

Abstract
The chemokine CXCL12 and its shared seven-transmembrane receptors CXCR4 and CXCR7 regulate diseases including cancer, atherosclerosis, autoimmunity, and HIV infection, making these molecules promising drug targets. These molecules also control key processes in normal development and physiology, suggesting the need to selectively modulate CXCR4 and/or CXCR7 functions and signaling to reduce potential complications of long-term therapy. We previously identified two peptides that functioned as allosteric agonists driving CXCR4-dependent chemotaxis, providing key structural information to design a small number of additional peptides to investigate determinants of CXCL12 interactions and signaling through CXCR4 and CXCR7. In the current study, we show that the previously identified peptides only minimally activated CXCR4 signaling through the cytosolic adapter protein β-arrestin 2 and do not initiate signaling to ERK1/2. By comparison, peptides with diverse N-terminal amino acid sequences effectively activated CXCR7 signaling to β-arrestin 2. One peptide, designated as GSLW based on its N-terminal amino acids, activated CXCR7 signaling and potentiated CXCL12-CXCR7 signaling without blocking the scavenger function of CXCR7 to internalize CXCL12. These results advance our understanding of CXCR7 ligand recognition and signaling, and provide structural information to target allosteric binding sites on this receptor as chemical probes and potential therapeutic agents.
Keywords: chemokine receptor, chemokine, luciferase, bioluminescence, peptide
1. Introduction
Chemokine receptors CXCR4 and CXCR7, members of the larger family of seven-transmembrane (G protein coupled) receptors, are indispensable for normal development of the central nervous and cardiovascular systems in addition to bone marrow stem and progenitor cells [1–5]. These receptors also are central to normal physiology, including trafficking of hematopoietic cells and cell cycle regulation of neural stem cells [6, 7]. CXCR4 and CXCR7 regulate fundamental biologic processes including proliferation, cell survival, and chemotaxis, which contribute to essential functions of these receptors in multiple aspects of normal development and physiology [8]. CXCR4 and/or CXCR7 are associated with a wide spectrum of diseases, such as HIV, cancer, stroke, and autoimmunity, and these receptors also mediate trafficking of stem cell populations to repair damage tissues [9–14]. Key functions of CXCR4 and/or CXCR7 in normal and pathologic conditions emphasize the need to understand both independent and integrated functions of these receptors to develop therapeutic strategies for improving and optimizing treatments. CXCR4 and CXCR7 are promising therapeutic targets in a number of different diseases, motivating ongoing efforts to develop drugs targeting these receptors with the potential for widespread benefits to patients.
CXCR4 and CXCR7 share chemokine CXCL12 (SDF-1) as a common ligand. CXCL12 binding to CXCR4 drives downstream signaling pathways typical of seven transmembrane (G protein coupled) receptors, including activation of G proteins, mitogen activated protein kinases (MAPK), calcium flux, and recruitment of the cytosolic scaffolding protein β-arrestin 2 [15]. By comparison, biologic functions of CXCR7, which binds CXCL12 with 10-fold greater affinity than CXCR4, remain poorly defined and are possibly cell-type specific [16, 17]. CXCR7 acts as a scavenger, or decoy, receptor for CXCL12, removing this chemokine from the extracellular space and degrading it [18, 19]. By controlling levels of CXCL12, CXCR7 establishes chemotactic gradients and limits desensitization of CXCR4. CXCL12 binding to CXCR7 also recruits the cytosolic adapter protein β-arrestin 2 to the receptor. Studies by our group and others show that CXCR7 functions in at least some cell types as a G-protein independent, β-arrestin 2-biased receptor that drives mitogen activated protein kinase (MAPK) pathways from endosomes [20, 21]. Currently available small molecules targeting CXCR7 not only function as orthosteric inhibitors to block CXCL12 binding and internalization but also activate recruitment of β-arrestin 2 to the receptor [22–24]. Such CXCR7-targeted compounds limit the scavenger function of CXCL12, resulting in higher extracellular levels of CXCL12 that may desensitize CXCR4 signaling and chemotaxis. However, these compounds potentially also may initiate CXCR7 signaling in cells, producing unexpected side effects. Identifying molecules that selectively regulate β-arrestin 2 recruitment to CXCR7 versus CXCL12 scavenging will help dissect mechanisms of action and functional outputs of CXCR7. Molecules that regulate defined functions of CXCR7 also may provide leads for new drugs targeting this receptor.
