

Introduction
The last century represents an exciting era for the chemical evolution of transition-metal catalyzed reactions,1–3 and among these, the chemistry of palladium,4–6 ruthenium,7–10 and rhodium11,12 etc. has attracted considerable attention in the organic community. For example, the palladium-catalyzed carbon-carbon (C-C) and carbon-heteroatom (C-X) bond formations have been widely used in the synthesis of complex natural products,13 bioactive molecules,14 and organic materials.15–17 In contrast, the use of copper metal seemed to lag behind the trend of development in this area, though the copper-catalyzed C-C, C-O, and C-N bond formations belong to one of the oldest reactions, namely the Ullmann reaction.
The pioneering works of Ullmann and Goldberg form the basis of modern copper-mediated chemistry (Table 1). As early as 1901, Ullmann reported the first copper-mediated coupling reaction,18 in which two aryl iodides were coupled to form the biaryl product by consuming one equivalent of Cu. Later, both Ullmann and Goldberg found that copper could be used in the formation of aryl C-N19,20 and C-O21 bonds. In 1929, Hurtley22 found that diketones and malonates could be coupled with o-bromobenzoic acid in the presence of catalytic copper-bronze or copper acetate and sodium, as the base. But the generally harsh conditions (>200°C), high copper catalyst loading, and poor functional group tolerance had limited the applications of Ullmann and Ullmann-type reactions, since they were discovered.
Table 1.
Ullmann, Goldberg, and Hurtley Reactions
With increasing understanding of the mechanisms of transition-metal mediated reactions and the development of novel ligands, significant advances have been achieved in copper-catalyzed coupling reactions of aryl halides with O-, N- and S-nucleophiles over the past decade. The application of efficient ligands, such as 1-naphthoic acid,23 8-hydroxyquinoline,24 2,2,6,6-tetramethylheptane-3,5-dione,25 1,10-phenanthroline,26 amino acids,27 diimine ligands,28 and β-ketoester,29 among others, allowed the reaction to be conducted under mild conditions with desirable yields and excellent functional group tolerance.
Several reviews concerning the Ullmann and Ullmann-type reactions have been reported due to the rapid development of copper-catalyzed coupling reactions. For example, Beletskaya and Cheprakov30 gave a good summary of copper-catalyzed formation of different bonds (C-C, C-O, C-N, C-S, C-P, etc.). Other reviews include applications of the Ullmann reaction in the synthesis of bioactive natural products,31 anti-cancer agents,32 and alkaloids.33 Ma and coworkers reviewed the developments and applications of amino acid-based ligands in copper-catalyzed coupling reactions.27, 34 Comparison of palladium- and copper-catalyzed coupling reactions was described by Senra35 and Koenig.36 The development of copper-catalyzed formations of C-N, C-O, and C-C bonds, since 2004, was highlighted by Monnier and Taillefer.37,38 The review for copper-promoted C-N and C-O formations, which covered the references from 2000 to 2008, has been discussed by Das et al.39 The copper-catalyzed reactions using water as the solvent, were reviewed by Marinelli.40
The present review includes two main sections. The first focuses on the recent development of synthetic methodologies related to new or advanced catalytic systems and other “green” technologies, including ligand-free systems, heterogeneous catalysts, and microwave and ultrasound-assisted reactions. The second part emphasizes recent applications of Ullmann reactions in the synthesis of heterocycles, drug-like molecules, and natural products. Due to the rapid development and wide application of Ullmann reaction in organic synthesis, medicinal chemistry, and natural products, this review will primarily focus on papers recently published since 2008.
I. Recent Developments of Ullmann-type (C-O, C-N, C-S) Reactions
The formation of C-O, C-N, and C-S bonds is one of the most attractive methods for organic chemists. The successful Pd-mediated C-X bond formation provided a powerful tool to access a wide range of pharmaceutical molecules and bioactive natural products. However, costly palladium reagents and general air-sensitive system limited its applications in organic synthesis, process chemistry, and industrial manufacturing. These disadvantages required exploration of new metal-catalyzed reagents and systems to conduct these reactions. The inexpensive metal copper attracted increasing attention after the pioneering work of Buchwald and his group23, 41 in this area. The combination of copper salts and efficient ligands significantly improved the reaction yields and compatibilities, probably due to the increased solubility and decreased aggregation of copper salts in the presence of the ligands. These ligands could be mainly classified as O,O-, N,N-, and N,O-ligands according to their corresponding chemical structures (Figure 1).
Figure 1.

O,O-, N,N-, and N,O-Ligands
Several copper complexes used in Ullmann coupling reactions are shown in Figure 2.
Figure 2.

Copper complexes
In addition, with the development of sustainable, environmentally benign, and cost-effective chemistry, “green” concept and techniques in organic synthesis have become widely accepted and applied in recent years.42,43 Recent and representative green synthetic methodologies, such as ligand-free systems, reusable catalysts, and microwave or ultrasound irradiation, will also be highlighted in this review.
1. New Development of Ligands and Copper Complexes
a. C-O Bond Formation
In 2009, Yang et al.44 reported an effective glyoxal bis(phenylhydrazone) ligand (L15) for the catalytic Ullmann O-arylation of various substituted phenols and aryl bromides under mild reaction conditions (Table 2, Entry 1).
Table 2.
Development of the Ullmann O-Arylation
| Entry | Copper source | Ligand | Substrates | Conditions | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|
| Ar-X | Nucleophile | ||||||
| 1 | CuI (10 mol%) | L15 | Aryl-Br | Phenols | K3PO3, CH3CN 60°C, 12 h | 60–92 | 44 |
| 2 | CuBr (10 mol%) | L23 | (Hetero)Aryl-I | Phenols | Cs2CO3, DMSO 90°C | 95–97 | 45 |
| (Hetero)Aryl-Br | 50–98 | ||||||
| (Hetero)Aryl-Cl | Cs2CO3, DMSO 120°C | 19–97 | |||||
| 3 | CuI (5–10mol%) | L25 | (Hetero)Aryl-I | Phenols | K3PO4, DMSO 80–90°C | 68–96 | 49 |
| (Hetero)Aryl-Br | |||||||
| 4 | CuI (1 mol%) | L24 | Aryl-I | Phenols | Cs2CO3, CH3CN 80°C, 24 h | 32–99 | 50 |
| 5 | CuI (5 mol%) | L11 | Aryl-I, Br | Aliphatic alcohol | Cs2CO3, toluene 80°C, 12 h | 59–99 | 51 |
| 6 | Cu(OTf)2 (20 mol%) | L16 | Aryl-I, Br | Phenols | Cs2CO3, dioxane 110°C | 60–90 | 52 |
| 7 | CuI (5 mol%) | L18 | Aryl-I, Br | Phenols | K3PO4, DMF 110°C | 15–91 | 53 |
| 8 | CuI (10 mol%) | L25 | Aryl-I, Br | 3-Aminophenols | K3PO4, DMSO 80–90°C | 54–91 | 54 |
| L17 | Aryl-I | 4-Aminophenols | K2CO3, butyronitrile 70°C | 38–67 | |||
| 9 | CuI (10 mol%) | L10 or L29 | Aryl-I | Phenols | KF/CP, DMSO 110°C | 63–95 | 55 |
| 10 | C1 (1–5 mol%) | Aryl-I, Br | Phenols | K3PO4, DMF 110°C | 42–92 | 56 | |
| Aliphatic alcohol | K3PO4, solvent-free 110°C | 70–92 | |||||
| 11 | CuI (10 mol%) | L1 | Aryl-I | CsOH | DMSO/H2O, 130°C | 70–95 | 57 |
| L14 | Aryl-Br | 1,4-dioxane/H2O, NaI, 130°C | 70–90 | ||||
| 12 | Cu2O (5 mol%) | L28 | Aryl-I, Br, Cl | CsOH | n-Bu4N+Br− H2O, 100°C | 45–96 | 58 |
(2-Pyridyl)acetone45 (L23), as a new supporting ligand, was used in the copper-catalyzed Ullmann O-arylation of diverse (hetero)aryl halides (X = I, Br, Cl) with different phenols (Table 2, Entry 2). Until then, there were only a few examples46–48 about the successful copper-catalyzed coupling of chlorobenzene with phenols. The electron-deficient aromatic chlorides gave diaryl ethers in moderate to excellent yields at high temperature (120°C); however, acceptable yields were obtained only with electron-rich aryl chlorides in the presence of excess substrates.
In 2010, Buchwald and Maiti49 described a simple and effective protocol for the Ullmann coupling between sterically hindered phenols and aryl halides (X = I; Br). Inexpensive picolinic acid (L25) was employed as the ligand in this method, which tolerated a variety of functional groups (Table 2, Entry 3).
An efficient Cu2O/1H-imidazole-4-carboxylic acid (L24) catalytic system for the O-arylation of phenols and iodoarenes was reported by Cheng and Hsieh.50 The reaction afforded an array of substituted diaryl ethers under mild conditions. The low catalytic loading (1 mol%) and economical copper source increased the possibility for the industrial application (Table 2, Entry 4).
In 2008, Buchwald et al.51 described mild conditions for the Ullmann O-arylation of aryl halides (X = I, Br) and aliphatic alcohols (Table 2, Entry 5). The relatively low catalyst loading and good functional group tolerance allowed easy access to a wide range of arylalkyl ethers.
Sekar et al.52 presented a simple and effective Cu(OTf)2/1,1′-binaphthyl-2,2′-diamine (BINAM, L16) catalytic system for the synthesis of a large number of diaryl ethers (Table 2, Entry 6).
Methenamine (L18), a commercially available and inexpensive material, was found to be efficient in the Ullmann etherification by Qian’s team.53 This methodology was applicable to a variety of phenols and aryl halides under mild conditions; twenty-eight compounds were obtained in moderate to excellent yields, except for p-acylphenol (Table 2, Entry 7). Notably, methenamine, a low molecular weight polymer of ammonia and formaldehyde, is also an antibiotic and undergoes acidic hydrolysis to give the active component formaldehyde with antimicrobial property.
Buchwald and Maiti54 described the selective formation of the C-O bond in the copper-catalyzed reaction of aryl halides with 3-amino- and 4-aminophenols by employing picolinic acid (L25) and trans-N,N′-dimethyl-1,2-cyclohexanediamine (CyDMEDA, L17), as the ligand, respectively (Table 2, Entry 8).
In 2011, commercially available potassium fluoride/clinoptilolite (KF/CP) was found to be an effective base in the Ullmann O-arylation of aryl iodide and phenols under the CuI/L10 or L29 catalytic system (Table 2, Entry 9).55 The base was also efficient in the SNAr reaction of activated aryl fluorides and phenols without catalyst.
In 2008, Hu et al.56 reported an air-stable copper(I)-bipyridyl complex, (C1, Figure 2) which exhibited high catalytic capability in the Ullmann O-arylation of both phenols and aliphatic alcohols with aryl halides (Table 2, Entry 10).
Taillefer and coworkers57 described a simple and efficient method for the copper-catalyzed synthesis of phenols by employing hydroxide salts (Table 2, Entry 11). The reaction solvent was essential to the selectivity between the phenol product and the diaryl ether side-product, which was formed by further Ullmann etherification of the in situ produced phenol with aryl iodide. The screened ligands dibenzoylmethane (L1) and N,N′-dimethylenediamine (L14) were found to be effective with aryl iodides and aryl bromides, respectively, but the ligand loading was high (up to 50 mol%).
In 2010, Fu and Yang58 reported a similar protocol for the synthesis of substituted phenols via copper-catalyzed, Ullmann-type reaction by employing pyridine-2-aldoxime (L28), as the ligand, and water, as the solvent (Table 2, Entry 12). Interestingly, the acid group of 2-chlorobenzoic acid greatly facilitated the hydroxylation process; while other aryl chlorides remained unreactive under this catalytic system. In 2010, Ma et al. also observed the ortho-effect in the ligand-free Ullmann O-arylation of o-chlorotrifluoroacetanilides with phenols;59 this CuBr/L22 catalytic system was also efficient with sterically hindered phenols.
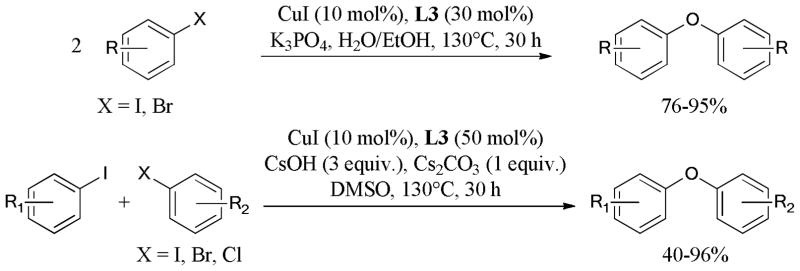
Based on its previous work, the Taillefer group60 described a new protocol allowing the synthesis of symmetrical and unsymmetrical diaryl ethers from aryl halides and simple oxygen source, such as H2O or hydroxide salts (Scheme 1). The reaction proceeded to give phenols initially, followed by faster etherification of aryl halides with the formed phenols. The unsymmetrical coupling of different aryl halides accompanied the formation of symmetrical diaryl ethers, especially for the reactive aryl iodide.
Scheme 1.
In 2013, a highly efficient and regioselective Ullmann reaction of 2,x-dihalopyridines with phenols was described by Chen et al.61 The corresponding 2-aryloxypyridines were obtained in good to high yields under the CuI/TMEDA catalytic system (Scheme 2).
Scheme 2.
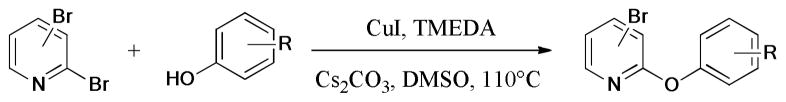
b. C-N Bond Formation
In 2009, the Wan group62 reported an efficient CuO/oxalyl dihydrazide/ketone system for the Ullmann amination of aryl halides in water. The most reliable ketone was found to be hexane-2,5-dione (Table 3, Entry 1). Both aryl bromides and iodides could be aminated by a variety of anilines, aliphatic amines, and imidazoles under microwave irradiation or conventional heating. The reaction could even proceed smoothly at room temperature by increasing the catalyst loading to 20 mol% and prolonging the reaction time to 96 h, except for the imidazole substrates. But this method required high loading of oxalyl dihydrazide (L34, 50 mol%) and hexane-2,5-dione (L4, 1 equiv.) additives.
Table 3.
Development of the Ullmann N-Arylation
| Entry | Copper source | Ligand | Substrates | Conditions | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|
| Ar-X | Nucleophile | ||||||
| 1 | CuO (5 mol%) | L34 and L4 | Aryl-I, Br | Anilines | KOH/TBAB, H2O MW, 120°C, 5 min or KOH/TBAB, H2O 90°C, 8 h | 45–78 | 62 |
| Aliphatic amines | 50–91 | ||||||
| Imidazoles | KOH/TBAB, H2O MW, 140°C, 5 min | 52–89 | |||||
| 2 | CuI (5 mol%) | L32 | Aryl-I, Br | Anilines | KOH/TBAB, H2O MW, 130°C, 5 min or KOH/TBAB, H2O 130°C, 5 min | 47–83 | 63 |
| Aliphatic amines | 54–85 | ||||||
| Aryl-I | Pyrazole | KOH/TBAB, H2O MW, 130°C, 5 min | 54–78 | ||||
| 3 | CuO (5 mol%) | L35 | Aryl-I, Br | Anilines | KOH/TBAB, H2O MW, 130°C, 5 min | 43–85 | 64 |
| Aliphatic amines | 18–87 | ||||||
| 4 | Cu2O (5 mol%) | L27 | 4-Iodotoluene | Pyrazole | Cs2CO3, CH3CN 100°C, 22 h | 80 | 65 |
| L13 | 1,2,4-Triazole | C2CO3, DMF 100°C, 22 h | 80 | ||||
| 5 | CuI (10 mol%) | L33 | Aryl-Br Pyridyl-Br |
N-Heterocycles | Cs2CO3, DMSO 120°C | 60–95 | 66 |
| 6 | CuCl (10 mol%) | L30 | Aryl-I, Br, Cl | N-Heterocycles/alkyl amines | NaOH, n-Bu4N+Br− H2O, 100°C | 62–97 | 67 |
| 7 | CuI (10 mol%) | L31 | Aryl-I | Guanidine nitrate | K3PO4, MeCN 120°C | 19–92 | 68 |
| 8 | CuI (10 mol%) | L19 | Aryl-I, Br | Substituted-amidine·HCl | Cs2CO3, DMF 110–120°C | 64–94 | 69 |
| 9 | CuI (10 mol%) | None or L14 | o-Functional aryl-I, Br | NaN3 | Cs2CO3/K2CO3 EtOH, 95°C | 43–95 | 70 |
| 10 | Cu(acac)2 (10 mol%) | L2 or L3 | Aryl-I, Br Heteroaryl-Br |
NH3·H2O | Cs2CO3, DMF 60–90°C | 23–98 | 71 |
On the basis of their previous work, Wan et al.63 subsequently developed a novel and improved ligand pyrrole-2-carbohydrazide (L32) for the Cu-catalyzed amination of aryl halides with amines in water (Table 3, Entry 2). The reaction time could be reduced to 5 min under microwave irradiation. Interestingly, the N2,N2′-disubstituted oxalic acid bishydrazide derivative (L35)64 was also an effective ligand, for the Ullmann reaction, which was then reported by the same group (Table 3, Entry 3).
In 2010, Wu et al.65 reported an Ullmann coupling reaction of 4-iodotoluene with pyrazole and 1,2,4-triazole by employing ligands L27 and L13 (Table 3, Entry 4).
The copper-catalyzed arylation of oxadiamines and polyamines for the synthesis of N,N′-diaryl derivatives was studied by Beletskaya and her students.72 The yields of the target products and the selectivity of the arylation of the amino groups were strongly dependent on the nature of oxadiamines, polyamines, and aryl halides, as well as on the reaction conditions, such as ligands, solvents, and bases. The best results were achieved by using CuI/proline/Cs2CO3/MeCN or EtCN system for the N,N′-diarylation of tetraamines and CuI/α-acetylcyclohexanone or α-isobutyrylcyclohexanone/Cs2CO3/DMF system for the N,N′-diarylation of oxadiamines.
