16S rRNA reads), this analysis indicates that PICRUSt may actually outperform metagenomic sequencing for read depths below ~72,000 total sequences per sample. Although most metagenomes exceed this threshold, it is worth noting that 16.7% (411/2,462) of bacterial and archaeal WGS samples in MG-RAST as of November 2012 are reported as containing fewer than 72,000 sequences. Our results clearly demonstrate the value of deep metagenomic sequencing, but also show that the number of sequences recovered per sample in a typical 16S survey (including those using Sanger sequencing) is more than sufficient to generate high-quality predictions from PICRUSt.
Figure 4.

Accuracy of PICRUSt prediction compared with shotgun metagenomic sequencing at shallow sequencing depths. Spearman correlation (y-axis) between either PICRUSt predicted metagenomes (blue lines) or shotgun metagenomes (dashed red lines) using 14 soil microbial communities subsampled to the specified number of annotated sequences (x-axis). This rarefaction reflects random subsets of either the full 16S OTU table (blue) or the corresponding gene table for the sequenced metagenome (red). Ten randomly chosen rarefactions were performed at each depth to indicate the expected correlation obtained when assessing an underlying true metagenome using either shallow 16S rRNA gene sequencing with PICRUSt prediction or shallow shotgun metagenomic sequencing. The data label describes the number of annotated reads below which PICRUSt-prediction accuracy exceeds metagenome sequencing accuracy. Note that the plotted rarefaction depth reflects the number of 16S or metagenomic sequences remaining after standard quality control, dereplication, and annotation (or OTU picking in the case of 16S sequences), not the raw number returned from the sequencing facility. The number of total metagenomic reads below which PICRUSt outperforms metagenomic sequencing (72,650) for this dataset was calculated by adjusting the crossover point in annotated reads (above) using annotation rates for the soil dataset (17.3%) and closed-reference OTU picking rates for the 16S rRNA dataset (68.9%). The inset figure illustrates rapid convergence of PICRUSt predictions given low numbers of annotated reads (blue line).
Functional and phylogenetic determinants of PICRUSt accuracy
We further tested and optimized the genome prediction step of PICRUSt using additional information from sequenced reference genomes (Supplemental Online Results, Supplemental Figs. 5-9). The prediction accuracy of PICRUSt was largely consistent across diverse taxa throughout the phylogenetic tree of archaea and bacteria (Fig. 5). Notably, PICRUSt predictions were as accurate for archaeal (mean=0.94 +/- 0.04 s.d., n=103) as for bacterial genomes (mean=0.95 +/- 0.05 s.d., n=2,487). Most of the variation seen across groups was due to differences in their representation by sequenced genomes. For example, of the 40 taxonomic families that had an associated accuracy less than 0.80, each of these families had at most six sequenced members, whereas the 53 families with a predicted accuracy greater than 0.95 had on average 30 sequenced representatives. This coincides with our findings that the accuracy of PICRUSt at both the genome and metagenome level depends on having closely sequenced relatives with accurate annotations.
Figure 5.

PICRUSt prediction accuracy across the tree of bacterial and archaeal genomes. Phylogenetic tree produced by pruning the Greengenes 16S reference tree down to those tips representing sequenced genomes. Height of the bars in the outermost circle indicates the accuracy of PICRUSt for each genome (accuracy: 0.5-1.0) colored by phylum, with text labels for each genus with at least 15 strains. PICRUSt predictions were as accurate for archaeal (mean=0.94 +/- 0.04 s.d., n=103) as bacterial genomes (mean=0.95 +/- 0.05 s.d., n=2487).