Previous studies have defined the amino (N)-terminal amino acids of CXCL12 as critical determinants of binding and signaling through CXCR4. While full-length CXCL12 (68 amino acids) activates CXCR4, truncation of the N-terminus by 2–6 amino acids by enzymes such as dipeptidyl peptidase-IV (CD26) abrogates receptor signaling and switches CXCL12 from an agonist to an antagonist [25]. Amino acids 1–17 of CXCL12 are sufficient to activate CXCR4 signaling with greatly reduced potency, and amino acids 9–17 also function as weak agonists of this receptor [26, 27]. By comparison, little is known about critical amino acids required for CXCL12 binding to CXCR7. Conversion of the three arginine residues in the first 20 amino acids of CXCL12 to citrulline, which reduces total positive charge of the chemokine, eliminates binding to CXCR7 [28]. However, effects of specific N-terminal amino acids and fragments of CXCL12 on CXCR7 binding and activation remain to be defined.
We previously screened a library of 160,000 17-mer peptides with randomized amino acids at positions 1–4 and amino acids 5–17 from wild-type CXCL12 to identify structural determinants of CXCL12-CXCR4 interactions as potential lead molecules for new therapeutic agents [29]. Two peptides from this screen with amino acids ASLW and RSVM at positions 1–4 functioned as allosteric agonists for CXCR4. Based on chemotaxis of CXCR4+ cells, we determined that RSVM and ASLW acted as partial and super agonists, respectively. Studies with these peptides established the presence of alternative ligand-binding sites on CXCR4 that could be exploited to regulate CXCR4 while avoiding side effects of directly blocking the CXCL12 binding site.
In the current study, we further investigated effects of peptides ASLW and RSVM on CXCR4 signaling pathways. We also analyzed effects of these two peptides and a limited number of similar 17-mer peptides using alanine substitution for the first four residues (glycine for the Ala in ASWL) on CXCR7 binding, function, and activation of downstream signaling to β-arrestin 2. Unlike CXCR4 cells in which peptides without wild-type amino acids at the N-terminus only minimally stimulated recruitment of β-arrestin 2, several 17-mer peptides activated CXCR7-β-arrestin 2 at least as well as modest concentrations of full-length, wild-type CXCL12. In particular, one 17-mer peptide with amino acids GSLW at positions 1–4 only partially blocked CXCL12 binding to CXCR7, did not alter CXCR7-dependent scavenging, and potentiated CXCL12-CXCR7 signaling to β-arrestin 2. These studies show that CXCR7 binds peptides with diverse N-terminal amino acids and identifies allosteric regulation of CXCR7. The GSLW peptide establishes proof-of-concept for discovering molecules that modulate selective functions of CXCR7 as chemical probes and potential therapeutic agents.
2. Materials and Methods
2.1. Cells
We recently described MDA-MB-231 breast cancer cells stably expressing click beetle luciferase complementation reporters for CXCR4 and CXCR7 interaction with β-arrestin 2 [30]. We also used MDA-MB-231 cells stably transduced with CXCR7 fused to GFP (231-CXCR7), 293T cells expressing CXCL12-α fused to Gaussia luciferase (CXCL12-GL), and 293T cells with CXCR4-GFP [31, 32]. We purchased parental MDA-MB-231 cells from the ATCC. U343 glioblastoma cells were a gift from ChemoCentryx. We cultured all cells in DMEM with 10% serum, 1% glutamine, and 0.1% penicillin/streptomycin (Life Technologies, Grand Island, NY, USA).
2.2. Peptides
Peptides were synthesized to contain amino acids 1–4 from wild-type CXCL12 or selected amino acids substituted at these positions (Bio Basic, Inc., Ontario, CA). For all peptides, amino acids from 5–17 correspond to the sequence of wild-type CXCL12. Sequences of peptides are listed in Table 1. We prepared 5 mM stocks of all peptides in water with 1% acetic acid (Sigma-Aldrich, St. Louis, MO, USA).
Table 1. Peptide Sequences.
All 17-mer peptides have amino acids LSYRCPCRFFESH at positions 5–17.
| Peptide | Amino acids 1-4 |
|---|---|
| 1 (wild-type) | KPVS |
| 2 | RSVM |
| 3 | ASLW |
| 4 | ASVM |
| 5 | RAVM |
| 6 | RSAM |
| 7 | RSVA |
| 8 | GSLW |
| 9 | AALW |
| 10 | ASAW |
| 11 | ASLA |
2.3. β-arrestin complementation assay
We performed click beetle luciferase complementation assays for interaction of CXCR4 or CXCR7 with β-arrestin 2 as described previously [30, 31]. Briefly, we plated 1.5 x 104 231-CXCR4/β-arrestin 2 or 231-CXCR7/β-arrestin 2 cells in black wall 96 well plates. We incubated cells in phenol red free DMEM medium (Life Technologies, Grand Valley, NY, USA) with 0.2% probumin BSA (EMD Millipore, Billerica, MA, USA) for 30 minutes before adding 150 μg/ml luciferin (Promega, Madison, WI, USA) for five minutes followed by concentrations of CXCL12-α (R&D Systems) or peptides listed in figures (n = 4 per condition). We treated control wells with vehicle alone. After adding CXCL12 or peptides, we acquired bioluminescence images every three minutes for 45 minutes using an IVIS 100 instrument (Perkin-Elmer). We quantified data as photon flux (photons/second/cm2/sr) using LivingImage software (Perkin-Elmer). For time course assays, we normalized photon flux values for cells treated with CXCL12 or a peptide to corresponding cells treated only with vehicle at matched time points. We graphed these normalized data as fold change relative to control.