In 2011, Wang et al.66 demonstrated an efficient Ullmann reaction of aryl bromides with N-heterocycles catalyzed by the combination of CuI and acylhydrazine- or acylhydrazone-type ligands. The facile access to the modifications of the acylhydrazine and acylhydrazone units allowed the optimization of catalytic activity and selectivity. The coupling reaction gave the corresponding products in moderate to high yields using L33, as the ligand (Table 3, Entry 5).
The CuCl/L30 catalytic system was found to be efficient in the Ullmann N-arylation of aryl halides with N-heterocycles and alkylamines (Table 3, Entry 6).67
The Ullmann reaction of aryl iodides and guanidine nitrate was successfully performed under the CuI/L31 catalytic system (Table 3, Entry 7).68 The desired N,N′-diarylguanidines were obtained in 19–92% yields.
The Ullmann coupling of aryl halides (X = I, Br) and substituted amidine hydrochlorides provided a practical methodology for the synthesis of substituted anilines (Table 3, Entry 8).69 The reaction gave the corresponding products in moderate to good yields under optimized conditions (CuI/L19/Cs2CO3/DMF).
In 2010, Qiao et al. reported a simple protocol for the copper-catalyzed synthesis of substituted aromatic amines using NaN3, as the amine source (Table 3, Entry 9).70 Control experiments showed ortho-functional groups (COOH, CONH2, NHCOR) played an important role in the catalytic cycles. Notably, the ortho-effect was also observed in the Ullmann coupling of o-halobenzoic acid and alkylamines;73 the reaction was carried out at ambient temperature, using CuI/L9 as the catalyst.
In 2009, Taillefer and Xia71 reported a practical and economical synthesis of anilines by employing aryl halides and ammonia, as the starting materials. The inexpensive ligands (L2/L3) and nitrogen source, together with the mild conditions, provided a possibility for industrial scale production (Table 3, Entry 10).
In a recent mini-review by Enthaler,74 several protocols using ammonia as the starting material for the synthesis of anilines via copper-mediated coupling reaction were discussed (Scheme 3). In general, these protocols could be performed at low reaction temperatures suitable for potential industrial applications, but the high catalyst loadings remain a challenge for use in commercial processes.
Scheme 3.
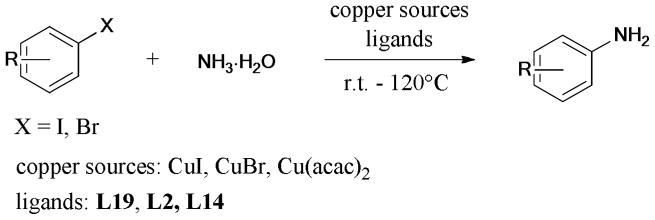
c. C-S Bond Formation
In 2009, Li et al.75 reported a simple and highly efficient copper-catalyzed S-arylation of thiophenols and aryl halides (X = I, Br). This method gave moderate to excellent yields of diaryl sulfides with a wide range of functional groups by using 1,2,3,4-tetrahydro-8-hydroxyquinoline (L26), as the ligand. The reactions of activated aryl iodides could be conducted even at room temperature (Table 4, Entry 1).
Table 4.
Development of the Ullmann S-Arylation
| Entry | Copper source | Ligand | Substrates | Conditions | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|
| Ar-X | Nucleophile | ||||||
| 1 | CuBr (10 mol%) | L26 | Aryl-I, Br | Thiophenols | K2CO3, DMSO 80°C, 24 h | 62–99 | 75 |
| 2 | CuI (10 mol%) | L8 | Vinyl/Aryl/Heteroaryl-I | Aromatic/Aliphatic/Heterocyclic thiols | K3PO4, DMF 30–60°C | 84–98 | 76 |
| Vinyl/Aryl-Br | K3PO4, DMF 70–110°C | 84–93 | |||||
| 3 | Cu(OTf)2 (20 mol%) | L16 | Aryl-I, Br, OTs | Thiophenols/Aliphatic thiols | Cs2CO3, DMF 110°C | 53–98 | 77 |
| 4 | Cu2O (5 mol%) | L5 | Aryl-I, Br, Cl Heteroaryl-I, Br |
Aromatic/Aliphatic/Heterocyclic thiols | Cs2CO3, DMSO 80°C, Ar | 51–96 | 78 |
| 5 | CuI (10 mol%) | L19 | Aryl-I Heteroaryl-I |
Thiourea | 1) t-BuONa, DMSO 90°C 2) HCl (aq) |
81–97 | 79 |
The combination of CuI and cis-1,2-cyclohexanediol76 (L8) was described as a general, mild, and efficient catalytic system for the synthesis of various sulfides bearing arylvinyl, diaryl, heteroaryl, and alkyl motifs. The notably mild reaction conditions enabled a wide range of functional groups to be present in both reaction substrates. However, vinyl chloride, vinyl tosylate, vinyl trifluoromethanesulfonate, and potassium vinyltrifluoroborate were found to be completely inert in this catalytic system. Notably, coupling reactions of E- and Z-vinyl iodides with RSH gave their corresponding products with the retention of the stereochemistry (Table 4, Entry 2).
The Cu(OTf)2/BINAM (L16) catalytic system, used in the Ullmann O-arylation, was also effective in the synthesis of a variety of diaryl and arylalkyl thioethers.77 Highly activated aryl chlorides and tosylates provided the corresponding thioether products without a catalyst, suggesting nucleophilic addition elimination mechanism (Table 4, Entry 3).
In 2009, Feng et al.78 reported a copper-catalyzed Ullmann coupling of various alkyl, aryl, and heteroaryl thiols with aryl and heteroaryl halides (X = I, Br, Cl). Notably, this catalytic system was also effective with highly activated aryl chlorides, such as p-acetyl, p-cyano, p-nitro, and p-trifluoromethylchlorobenzenes, although the yields were relatively low (52–82%) (Table 4, Entry 4).
Qi’s group79 recently described a general and economical one-pot synthesis of substituted thiophenols via copper-catalyzed C-S formation of aryl and heteroaryl iodides and thiourea, followed by the treatment of aqueous hydrochloric acid (Table 4, Entry 5).
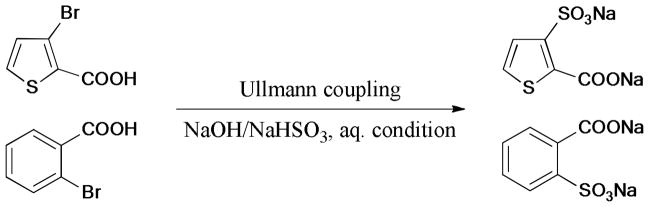
In 2011, Ramaswamy et al.80 reported an Ullmann-type reaction of halothiophenecarboxylic acids and halobenzoic acid with sodium bisulfite (Scheme 4). The proposed oxidative addition and reductive elimination mechanism were supported by the following observations: 1) The reactivity order ArBr 〉〉 ArCl, and the cyclic voltammetric data on cathodic potential was consistent with the nucleofugicity of the halide; 2) Couplings were favored by the presence of electron-withdrawing groups; 3) Coupling did not occur without a copper catalyst; 4) Radical inhibitors, such as butylated hydroxytoluene (BHT) did not suppress the reaction, and the results showed that ortho-carboxylic acid groups accelerated significantly the rate of oxidative addition process of the copper complex to the C-Br bond.
Scheme 4.
d. More Than One-type Bond Formation
The ligands, mentioned above, are largely applicable to one specific type of bond formation (C-O, C-N, or C-S). As more novel and advanced ligands were explored and developed, several versatile catalytic systems for different bond formations were established over the past few years. This review highlights some recent examples of these highly active ligands.
In 2006, Fu et al.46 disclosed a new and efficient copper-catalyzed reaction for the formations of C-N, C-O, and C-P bonds (Table 5, Entry 1). A broad array of N-, O-, and P-arylated products was synthesized in good to excellent yields by using the CuI/pyrrolidine-2-phosphonic acid phenyl monoester (PPAPM, L21) catalytic system. The aryl chloride substrates gave lower yields due to decreased reactivity.
Table 5.
Versatile Ligands for the Ullmann Reaction
| Entry | Bond type | Copper source | Ligand | Substrates | Conditions | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Ar-X | Nucleophile | |||||||
| 1 | C-N | CuI (10 mol%) | L21 | Aryl-I, Br, Cl | Anilines/Alkyl amines/Amides/Hydrazine/N-Heterocycles | K3PO4, DMF 90–120°C | 32–98 | 46 |
| C-O | Aryl-I, Br, Cl | Phenols/Alkyl alcohols | Cs2CO3, DMF 110°C | 20–98 | ||||
| C-P | Ary-I, Br | H-phosphonates/Hy pophosphite | Cs2CO3, toluene/DMAP, DMF 110°C | 20–85 | ||||
| 2 | C-N | CuI (10 mol%) | L6 | Aryl-I | Amides | Cs2CO3, 110°C DMF/dioxane | 73–98 | 81 |
| C-S | Thiophenols | 91–98 | ||||||
| C-O | Phenols | Cs2CO3, 110°C dioxane | 81–87 | |||||
| 3 | C-N | CuBr (10 mol%) | L5 | Aryl-I, Br | Amides/N-Heterocycles | Cs2CO3, DMSO r.t.−75°C | 72–96 | 29 |
| C-O | Phenols | Cs2CO3, DMSO 60–80°C | 72–97 | |||||
| C-S | Aryl-I | Aromatic/Alkyl thiols | 81–97 | |||||
| 4 | C-N | CuI (5 mol%) | L12 | E)-Vinyl-I, Br | N-Heterocycles/Phenols | Cs2CO3, DMF 60–80°C | 81–95 | 82 |
| C-O | (Z)-Vinyl-I, Br | N-Heterocycles/Phenols | Cs2CO3, DMF r.t. or 40°C | 81–98 | ||||
| 5 | C-N | CuI (10 mol%) | L36 | Aryl-I, Br | N-Heterocycles/Aliphatic amines | Cs2CO3, DME 80°C | 62–97 | 83 |
| C-O | Aryl-I, Br | Phenols | 70–98 | |||||
| C-S | Aryl-I | Thiophenols | 75–95 | |||||
1,1,1-tris(Hydroxymethyl)ethane81 (L6) and ethyl 2-oxocyclohexanecarboxylate29 (L5) were then found to be efficient in the copper-catalyzed Ullmann reaction of aryl halides with O-, N-, and S-nucleophiles (Table 5, Entry 2–3).
A new and practical ligand, 2-pyridin-2-yl-1H-benzoimidazole82 (L12), was reported in the coupling reactions of vinyl halides (X = I, Br) with N-heterocycles and phenols (Table 5, Entry 4). A broad range of N-vinyl heterocycles and arylvinyl ethers was achieved in good to excellent yields with the retention of the stereochemistry under mild conditions.
In 2011, Chen et al.83 developed a novel type of N,N′-dioxide ligand (L36), which was efficient for the Ullmann coupling of aryl halides with diverse O-, N-, and S-nucleophilic reagents (Table 5, Entry 5).
2. Green Synthetic Methodology
In a simple term, the principles of green chemistry require low “cost” and high “benefit” in organic reactions. As for the Ullmann reaction, it means that less expensive ligands or ligand- or additive-free conditions, reusable catalysts, and shorter reaction time should be selected. In this context, great efforts have been devoted to developing greener and more sustainable synthetic methods for copper-mediated Ullmann coupling reactions.
a. Ligand- or Additive-free Conditions
Ligand-free, Ullmann-type coupling reactions attracted considerable attention due to the advantage of economical and green conditions.
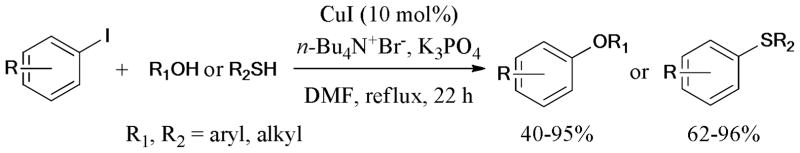
Chan et al.84 developed a catalytic and efficient ligand-free condition (CuI, n-Bu4N+Br−, DMF, K3PO4, reflux, 22 h) for the Ullmann O-arylation of various substituted phenols and aliphatic alcohols with aryl iodides (Scheme 5). This reaction system was also found to be effective in the Ullmann S-arylation85 of aryl iodides with aromatic and aliphatic thiols.
Scheme 5.
In 2007, Correa and Bolm86 reported a catalytic and ligand-free Ullmann N-arylation (Scheme 6) of various N-heterocycles with aryl halides (X = I, Br, Cl). Cu2O was employed, as the catalyst. Unfortunately, aniline and benzylamine proved to be unsuccessful substrates for this protocol. A much milder ligand-free condition at 35–40°C and using 20 mol% CuI, as the catalyst, was established for a similar scope of substrates.87
Scheme 6.
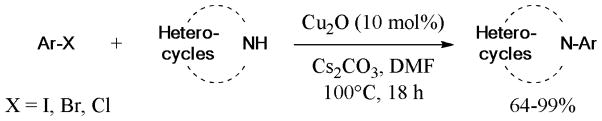
Following on its previous work, in 2010, Bolm’s group88 further developed another Cu powder/CsOAc/DMSO system for the ligand-free Ullmann coupling reaction (Scheme 7). The heteroaryl chloride exhibited low reactivity under the same reaction condition. Interestingly, the reaction with electron-rich 3-bromothiophene gave a moderate yield of the desired product; in contrast, the use of 2-bromothiophene, as the substrate, was unsuccessful.
Scheme 7.

In 2011, the ligand- and solvent-free Ullmann reaction of aryl halides with alkyl amines was reported by Wei’s group.89 The reaction gave the corresponding anilines in moderate to excellent yields under the catalysis of 5 mol% copper powder (Scheme 8).
Scheme 8.
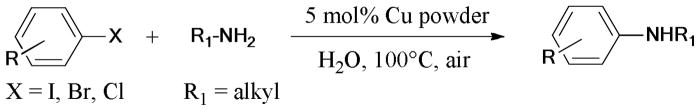
The CuCl/n-Bu4N+OH− (40% aq.) catalytic system was found to be an efficient ligand-free protocol for the Ullmann C-N bond formation of aryl halides (X = I, Br) and alkylamines or N-heterocycles,90 the yield was dramatically reduced when the reaction was not carried under an inert atmosphere.
In 2010, Punniyamurthy et al.91 revealed a ligand-free protocol of copper-catalyzed coupling of aryl iodides with amides or imidazoles (Scheme 9).
Scheme 9.
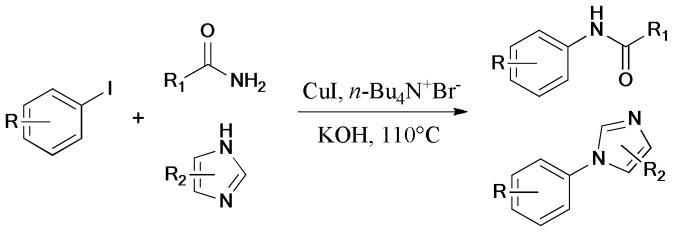
In 2011, Antilla et al.92 disclosed a ligand-free protocol for the Ullmann coupling reaction of aryl iodides with amidines or benzamidines (Scheme 10).
Scheme 10.
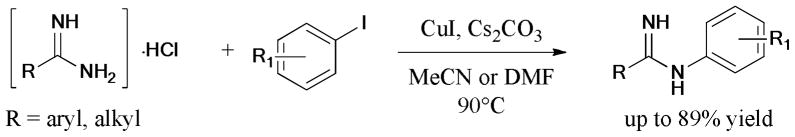
Punniyamurthy et al.93 reported the ligand-free synthesis of substituted 2-(arylthio)arylcyanamides via the cascade intra- and intermolecular Ullmann C-S coupling reaction of 2-(iodoaryl)thioureas with aryl iodides (Scheme 11).
Scheme 11.
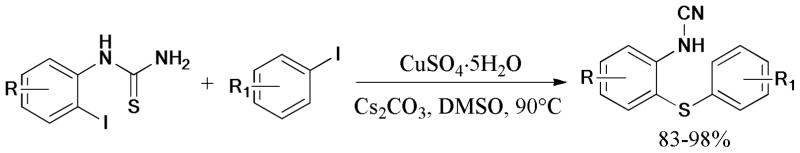
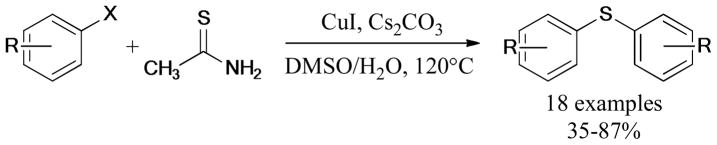
In 2011, a novel method for the preparation of symmetric diaryl thioethers was developed by Zhao and coworkers94 via double Ullmann S-arylation of aryl halides with thioacetamide under ligand-free conditions (Scheme 12).
Scheme 12.
Additional examples for the application of ligand-free Ullmann reaction in the synthesis of heterocycles will be discussed in the section II.1.
b. Recyclable Heterogeneous Catalysts
In most homogeneous catalytic systems, the copper catalysts are usually discarded following the reaction and the catalyst loading is high. Thus, the development of inexpensive, ligand-free, and recyclable catalysts remains a very active area of research. In addition, heterogeneous catalysts offer the advantage of easy separation from the organic solvents. The combination of polymers,95 charcoal,96 cellulose,97 zeolites,98 alumina,99 and silica gel100 with copper salt allowed facile access to heterogeneous catalysts.
In 2012, Jain et al.101 described a recyclable and efficient heterogeneous copper(II) trans-bis-(glycinato) complex catalyst for the synthesis of diaryl ethers via Ullmann-type reaction (Scheme 13). The catalyst could be easily prepared from copper(II) acetate and glycine.102 The reaction gave moderate to high yields of diaryl ethers under mild conditions. The presence of ortho-substituted and electron-withdrawing groups of the phenol substrates had a negative effect on this reaction. The authors also studied the reactivity of the recycled catalyst and found that it retained high activity for the coupling reaction, even after 7 runs.
Scheme 13.
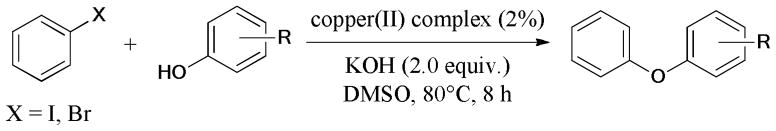
A series of supported CuO catalysts were prepared and characterized by Li and coworkers.103 Among them, CuO/γ-Al2O3 proved to be the most effective catalyst for the Ullmann O-arylation of iodobenzene. The recycle experiment showed that the catalytic activity of the recycled catalyst remained the same after 3 cycles.