Analysis of PICRUSt predictions across functional groups (Fig. 6; Supplemental Fig. 10) revealed that, as a positive control, core or ‘housekeeping’ functions, such as genetic information processing, were best predicted (mean accuracy=0.99 +/- 0.03 s.d.). Conversely, gene families that are variable across genomes and more likely to be laterally transferred, such as those in environmental information processing, had slightly lower accuracy (mean accuracy=0.95 +/- 0.04 s.d.). The subcategories of this group predicted least well were membrane-associated and therefore expected to change rapidly in abundance in response to environmental conditions35. Such functional categories also typically show large differences in relative abundance between similar communities (e.g. metal cation efflux36 and nickel/peptide transporters19) and are enriched for lateral gene transfer21, 37. However, even these more challenging functional groups were well predicted by PICRUSt (min. accuracy=0.82), suggesting that our inference of gene abundance across various types of functions is reliable.
Figure 6.

Variation in inference accuracy across functional modules within single genomes. Results are colored by functional category, and sorted in decreasing order of accuracy within each category (indicated by triangular bars, right margin). Note that all accuracies were >0.80, and therefore the region 0.80-1.0 is displayed for clearer visualization of differences between modules.
Biological insights from the application of PICRUSt
As a final illustration of PICRUSt’s computational efficiency and ability to generate biological insights, we applied PICRUSt to three large 16S rRNA datasets. In the first example, all 6,431 16S samples from the HMP were analyzed to predict metagenomes using PICRUSt, requiring <10 minutes of runtime on a standard desktop computer. One of the many potential applications of such data is in functionally explaining shifts in microbial phylogenetic distributions between distinct habitats. Previous culture-based studies had detected higher frequencies of aerobic bacteria in the supragingival plaque relative to subgingival plaque38, and an analysis of HMP 16S rRNA sequences detected taxonomic differences between these two sites39. Analysis of the PICRUSt-predicted HMP metagenomes revealed an enrichment in the metabolic citrate cycle (M00009) genes in supragingival plaque samples in comparison to subgingival plaque (p<1e-10; Welch’s t-test with Bonferroni correction), supporting previous claims that aerobic respiration is more prevalent in the supragingival regions38.
In the second example, we applied PICRUSt to generate functional predictions for ecologically-critical microbial communities associated with reef-building corals. The system under study is subject to an experimental intervention simulating varying levels of eutrophication and overfishing40. One hypothesis to explain the role of algae in the global decline of coral populations posits that eutrophication favors algal growth, which in turn increases dissolved organic carbon (DOC) loads. DOC favors overgrowth of fast-growing opportunist microbes on the surface of coral, outcompeting more-typical commensal microbes, depleting O215 and ultimately causing coral disease or death. This is known as the dissolved organic carbon, disease, algae and microbes model41 (although direct algal toxicity through secreted allelochemicals also appears to play a role42). To shed light on this hypothesis using PICRUSt, we predicted metagenomes for 335 coral mucus samples collected in situ from corals in experimental plots with varying levels of algal cover (Supplemental Fig. 11). Consistent with algae-driven increases in opportunistic pathogen loads, genes in the secretion system were perfectly correlated with relative algal cover (Spearman r=1.0, p=0.0), with 46% enrichment in corals from high- vs. low- algal cover plots. Algal cover also produced significant variation in ribosomal biogenesis genes (ANOVA raw p=1.6 × 10-4; Bonferroni-corrected: 0.049; FDR q= 0.0047), indicating an effect on generally faster-growing organisms. This variation was strongly correlated with relative algal cover across plots and timepoints (Spearman r=0.90, p=0.037) and represented a 25% increase in this gene category between corals in plots with the highest versus lowest algal cover. Further evidence that supported a decrease in typical consumers of coral mucus carbohydrates in favor of fast-growing opportunists was provided by significant depletion of two categories of carbohydrate metabolism genes (Spearman r=-1.0; p=0.0 “Galactose metabolism”; Spearman r=-0.90, p=0.037 “Ascorbate and alderate metabolism”). As the weighted NSTI in this case was 0.12 (+/- 0.02 s.d.), these results suggest that PICRUSt may provide biologically actionable hypotheses even in challenging environments with fewer available reference genomes.