2.4. Fluorescence microscopy
We plated 293T cells at 1 x 105 cells per well in 12 well plates and transfected cells the next day with 100 ng each of plasmids for CXCR7-GFP and β-arrestin 2-mCherry [22]. Cells were moved to 35-mm dishes (Thermo Fisher Scientific, Waltham, MA, USA) for confocal microscopy performed two days after transfection. Prior to imaging, we incubated cells for one hour in phenol red free DMEM with 0.2% probumin and then added 300 ng/ml CXCL12 or 300 μM of selected peptides listed in the figure legend for 30 minutes. We imaged cells by confocal fluorescence microscopy on an Olympus Fluoview1000MPE Twin system using a 60X, 1.1 NA water immersion objective and sequential line scanning with 488 nm and 559 nm lasers. We merged green and red images with Olympus Fluoview software.
2.5. Competitive binding assay for CXCL12-GL and cell surface CXCR7
We measured binding of CXCL12-GL to cell surface CXCR7 in the absence or presence of full-length CXCL12 or various peptides as reported previously [33]. Briefly, we plated parental MDA-MB-231 cells or MDA-MB-231 cells stably transduced with CXCR7-GFP (231-CXCR7) at 2 x 104 cells per well in 96 well black wall plates. We changed cells to phenol red free medium with 0.2% probumin for 30 minutes and incubated plates on ice for 30 minutes before adding 300 ng/ml CXCL12 or 300 μM peptide to each well for 45 minutes on ice (n = 4 per condition). We then added CXCL12-GL (≈10 ng/ml) to each well for an additional 45 minutes on ice before quantifying cell-associated CXCL12-GL by bioluminescence imaging with coelenterazine (Promega, Madison, WI, USA).
2.6. Uptake of CXCL12-GL
We determined intracellular accumulation of CXCL12-GL in parental 231 and 231-CXCR7 cells as described [19]. We plated cells as detailed for the competitive binding assay described above. We incubated cells with ≈ 10 ng/ml CXCL12-GL in the presence of 300 ng/ml CXCL12, 300 μM peptide, or vehicle control for 60 minutes at 37°C. At the end of the incubation period, we acid-washed cells twice to remove extracellular, cell-associated chemokine measuring internalized CXCL12-GL in all cells by bioluminescence imaging (n = 4 per condition).
2.7. Western blotting
We cultured 293T-CXCR4 or U343 cells overnight in serum free medium with 0.2% bovine serum albumin. We incubated 293T-CXCR4 cells with 300 ng/ml CXCL12, 300 μM of selected peptides, or vehicle only for five minutes before lysing cells in RIPA buffer with added protease and phosphatase inhibitors (Thermo Fisher Scientific, Rockford, IL, USA). For U343 cells, we treated cells with 100 ng/ml CXCL12, 100 μM selected peptides, both CXCL12 and peptide, or vehicle control for 20 minutes before lysing cells. We used Western blotting to determine levels of phosphorylated ERK1/2 or JNK as described (Cell Signaling Technology, Beverly, MA, USA) [21]. We stripped and re-probed blots for total ERK1/2 or JNK as well as GAPDH as loading controls. We quantified band intensities by ImageJ and expressed data as relative intensity of phosphorylated ERK1/2 divided by total ERK1/2 and GAPDH for the U343 Western blot.
2.8. Statistics
We graphed data as mean values ± SEM (n = 4 per condition). Experiments were performed 2–3 times each. We determined statistically significant differences by unpaired t-test with p < 0.05 defining significance (GraphPad Prism).
3. Results
3.1. Allosteric peptide agonists minimally activate CXCR4 signaling
We previously identified two allosteric peptide agonists of CXCR4 through a high-throughput cDNA library screen in yeast [29]. These 17-mer peptides had amino acids RSVM or ASLW replacing the initial four amino acids of wild-type CXCL12-α, followed by amino acids 5–17 from the wild-type chemokine. Using chemotaxis as an assay for CXCR4 activity, we previously determined that RSVM and ASLW functioned as a partial agonist and superagonist, respectively. However, we did not analyze effects of these peptides on activation of downstream effector pathways in CXCL12-CXCR4 signaling.