The combination of CuFe2O4 and 1,10-phenanthroline (L10) was determined to be an efficient catalytic system for the Ullmann alkoxylation of aryl halides and aliphatic alkyl alcohols.104 The recycle experiment suggested the used catalyst retained high activity by simple grinding with an agate mortar, even in the seventh run.
As reported by Mulla and coworkers,105 heterogeneous recyclable copper fluorapatite (CuFAP) could be used as an efficient catalyst for the Ullmann O-arylation of aryl halides (X = I, Br) with the potassium salt of substituted phenols in the presence of N-methyl 2-pyrrolidone (NMP), as the solvent, at 120°C.
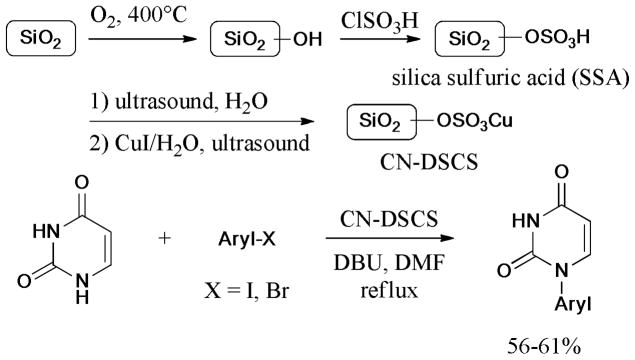
In 2011, Rad and coworkers106 designed and synthesized a novel and efficient silica-supported heterogeneous catalyst (Scheme 14), namely, copper nanoparticle-doped silica cuprous sulfate (CN-DSCS) for the Ullmann N-arylation of nucleobases and N-heterocycles with aryl halides (X = I, Br). The screening of bases and solvents revealed that 1,8-diazabicycloundec-7-ene (DBU) and DMF were the most suitable conditions for this protocol. Subsequently, a series of N-aryl-nucleobases was achieved in moderate to good yields under the optimized conditions.
Scheme 14.
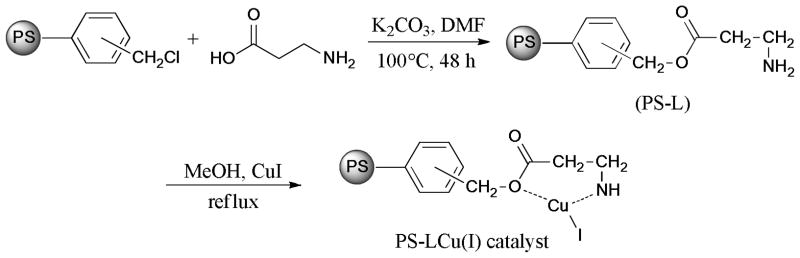
In 2011, a new polymer-supported copper(I) complex (Scheme 15) was synthesized and characterized by Islam et al.107 The Ullmann coupling of aryl halides and substituted anilines gave the triarylamine products. The catalyst could be easily recycled by filtration, washed, dried under vacuum, and then conducted in the next run under optimized conditions. The reused catalyst maintained its catalytic potency in this reaction.
Scheme 15.
Recently, an alumina-supported Cu(II) complex108 was synthesized from CuSO4 and basic alumina by stirring them in water followed by removal of the water under reduced pressure at 120°C (Scheme 16). This catalyst complex exhibited excellent catalytic activity in the Ullmann coupling of various aromatic and alkyl thiols, phenols, and aliphatic amines with aryl halides (X = I, Br). Interestingly, the coupling reaction of aryl iodides or bromides was controlled by the base selected, this protocol allowed facile access to unsymmetrical bis-thioethers. The recyclable CuO on alumina was found to be effective in the catalytic formation of aryl C-O bond.109
Scheme 16.
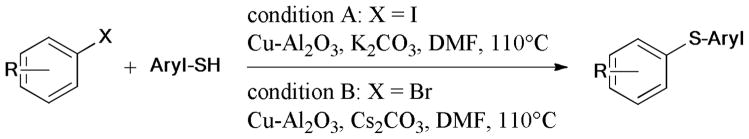
In 2012, Li et al.110 disclosed a newly developed heterogeneous catalytic system for the Ullmann O-arylation, using metal-organic framework (MOF) material-supported CuI as the catalyst. Very recently, the MOF-119111 was synthesized and used as a reusable catalyst in the Ullmann O-arylation.
Nanomaterials have been widely explored and used, as catalysts, for organic synthesis112–117 due to their high surface area. The use of copper nanoparticles, as catalysts for Ullmann reaction, were summarized in a recent review.118
Nano CuO particles48 demonstrated enhanced catalytic activity in the Ullmann coupling of aryl halides (X = I, Br, Cl) with phenols under ligand-free conditions than regular CuO powder.
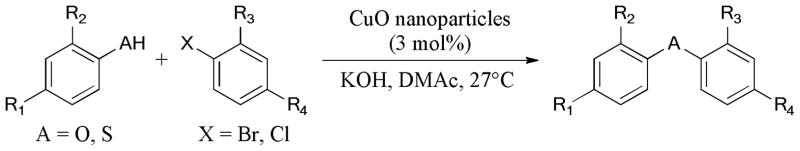
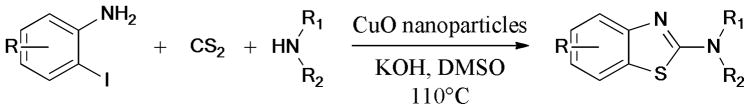
Recently, a practical Ullmann C-O and C-S bond formation using CuO nanoparticles as catalysts, was described by Karvembu and Babu.119 (Scheme 17) The corresponding diaryl ethers and sulfides were easily obtained under mild conditions (3 mol% CuO nanoparticles/KOH/DMAc/27°C).
Scheme 17.
In 2009, the CuO nanoparticles were reported as an efficient catalyst for the formation of C-N, C-O, and C-S bonds.120 In this study, a broad range of amides, amines, imidazoles, phenols, alcohols, and thiols were used to react with aryl iodides under mild conditions; and to evaluate the scope and generality of this reaction system. Notably, the CuO nanoparticles retained high catalytic activity, even after 3 runs.
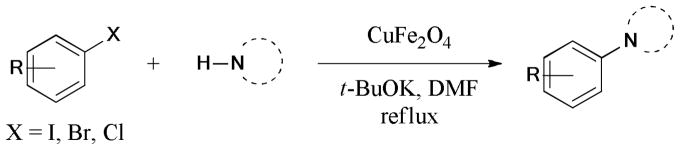
The CuFe2O4 nanoparticles were found to be an effective catalyst for the Ullmann reaction of aryl halides and N-heterocycles (Scheme 18).121 The catalyst could be easily removed from the reaction mixture by a magnetic separator due to its magnetic property. Similar catalytic behavior was observed, even after three consecutive cycles. This reusable catalyst was also found to be efficient in the catalysis of the Ullmann C-S bond formation.122
Scheme 18.
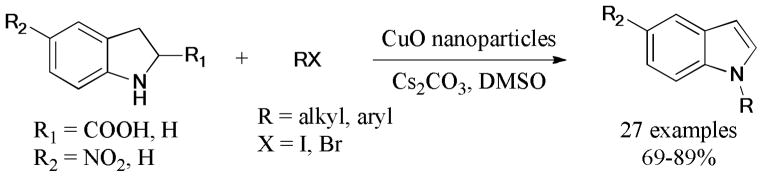
In 2012, the CuO nanoparticles-catalyzed ligand-free Ullmann reaction of indoline/indoline carboxylic acid with aryl/alkyl halides was disclosed by Nageswar and coworkers.123 The corresponding 1-substituted indole derivatives were easily achieved via aromatization under these catalytic conditions (Scheme 19).
Scheme 19.
With the catalysis of CuI nanoparticles, the Ullmann C-O and C-N bond formation of aryl chlorides with phenols, N-heterocycles, or alkylamines gave the desired products in good to excellent yields.124 The catalyst loading was reduced to 1.25 mol% and the catalyst could be reused for several runs without the loss of catalytic activity.
The Ullmann coupling reaction of phenols with aryl halides (X = I, Br, Cl) using Cu2O nanocubes as the catalyst was described by Park and coworkers.125 It is noteworthy that the catalyst loading was reduced to as low as 0.1 mol%.
In 2012, Obora et al. reported the preparation of single nano-sized Cu nanoparticles via the DMF reduction method.126 With the catalysis of these highly effective nanoparticles, the highest turnover number reached 2.2×104 when the catalyst loading was reduced to 1×10−3 mol% in the Ullmann O-arylation of aryl halides (X = I, Br) and phenols.
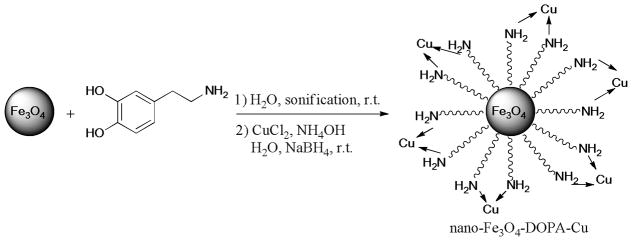
The nano-ferrite-dopamine-supported copper complex (nano-Fe3O4-DOPA-Cu) was reported as an efficient catalyst for the Ullmann S-arylation of aryl halides (X = I, Br) with thiolphenols (Scheme 20).127 The catalyst can be easily separated from the reaction mixture by a magnetic separator on the basis of its magnetic property.
Scheme 20.
c. Microwave/ultrasound-assisted Synthesis
The most notable advantages of microwave-assisted organic synthesis (MAOS)128–132 include faster and easier heating of the reaction, shorter reaction time, and high-throughput chemistry. In some cases, the reaction yields and selectivity can also be greatly improved under microwave irradiation.
Macrocyclic diaryl ether derivatives133 represent an important class of naturally occurring diarylheptanoids and show a wide range of biological activities. To improve the efficiency of macrocyclization, in 2012, Sun et al. developed an efficient and modular microwave-assisted macrocyclization of diaryl ethers via copper-catalyzed intra- and/or bimolecular Ullmann coupling; and investigated the scope and generality of a number of substrates with different linkers, ring sizes, and substitution patterns (Scheme 21).134 The structure of the intra- and bimolecular cyclic products was confirmed by X-ray crystallography.
Scheme 21.

In 2012, the intramolecular Ullmann macrocyclization was demonstrated by James and coworkers under high concentration conditions (up to 0.1 M).135 The highest macrocyclization efficiency (Emac) value136 for these reactions was 7.86, indicating that the macrocyclization process is highly efficient (Scheme 22).
Scheme 22.
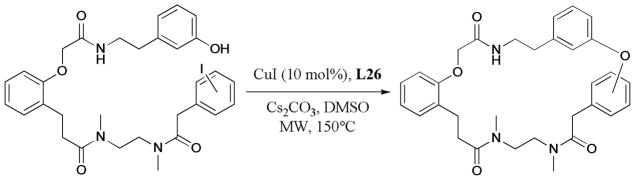
In 2007, Müller and Baqi137 described the microwave-assisted Ullmann synthesis of anilinoanthraquinone derivatives. The reaction gave the corresponding products in moderate to good yields in 2–20 minutes (Scheme 23).
Scheme 23.
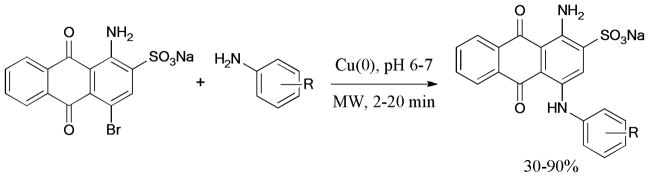
In 2011, Bolm et al.138 reported a solvent- and ligand-free Ullmann coupling of halopyridines with N-nucleophiles under microwave irradiation (Scheme 24). In this work, N-heterocycles such as pyrazole, imidazole, pyrrole, and indole reacted well with halopyridines to give the corresponding products in moderate to good yields; however, benzyl and arylamines were found to be poor substrates under the reported conditions.
Scheme 24.
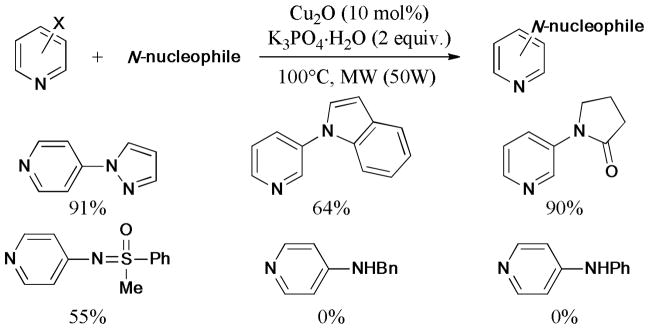
In 2012, Su’s group139 described microwave-assisted Ullmann reaction of aryl halide (X = I, Br, Cl) with N-heterocycles by using calix[4]arene [Emim][Pro] ionic liquid as both the ligand and surfactant (Scheme 25). A variety of N-heterocycle substrates were used in the reaction to give the corresponding products in good to excellent yields; among them, the π-electron-rich N-heterocycles showed lower reactivity in this catalytic system.
Scheme 25.
An efficient and ligand-free protocol for the Ullmann synthesis of N-aryl-1H-imidazoles was described by Zhang et al. (Scheme 26).140 The reaction of aryl bromides and imidazole or N,N′-carbonyldiimidazole gave the corresponding products in moderate to good yields, under microwave irradiation.
Scheme 26.
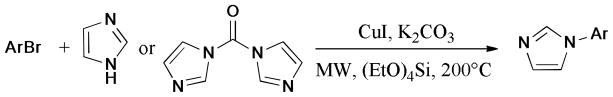
The PEG3400-Cu2O-Cs2CO3 was reported as a recyclable and ligand-free catalytic system for the Ullmann arylation of aryl halides with indole and benzimidazole under microwave irradiation.141
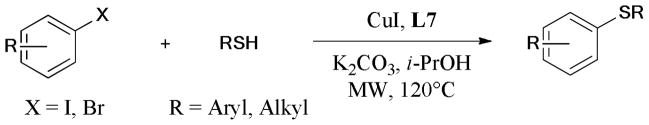
Microwave-assisted Ullmann C-S bond formation was reported by Bagley and coworkers (Scheme 27).142,143 The reaction of aryl-X (X = I, Br) and aryl/alkyl-SH gave the corresponding sulfide in moderate to good yields under the optimized conditions (CuI/L7/K2CO3/i-PrOH/microwave).
Scheme 27.
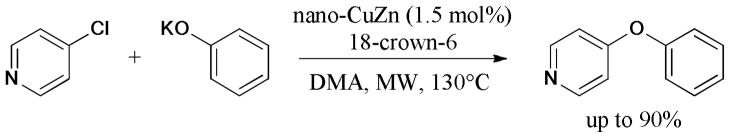
The base-free Ullmann O-arylation was reported by Schouten and coworkers.144 The combination of nano-Cu, -CuZn, and -CuSn as the catalysts and microwave heating led to improved turnovers and reaction yields. The additive 18-crown-6 ether also enhanced the reaction activity (Scheme 28). In 2012, the same group designed a continuous-flow milli-sized tubular reactor for the Ullmann O-arylation of phenol and 4-chloropyridine.145 Both low microwave power and Cu/ZnO-coated internal walls of the tubular milli-reactor were found to be beneficial to increase the yield.
Scheme 28.
Several additional examples62–64 related to microwave-assisted Ullmann C-N bond formation have been included in the section I.1.b (Table 3, Entry 1–3).
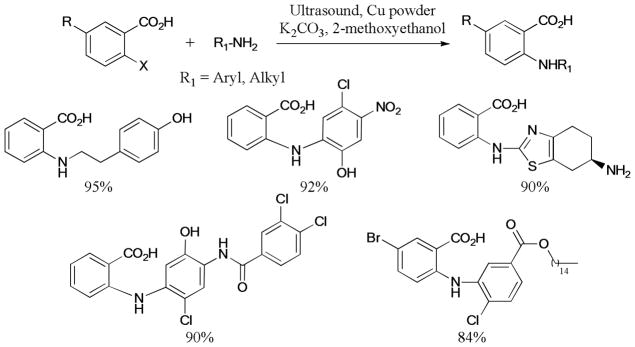
Ultrasonic irradiation was also found to be effective to improve the Ullmann reactions. For example, in 2012, Anandan et al.146 described the ultrasound-assisted Ullmann N-coupling of 2-halobenzoic acid with complex aryl- and alkylamines to give the corresponding products in good to excellent yields (Scheme 29).
Scheme 29.
3. Mechanistic Study
Although various efficient catalytic systems for Ullmann and Ullmann-type reactions have been developed over the last decade, the exact mechanism for these processes still remains elusive. In general, two potential routes have been proposed in the literature. One involves the Cu(III) intermediate via an oxidative addition and reductive elimination mechanism,147–151 while the other proceeds through the single-electron transfer (SET) mechanism.152–155 Overall, it appears that the reaction mechanism may depend mainly on the specific reaction conditions.
The copper(I) phenoxide complexes containing ancillary nitrogen-donor ligands were synthesized and structurally identified by Hartwig’s group151 in 2010 (Scheme 30). The crystal structure of the [(Me2phen)2Cu][Cu(OPh)2] complex revealed that this copper complex existed as an ionic form in the solid state. In this work, the status of the complexes in DMSO was also studied by the measurement of the molar conductivity of the solutions. The values of 1.0 mM solution containing complexes C2, C3, C4 were 37.1, 27.0, 31.9 Ω−1cm2mol−1, respectively. These data suggested the copper complexes exist mainly in an ionic form in a polar solvent.
Scheme 30.

The following study of the reactions using a stoichiometric amount of C2 (Figure 2) with o-(allyloxy)iodobenzene provided a supporting evidence for a redox route rather than a radical mechanism (Scheme 31). Typically, the corresponding aryl radical of o-(allyloxy)iodobenzene was recognized to undergo rapid cyclization to form a [3-(2,3-dihydrobenzofuran)]methyl radical, subsequently trapped by the aryloxide ion (ArO−) group. But none of the cyclized products was observed in this case.
Scheme 31.