Finally, we assessed 993 samples from time courses covering ~16 weeks each from the vaginal microbiomes of 34 individual subjects43. These samples have been previously analyzed only in the context of longitudinal changes in microbial taxonomic composition over time; PICRUSt provided insights into what additional putative microbial pathway changes might explain or accompany this compositional variation. The first observation this enabled was a comparison of community beta-diversity within subjects over time, contrasting the degree of similarity of microbial composition over time with the similarity of the accompanying inferred metagenomes. In all cases, the mean Bray-Curtis diversity using KOs predicted by PICRUSt was more stable over time than when using OTU composition (Supplemental Fig. 12). To our knowledge, this provides the first longitudinal results mirroring the functional stability in metagenomes that has been observed cross-sectionally22, 44. Second, we identified seven KEGG modules that had significant differences in mean abundances in samples taken during menses (Supplemental Fig. 13). The KEGG module with the largest significant increase in mean proportion during menses was “M00240: Iron complex transport system”, suggesting a shift in the microbiome that might be explained by pathways that utilize the iron-rich environment provided during menstruation.
Discussion
The application of PICRUSt to diverse metagenomic data sets shows that the phylogenetic information contained in 16S marker gene sequences is sufficiently well correlated with genomic content to yield accurate predictions when related reference genomes are available. Our validation results support widespread application of PICRUSt to 16S datasets containing as few as a few hundred sequences per sample, provided that NSTI or a similar measure is used to quantify the expected prediction accuracy. Although PICRUSt’s predictive approach neither precludes nor outperforms deep metagenomic sequencing, it can predict and compare probable functions across many samples from a wide range of habitats at a small fraction of the cost of such sequencing. This approach thus opens up new avenues for tiered, more cost-effective study designs and provides functional insights into the tens of thousands of existing samples for which only 16S data is available.
To best leverage the strengths both of (meta)genomic sequencing and of PICRUSt, we recommend its incorporation into marker gene studies using a deliberate, tiered approach. Because phylogenetic dissimilarity among environmental organisms and sequenced genomes (as captured by NSTI) affects PICRUSt accuracy, NSTI values can be calculated from preliminary 16S rRNA data to assess whether reference genome coverage is sufficiently dense to allow for accurate PICRUSt prediction. If adequate reference genomes are not available, additional genome sequences can be collected to fill in phylogenetic “gaps” in the reference database and allow for accurate prediction. This can be performed either through traditional culture-based techniques, single-cell genomic approaches or deep metagenomic sequencing of samples targeted based on 16S data. If NSTI appears sufficient but additional controls are desired, a preliminary set of paired 16S rRNA and shotgun metagenomic samples can be compared using PICRUSt’s built-in tools to empirically test prediction accuracy on the sample types of interest. On the basis of such validations from select samples, PICRUSt can then be used to extend approximate functional information from a few costly metagenomes to much larger accompanying 16S rRNA gene sequence collections.
However, the limitations of this approach must be considered in interpreting PICRUSt predictions. For example, only 16S marker gene sequences corresponding to bacterial and archaeal genomes are currently included; thus this version of the system does not infer viral or eukaryotic components of a metagenome. PICRUSt’s ability to detect patterns also depends on the input data used: the software cannot distinguish variation at the strain level if the marker gene sequence used is identical among strains, and it cannot detect genes families (or summarize them into pathways) if those genes are not included in the input genomic data used, or if pathway annotations are currently poor (e.g. for acetogenesis genes). However, because PICRUSt can accept trees produced by alternative marker genes or gene/pathway annotations, users have the flexibility to customize the tool to meet the needs of their system. Although high overall accuracy was obtained despite microbial lateral gene transfer and other processes of gene gain and loss, gene families or pathways (e.g. methane oxidation) with highly variable distribution throughout the tree of life can still lead to incorrect predictions in individual cases. PICRUSt thus provides confidence intervals for each functional abundance prediction that reflect the degree of variation in that function among sequenced phylogenetic neighbors of predicted (meta)genomes, with wide confidence intervals indicating a high degree of uncertainty (Supplemental Fig. 7). If individual gene abundances (rather than aggregate patterns) are of interest, users can choose to either discard predictions with low confidence, or confirm them experimentally.