CXCL12 binding to CXCR4 results in recruitment of the cytosolic adapter protein β-arrestin 2, inducing internalization of the ligand-bound receptor and initiating β-arrestin 2-dependent signaling from endosomes. To quantify association of CXCR4 with β-arrestin 2, we used a click beetle luciferase complementation assay developed by our group in which CXCR4 and β-arrestin 2 are fused to amino (N)- and carboxy (C)-terminal luciferase fragments, respectively [34]. Interaction of CXCR4 with β-arrestin 2 reconstitutes luciferase activity, producing a bioluminescent readout of CXCL12-CXCR4 signaling to β-arrestin 2.
We treated MDA-MB-231 breast cancer cells stably expressing the CXCR4-β-arrestin 2 luciferase complementation reporter with full-length CXCL12-α, a peptide with the first 17 amino acids of wild-type CXCL12 (peptide 1), RSVM and ASLW 17-mer peptides (peptides 2, 3), or one of nine other 17-mer peptides with various combinations of amino acids in the first four positions (peptides 3–11 in Table 1). Bioluminescence imaging in intact cells showed greatest induction of luciferase activity from association of CXCR4 and β-arrestin 2 in cells treated with full-length CXCL12-α. Relative to control cells, full-length CXCL12-α increased luciferase activity by greater than 3-fold after 20 minutes, followed by a gradual decline (p < 0.005) (Fig 1). The wild-type 17-mer peptide was the next most potent inducer of β-arrestin 2 recruitment with a two-fold increase in bioluminescence after 45 minutes (p < 0.01). For the wild-type and all other 17-mer peptides, the kinetics of β-arrestin 2 recruitment were delayed relative to full-length CXCL12 with luciferase activity increasing through 45 minutes. RSVM, ASLW, and five other peptides (peptides 4, 8, 9, 10, 11) produced modest (1.3–1.5-fold), yet statistically significant increases in CXCR4-β-arrestin 2 interaction as compared with cells treated with vehicle control (p < 0.05). These peptides contained the following first four amino acids: 4, ASVM; 8, GSLW; 9, AALW; 10, ASAW; and 11, ASLA. By comparison, peptides 5 (RAVM), 6 (RSAM), and 7 (RSVA) did not mediate recruitment of β-arrestin 2 to CXCR4. These data show that peptides with a variety of amino acid motifs with positive or neutral charge can activate CXCR4 signaling to β-arrestin 2, although none of these mutant sequences are as effective as the wild-type first four amino acids.
Figure 1. Recruitment of β-arrestin 2 to CXCR4 in response to peptides.
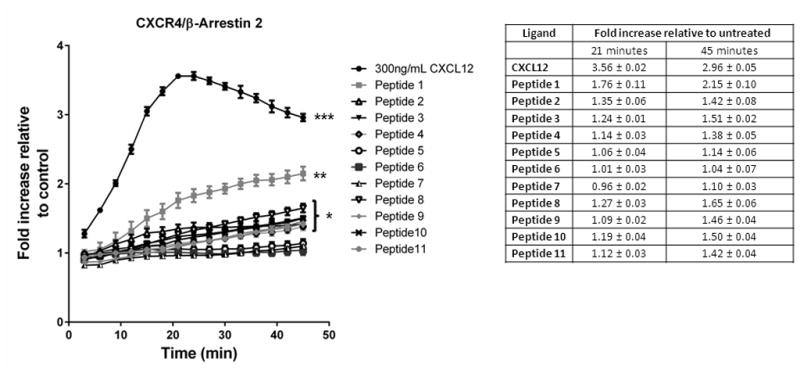
MDA-MB-231 breast cancer cells expressing a click beetle luciferase complementation reporter for CXCR4 interaction with β-arrestin 2 were incubated with 300 ng/ml CXCL12, 300 μM 17-mer peptide, or vehicle control. We acquired bioluminescence images every 3 minutes for 45 minutes. Graph shows mean values ± SEM for fold increase in bioluminescence relative to cells treated with vehicle control (n = 4 per condition). *** denotes p < 0.005; **, p < 0.01; and *, p < 0.05 relative to vehicle control.
CXCL12-CXCR4 signaling also results in phosphorylation of the mitogen activated protein kinases ERK1/2, contributing to known growth promoting effects of this pathway [35]. For these assays, we focused on a subset of peptides that most effectively stimulated recruitment of β-arrestin 2 to CXCR4. We treated 293T cells expressing CXCR4 with full-length CXCL12 or peptides 2, 3, 8, or 10 for 5 minutes and then harvested cells for Western blot. While we detected phosphorylation of ERK1/2 in cells treated with full-length CXCL12, none of the tested peptides activated this known CXCR4 signaling pathway (Fig 2). These data show that selected 17-mer peptides modestly initiate recruitment of β-arrestin 2 to CXCR4 without signaling to ERK1/2, further demonstrating that these peptides interact with CXCR4 in a manner distinct from natural ligand CXCL12.
Figure 2. Regulation of CXCR4 signaling to ERK1/2 by peptides.