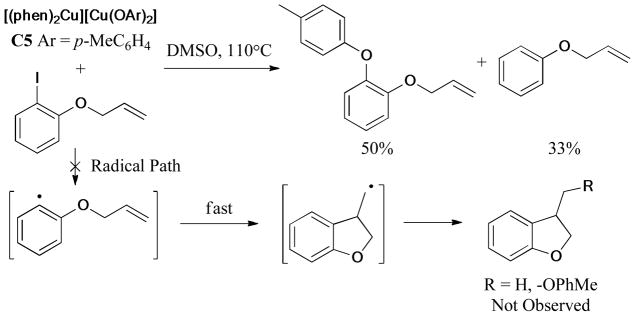
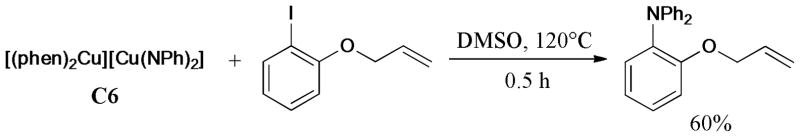
Furthermore, several Cu(I)-amido complexes were synthesized and identified by Hartwig and Giri150 in the same year (Scheme 32). The reaction of C6 with o-(allyloxy)iodobenzene gave the N-arylated product in 60% yield, without the formation of the cyclization product. The subsequent DFT calculation also supported the mechanism proceeding through Cu(III) intermediates.
Scheme 32.
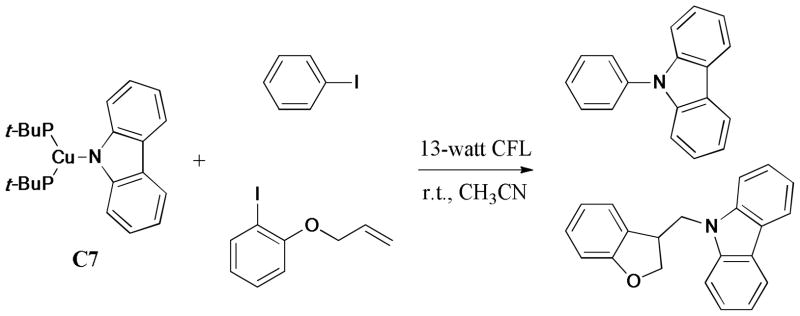
In contrast, Fu et al.156 recently presented evidence for the viability of a radical pathway (Scheme 33), which demonstrated the Ullmann C-N chemistry can proceed by the radical route following irradiation. In this report, reaction of the stoichiometric (t-Bu3P)2Cu(carbazolide) (C7) with iodobenzene gave the N-arylated product under the exposure to the 100-W mercury lamp at −40°C. Interestingly, the photoinduced coupling of C7 with o-(allyloxy)iodobenzene gave a cyclic product instead of the N-arylated product. The subsequent experimental data also indicated the formation of the aryl radical under this light-induced system.
Scheme 33.
II. Recent Synthetic Applications in Heterocycles, Medicinal Chemistry, and Natural Products
The aromatic C-O, C-N, and C-S bonds are widely present in heterocycles, natural products, and drug-like molecules. Although the palladium-catalyzed coupling reaction is an efficient method for the synthesis of these compounds, the inherent drawbacks, such as toxicity and high cost, could limit its application in medicinal chemistry and pharmaceutical industry. In contrast, the recent developments and improvements of copper-mediated Ullmann reactions provide an alternative and practical choice. In this section, we highlight some recent and representative applications of Ullmann reaction in heterocycles, medicinal chemistry, and natural products.
1. Heterocycles
a. C-O Formation
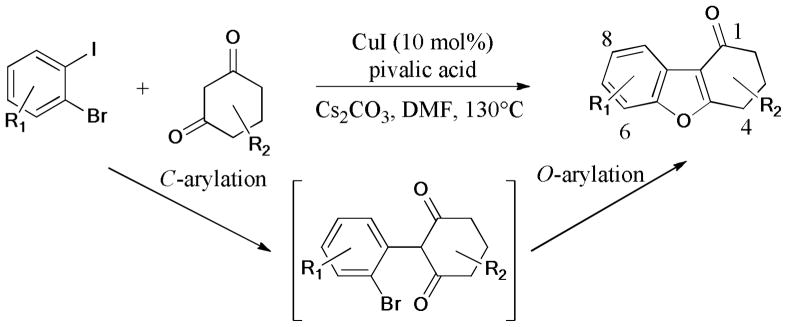
The dibenzofuran heterocyclic ring system157–159 is commonly found in a variety of natural products and biologically active molecules. A simple method for the preparation of 3,4-dihydrodibenzo[b,d]furan-1(2H)-ones is via copper-catalyzed sequential intermolecular C-C bond and intramolecular C-O bond formations, which were recently revealed by Beifuss and coworkers (Scheme 34).160 The structure of the product (R1 = 8-Me; R2 = H) was confirmed by X-ray crystal analysis. The authors proposed that C-C bond formation occurred chemoselectively at the C-I bond in the first step, followed by the intramolecular O-arylation of the C-Br bond.
Scheme 34.
A highly efficient intramolecular copper-catalyzed O-vinylation of various ketones was introduced by Li’s group.161 The corresponding polysubstituted furans were obtained in good to excellent yields, under the CuI/L11 catalytic system (Scheme 35). It is noteworthy that the same strategy was also applied in the synthesis of five- and six-membered enol lactones by the same group.162 The O-vinylation of carboxylic acids gave the desired products in 58–98% yields.
Scheme 35.
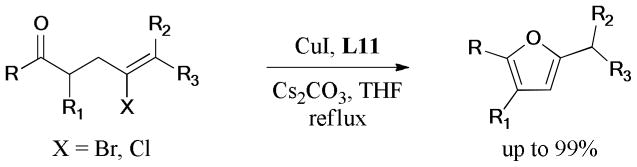
The 1,4-benzoxazine and 1,4-benzodioxane motifs are known to have diverse biological activities163,164 and thus attract considerable attention for organic and medicinal chemists.165–167 An efficient protocol for the synthesis of 1,4-benzoxazine and 1,4-benzodioxane derivatives via one-pot ring opening/Ullmann cyclization of aziridines and epoxides was described by Ranu and coworkers (Scheme 36).168 This method could also be used in the one-pot synthesis of 1,4-benzoxazine thioethers, following sequential formation of C-O, C-N, and C-S bonds in the presence of Al2O3-supported Cu(II) catalyst.108, 169
Scheme 36.
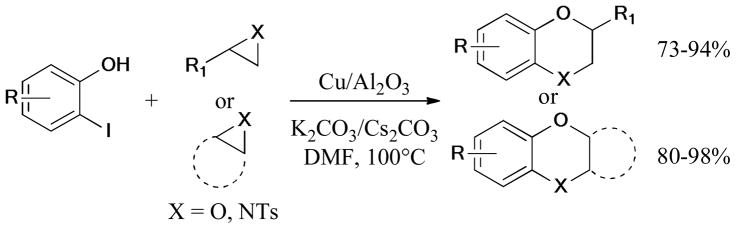
In 2009, a general and practical protocol for the synthesis of 2-aminobenzimidazoles, 2-aminobenzothiazoles, and benzoxazoles was reported by Punniyamurthy and coworkers (Scheme 37).170 The intramolecular and ligand-free cyclization of o-bromoaryl derivatives was catalyzed by CuO nanoparticles to give the corresponding products in high yields.
Scheme 37.
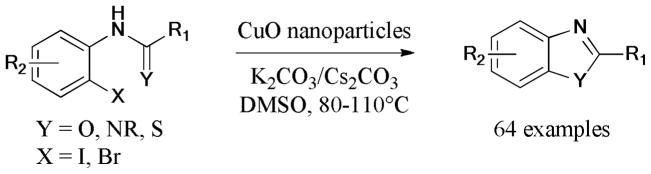
In 2012, Snieckus’s group171 envisaged a domino reaction between 2-iodobenzamides and 2-bromophenols to form the corresponding dibenzoxazepinones based on their previous work on Ullmann reactions (Scheme 38).172 The reaction proceeded first by the Ullmann etherification, followed by an unusual Smiles rearrangement, and finally cyclization to give the corresponding products.
Scheme 38.
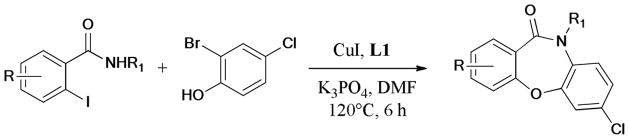
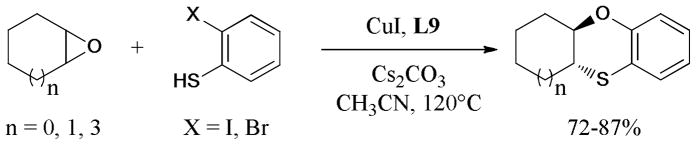
In 2011, Korupalli et al.173 demonstrated a new and efficient synthesis of 1,4-benzoxathiine skeleton from easily available epoxides and o-halothiophenols (Scheme 39). By using CuI-BINOL (L9) complex, as catalyst, the domino SN2 epoxide ring-opening reaction, followed by the Ullmann O-arylation, gave the corresponding products in moderate to good yields.
Scheme 39.
In 2011, Martin and Sahn174 developed an useful synthetic method for the preparation of O-aryl conformationally-constrained benzoxazocines via one-pot intra-and intermolecular Ullmann reactions using 3,4,7,8-tetramethyl-1,10-phenanthroline (L11), as the efficient ligand (Scheme 40).
Scheme 40.
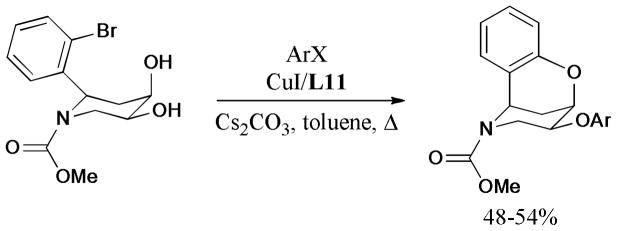
b. C-N Formation
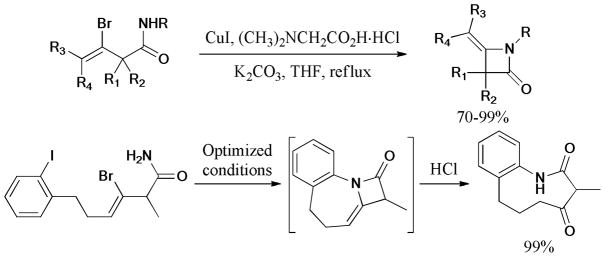
In 2008, Li and Zhao175 reported the synthesis of 4-alkylidene-2-azetidinones via copper-catalyzed intramolecular N-vinylation of amides and vinyl bromides (Scheme 41). Notably, the 4-exo ring product was the only product formed from the competition with the formation of 5-exo, 6-exo, and 6-endo cycles. Furthermore, the combination of double intramolecular Ullmann and ring opening reaction under acidic conditions provided a highly efficient and practical method for the synthesis of medium-sized lactams.
Scheme 41.
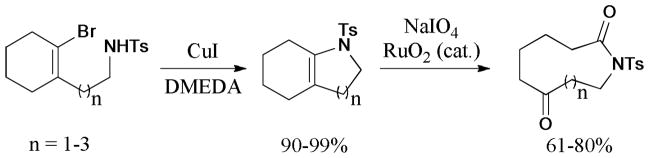
An efficient protocol for the synthesis of five- to eight-membered heterocyclic enamines in both exo and endo modes was described by the same group in 2008.176 The intramolecular Ullmann reaction was catalyzed by the CuI/DMEDA (L14) system to give the corresponding products in good to excellent yields. The subsequent oxidative C=C bond cleavage provided facile access to the 9- to 12-membered lactams (Scheme 42).
Scheme 42.
The Ullmann N-arylation of aryl halides with N-Boc-substituted hydrazine was established by using CuI/1,10-phenanthroline (L10) catalytic system.177,178 On the basis of this work, Breton and Lepore179 designed a copper-catalyzed protocol for the one-pot synthesis of 2,3-dihydro-1H-indazoles using L10 as the ligand in 2011 (Scheme 43). Copper-catalyzed reaction of o-halophenyl aldehydes or ketones with monosubstituted hydrazines was reported in the synthesis of 1-aryl-1H-indazoles by Ding’s team.180
Scheme 43.
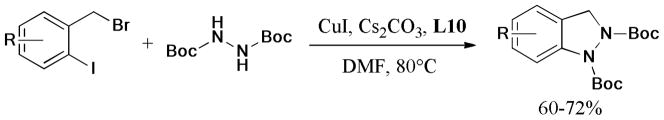
A similar strategy181 was applied in the one-pot synthesis of imidazobenzothiazine and pyrimidobenzothiazine derivatives (Scheme 44). The domino reaction of 2-mercaptoimidazoles and o-bromobenzyl bromides gave moderate to excellent yields of the corresponding products catalyzed by the CuI/L-proline (L19) system.
Scheme 44.
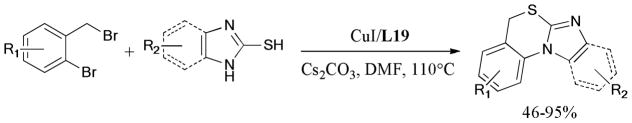
The one-pot synthesis of benzimidazoles via the domino reaction of o-haloacetoanilides and substituted amidine hydrochlorides was described by Fu and coworkers.182 The ligand-free protocol gave the desired products in moderate to good yields (Scheme 45).
Scheme 45.
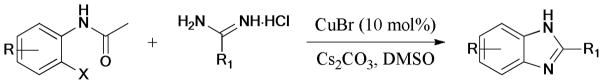
The synthesis of 2,4-disubstituted imidazolones using readily available 2-bromo-3-alkylacrylic acids and amidine hydrochlorides as starting materials was described by the same group.183 The ligand-free protocol gave the corresponding products in 44–94% yields (Scheme 46).
Scheme 46.
1,4-Benzoxazine and thiazine fragments are well-known motifs in many biological active compounds.184–187 In 2011, Karchava et al.188 reported the synthesis of N-substituted 4H-1,4-benzoxazine- and 4H-1,4-benzothiazine-2-carboxylates using the copper-catalyzed Ullmann cyclization, as the key step. The ligand-free coupling reaction tolerated diverse functional groups, such as alkoxy, fluoro, bromo, and cyclopropyl groups (Scheme 47).
Scheme 47.
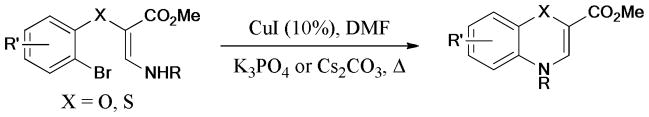
The quinazolinone skeleton is widely present in natural products189,190 and biologically active molecules.191–193 In 2009, Fu’s group194 reported a ligand-free protocol for the synthesis of quinazolinone derivatives via copper-catalyzed Ullmann coupling of 2-halobenzoic acid with substituted amidines and guanidines under very mild conditions (Scheme 48). The ortho-effect of the carboxylic acid group greatly facilitated the copper-catalyzed formation of the C-N bond.
Scheme 48.
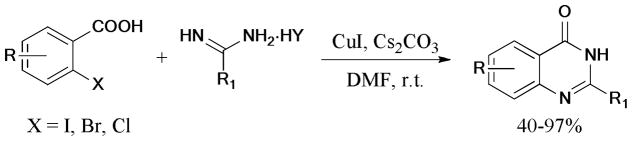
A similar strategy was employed in the synthesis of 4-aminoqunazoline and 2,4-diaminoquinazoline derivatives.195 The reaction of substituted 2-bromobenzonitriles and amidines or guanidines gave the corresponding products in moderate to good yields under the CuI/DMEDA/K2CO3/DMF catalytic system.
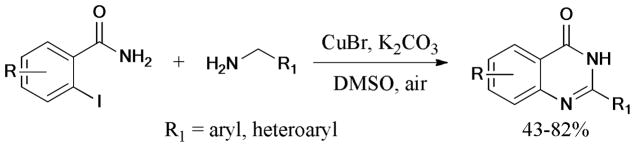
A ligand-free protocol for the synthesis of quinazolinones via sequential Ullmann N-arylation, aerobic oxidation, intramolecular nucleophilic addition and aromatization using CuBr, as the catalyst, was established by Fu and coworkers (Scheme 49).196 Twenty-three examples with various functional groups were presented in moderate to good yields. In 2012, the same group197 reported a combination of the Ullmann N-arylation and aerobic oxidative intramolecular C-H amidation; this methodology allowed facile access to a wide range of imidazo/benzoimidazoquinazolinone derivatives. The one-pot protocol gave the corresponding products in good to excellent yields using 2-halo-N-alkylbenzamides, imidazole, and benzimidazole derivatives as the starting materials and CuI/L-proline (L19), as the catalytic system. A similar strategy was also used in the synthesis of pyrimido[4,5-b]carbazolones.198
Scheme 49.
In 2010, Fu et al.199 described an efficient synthesis of quinazoline derivatives via sequential Ullmann C-N formation, cyclization, and aerobic oxidized aromatization (Scheme 50). The ligand-free protocol was catalyzed by CuI to give the corresponding products in moderate to good yields.
Scheme 50.
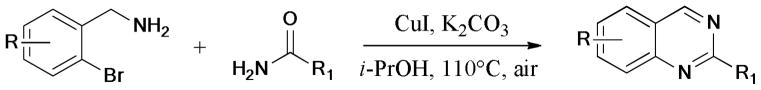
The cascade reaction of o-iodobenzaldehydes and amidine hydrochlorides under ligand-free Ullmann conditions offered facile access to a wide range of quinazolines.200 The quinazoline derivatives could also be synthesized via the domino reaction of amidine hydrochlorides with 2-halobenzaldehydes or 2-halophenylketones under the CuI/L19 catalytic system;201 notably, this catalytic protocol was used in the synthesis of quinozolinones as well.
The one-pot synthesis of 1,2,4-benzothiadiazine 1,1-dioxide derivatives via the Ullmann N-arylation of 2-halobenzenesulfonamides with amidines was developed by Fu and coworkers.202 Comparing to previous methods, this ligand-free protocol was more convenient and economical. Fifteen examples were reported in 65–81% yields (Scheme 51).
Scheme 51.