We anticipate several experimental and computational improvements that will further refine the predictive accuracy of PICRUSt. In addition to extending genome coverage and metagenome calibration as above, PICRUSt predictions could also likely be improved by including habitat information in a predictive model. This may provide additional predictive power in that some genes might correlate strongly with environmental parameters as well as phylogenetic similarity to reference organisms9, 16. Modification of prediction methods that incorporate information from partial genome sequences could expand the sensitivity of predictions in under-studied environments by including additional reference gene content information. Finally, as reference genome sequence databases continue to expand and incorporate isolates from ever more diverse environments, the prediction accuracy of PICRUSt will improve by default over time. Predictive metagenomics thus holds the promise of uniting completed genome sequences, 16S rRNA gene studies and shotgun metagenomes into a single quantitative approach for assessing community function.
Methods
Reference genomes and 16S data used by PICRUSt
PICRUSt requires a phylogenetic tree of marker genes that includes both tips with known data (e.g. complete reference genomes) and unknown tips (e.g. environmental sequences). Although any type of marker gene tree could be used with PICRUSt, the 16S ‘tax2tree’ version of Greengenes45 was downloaded and used for all presented research. Similarly, PICRUSt can make inferences about any type of continuous trait, but for this research we used the popular KEGG26 and COG27 databases for annotations. Specifically, we obtained all KEGG Ortholog (KO) and COG annotations from v3.5 of IMG25 to produce a table of 6,885 KO and 4715 COG abundances for 2,590 genomes that had identifiers in the Greengenes reference tree. The number of copies of the 16S gene in each of these genomes was also obtained from IMG.
The PICRUSt algorithm
PICRUSt begins by formatting the marker phylogenetic tree and functional annotation file in preparation for ancestral state reconstruction. This includes creation of internal node labels in the tree, matching tree tips with reference genomes to the annotation file and creating a pruned version of the tree that contains only tips with corresponding reference genomes. An ancestral state reconstruction method is then applied to the pruned tree. This provides predicted values for each of the KOs (and the additional 16S copy number trait) for all internal nodes in the pruned tree. Four different ancestral state reconstruction methods were tested including Wagner Parsimony from the COUNT package (v11.0502)46 and ACE ML, ACE REML and ACE PIC of the APE R library (v2.8)47. The next step makes predictions for all tips in the reference tree that do not have corresponding genomes using the inferences for the internal nodes from the ancestral state reconstruction step. A prediction of gene content is generated using an average of the contents of extant and inferred ancestral genomes, weighted exponentially by the reciprocal of phylogenetic distance. This causes very closely related existing or ancestral genomes to be counted much more heavily than more distant relatives, and it is also consistent with previous research suggesting an exponential relationship between 16S phylogenetic distance and gene content conservation9. (Confidence intervals on this prediction are also optionally calculated when using any of the ACE methods (Supplemental Fig. 7).) It is important to note that the prediction of gene content for tips in the trees without reference genomes is an estimate only, and that although our method does model gene gain and loss, some instances of gain or loss or laterally transferred genes will be poorly predicted (with broad confidence intervals as a result). This is rare in practice, however, as validated at the genome and metagenome level by comparing our predictions with the known gene contents from actual sequencing (see below). This genome prediction step only needs to be precomputed once, resulting in a pre-calculated file that is provided with the PICRUSt package containing predicted genome contents for all tips in the marker reference tree.
For metagenome prediction, PICRUSt takes an input OTU table that contains identifiers that match tips from the marker gene (e.g. greengenes identifiers) with corresponding abundances for each of those OTUs across one or more samples. First, PICRUSt normalizes the OTU table by the 16S copy number predictions so that OTU abundances more accurately reflect the true abundances of the underlying organisms. The metagenome is then predicted by looking up the pre-calculated genome content for each OTU, multiplying the normalized OTU abundance by each KO abundance in the genome and summing these KO abundances together per sample. The prediction yields a table of KO abundances for each metagenome sample in the OTU table. For optional organism-specific predictions, the per-organism abundances are retained and annotated for each KO.