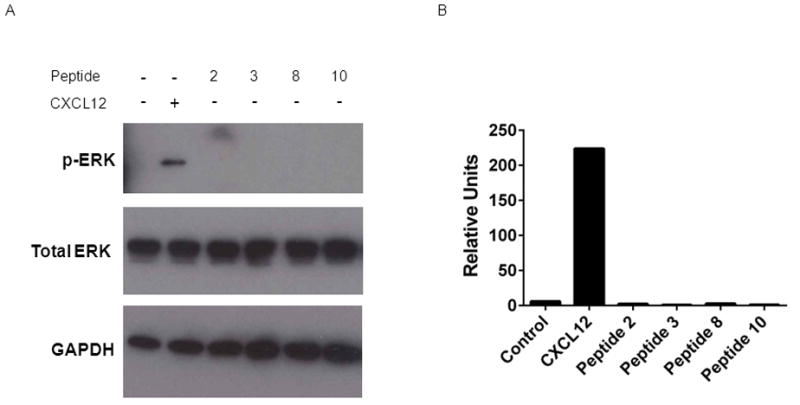
A. 293T cells expressing CXCR4 were treated for 5 minutes with 100 ng/ml CXCL12, 300 μM 17-mer peptides, or vehicle control. Total cell lysates were probed for phosphorylated ERK1/2. Membranes then were stripped and re-probed for total ERK1/2 and GAPDH as loading controls. B. Graph shows quantified data for phosphorylated ERK1/2 normalized to total ERK1/2 and GAPDH.
3.2. Diverse N-terminal motifs drive CXCR7-β-arrestin 2 interaction
In addition to CXCR4, CXCL12 binds to a second chemokine receptor, CXCR7. CXCL12 binding to CXCR7 also recruits β-arrestin 2 to the activated receptor, initiating β-arrestin 2-dependent signaling. We quantified effects of various peptides to activate CXCR7-β-arrestin 2 using a luciferase complementation system analogous to that described for CXCR4.
Unlike results with CXCR4 and β-arrestin 2, all tested peptides significantly increased bioluminescence from CXCR7 association with β-arrestin 2 relative to vehicle control. Signal increased progressively through 45 minutes, consistent with prior studies showing prolonged association of CXCR7 and β-arrestin 2 (Fig 3) [22]. Remarkably, by the 45 minute time point, all 17-mer peptides except peptides 10 and 11 stimulated recruitment of β-arrestin 2 to CXCR7 at least as effectively as 300 ng/ml full-length CXCL12. In addition to peptide 1 with wild-type amino acids at the N-terminus and the CXCR4 allosteric peptide 3 (ASLW), peptides 5 (RAVM), 8 (GSLW), and 9 (AALW) generated significantly greater association of CXCR7 and β-arrestin 2 than full-length CXCL12 (p < 0.05).
Figure 3. Activation of CXCR7-β-arrestin 2 interaction by peptides.
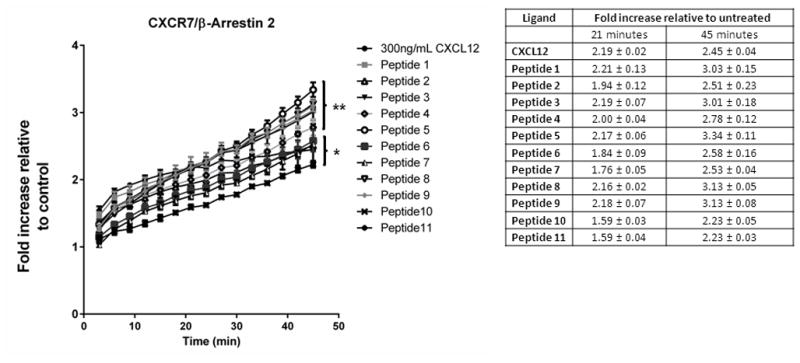
MDA-MB-231 breast cancer cells expressing a click beetle luciferase complementation reporter for CXCR7 interaction with β-arrestin 2 were incubated with 300 ng/ml CXCL12, 300 μM 17-mer peptide, or vehicle control. We acquired bioluminescence images every 3 minutes for 45 minutes. Graph shows mean values ± SEM for fold increase in bioluminescence relative to cells treated with vehicle control (n = 4 per condition). **, p < 0.01; and *, p < 0.05 relative to vehicle control.
To further investigate effects of selected peptides on recruitment of β-arrestin 2 to CXCR7, we treated luciferase complementation cells with three different concentrations of selected peptides (1, 3, 5, 8, 9, and 10) (Fig 4). All peptides except peptide 3 produced bioluminescence significantly above control after 45 minutes of incubation at 25 μM, the lowest concentration tested (p < 0.05) (Fig 4). We observed concentration-dependent increases in bioluminescence from all peptides, although differences between 25 and 100 μM were not significant for peptides 1 and 10 (p < 0.05). Treating cells with 300 μM of peptide increased bioluminescence by at least three-fold above control after 45 minutes, which was comparable to or greater than that produced by 300 ng/ml CXCL12.