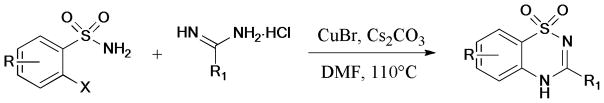
Based on the CuCl/dimethylethylenediamine (L14) catalyst system, an efficient and low catalyst loading synthesis of chiral quinoxalinones was described by Tanimori et al.203 (Scheme 52). A number of optically pure quinoxalin-2-ones were prepared from readily available 2-haloanilines and amino acids.
Scheme 52.
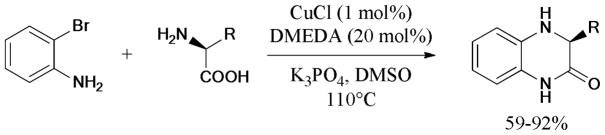
In 2011, Balalaie et al.204 developed a practical protocol for the synthesis of indolo[1,2-α]quinoxalinones via a sequential 4-component Ugi reaction and subsequent Ullmann intramolecular N-arylation catalyzed by the CuI/L-proline (L19) system (Scheme 53).
Scheme 53.
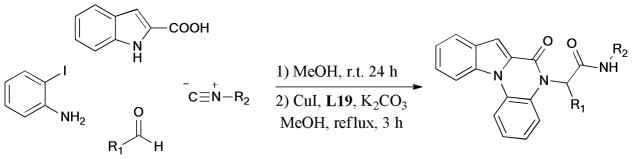
One-pot synthesis of furo[2,3-b]indole derivatives via multiple components reaction and subsequent Ullmann coupling reaction was reported by Ji and coworkers.205 The cascade reaction gave the corresponding products in 61–90% yields (Scheme 54).
Scheme 54.
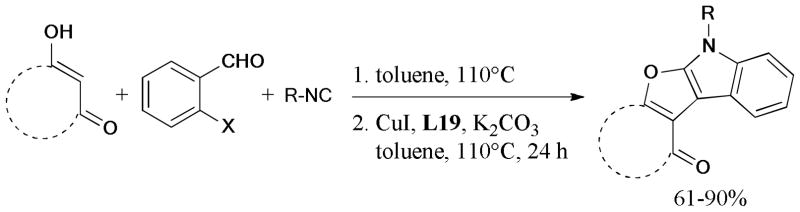
In 2012, a facile tandem reaction for the synthesis of 5,12-dihydroindolo[2,1-b]quinazoline derivatives via copper-catalyzed Ullmann-type intermolecular C-C and intramolecular C-N coupling reactions using trans-4-OH-L-proline (L20), as the ligand, was reported by Dong and coworkers206 (Scheme 55).
Scheme 55.
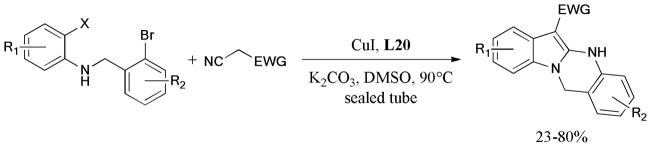
In 2010, a simple and practical method for the copper-catalyzed synthesis of benzimidazo[1,2-b]isoquinolin-11-one derivatives was developed by Fu and coworkers.207 The corresponding products were formed in moderate to good yields under ligand-free conditions (Scheme 56).
Scheme 56.
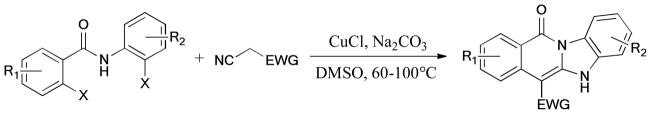
Cai et al.208 presented a general and efficient cascade reaction for the synthesis of aza-fused polycyclic quinolines by using the CuI/L-proline (L19) catalytic system (Scheme 57). Jin et al.209 found that CuI/1,10-phenanthroline (L10) was also effective in the synthesis of benzimidazo[1,2-α]quinolines.
Scheme 57.
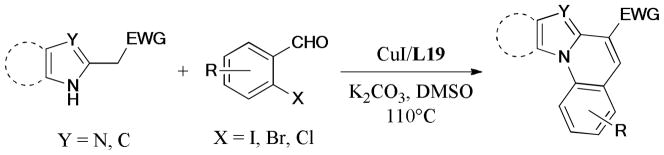
The 11-oxo-dibenzodiazepine moiety is an important heterocycle frequently found in drug-like compounds.210,211 A general method for the synthesis of 5-substituted 11-oxo-dibenzodiazepines via copper-catalyzed Ullmann inter- and intraaminations under very mild conditions was described by Ma and coworkers in 2011.212 Fourteen examples bearing various functional groups were obtained in moderate to good yields (Scheme 58).
Scheme 58.
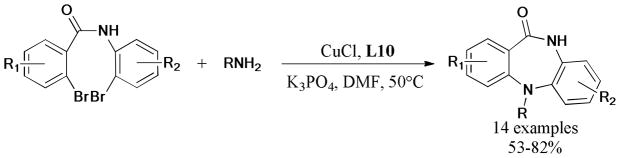
The cascade reaction of copper-catalyzed click reaction of sodium azide with alkynes and subsequent Ullmann N-arylation provided a simple and efficient method for the synthesis of [1,2,3]-triazolo[1,5-α]quinoxalin-4(5H)-ones (Scheme 59).213 The ligand-free protocol gave the tricyclic products in moderate to good yields and was superior to previously reported synthetic methods,214,215 which suffered from multisteps, low efficiency, and poor functionality tolerance.
Scheme 59.
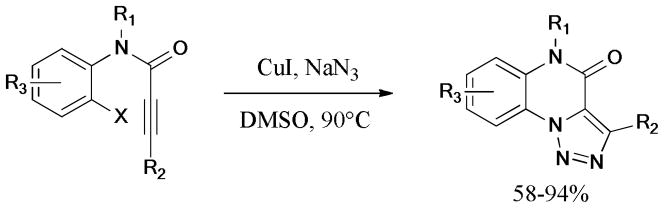
In 2012, a four-step cascade reaction, including Ullmann reaction, aza-Claisen rearrangement, 6π-electrocyclization, and intramolecular rearrangement was disclosed by Wu and coworkers.216 These highly efficient sequential reactions were catalyzed by the Cu(OAc)2/L10 system to give the desired benzoindoline derivatives in moderate to good yields. A proposed mechanism is showed in Scheme 60.
Scheme 60.
c. C-S Formation
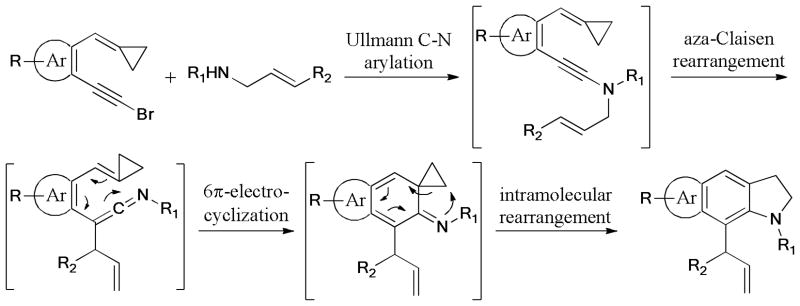
The intramolecular Ullmann C-S coupling of thiols with vinyl halides (X = Br, Cl) has been catalyzed by CuI/K3PO4·3H2O system.217 This ligand-free protocol favored the formation of the 4-exo ring rather than 5-exo, 6-exo, and 6-endo modes in the competition experiment (Scheme 61).
Scheme 61.
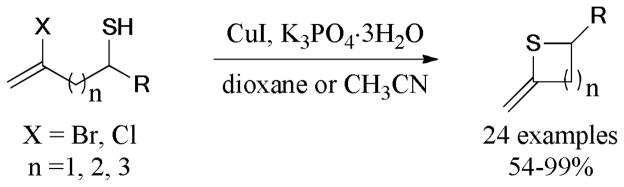
In 2010, Ma et al.218 described a simple and practical method for the synthesis of substituted phenothiazines by using CuI/L-proline (L19) as the catalytic system via efficient Ullmann S-arylation219,220 and N-arylation221 (Scheme 62). This protocol overcomes the drawbacks of harsh conditions and poor selectivity over traditional methods.222–225
Scheme 62.
The cascade three-component reaction of 2-haloanilines, carbon disulfide, and N-nucleophiles was reported as an effective method for the construction of 2-N-substituted benzothiaoles (Scheme 63).226 The ligand-free protocol was catalyzed by reusable copper oxide nanoparticles.
Scheme 63.
2. Medicinal Chemistry
a. C-O Formation
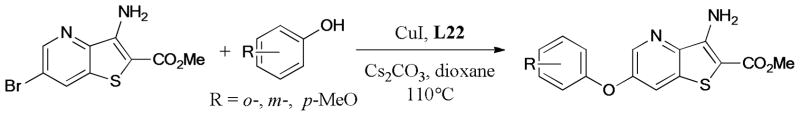
The Ullmann O-arylation is extensively employed in the synthesis of drug-like molecules. In 2012, three ether derivatives containing a thieno[3,2-b]pyridyl ring system were prepared by Queiroz et al.227 via the Ullmann O-arylation (Scheme 64). The anti-cancer inhibitory activities of these compounds against a panel of human tumor cell lines including MCF-7 (breast adenocarcinoma), A375-C5 (melanoma), and NCI-H460 (non-small cell lung cancer) were biologically evaluated; such evaluation revealed that the ether derivatives with o- and m-MeO group showed promising anti-cancer activity.
Scheme 64.
In 2012, Luo et al.228 reported the synthesis and evaluation of a series of diaryl ethers as potential antitubercular agents (Scheme 65, Left). The same year, several xanthone derivatives as nerve conduction blockers (Scheme 65, Right) were prepared by Pinto et al.229 via Ullmann and Friedel-Craft reactions.
Scheme 65.

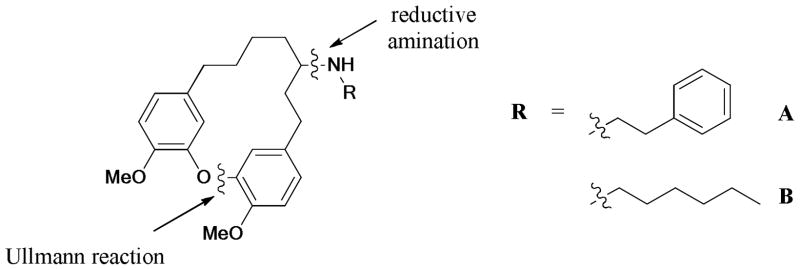
A small chemical library of diarylheptanoid engelhardione and derivatives bearing the oxime, N- and O-alkylated functionalities were designed and synthesized by Sun and coworkers.230 Antibacterial evaluation revealed that the reductive amination derivatives A and B showed moderate activities with minimum inhibitory concentrations (MIC) values of 12.5–25 μg/mL against Mycobacterium tuberculosis and Gram-positive pathogens, as well as anti-Gram-negative activity against an efflux impaired E. coli strain (Scheme 66).
Scheme 66.
In 2012, Natarajan et al.231 reported a series of macrocyclic diarylether heptanoids (MDEH), including natural product platycarynol. A similar strategy was applied to the construction of the diaryl ether moiety. These compounds were evaluated for their inhibitory activity against nuclear factor-κB (NF-κB), as well as anti-cancer and synergistic effect against pancreatic cancer cell line.
b. C-N Formation
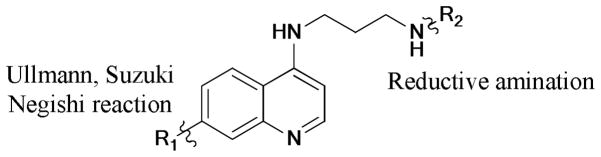
Chloroquine (CQ), one of the quinine drug class, has been widely used for antimalarial therapy.232,233 However, due to the emergence of CQ-resistant strains of P. falciparum,234 the most lethal form of malaria, there is an urgent need to develop new quinine drugs with improved antimalarial activity and low cross-resistance profile. Recently, a chemical library of 7-substituted 4-aminoquinoline was synthesized by Guy’s group235 via Ullmann, Suzuki, and Negishi coupling strategies. All the compounds were tested against CQ-sensitive (3D7) and -resistant (K1) strains of P. falciparum, as well as four mammalian cell lines (HepG2, HEK293, Raji, and BJ) in concentration-response experiments (Scheme 67). Biological data demonstrated that less lipophilic compounds performed higher bioactivities, especially for the biaryl series.
Scheme 67.
A series of substituted dihydroquinozalin-2-ones were synthesized by Tanimori et al.236 via copper-catalyzed coupling reaction of 2-haloanilines and α-amino acids. Some of the synthesized compounds exhibited moderate cytotoxic activity against HaLaS3 cell lines (Scheme 68).
Scheme 68.
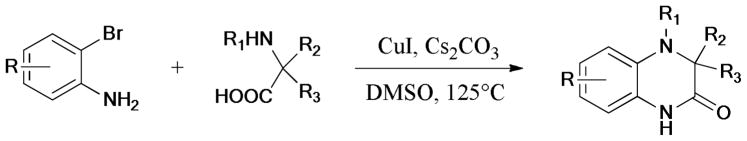
A series of polysubstituted pyrazole derivatives was synthesized via the Ullmann N-arylation by Deprez-Poulain and coworkers237 (Scheme 69). The authors found that the 3-acetylamino substituent on the pyrazole ring played an important role in the regioselectivity of the C-N bond formation. The coupling reaction of 3-acetylamino-pyrazole with iodobenzene gave the N1-regioisomer exclusively; however, a mixture of N1- and N2-isomers was obtained when 3-aminopyrazole was used as the starting material. These pyrazole derivatives were evaluated as angiotensin-2 receptor type 1 (AT1) antagonists. Interestingly, 5-((2′-(1H-tetrazol-5-yl)-[1,1′-biphenyl]-4-yl)-amino)-1-butyl-1H-pyrazole-4-carboxylic acid exhibited potent binding affinity on AT1 receptor with an IC50 value as low as 2.6 nM.
Scheme 69.

A library of 55 anthraquinone derivatives were prepared via microwave-assisted Ullmann coupling by Müller’s group.238 Some compounds exhibited selective inhibition activity against ecto-5′-nucleotidase. In 2012, a convergent synthesis of MG 50-3-1 was reported by the same group239 by using the same strategy. Notably, the improved synthetic route dramatically increased the overall yield of MG 50-3-1, which exhibited excellent inhibition activity (IC50 = 4.6 nM) toward the P2Y-type receptor, a purinergic receptor in the G protein-coupled receptor family (Scheme 70).
Scheme 70.

SNX-5422, a novel and efficient heat shock protein 90 (Hsp90) inhibitor, is a clinical candidate for cancer treatment. To support their clinical studies, Venkatraman and coworkers very recently reported an improved process chemistry route for SNX-5422.240 The efficiency of the new synthetic route was dramatically improved by using 2-fluoro-4-bromobenzonitrile, as the starting material, followed by the kilogram-scale and regioselective Ullmann coupling of the corresponding aryl bromide with N-heterocycles (Scheme 71).
Scheme 71.

In 2012, a series of thioxanthone derivatives were synthesized by Sousa’s group241 via the copper-catalyzed Ullmann reaction of various amines and chloro-substituted thioxanthone (Scheme 72). These compounds were then screened for inhibitory activities against P-glycoprotein (P-gp) and tumor cell growth; six of these compounds exhibited inhibitory activity (GI50 < 10 μM) against the K562 cell line.
Scheme 72.
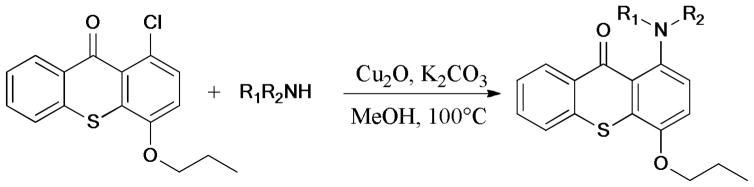
c. C-S Formation
In 2009, the synthesis of the clinical candidate VX-745, a p38α MAPK inhibitor in Werner syndrome cells, was reported by Bagley and coworkers242 (Scheme 73). As a key step, microwave-assisted Ullmann C-S bond formation was studied extensively. The model reaction showed that reliable results were achieved by using CuI (5 mol%) and trans-1,2-cyclohexanediol (L7) as ligand, under basic conditions and microwave irradiation. A notable decrease of the yield was observed without microwave irradiation.
Scheme 73.
3. Natural Products
a. C-O Formation
A series of diaryl ether natural products were synthesized by Bräse and Jung243 via solid-supported Ullmann reaction.
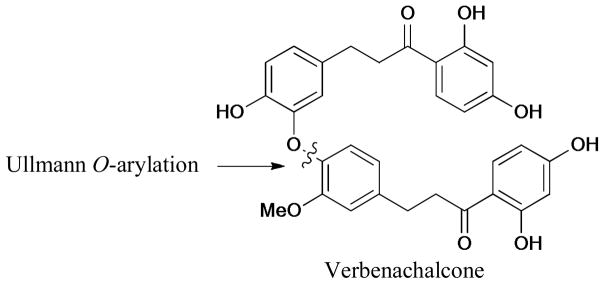
In 2010, Williams and Dandepally244 reported the scalable total synthesis of verbenachalcone, which was isolated from the aerial parts of Verbena littoralis. The Ullmann O-arylation was employed in the construction of diaryl ether moiety and the overall yield was 28% for eight linear steps (Scheme 74).
Scheme 74.
The Ullmann reaction of dihalo aryl moiety and methanol was used in the synthesis of tetracyclic core skeleton of natural product landmycine A.245
Bulbophylol-B246 was isolated from Bulbophyllum kwangtungense Schltr (Orchidaceae) and exhibited versatile biological activities. This natural product was synthesized by employing Wittig and Ullmann reactions as key steps.247 The intramolecular C-O formation was performed by using 2 equiv. of copper catalyst to give the 7-membered ring product in 89% yield (Scheme 75).
Scheme 75.
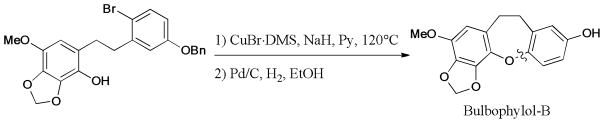
In addition, the Ullmann coupling reaction provides a practical method for the synthesis of naturally occurring macrocyclic diarylethers. In 2011, Sun and Shen248 reported the first total synthesis of the published structure of engelhardione, which was isolated from the roots of Engelhardia roxburghiana (Juglandaceae)249 (Scheme 76). This work ultimately led to the structural revision of this macrocyclic natural product. The intramolecular Ullmann coupling was employed as the key step for the formation of macrocyclic ether bond.