Paired 16S and metagenome validations and metagenome predictions from 16S data
Several microbiome studies that included both 16S sequencing and WGS metagenome sequencing for the same samples were used to test the accuracy of PICRUSt. These included 530 paired human microbiome samples22, 39 paired mammal gut samples24, 14 paired soil samples34, 10 paired hypersaline microbial mats23, 24 and two even/staggered synthetic mock communities from the HMP33. We additionally used PICRUSt to make predictions on three 16S-only microbiome studies, specifically 6,431 HMP samples (http://hmpdacc.org/HMQCP), 993 vaginal time course samples43 and 335 coral mucus samples(http://www.microbio.me/qiime/; Study ID 1854).
For 16S data, PICRUSt-compatible OTU tables were constructed using the closed-reference OTU picking protocol in QIIME 1.5.0-dev (pick_reference_otus_through_otu_table.py) against Greengenes+IMG using ‘uclust’48. For paired metagenomes, WGS reads were annotated to KOs using v0.98 of HUMAnN30. Expected KO counts for the HMP mock communities were obtained by multiplying the mixing proportions of community members by the annotated KO counts of their respective reference genomes in IMG. PICRUSt was used to predict the metagenomes using the 16S-based OTU tables, and predictions were compared to the annotated WGS metagenome across all KOs using Spearman rank correlation. In addition, KOs were mapped to KEGG Module abundances, following the conjugative normal form as implemented in HUMAnN script “pathab.py” for the HMP and vaginal datasets to compare modules and pathways. Bray-Curtis distances (for Beta-diversity comparison between OTU or PICRUSt KO abundances across samples) were calculated using as implemented in the QIIME “beta_diversity.py” script. The PCA plot and identification of KEGG modules with significant mean proportion differences for both the HMP and vaginal datasets was created using STAMP v2.036.
The Nearest Sequenced Taxon Index (NSTI) was developed as an evaluation measure describing the novelty of organisms within an OTU table with respect to previously sequenced genomes. For every OTU in a sample, the sum of branch lengths between that OTU in the Greengenes tree to the nearest tip in the tree with a sequenced genome is weighted by the relative abundance of that OTU. All OTU scores are then summed to give a single NSTI value per microbial community sample. PICRUSt calculates NSTI values for every sample in the given OTU table, and we compared NSTI scores and PICRUSt accuracies for all of the metagenome validation datasets.
In the metagenome rarefaction analysis (Fig. 4), a given number of counts were randomly selected from either the collection of microbial OTUs for each sample (i.e. the 16S rRNA OTU table) or the collection of sequenced genes in that sample using the multiple_rarefactions.py script in QIIME 1.5.0-dev29. To estimate the number of raw reads at which PICRUSt outperforms metagenomic sequencing the annotated shotgun reads were transformed to total sequenced reads by dividing by the mean annotation rates from the original manuscript (17.3%), while 16S rRNA reads were transformed using the success rate for closed-reference OTU picking at a 97% 16S rRNA identity threshold (68.9%). Both the subsampled metagenome and the PICRUSt predictions from the subsampled OTU table were compared for accuracy using Spearman rank correlation versus the non-subsampled metagenome.
Single-genome, phylogenetic and pathway-specific validations
The accuracy of metagenomic prediction depends on accurate prediction of the gene families (e.g. KOs) present in unsequenced organisms. The accuracy of this gene content prediction step was assessed by using fully sequenced genomes (in which gene content is known) as controls. A test dataset was generated for each sequenced genome in IMG in which that genome was excluded from the reference gene by genome table. PICRUSt was then used to infer the content of the excluded genome. Subsequently, this predicted gene content was compared against the actual gene content, i.e. the sequenced genome annotations. The results were compared using Spearman rank correlation for the actual versus estimated number of gene copies in each gene family or using accuracy and/or balanced accuracy for presence/absence evaluations. These results are presented as the ‘genome holdout’ dataset. In addition to using this dataset to calculate the accuracy of each genome, it was also used to calculate the accuracy per functional gene category. This was done by first mapping KOs to KEGG Modules (described above) for each genome (for both real and PICRUSt predictions) and then comparing each module across all genomes. For visualization, the accuracy of each module was mapped into more general functional categories using the BRITE hierarchy26.