Figure 4. Concentration-dependent recruitment of β-arrestin 2 to CXCR7 by selected peptides.
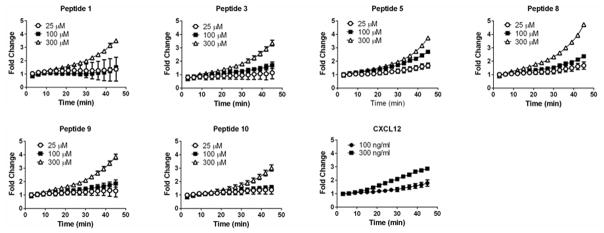
We incubated CXCR7-β-arrestin 2 luciferase complementation reporter cells with 25, 100, or 300 μM of peptides 1, 3, 5, 8, 9, or 10 and quantified bioluminescence over time as described in figure 3. We also incubated parallel cultures of cells with 100 or 300 ng/ml CXCL12. Graph displays mean values ± SEM for fold increase in bioluminescence relative to cells treated with vehicle control (n = 4 per condition).
We corroborated luciferase complementation data by analyzing co-localization of CXCR7 and β-arrestin 2 on endosomes by fluorescence microscopy following treatment with selected peptides. We and others previously have demonstrated that CXCL12 drives association of CXCR7 and β-arrestin 2 using fusions of each protein to fluorescent proteins [20, 22]. Treatment with peptides 1, 3, 5, or 8 for 30 minutes promoted formation of endosomes with both CXCR7-GFP and β-arrestin-mCherry (Fig 5). The extent of co-localization between CXCR7-GFP and β-arrestin 2-mCherry produced by various peptides was comparable to that produced by CXCL12 as assessed qualitatively. Overall, these results suggest that a wide range of N-terminal amino acids activate CXCR7 and that truncating the C-terminus of CXCL12 has much less effect on CXCR7 signaling to β-arrestin 2 as compared with CXCR4.
Figure 5. Ligand-dependent co-localization of CXCR7 and β-arrestin 2 on endosomes.
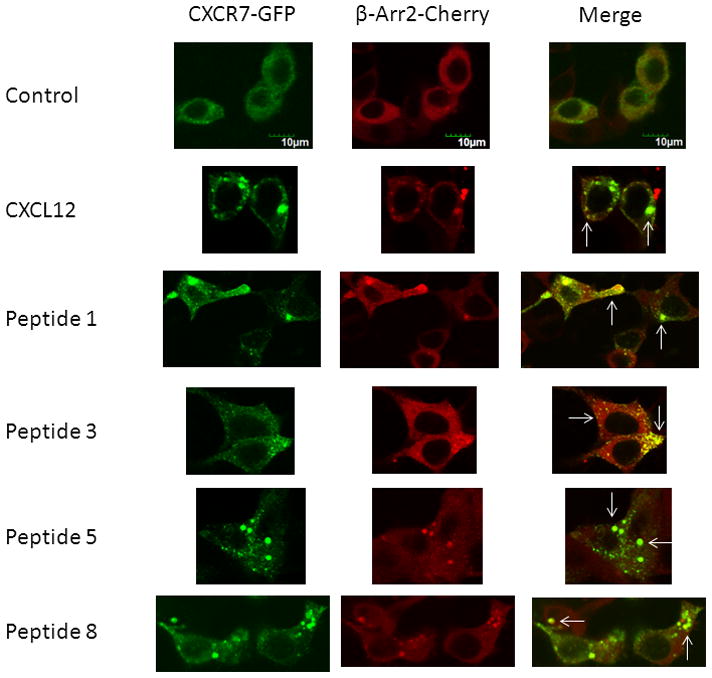
293T cells transiently transfected with CXCR7-GFP and β-arrestin 2-mCherry were treated for 30 minutes with vehicle only, 300 ng/ml CXCL12, or 300 μM of listed peptides. Images show separate green, red, and merged images from confocal microscopy. White arrows highlight some endosomes with co-localization of CXCR7-GFP and β-arrestin 2-mCherry. Scale bar denotes 10 μm.
3.3. Effects of peptides on CXCL12 binding to CXCR7 and internalization
To further investigate interactions of peptides with CXCR7, we performed a receptor binding assay to determine to what extent full-length CXCL12-α or one of the 17-mer peptides block binding of CXCL12-α fused to Gaussia luciferase (CXCL12-GL) to MDA-MB-231 cells stably transduced with CXCR7. We incubated cells on ice with 300 ng/ml CXCL12 or 300 μM of a single peptide and then quantified binding of CXCL12-GL by bioluminescence imaging. We previously demonstrated that fusing Gaussia luciferase to CXCL12 does not alter binding or signaling properties relative to the unfused chemokine [32]. As a negative control, we also quantified binding of CXCL12-GL to parental MDA-MB-231 cells that do not express CXCR7 [36].