Scheme 76.
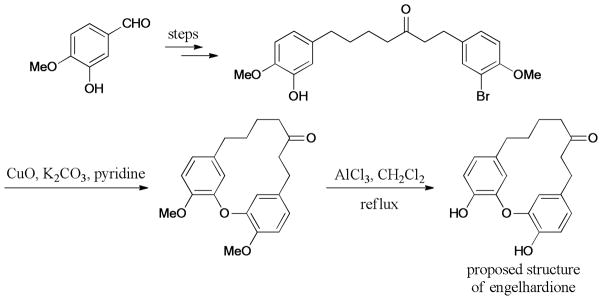
In 2012, Salih and Beaudry250 synthesized a focused set of naturally occurring diarylheptanoids including myricatomentogenin, jugcathanin, acerogenin L, and acerogenin C (Scheme 77). The synthesized samples were used to measure their optical activities and free energy of activation for racemization. In 2013, the Beaudry group reported the total synthesis of the garuganin and garugamblin diarylether heptanoids using an intramolecular Ullmann coupling.251,252
Scheme 77.
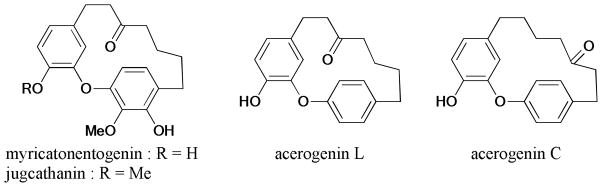
Very recently, Reddy et al.253 reported the synthesis and antitubercular evaluation of isomeric corniculatolides (Scheme 78). The diaryl ethers were prepared through the SNAr process using highly activated aryl fluoride. Unfortunately, none of these compounds showed notable inhibitory activity against Mycobacterium tuberculosis (H37Rv).
Scheme 78.
In 2009, Fürstner et al.254 reported the total synthesis of aspercyclides. The diaryl ether moiety was prepared via the Ullmann O-arylation (Scheme 79).
Scheme 79.
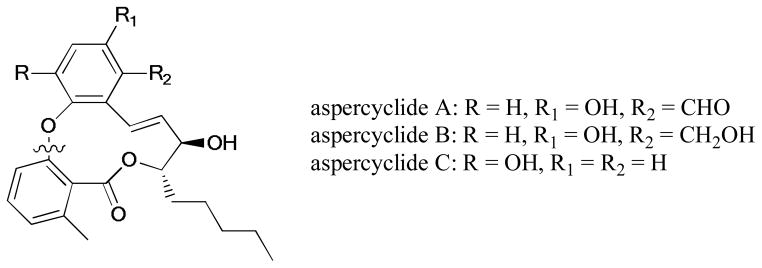
The first total synthesis of Marchantin C was introduced by Speicher and Holz.255 This bisbibenzyl natural product exhibited diverse bioactivities256,257 including cytotoxicity in KB cell lines and α-glucosidase inhibition. The Ullmann reaction of the phenol intermediate with 3-bromobenzaldehyde gave the precursor of the bisbibenzyl macrocycle. Later, another route for this compound and its derivatives was disclosed by Lou and coworkers258 (Scheme 80).
Scheme 80.

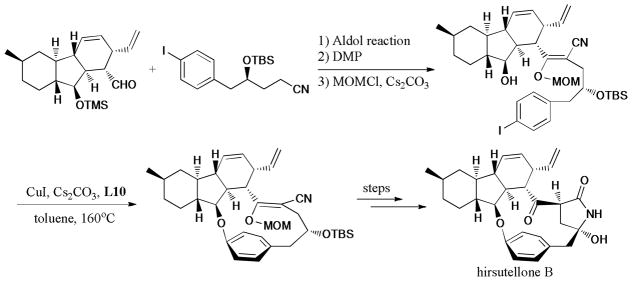
Hirsutellone B,259,260 a member of a novel family of decahydrofluorene class bioactive natural products, was recently discovered. The direct construction of the strained 13-membered macrocycle poses a challenge for organic chemists. Following the first total synthesis of hirsutellones B reported by Nicolaou and coworkers in 2009.261 Uchiro et al.262 reported the synthesis of hirsutellone B via a successful application of Ullmann coupling reaction in the formation of the 13-membered ring in 2011 (Scheme 81).
Scheme 81.
b. C-N Formation
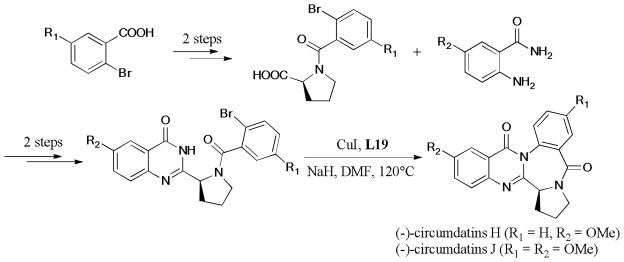
Circumdatins263,264 belong to a class of marine natural products isolated from fungi of Aspergillus. Recently, Circumdatins H and J265 were isolated from Aspergillus ochraceus and Aspergillus ostianus, respectively. Circumdatin H exhibited good inhibitory activity of the mammalian mitochondrial respiratory chain. In 2010, Kshirsagar and Argade266 reported the total synthesis of Circumdatins H and J. (Scheme 82). Optimal conditions of the final step of the Ullmann coupling were achieved using L-proline, as the ligand, and NaH, as the base. The methoxy group played an important role in the maintenance of chirality under this basic condition.
Scheme 82.
Recently, Ichikawa et al.267,268 described the total synthesis of pacidamycin D and 3′-hydroxypacidamycin D. The Ullmann coupling reaction of the Z-oxyvinyl iodides and tetrapeptide carboxamide was introduced in the late stages of the synthetic route. The corresponding Z-oxyacyl enamide product was achieved in 69% and 86% yields under the optimized conditions (CuI, L14, Cs2CO3, THF, 70°C). Subsequent antibacterial evaluation revealed that pacidamycin D and 3′-hydroxypacidamycin D exhibited similar antibacterial activity against P. aeruginosa strains as well as comparable inhibitory activity against MraY enzyme (Scheme 83).
Scheme 83.

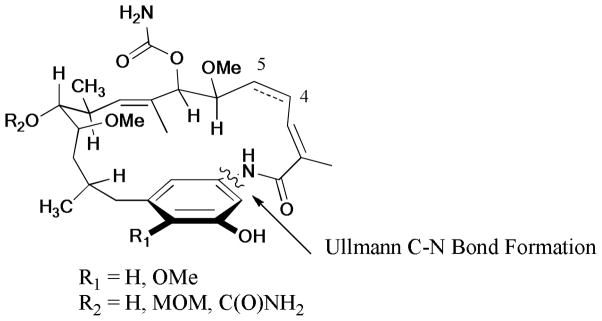
The Ullmann C-N coupling strategy was successfully employed in the synthesis of reblastatin (R1 = OMe, R2 = H, C4-C5 = H4), autolytimycin (R1 = R2 = H, C4-C5 = H4), and their analogues.269 These compounds were evaluated as potential Hsp90 inhibitors; reblastatin exhibited the most potent inhibitory activity with the lowest Kd value of 7.34 nM (Scheme 84).
Scheme 84.
Conclusion
Ullmann coupling reaction has become a powerful and essential tool in organic synthesis and drug discovery. Copper-catalyzed Ullmann reactions were well developed recently by employing novel ligands and ancillary synthetic tools. Herein, we have reviewed recent advances and applications of the copper-catalyzed Ullmann chemistry in the synthesis of heterocycles, drug-like molecules, and natural products. Among many exciting and rapid developments of the Ullmann coupling reactions, we believe, green synthetic methodologies, such as metal-, ligand-, and additive-free conditions, recyclable heterogeneous catalysts, and microwave-assisted synthesis will continue to have a significant impact on this field. We also noted that the reactions with low catalyst loading (<1 mol%) are relatively rare.270 Furthermore, aryl chlorides and tosylates are largely excluded as the active substrates in most cases. These challenges necessitate the synthetic organic community to further explore and develop novel and efficient ligands and catalytic systems to expand the scope and generality of the century-old Ullmann chemistry.
Acknowledgments
We thank financial support from the National Institutes of Health grant (R15AI092315).
References
- 1.Kohei T, Miyaura N. Cross-Coupling Reactions. Springer; Berlin Heidelberg: 2002. [Google Scholar]
- 2.Söderberg BCG. Coord Chem Rev. 2006;250:2411. [Google Scholar]
- 3.Omae I. Coord Chem Rev. 2011;255:139. [Google Scholar]
- 4.Tsuji J. Palladium Reagents and Catalysts: New Perspectives for the 21st Century. John Wiley & Sons, Ltd; 2005. [Google Scholar]
- 5.Wu W, Jiang H. Acc Chem Res. 2012;45:1736. doi: 10.1021/ar3000508. [DOI] [PubMed] [Google Scholar]
- 6.Ackermann L, Lygin AV, Hofmann N. Org Lett. 2011;13:3278. doi: 10.1021/ol201244s. [DOI] [PubMed] [Google Scholar]
- 7.Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338. doi: 10.1021/ja801213x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugita N, Hayashi S, Hino F, Takanami T. J Org Chem. 2012;77:10488. doi: 10.1021/jo302122f. [DOI] [PubMed] [Google Scholar]
- 9.Zbieg JR, Moran J, Krische MJ. J Am Chem Soc. 2011;133:10582. doi: 10.1021/ja2046028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marx VM, Herbert MB, Keitz BK, Grubbs RH. J Am Chem Soc. 2012;135:94. doi: 10.1021/ja311241q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seiser T, Roth OA, Cramer N. Angew Chem Int Ed. 2009;48:6320. doi: 10.1002/anie.200903189. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Li Y, Zhang XS, Chen K, Wang X, Shi ZJ. J Am Chem Soc. 2011;133:15244. doi: 10.1021/ja205228y. [DOI] [PubMed] [Google Scholar]
- 13.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 14.Torborg C, Beller M. Adv Synth Catal. 2009;351:3027. [Google Scholar]
- 15.Lightowler S, Hird M. Chem Mater. 2004;16:3963. [Google Scholar]
- 16.Zhang HH, Xing CH, Hu QS. J Am Chem Soc. 2012;134:13156. doi: 10.1021/ja302745t. [DOI] [PubMed] [Google Scholar]
- 17.Zhan Y, Cao K, Xue P, Lu R. Tetrahedron Lett. 2013;54:594. [Google Scholar]
- 18.Ullmann F, Bielecki J. Ber Dtsch Chem Ges. 1901;34:2174. [Google Scholar]
- 19.Ullmann F. Ber Dtsch Chem Ges. 1903;36:2382. [Google Scholar]
- 20.Goldberg I. Ber Dtsch Chem Ges. 1906;39:1691. [Google Scholar]
- 21.Ullmann F, Sponagel P. Ber Dtsch Chem Ges. 1905;38:2211. [Google Scholar]
- 22.Hurtley WRH. J Chem Soc. 1929:1870. [Google Scholar]
- 23.Marcoux JF, Doye S, Buchwald SL. J Am Chem Soc. 1997;119:10539. [Google Scholar]
- 24.Fagan PJ, Hauptman E, Shapiro R, Casalnuovo A. J Am Chem Soc. 2000;122:5043. [Google Scholar]
- 25.Buck E, Song ZJ, Tschaen D, Dormer PG, Volante RP, Reider PJ. Org Lett. 2002;4:1623. doi: 10.1021/ol025839t. [DOI] [PubMed] [Google Scholar]
- 26.Wolter M, Nordmann G, Job GE, Buchwald SL. Org Lett. 2002;4:973. doi: 10.1021/ol025548k. [DOI] [PubMed] [Google Scholar]
- 27.Ma D, Cai Q. Acc Chem Res. 2008;41:1450. doi: 10.1021/ar8000298. [DOI] [PubMed] [Google Scholar]
- 28.Cristau HJ, Cellier PP, Hamada S, Spindler JF, Taillefer M. Org Lett. 2004;6:913. doi: 10.1021/ol036290g. [DOI] [PubMed] [Google Scholar]
- 29.Lv X, Bao W. J Org Chem. 2007;72:3863. doi: 10.1021/jo070443m. [DOI] [PubMed] [Google Scholar]
- 30.Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004;248:2337. [Google Scholar]
- 31.Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054. doi: 10.1021/cr8002505. [DOI] [PubMed] [Google Scholar]
- 32.Bedos-Belval F, Rouch A, Vanucci-Bacque C, Baltas M. MedChemComm. 2012;3:1356. [Google Scholar]
- 33.Evano G, Toumi M, Coste A. Chem Commun. 2009:4166. doi: 10.1039/b905601g. [DOI] [PubMed] [Google Scholar]
- 34.Ma D, Jiang Y. Chimia. 2011;65:914. doi: 10.2533/chimia.2011.914. [DOI] [PubMed] [Google Scholar]
- 35.Senra JD, Aguiar LCS, Simas ABC. Curr Org Synth. 2011;8:53. [Google Scholar]
- 36.Fischer C, Koenig B. Beilstein J Org Chem. 2011;7:59. doi: 10.3762/bjoc.7.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monnier F, Taillefer M. Angew Chem Int Ed. 2008;47:3096. doi: 10.1002/anie.200703209. [DOI] [PubMed] [Google Scholar]
- 38.Monnier F, Taillefer M. Angew Chem Int Ed. 2009;48:6954. doi: 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- 39.Das P, Sharma D, Kumar M, Singh B. Curr Org Chem. 2010;14:754. [Google Scholar]
- 40.Marinelli F. Curr Org Synth. 2012;9:2. [Google Scholar]
- 41.Klapars A, Antilla JC, Huang X, Buchwald SL. J Am Chem Soc. 2001;123:7727. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- 42.Anastas PT, Kirchhoff MM. Acc Chem Res. 2002;35:686. doi: 10.1021/ar010065m. [DOI] [PubMed] [Google Scholar]
- 43.Horváth IT, Anastas PT. Chem Rev. 2007;107:2169. doi: 10.1021/cr078380v. [DOI] [PubMed] [Google Scholar]
- 44.Liu YH, Li G, Yang LM. Tetrahedron Lett. 2009;50:343. [Google Scholar]
- 45.Zhang Q, Wang D, Wang X, Ding K. J Org Chem. 2009;74:7187. doi: 10.1021/jo9012157. [DOI] [PubMed] [Google Scholar]
- 46.Rao H, Jin Y, Fu H, Jiang Y, Zhao Y. Chem-Eur J. 2006;12:3636. doi: 10.1002/chem.200501473. [DOI] [PubMed] [Google Scholar]
- 47.Xia N, Taillefer M. Chem-Eur J. 2008;14:6037. doi: 10.1002/chem.200800436. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Zhang Z, Wang Y, Zheng X, Wang Z. Eur J Org Chem. 2008:5112. [Google Scholar]
- 49.Maiti D, Buchwald SL. J Org Chem. 2010;75:1791. doi: 10.1021/jo9026935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng AY, Hsieh JC. Tetrahedron Lett. 2012;53:71. [Google Scholar]
- 51.Altman RA, Shafir A, Choi A, Lichtor PA, Buchwald SL. J Org Chem. 2008;73:284. doi: 10.1021/jo702024p. [DOI] [PubMed] [Google Scholar]
- 52.Naidu AB, Raghunath OR, Prasad DJC, Sekar G. Tetrahedron Lett. 2008;49:1057. [Google Scholar]
- 53.Qian C, Zong Q, Fang D. Chin J Chem. 2012;30:199. [Google Scholar]; Chem Abstr. 2012;156:477259. [Google Scholar]
- 54.Maiti D, Buchwald SL. J Am Chem Soc. 2009;131:17423. doi: 10.1021/ja9081815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khalilzadeh MA, Hosseini A, Pilevar A. Eur J Org Chem. 2011:1587. [Google Scholar]
- 56.Niu J, Zhou H, Li Z, Xu J, Hu S. J Org Chem. 2008;73:7814. doi: 10.1021/jo801002c. [DOI] [PubMed] [Google Scholar]
- 57.Tlili A, Xia N, Monnier F, Taillefer M. Angew Chem Int Ed. 2009;48:8725. doi: 10.1002/anie.200903639. [DOI] [PubMed] [Google Scholar]
- 58.Yang D, Fu H. Chem-Eur J. 2010;16:2366. doi: 10.1002/chem.200903468. [DOI] [PubMed] [Google Scholar]
- 59.Hao C, Zhang Y, Jiang Y, Ma D. Chin J Chem. 2010;28:1645. [Google Scholar]; Chem Abstr. 2011;154:87890. [Google Scholar]
- 60.Tlili A, Monnier F, Taillefer M. Chem-Eur J. 2010;16:12299. doi: 10.1002/chem.201001373. [DOI] [PubMed] [Google Scholar]
- 61.Zhou Q, Zhang B, Du T, Gu H, Ye Y, Jiang H, Chen R. Tetrahedron. 2013;69:327. [Google Scholar]
- 62.Zhu X, Su L, Huang L, Chen G, Wang J, Song H, Wan Y. Eur J Org Chem. 2009:635. [Google Scholar]
- 63.Xie J, Zhu X, Huang M, Meng F, Chen W, Wan Y. Eur J Org Chem. 2010:3219. [Google Scholar]
- 64.Meng F, Wang C, Xie J, Zhu X, Wan Y. Appl Organomet Chem. 2011;25:341. [Google Scholar]
- 65.Yang Y, Jin YS, Hu HG, Zhao QJ, Zou Y, Wu QY. Asian J Chem. 2010;22:7435. [Google Scholar]; Chem Abstr. 2011;154:109519. [Google Scholar]
- 66.Li L, Zhu L, Chen D, Hu X, Wang R. Eur J Org Chem. 2011:2692. [Google Scholar]
- 67.Li X, Yang D, Jiang Y, Fu H. Green Chem. 2010;12:1097. [Google Scholar]
- 68.Cortes-Salva M, Nguyen BL, Cuevas J, Pennypacker KR, Antilla JC. Org Lett. 2010;12:1316. doi: 10.1021/ol1002175. [DOI] [PubMed] [Google Scholar]
- 69.Gao X, Fu H, Qiao R, Jiang Y, Zhao Y. J Org Chem. 2008;73:6864. doi: 10.1021/jo800818e. [DOI] [PubMed] [Google Scholar]