The accuracy of PICRUSt across different taxonomic groups in the phylogenetic tree of bacteria and archaea was visualized using GraPhlAn v0.9 (http://huttenhower.sph.harvard.edu/graphlan). The phylogenetic tree for display was constructed by pruning the Greengenes tree down to tips with corresponding genomes as above, with taxonomic labels at the phylum and genus level obtained for each genome from NCBI Taxonomy49.
We expected that the accuracy of PICRUSt’s predictions would decrease when large phylogenetic distances separated the organism of interest and the nearest sequenced reference genome(s). To test this expectation, ‘distance holdout’ datasets were constructed. These datasets were constructed in the same manner as ‘genome holdout’ datasets described above, except that all genomes within a particular phylogenetic distance (on the 16S tree) of the test organism were excluded from the reference dataset. For example, when predicting Escherichia coli MG1655, a distance holdout of 0.03 substitutions/site would exclude not only that genome, but also all other E. coli strains. These tests were conducted at phylogenetic distances ranging from 0.0 to 0.50 substitutions/site in the full-length 16S rRNA gene, in increments of 0.03 substitutions/site.
Finally, we tested the effects of local inaccuracy in tree construction on PICRUSt’s performance. These ‘tree randomization holdouts’ were constructed the same as the ‘genome holdout’ dataset (above), except that in addition to excluding one genome, the labels of all organisms within a specified phylogenetic distance of the test organism were randomized on the 16S tree. For example, our ‘tree randomization holdout’ targeting E.coli with a distance of 0.03 scrambled the phylogeny of all reference E.coli strains around the tip to be predicted, while leaving the rest of the tree intact. These tests were conducted at phylogenetic distances ranging from 0.0 to 0.50 substitutions/site in the 16S rRNA gene, in increments of 0.03 substitutions/site.
Supplementary Material
Acknowledgments
We would like to thank Adam Robbins-Pianka and Nicola Segata, along with all members of the Knight, Beiko, Vega Thurber, Caporaso and Huttenhower labs, for their assistance during PICRUSt conception and development. This work was supported in part by the Canadian Institutes of Health Research (MGIL,RGB), the Canada Research Chairs program (RGB), NSF OCE #1130786 (RVT,DB), the Howard Hughes Medical Institute (RK), NIH P01DK078669, U01HG004866, R01HG004872 (RK), the Crohn’s and Colitis Foundation of America (RK), the Sloan Foundation (RK), NIH 1R01HG005969 (CH), NSF CAREER DBI-1053486 (CH), and ARO W911NF-11-1-0473 (CH).