In the absence of competitive inhibitor, 231-CXCR7 cells bound approximately two-fold more CXCL12-GL than control 231 cells (Fig 6). Treatment with CXCL12 or nine of the 11 peptides reduced bound CXCL12-GL to levels equal to or below that quantified for 231 control cells. Reduction in signal below the level of control cells may be due to competition for CXCL12-GL binding to cell surface glycosaminoglycans such as heparan sulfate [37]. By comparison, CXCL12-GL binding to 231-CXCR7 cells treated with peptides 8 (GSLW) or 9 (AALW) remained significantly greater than 231 control cells (p < 0.01 and p < 0.05, respectively). Peptides 8 and 9 inhibited binding of CXCL12-GL to CXCR7 by ≈ 50% and 80%, respectively. These two peptides also produced recruitment of β-arrestin 2 to CXCR7 that slightly exceeded full-length CXCL12. Results from competition binding suggest that peptide 8 and to a lesser extent peptide 9 do not interact with CXCR7 exclusively at the binding site for CXCL12.
Figure 6. Competition of peptides with CXCL12 for binding to CXCR7.
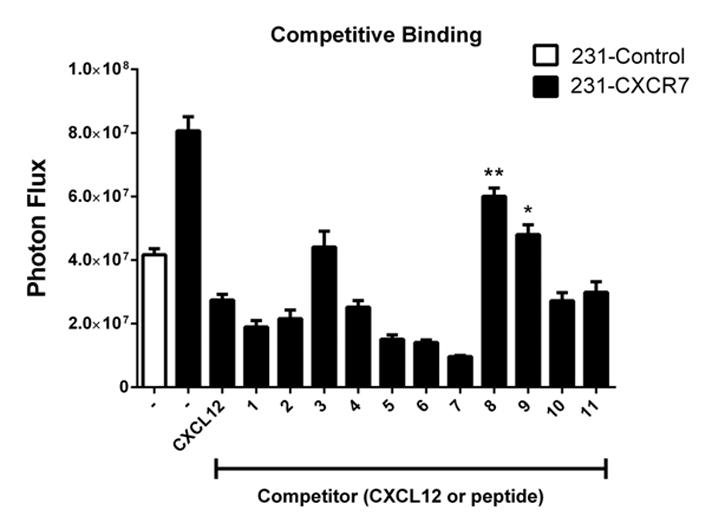
MDA-MB-231 cells stably transduced with CXCR7 were incubated on ice with 300 ng/ml CXCL12, 300 μM peptide, or vehicle control prior to adding ≈ 10 ng/ml CXCL12 fused to Gaussia luciferase (CXCL12-GL). 231-control cells without CXCR7 also were incubated with vehicle control before adding CXCL12-GL to account for CXCR7-independent binding to cells. Cells remained on ice for 45 minutes and then were washed with PBS before quantifying cell-associated CXCL12-GL. Graph shows mean values + SEM for photon flux (n = 4 per condition). **, p < 0.01 and *, p < 0.05 relative to 231-control cells.
To further investigate effects of various peptides on CXCL12-CXCR7 interactions, we quantified uptake of CXCL12-GL in 231-CXCR7 cells at 37°C. We and others have shown that CXCR7 functions as a scavenger receptor to internalize CXCL12 from the extracellular space [18, 19, 38]. We incubated 231-CXCR7 cells or parental 231 cells with CXCL12-GL in the absence or presence of CXCL12, peptide, or vehicle control for 60 minutes and then quantified intracellular CXCL12-GL by bioluminescence. 231-CXCR7 cells accumulated ≈ three-fold more CXCL12-GL than control 231 cells (Fig 7). Full length CXCL12 and the wild-type 17-mer peptide (peptide 1) completely blocked CXCR7-dependent uptake of CXCL12-GL, as did peptides 5 (RAVM) and 10 (ASAW). Peptides 3 (ASLW), 6 (RSAM), 7 (RSVM), and 9 (AALW) also significantly decreased uptake of CXCL12 relative to 231-CXCR7 cells incubated with vehicle control. However, we identified four peptides that did not significantly reduce accumulation of CXCL12-GL in 231-CXCR7 cells (peptide 2, RSVM; peptide 4, ASVM; peptide 8, GSLW; and peptide 11, ASLA). Intracellular CXCL12-GL was higher in 231-CXCR7 incubated with peptides 8 or 11 and CXCL12-GL as compared with 231-CXCR7 cells treated only with vehicle control, although differences were not statistically significant. We note that peptide 8 also produced least inhibition of CXCL12-GL binding to 231-CXCR7 cells. Peptides 2, 4, and 11 were much less effective inhibitors of CXCL12-GL internalization as compared with CXCL12-CXCR7 binding (Fig 6), potentially because we performed internalization assays by adding peptides and CXCL12-GL simultaneously to cells at 37°C rather than pre-incubating peptides with cells on ice prior to addition of CXCL12-GL in the competition assays. These data further indicate that activation of CXCR7 signaling to β-arrestin 2 by peptide 8 occurs via an allosteric effect.