- 70.Zhao H, Fu H, Qiao R. J Org Chem. 2010;75:3311. doi: 10.1021/jo100345t. [DOI] [PubMed] [Google Scholar]
- 71.Xia N, Taillefer M. Angew Chem Int Ed. 2009;48:337. doi: 10.1002/anie.200802569. [DOI] [PubMed] [Google Scholar]
- 72.Anokhin MV, Averin AD, Beletskaya IP. Eur J Org Chem. 2011:6240. [Google Scholar]
- 73.Zeng L, Fu H, Qiao R, Jiang Y, Zhao Y. Adv Synth Catal. 2009;351:1671. [Google Scholar]
- 74.Enthaler S. ChemSusChem. 2010;3:1024. doi: 10.1002/cssc.201000145. [DOI] [PubMed] [Google Scholar]
- 75.Feng Y, Wang H, Sun F, Li Y, Fu X, Jin K. Tetrahedron. 2009;65:9737. [Google Scholar]
- 76.Kabir MS, Lorenz M, Van Linn ML, Namjoshi OA, Ara S, Cook JM. J Org Chem. 2010;75:3626. doi: 10.1021/jo1004179. [DOI] [PubMed] [Google Scholar]
- 77.Prasad DJC, Naidu AB, Sekar G. Tetrahedron Lett. 2009;50:1411. [Google Scholar]
- 78.Xu HJ, Zhao XY, Deng J, Fu Y, Feng YS. Tetrahedron Lett. 2009;50:434. [Google Scholar]
- 79.Qiao S, Xie K, Qi J. Chin J Chem. 2010;28:1441. [Google Scholar]; Chem Abstr. 2010;153:505432. [Google Scholar]
- 80.Ramaswamy GK, Mohanasundaram T, Velayutham M, Kuppuswamy BK. J Chin Chem Soc. 2011;58:884. [Google Scholar]; Chem Abstr. 2012;156:533551. [Google Scholar]
- 81.Chen YJ, Chen HH. Org Lett. 2006;8:5609. doi: 10.1021/ol062339h. [DOI] [PubMed] [Google Scholar]
- 82.Kabir MS, Lorenz M, Namjoshi OA, Cook JM. Org Lett. 2010;12:464. doi: 10.1021/ol9026446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang H, Xi C, Miao Z, Chen R. Eur J Org Chem. 2011:3353. [Google Scholar]
- 84.Chang JWW, Chee S, Mak S, Buranaprasertsuk P, Chavasiri W, Chan PWH. Tetrahedron Lett. 2008;49:2018. [Google Scholar]
- 85.Buranaprasertsuk P, Chang JWW, Chavasiri W, Chan PWH. Tetrahedron Lett. 2008;49:2023. [Google Scholar]
- 86.Correa A, Bolm C. Adv Synth Catal. 2007;349:2673. [Google Scholar]
- 87.Zhu L, Li G, Luo L, Guo P, Lan J, You J. J Org Chem. 2009;74:2200. doi: 10.1021/jo802669b. [DOI] [PubMed] [Google Scholar]
- 88.Liu ZJ, Vors JP, Gesing ERF, Bolm C. Adv Synth Catal. 2010;352:3158. [Google Scholar]
- 89.Jiao J, Zhang XR, Chang NH, Wang J, Wei JF, Shi XY, Chen ZG. J Org Chem. 2011;76:1180. doi: 10.1021/jo102169t. [DOI] [PubMed] [Google Scholar]
- 90.Xu HJ, Zheng FY, Liang YF, Cai ZY, Feng YS, Che DQ. Tetrahedron Lett. 2010;51:669. [Google Scholar]
- 91.Ali MA, Saha P, Punniyamurthy T. Synthesis. 2010:908. [Google Scholar]
- 92.Cortes-Salva M, Garvin C, Antilla JC. J Org Chem. 2011;76:1456. doi: 10.1021/jo102235u. [DOI] [PubMed] [Google Scholar]
- 93.Ramana T, Saha P, Das M, Punniyamurthy T. Org Lett. 2010;12:84. doi: 10.1021/ol9024088. [DOI] [PubMed] [Google Scholar]
- 94.Tao C, Lv A, Zhao N, Yang S, Liu X, Zhou J, Liu W, Zhao J. Synlett. 2011:134. [Google Scholar]
- 95.Girard C, Önen E, Aufort M, Beauvière S, Samson E, Herscovici J. Org Lett. 2006;8:1689. doi: 10.1021/ol060283l. [DOI] [PubMed] [Google Scholar]
- 96.Lipshutz BH, Taft BR. Angew Chem Int Ed. 2006;45:8235. doi: 10.1002/anie.200603726. [DOI] [PubMed] [Google Scholar]
- 97.Reddy KR, Kumar NS, Sreedhar B, Kantam ML. J Mol Catal A: Chem. 2006;252:136. [Google Scholar]
- 98.Chassaing S, Kumarraja M, Sani Souna Sido A, Pale P, Sommer J. Org Lett. 2007;9:883. doi: 10.1021/ol0631152. [DOI] [PubMed] [Google Scholar]
- 99.Kantam ML, Jaya VS, Sreedhar B, Rao MM, Choudary BM. J Mol Catal A: Chem. 2006;256:273. [Google Scholar]
- 100.Likhar PR, Roy S, Roy M, Kantam ML, De RL. J Mol Catal A: Chem. 2007;271:57. [Google Scholar]
- 101.Verma S, Kumar N, Jain SL. Tetrahedron Lett. 2012;53:4665. [Google Scholar]
- 102.Wang L, Liu L, Jia D, Cao Y, Xin X. Chin Sci Bull. 2005;50:758. [Google Scholar]; Chem Abstr. 2006;144:303876. [Google Scholar]
- 103.Ling P, Li D, Wang X. J Mol Catal A: Chem. 2012;357:112. [Google Scholar]
- 104.Yang S, Xie W, Zhou H, Wu C, Yang Y, Niu J, Yang W, Xu J. Tetrahedron. 2013;69:3415. [Google Scholar]
- 105.Mulla SAR, Inamdar SM, Pathan MY, Chavan SS. Tetrahedron Lett. 2012;53:1826. [Google Scholar]
- 106.Rad MNS, Behrouz S, Doroodmand MM, Moghtaderi N. Synthesis. 2011:3915. [Google Scholar]
- 107.Islam SM, Mondal S, Mondal P, Roy AS, Tuhina K, Mobarok M. Inorg Chem Commun. 2011;14:1352. [Google Scholar]
- 108.Bhadra S, Sreedhar B, Ranu BC. Adv Synth Catal. 2009;351:2369. [Google Scholar]
- 109.Swapna K, Murthy SN, Jyothi MT, Nageswar YVD. Org Biomol Chem. 2011;9:5978. doi: 10.1039/c1ob05411b. [DOI] [PubMed] [Google Scholar]
- 110.Wang M, Yuan B, Ma T, Jiang H, Li Y. RSC Advances. 2012;2:5528. [Google Scholar]
- 111.Phan NTS, Nguyen TT, Nguyen CV, Nguyen TT. Appl Catal, A. 2013;457:69. [Google Scholar]
- 112.Tamura M, Fujihara H. J Am Chem Soc. 2003;125:15742. doi: 10.1021/ja0369055. [DOI] [PubMed] [Google Scholar]
- 113.Chung MK, Schlaf M. J Am Chem Soc. 2004;126:7386. doi: 10.1021/ja049386u. [DOI] [PubMed] [Google Scholar]
- 114.Hou Z, Theyssen N, Brinkmann A, Leitner W. Angew Chem Int Ed. 2005;44:1346. doi: 10.1002/anie.200461493. [DOI] [PubMed] [Google Scholar]
- 115.Cho JK, Najman R, Dean TW, Ichihara O, Muller C, Bradley M. J Am Chem Soc. 2006;128:6276. doi: 10.1021/ja057480k. [DOI] [PubMed] [Google Scholar]
- 116.Proch S, Mei Y, Villanueva JMR, Lu Y, Karpov A, Ballauff M, Kempe R. Adv Synth Catal. 2008;350:493. [Google Scholar]
- 117.Su FZ, Liu YM, Wang LC, Cao Y, He HY, Fan KN. Angew Chem Int Ed. 2008;47:334. doi: 10.1002/anie.200704370. [DOI] [PubMed] [Google Scholar]
- 118.Ranu BC, Dey R, Chatterjee T, Ahammed S. ChemSusChem. 2012;5:22. doi: 10.1002/cssc.201100348. [DOI] [PubMed] [Google Scholar]
- 119.Babu SG, Karvembu R. Tetrahedron Lett. 2013;54:1677. [Google Scholar]
- 120.Jammi S, Sakthivel S, Rout L, Mukherjee T, Mandal S, Mitra R, Saha P, Punniyamurthy T. J Org Chem. 2009;74:1971. doi: 10.1021/jo8024253. [DOI] [PubMed] [Google Scholar]
- 121.Panda N, Jena AK, Mohapatra S, Rout SR. Tetrahedron Lett. 2011;52:1924. [Google Scholar]
- 122.Swapna K, Murthy SN, Jyothi MT, Nageswar YVD. Org Biomol Chem. 2011;9:5989. doi: 10.1039/c1ob05597f. [DOI] [PubMed] [Google Scholar]
- 123.Reddy KHV, Satish G, Ramesh K, Karnakar K, Nageswar YVD. Tetrahedron Lett. 2012;53:3061. [Google Scholar]
- 124.Sreedhar B, Arundhathi R, Reddy PL, Kantam ML. J Org Chem. 2009;74:7951. doi: 10.1021/jo901462g. [DOI] [PubMed] [Google Scholar]
- 125.Kim JY, Park JC, Kim A, Kim AY, Lee HJ, Song H, Park KH. Eur J Inorg Chem. 2009;2009:4219. [Google Scholar]
- 126.Isomura Y, Narushima T, Kawasaki H, Yonezawa T, Obora Y. Chem Commun. 2012:3784. doi: 10.1039/c2cc30975k. [DOI] [PubMed] [Google Scholar]
- 127.Baig RBN, Varma RS. Chem Commun. 2012:2582. doi: 10.1039/c2cc17283f. [DOI] [PubMed] [Google Scholar]
- 128.Lidström P, Tierney J, Wathey B, Westman J. Tetrahedron. 2001;57:9225. [Google Scholar]
- 129.Larhed M, Moberg C, Hallberg A. Acc Chem Res. 2002;35:717. doi: 10.1021/ar010074v. [DOI] [PubMed] [Google Scholar]
- 130.Polshettiwar V, Varma RS. Acc Chem Res. 2008;41:629. doi: 10.1021/ar700238s. [DOI] [PubMed] [Google Scholar]
- 131.Roberts BA, Strauss CR. Microwave Assisted Organic Synthesis. Blackwell Publishing Ltd; 2009. [Google Scholar]
- 132.Odell LR, Larhed M. Catalytic Methods in Asymmetric Synthesis. John Wiley & Sons, Inc; 2011. [Google Scholar]
- 133.Pitsinos EN, Vidali VP, Couladouros EA. Eur J Org Chem. 2011:1207. [Google Scholar]
- 134.Shen L, Simmons CJ, Sun D. Tetrahedron Lett. 2012;53:4173. doi: 10.1016/j.tetlet.2012.05.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Collins JC, Farley KA, Limberakis C, Liras S, Price D, James K. J Org Chem. 2012;77:11079. doi: 10.1021/jo302089f. [DOI] [PubMed] [Google Scholar]
- 136.Collins JC, James K. MedChemComm. 2012;3:1489. [Google Scholar]
- 137.Baqi Y, Müller CE. Org Lett. 2007;9:1271. doi: 10.1021/ol070102v. [DOI] [PubMed] [Google Scholar]
- 138.Liu ZJ, Vors JP, Gesing ERF, Bolm C. Green Chem. 2011;13:42. [Google Scholar]
- 139.Huang L, Jin C, Su W. Chin J Chem. 2012;30:2394. [Google Scholar]; Chem Abstr. 2013;158:158514. [Google Scholar]
- 140.Yang XD, Li L, Zhang HB. Helv Chim Acta. 2008;91:1435. [Google Scholar]
- 141.Colacino E, Villebrun L, Martinez J, Lamaty F. Tetrahedron. 2010;66:3730. [Google Scholar]
- 142.Bagley MC, Dix MC, Fusillo V. Tetrahedron Lett. 2009;50:3661. [Google Scholar]
- 143.Bagley MC, Fusillo V, Hills BEG, Mulholland AT, Newcombe J, Pentecost LJ, Radley EL, Stephens BR, Turrell CC. ARKIVOC. 2012:294. [Google Scholar]
- 144.Engels V, Benaskar F, Patil N, Rebrov EV, Hessel V, Hulshof LA, Jefferson DA, Vekemans JAJM, Karwal S, Schouten JC, Wheatley AEH. Org Process Res Dev. 2010;14:644. [Google Scholar]
- 145.Benaskar F, Patil NG, Engels V, Rebrov EV, Meuldijk J, Hulshof LA, Hessel V, Wheatley AEH, Schouten JC. Chem Eng J. 2012;207–208:426. [Google Scholar]
- 146.Raj MR, Arun K, Ashokkumar M, Anandan S. Org Prep Proced Int. 2012;44:271. [Google Scholar]
- 147.Weingarten H. J Org Chem. 1964;29:3624. [Google Scholar]
- 148.Cohen T, Cristea I. J Am Chem Soc. 1976;98:748. [Google Scholar]
- 149.Tye JW, Weng Z, Johns AM, Incarvito CD, Hartwig JF. J Am Chem Soc. 2008;130:9971. doi: 10.1021/ja076668w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Giri R, Hartwig JF. J Am Chem Soc. 2010;132:15860. doi: 10.1021/ja105695s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tye JW, Weng Z, Giri R, Hartwig JF. Angew Chem Int Ed. 2010;49:2185. doi: 10.1002/anie.200902245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Paine AJ. J Am Chem Soc. 1987;109:1496. [Google Scholar]
- 153.Jones GO, Liu P, Houk KN, Buchwald SL. J Am Chem Soc. 2010;132:6205. doi: 10.1021/ja100739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sperotto E, van Klink GPM, van Koten G, de Vries JG. Dalton Trans. 2010;39:10338. doi: 10.1039/c0dt00674b. [DOI] [PubMed] [Google Scholar]
- 155.Yu HZ, Jiang YY, Fu Y, Liu L. J Am Chem Soc. 2010;132:18078. doi: 10.1021/ja104264v. [DOI] [PubMed] [Google Scholar]
- 156.Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647. doi: 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]
- 157.Carotenuto A, Fattorusso E, Lanzotti V, Magno S. Eur J Org Chem. 1998:661. [Google Scholar]
- 158.Carney JR, Krenisky JM, Williamson RT, Luo J. J Nat Prod. 2002;65:203. doi: 10.1021/np010374l. [DOI] [PubMed] [Google Scholar]
- 159.Manniche S, Sprogøe K, Dalsgaard PW, Christophersen C, Larsen TO. J Nat Prod. 2004;67:2111. doi: 10.1021/np049843k. [DOI] [PubMed] [Google Scholar]
- 160.Aljaar N, Malakar CC, Conrad J, Strobel S, Schleid T, Beifuss U. J Org Chem. 2012;77:7793. doi: 10.1021/jo3014275. [DOI] [PubMed] [Google Scholar]
- 161.Chen L, Fang Y, Zhao Q, Shi M, Li C. Tetrahedron Lett. 2010;51:3678. [Google Scholar]
- 162.Sun C, Fang Y, Li S, Zhang Y, Zhao Q, Zhu S, Li C. Org Lett. 2009;11:4084. doi: 10.1021/ol9015578. [DOI] [PubMed] [Google Scholar]
- 163.Largeron M, Dupuy H, Fleury MB. Tetrahedron. 1995;51:4953. [Google Scholar]
- 164.Bourlot AS, Sánchez I, Dureng G, Guillaumet G, Massingham R, Monteil A, Winslow E, Pujol MD, Mérour JY. J Med Chem. 1998;41:3142. doi: 10.1021/jm970795t. [DOI] [PubMed] [Google Scholar]
- 165.Brown DW, Ninan A, Sainsbury M. Synthesis. 1997:895. [Google Scholar]
- 166.Massacret M, Lhoste P, Lakhmiri R, Parella T, Sinou D. Eur J Org Chem. 1999:2665. [Google Scholar]
- 167.Chen D, Shen G, Bao W. Org Biomol Chem. 2009;7:4067. doi: 10.1039/b906210f. [DOI] [PubMed] [Google Scholar]
- 168.Bhadra S, Adak L, Samanta S, Maidul Islam AKM, Mukherjee M, Ranu BC. J Org Chem. 2010;75:8533. doi: 10.1021/jo101916e. [DOI] [PubMed] [Google Scholar]
- 169.Bhadra S, Saha A, Ranu BC. J Org Chem. 2010;75:4864. doi: 10.1021/jo100755g. [DOI] [PubMed] [Google Scholar]
- 170.Saha P, Ramana T, Purkait N, Ali MA, Paul R, Punniyamurthy T. J Org Chem. 2009;74:8719. doi: 10.1021/jo901813g. [DOI] [PubMed] [Google Scholar]
- 171.Kitching MO, Hurst TE, Snieckus V. Angew Chem Int Ed Engl. 2012;51:2925. doi: 10.1002/anie.201106786. [DOI] [PubMed] [Google Scholar]
- 172.Kalinin AV, Bower JF, Riebel P, Snieckus V. J Org Chem. 1999;64:2986. doi: 10.1021/jo990114x. [DOI] [PubMed] [Google Scholar]
- 173.Korupalli C, Dandapat A, Prasad DJC, Sekar G. Org Chem Int. 2011:980765. [Google Scholar]
- 174.Sahn JJ, Martin SF. Tetrahedron Lett. 2011;52:6855. doi: 10.1016/j.tetlet.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhao Q, Li C. Org Lett. 2008;10:4037. doi: 10.1021/ol801545a. [DOI] [PubMed] [Google Scholar]
- 176.Lu H, Yuan X, Zhu S, Sun C, Li C. J Org Chem. 2008;73:8665. doi: 10.1021/jo8016617. [DOI] [PubMed] [Google Scholar]
- 177.Kim KY, Shin JT, Lee KS, Cho CG. Tetrahedron Lett. 2004;45:117. [Google Scholar]