References
- Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nature reviews Genetics. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen G, et al. An insect herbivore microbiome with high plant biomass-degrading capacity. PLoS genetics. 2010;6 doi: 10.1371/journal.pgen.1001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J, et al. Direct sequencing of the human microbiome readily reveals community differences. Genome biology. 2010;11:210. doi: 10.1186/gb-2010-11-5-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Beiko RG. Measures of phylogenetic differentiation provide robust and complementary insights into microbial communities. The ISME journal. 2013;7:173–183. doi: 10.1038/ismej.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R, et al. Unlocking the potential of metagenomics through replicated experimental design. Nature biotechnology. 2012;30:513–520. doi: 10.1038/nbt.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Huttenhower C. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS One. 2011;6:e24704. doi: 10.1371/journal.pone.0024704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nature genetics. 1999;21:108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaneveld JR, Lozupone C, Gordon JI, Knight R. Ribosomal RNA diversity predicts genome diversity in gut bacteria and their relatives. Nucleic acids research. 2010;38:3869–3879. doi: 10.1093/nar/gkq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RE, Higgs PG. Testing the infinitely many genes model for the evolution of the bacterial core genome and pangenome. Molecular biology and evolution. 2012;29:3413–3425. doi: 10.1093/molbev/mss163. [DOI] [PubMed] [Google Scholar]
- Martiny AC, Treseder K, Pusch G. Phylogenetic conservatism of functional traits in microorganisms. The ISME journal. 2013;7:830–838. doi: 10.1038/ismej.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome biology. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge BD, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barott KL, et al. Microbial to reef scale interactions between the reef-building coral Montastraea annularis and benthic algae. Proceedings Biological sciences / The Royal Society. 2012;279:1655–1664. doi: 10.1098/rspb.2011.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffron S, Rehrauer H, Pernthaler J, von Mering C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010;20:947–959. doi: 10.1101/gr.104521.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kembel SW, Wu M, Eisen JA, Green JL. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS computational biology. 2012;8:e1002743. doi: 10.1371/journal.pcbi.1002743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, et al. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- Meehan CJ, Beiko RG. Lateral gene transfer of an ABC transporter complex between major constituents of the human gut microbiome. BMC microbiology. 2012;12:248. doi: 10.1186/1471-2180-12-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher Y, et al. Lateral gene transfer and the origins of prokaryotic groups. Annual review of genetics. 2003;37:283–328. doi: 10.1146/annurev.genet.37.050503.084247. [DOI] [PubMed] [Google Scholar]
- Hemme CL, et al. Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. The ISME journal. 2010;4:660–672. doi: 10.1038/ismej.2009.154. [DOI] [PubMed] [Google Scholar]
- Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk Harris J, et al. Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. The ISME journal. 2013;7:50–60. doi: 10.1038/ismej.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, et al. Millimeter-scale genetic gradients and community-level molecular convergence in a hypersaline microbial mat. Molecular systems biology. 2008;4:198. doi: 10.1038/msb.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic acids research. 2012;40:D115–122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abubucker S, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy AC, Rigoutsos I. What’s in the mix: phylogenetic classification of metagenome sequence samples. Curr Opin Microbiol. 2007;10:499–503. doi: 10.1016/j.mib.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Haas BJ, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PV, et al. Analysis of membrane proteins in metagenomics: networks of correlated environmental features and protein families. Genome research. 2010;20:960–971. doi: 10.1101/gr.102814.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010;26:715–721. doi: 10.1093/bioinformatics/btq041. [DOI] [PubMed] [Google Scholar]
- Zuniga M, et al. Horizontal gene transfer in the molecular evolution of mannose PTS transporters. Molecular biology and evolution. 2005;22:1673–1685. doi: 10.1093/molbev/msi163. [DOI] [PubMed] [Google Scholar]
- Daniluk T, et al. Aerobic and anaerobic bacteria in subgingival and supragingival plaques of adult patients with periodontal disease. Advances in medical sciences. 2006;51(Suppl 1):81–85. [PubMed] [Google Scholar]
- Segata N, et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome biology. 2012;13:R42. doi: 10.1186/gb-2012-13-6-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton N, Jackson JB. Shifting baselines, local impacts, and global change on coral reefs. PLoS biology. 2008;6:e54. doi: 10.1371/journal.pbio.0060054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, et al. Indirect effects of algae on coral: algae-mediated, microbe-induced coral mortality. Ecology letters. 2006;9:835–845. doi: 10.1111/j.1461-0248.2006.00937.x. [DOI] [PubMed] [Google Scholar]
- Rasher DB, Stout EP, Engel S, Kubanek J, Hay ME. Macroalgal terpenes function as allelopathic agents against reef corals. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17726–17731. doi: 10.1073/pnas.1108628108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajer P, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132ra152. doi: 10.1126/scitranslmed.3003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME journal. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csuros M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–1912. doi: 10.1093/bioinformatics/btq315. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Federhen S. The NCBI Taxonomy database. Nucleic acids research. 2012;40:D136–143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.