- 178.Rivero MR, Buchwald SL. Org Lett. 2007;9:973. doi: 10.1021/ol062978s. [DOI] [PubMed] [Google Scholar]
- 179.Breton GW, Lepore AJ. Molecules. 2011;16:9553. doi: 10.3390/molecules16119553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gao M, Liu X, Wang X, Cai Q, Ding K. Chin J Chem. 2011;29:1199. [Google Scholar]; Chem Abstr. 2011;155:380206. [Google Scholar]
- 181.Wang R, Qian W, Bao W. Tetrahedron Lett. 2012;53:442. [Google Scholar]
- 82.Yang D, Fu H, Hu L, Jiang Y, Zhao Y. J Org Chem. 2008;73:7841. doi: 10.1021/jo8014984. [DOI] [PubMed] [Google Scholar]
- 183.Gong X, Yang H, Liu H, Jiang Y, Zhao Y, Fu H. Org Lett. 2010;12:3128. doi: 10.1021/ol1008813. [DOI] [PubMed] [Google Scholar]
- 184.Touzeau F, Arrault A, Guillaumet G, Scalbert E, Pfeiffer B, Rettori MC, Renard P, Mérour JY. J Med Chem. 2003;46:1962. doi: 10.1021/jm021050c. [DOI] [PubMed] [Google Scholar]
- 185.Fringuelli R, Milanese L, Schiaffella F. Mini-Rev Med Chem. 2005;5:1061. doi: 10.2174/138955705774933365. [DOI] [PubMed] [Google Scholar]
- 186.Schiaffella F, Macchiarulo A, Milanese L, Vecchiarelli A, Costantino G, Pietrella D, Fringuelli R. J Med Chem. 2005;48:7658. doi: 10.1021/jm050685j. [DOI] [PubMed] [Google Scholar]
- 187.Fringuelli R, Giacchè N, Milanese L, Cenci E, Macchiarulo A, Vecchiarelli A, Costantino G, Schiaffella F. Bioorg Med Chem. 2009;17:3838. doi: 10.1016/j.bmc.2009.04.051. [DOI] [PubMed] [Google Scholar]
- 188.Melkonyan F, Topolyan A, Karchava A, Yurovskaya M. Tetrahedron. 2011;67:6826. [Google Scholar]
- 189.Ma ZZ, Hano Y, Nomura T, Chen YJ. Heterocycles. 1997;46:541. [Google Scholar]
- 190.Deng Y, Xu R, Ye Y. J Chin Pharm Sci. 2000;9:116. [Google Scholar]; Chem Abstr. 2001;134:83482. [Google Scholar]
- 191.Witt A, Bergman J. Curr Org Chem. 2003;7:659. [Google Scholar]
- 192.Connolly DJ, Cusack D, O’Sullivan TP, Guiry PJ. Tetrahedron. 2005;61:10153. [Google Scholar]
- 193.Mhaske SB, Argade NP. Tetrahedron. 2006;62:9787. [Google Scholar]
- 194.Liu X, Fu H, Jiang Y, Zhao Y. Angew Chem Int Ed. 2009;48:348. doi: 10.1002/anie.200804675. [DOI] [PubMed] [Google Scholar]
- 195.Yang X, Liu H, Fu H, Qiao R, Jiang Y, Zhao Y. Synlett. 2010:101. [Google Scholar]
- 196.Xu W, Jin Y, Liu H, Jiang Y, Fu, Hua Org Lett. 2011;13:1274. doi: 10.1021/ol1030266. [DOI] [PubMed] [Google Scholar]
- 197.Xu H, Fu H. Chem Eur J. 2012;18:1180. doi: 10.1002/chem.201102794. [DOI] [PubMed] [Google Scholar]
- 198.Sreenivas DK, Ramkumar N, Nagarajan R. Org Biomol Chem. 2012;10:3417. doi: 10.1039/c2ob07179g. [DOI] [PubMed] [Google Scholar]
- 199.Wang C, Li S, Liu H, Jiang Y, Fu H. J Org Chem. 2010;75:7936. doi: 10.1021/jo101685d. [DOI] [PubMed] [Google Scholar]
- 200.Truong VL, Morrow M. Tetrahedron Lett. 2010;51:758. [Google Scholar]
- 201.Huang C, Fu Y, Fu H, Jiang Y, Zhao Y. Chem Commun. 2008:6333. doi: 10.1039/b814011a. [DOI] [PubMed] [Google Scholar]
- 202.Yang D, Liu H, Yang H, Fu H, Hu L, Jiang Y, Zhao Y. Adv Synth Catal. 2009;351:1999. [Google Scholar]
- 203.Tanimori S, Kashiwagi H, Nishimura T, Kirihata M. Adv Synth Catal. 2010;352:2531. [Google Scholar]
- 204.Balalaie S, Bararjanian M, Hosseinzadeh S, Rominger F, Bijanzadeh HR, Wolf E. Tetrahedron. 2011;67:7294. [Google Scholar]
- 205.Zhu X, Xu XP, Sun C, Chen T, Shen ZL, Ji SJ. Tetrahedron. 2011;67:6375. [Google Scholar]
- 206.Jiang M, Li J, Wang F, Zhao Y, Zhao F, Dong X, Zhao W. Org Lett. 2012;14:1420. doi: 10.1021/ol3001624. [DOI] [PubMed] [Google Scholar]
- 207.Lu J, Gong X, Yang H, Fu H. Chem Commun. 2010:4172. doi: 10.1039/c0cc00185f. [DOI] [PubMed] [Google Scholar]
- 208.Cai Q, Li Z, Wei J, Fu L, Ha C, Pei D, Ding K. Org Lett. 2010;12:1500. doi: 10.1021/ol1002225. [DOI] [PubMed] [Google Scholar]
- 209.Zhou B-W, Gao J-R, Jiang D, Jia J-H, Yang Z-P, Jin H-W. Synthesis. 2010:2794. [Google Scholar]
- 210.Wang L, Sullivan GM, Hexamer LA, Hasvold LA, Thalji R, Przytulinska M, Tao ZF, Li G, Chen Z, Xiao Z, Gu WZ, Xue J, Bui MH, Merta P, Kovar P, Bouska JJ, Zhang H, Park C, Stewart KD, Sham HL, Sowin TJ, Rosenberg SH, Lin NH. J Med Chem. 2007;50:4162. doi: 10.1021/jm070105d. [DOI] [PubMed] [Google Scholar]
- 211.Binaschi M, Boldetti A, Gianni M, Maggi CA, Gensini M, Bigioni M, Parlani M, Giolitti A, Fratelli M, Valli C, Terao M, Garattini E. ACS Med Chem Lett. 2010;1:411. doi: 10.1021/ml1001163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Diao X, Xu L, Zhu W, Jiang Y, Wang H, Guo Y, Ma D. Org Lett. 2011;13:6422. doi: 10.1021/ol202721h. [DOI] [PubMed] [Google Scholar]
- 213.Yan J, Zhou F, Qin D, Cai T, Ding K, Cai Q. Org Lett. 2012;14:1262. doi: 10.1021/ol300114w. [DOI] [PubMed] [Google Scholar]
- 214.Biagi G, Giorgi I, Livi O, Scartoni V, Betti L, Giannaccini G, Trincavelli ML. Eur J Med Chem. 2002;37:565. doi: 10.1016/s0223-5234(02)01376-4. [DOI] [PubMed] [Google Scholar]
- 215.Shen HC, Ding FX, Deng Q, Wilsie LC, Krsmanovic ML, Taggart AK, Carballo-Jane E, Ren N, Cai TQ, Wu TJ, Wu KK, Cheng K, Chen Q, Wolff MS, Tong X, Holt TG, Waters MG, Hammond ML, Tata JR, Colletti SL. J Med Chem. 2009;52:2587. doi: 10.1021/jm900151e. [DOI] [PubMed] [Google Scholar]
- 216.Li S, Li Z, Wu J. Adv Synth Catal. 2012;354:3087. [Google Scholar]
- 217.Zhao Q, Li L, Fang Y, Sun D, Li C. J Org Chem. 2008;74:459. doi: 10.1021/jo802235e. [DOI] [PubMed] [Google Scholar]
- 218.Ma D, Geng Q, Zhang H, Jiang Y. Angew Chem Int Ed. 2010;49:1291. doi: 10.1002/anie.200905646. [DOI] [PubMed] [Google Scholar]
- 219.Deng W, Zou Y, Wang Y-F, Liu L, Guo Q-X. Synlett. 2004:1254. [Google Scholar]
- 220.Zhang H, Cao W, Ma D. Synth Commun. 2007;37:25. [Google Scholar]
- 221.Zhang H, Cai Q, Ma D. J Org Chem. 2005;70:5164. doi: 10.1021/jo0504464. [DOI] [PubMed] [Google Scholar]
- 222.Dixit R, Dixit Y, Gautam DC, Gautam N. Phosphorus, Sulfur, Silicon Relat Elem. 2007;183:1. [Google Scholar]
- 223.Hauck M, Schönhaber J, Zucchero AJ, Hardcastle KI, Müller TJJ, Bunz UHF. J Org Chem. 2007;72:6714. doi: 10.1021/jo070922l. [DOI] [PubMed] [Google Scholar]
- 224.Madrid PB, Polgar WE, Toll L, Tanga MJ. Bioorg Med Chem Lett. 2007;17:3014. doi: 10.1016/j.bmcl.2007.03.064. [DOI] [PubMed] [Google Scholar]
- 225.Sailer M, Franz AW, Müller TJJ. Chem-Eur J. 2008;14:2602. doi: 10.1002/chem.200701341. [DOI] [PubMed] [Google Scholar]
- 226.Satish G, Reddy KHV, Ramesh K, Karnakar K, Nageswar YVD. Tetrahedron Lett. 2012;53:2518. [Google Scholar]
- 227.Queiroz MJRP, Dias S, Peixoto D, Rodrigues ARO, Oliveira ADS, Coutinho PJG, Vale-Silva LA, Pinto E, Castanheira EMS. J Photochem Photobiol A: Chem. 2012;238:71. [Google Scholar]
- 228.Yang Y, Wang Z, Yang J, Yang T, Pi W, Ang W, Lin Y, Liu Y, Li Z, Luo Y, Wei Y. Bioorg Med Chem Lett. 2012;22:954. doi: 10.1016/j.bmcl.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 229.Fernandes C, Oliveira L, Tiritan ME, Leitao L, Pozzi A, Noronha-Matos JB, Correia-de-Sá P, Pinto MM. Eur J Med Chem. 2012;55:1. doi: 10.1016/j.ejmech.2012.06.049. [DOI] [PubMed] [Google Scholar]
- 230.Shen L, Maddox M, Adhikari S, Bruhn DF, Kumar M, Lee RE, Hurdle JG, Lee RE, Sun D. J Antibiot. 2013 doi: 10.1038/ja.2013.21. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bryant VC, Kishore Kumar GD, Nyong AM, Natarajan A. Bioorg Med Chem Lett. 2012;22:245. doi: 10.1016/j.bmcl.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wiesner J, Ortmann R, Jomaa H, Schlitzer M. Angew Chem Int Ed. 2003;42:5274. doi: 10.1002/anie.200200569. [DOI] [PubMed] [Google Scholar]
- 233.Baird JK. N Engl J Med. 2005;352:1565. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- 234.Sidhu ABS, Verdier-Pinard D, Fidock DA. Science. 2002;298:210. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hwang JY, Kawasuji T, Lowes DJ, Clark JA, Connelly MC, Zhu F, Guiguemde WA, Sigal MS, Wilson EB, Derisi JL, Guy RK. J Med Chem. 2011;54:7084. doi: 10.1021/jm200636z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tanimori S, Nishimura T, Kirihata M. Bioorg Med Chem Lett. 2009;19:4119. doi: 10.1016/j.bmcl.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 237.Deprez-Poulain R, Cousaert N, Toto P, Willand N, Deprez B. Eur J Med Chem. 2011;46:3867. doi: 10.1016/j.ejmech.2011.05.056. [DOI] [PubMed] [Google Scholar]
- 238.Baqi Y, Lee S-Y, Iqbal J, Ripphausen P, Lehr A, Scheiff AB, Zimmermann H, Bajorath Jr, Müller CE. J Med Chem. 2010;53:2076. doi: 10.1021/jm901851t. [DOI] [PubMed] [Google Scholar]
- 239.Baqi Y, Müller CE. Molecules. 2012;17:2599. doi: 10.3390/molecules17032599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Duan S, Venkatraman S, Hong X, Huang K, Ulysse L, Mobele BI, Smith A, Lawless L, Locke A, Garigipati R. Org Process Res Dev. 2012;16:1787. [Google Scholar]
- 241.Palmeira A, Vasconcelos MH, Paiva A, Fernandes MX, Pinto M, Sousa E. Biochem Pharmacol. 2012;83:57. doi: 10.1016/j.bcp.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 242.Bagley MC, Davis T, Dix MC, Fusillo V, Pigeaux M, Rokicki MJ, Kipling D. J Org Chem. 2009;74:8336. doi: 10.1021/jo9017155. [DOI] [PubMed] [Google Scholar]
- 243.Jung N, Bräse S. Eur J Org Chem. 2009:4494. [Google Scholar]
- 244.Dandepally SR, Williams AL. Tetrahedron Lett. 2010;51:5753. [Google Scholar]
- 245.Yamaguchi S, Tanaka H, Yamada R, Kawauchi S, Takahashi T. Synlett. 2012:1327. [Google Scholar]
- 246.Wu B, He S, Pan Y-j. Planta Med. 2006;72:1244. doi: 10.1055/s-2006-947200. [DOI] [PubMed] [Google Scholar]
- 247.Lin J, Zhang W, Jiang N, Niu Z, Bao K, Zhang L, Liu D, Pan C, Yao X. J Nat Prod. 2008;71:1938. doi: 10.1021/np800226n. [DOI] [PubMed] [Google Scholar]
- 248.Shen L, Sun D. Tetrahedron Lett. 2011;52:4570. doi: 10.1016/j.tetlet.2011.06.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lin WY, Peng CF, Tsai IL, Chen JJ, Cheng MJ, Chen IS. Planta Med. 2005;71:171. doi: 10.1055/s-2005-837786. [DOI] [PubMed] [Google Scholar]
- 250.Salih MQ, Beaudry CM. Org Lett. 2012;14:4026. doi: 10.1021/ol301893t. [DOI] [PubMed] [Google Scholar]
- 251.Zhu ZQ, Beaudry CM. J Org Chem. 2013;78:3336. doi: 10.1021/jo302723n. [DOI] [PubMed] [Google Scholar]
- 252.Zhu ZQ, Salih MQ, Fynn E, Bain AD, Beaudry CM. J Org Chem. 2013;78:2881. doi: 10.1021/jo400157d. [DOI] [PubMed] [Google Scholar]
- 253.Raut GN, Chakraborty K, Verma P, Gokhale RS, Srinivasa Reddy D. Tetrahedron Lett. 2012;53:6343. [Google Scholar]
- 254.Pospíšil J, Müller C, Fürstner A. Chem-Eur J. 2009;15:5956. doi: 10.1002/chem.200802681. [DOI] [PubMed] [Google Scholar]
- 255.Speicher A, Holz J. Tetrahedron Lett. 2010;51:2986. [Google Scholar]
- 256.Harinantenaina L, Quang DN, Takeshi N, Hashimoto T, Kohchi C, Soma GI, Asakawa Y. J Nat Prod. 2005;68:1779. doi: 10.1021/np0502589. [DOI] [PubMed] [Google Scholar]
- 257.Harinantenaina L, Kida S, Asakawa Y. ARKIVOC. 2007;22 [Google Scholar]
- 258.Xi G-m, Sun B, Jiang H-h, Kong F, Yuan H-q, Lou H-x. Bioorg Med Chem. 2010;18:6725. doi: 10.1016/j.bmc.2010.07.055. [DOI] [PubMed] [Google Scholar]
- 259.Isaka M, Rugseree N, Maithip P, Kongsaeree P, Prabpai S, Thebtaranonth Y. Tetrahedron. 2005;61:5577. [Google Scholar]
- 260.Isaka M, Prathumpai W, Wongsa P, Tanticharoen M. Org Lett. 2006;8:2815. doi: 10.1021/ol060926x. [DOI] [PubMed] [Google Scholar]
- 261.Nicolaou KC, Sarlah D, Wu TR, Zhan W. Angew Chem Int Ed. 2009;48:6870. doi: 10.1002/anie.200903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Uchiro H, Kato R, Arai Y, Hasegawa M, Kobayakawa Y. Org Lett. 2011;13:6268. doi: 10.1021/ol202748e. [DOI] [PubMed] [Google Scholar]
- 263.Rahbaek L, Breinholt J. J Nat Prod. 1999;62:904. doi: 10.1021/np980495u. [DOI] [PubMed] [Google Scholar]
- 264.Dai J-R, Carté BK, Sidebottom PJ, Sek Yew AL, Ng S-B, Huang Y, Butler MS. J Nat Prod. 2000;64:125. doi: 10.1021/np000381u. [DOI] [PubMed] [Google Scholar]
- 265.Ookura R, Kito K, Ooi T, Namikoshi M, Kusumi T. J Org Chem. 2008;73:4245. doi: 10.1021/jo800348d. [DOI] [PubMed] [Google Scholar]
- 266.Kshirsagar UA, Argade NP. Org Lett. 2010;12:3716. doi: 10.1021/ol101597p. [DOI] [PubMed] [Google Scholar]
- 267.Okamoto K, Sakagami M, Feng F, Togame H, Takemoto H, Ichikawa S, Matsuda A. Org Lett. 2011;13:5240. doi: 10.1021/ol202124b. [DOI] [PubMed] [Google Scholar]
- 268.Okamoto K, Sakagami M, Feng F, Togame H, Takemoto H, Ichikawa S, Matsuda A. J Org Chem. 2012;77:1367. doi: 10.1021/jo202159q. [DOI] [PubMed] [Google Scholar]
- 269.Wrona IE, Gozman A, Taldone T, Chiosis G, Panek JS. J Org Chem. 2010;75:2820. doi: 10.1021/jo1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Larsson PF, Correa A, Carril M, Norrby PO, Bolm C. Angew Chem Int Ed. 2009;48:5691. doi: 10.1002/anie.200902236. [DOI] [PubMed] [Google Scholar]