Figure 7. Effects of peptides on CXCR7-dependent scavenging of CXCL12.
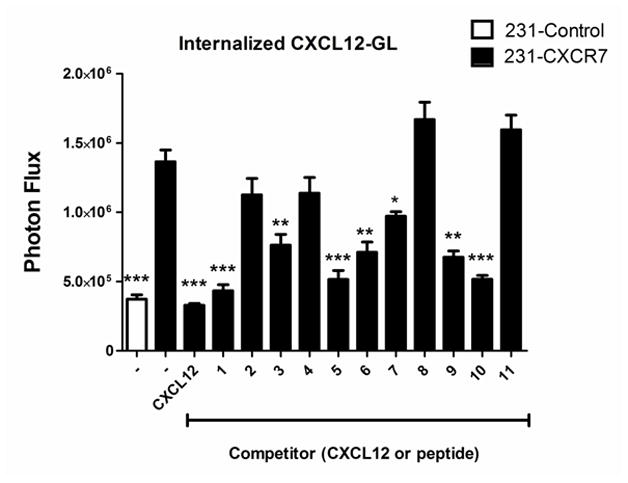
231-CXCR7 cells were incubated with 300 ng/ml CXCL12, 300 μM peptide, or vehicle control along with ≈ 10 ng/ml CXCL12-GL for one hour at 37°C. 231-control cells incubated with vehicle control and CXCL12-GL were used to measure CXCR7-independent uptake of CXCL12-GL. Cells were acid-washed to removed extracellular chemokine prior to quantifying internalized CXCL12-GL bioluminescence. Graph shows mean values + SEM for bioluminescence quantified as photon flux (n = 4 per condition). ***, p < 0.005; **, p < 0.01; *, p < 0.05 relative to uptake in 231-CXCR7 cells incubated with vehicle control.
3.4. Selected peptides potentiate CXCL12-dependent recruitment of β-arrestin 2 to CXCR7
We hypothesized that combined effects of selected peptides and CXCL12 would potentiate activation of CXCR7 signaling to β-arrestin 2 at reduced concentrations of either peptide or CXCL12 ligand. We selected peptides representative of different behaviors in the three different functional assays for CXCR7: high activation of CXCR7-β-arrestin 2 interaction only (peptide 5, RAVM); high activation of CXCR7 signaling to β-arrestin 2 with minimal effects on CXCL12-GL binding and internalization (peptide 8, GSLW); high activation of CXCR7 signaling to β-arrestin 2 with modest reductions in CXCL12 binding and inhibition of chemokine uptake (peptide 9, AALW); or relatively lower activation of CXCR7 signaling and complete inhibition of CXCL12-GL binding and uptake (peptide 10, ASAW). Based on studies shown in Fig 4, we selected 100 μM peptide as a concentration that produced detectable activation of β-arrestin 2 recruitment to CXCR7. We also used a concentration of CXCL12 (100 ng/ml) that only minimally activated CXCR7 signaling at the selected 30 minute time point. Interestingly, combining CXCL12 with either peptide 8 or peptide 9 significantly increased bioluminescence from CXCR7 and β-arrestin 2 above levels produced by either ligand alone (p < 0.01 and p < 0.05, respectively) (Fig 8). The combination of peptide 8 and CXCL12 generated ≈ 40% more signal than peptide 8 alone, suggesting allosteric activation of CXCR7 signaling.
Figure 8. Combined effects of selected peptides and CXCL12 on recruitment of β-arrestin 2 to CXCR7.
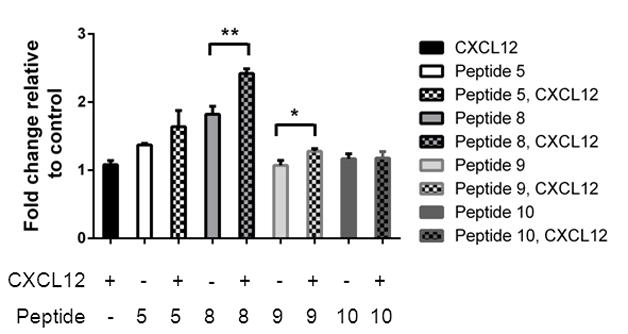
MDA-MB-231 CXCR7-β-arrestin 2 complementation cells were incubated with 100 ng/ml CXCL12, 100 μM listed peptide, or both for 30 minutes before measuring bioluminescence. Control cells were treated with vehicle only. Graph shows mean values + SEM for fold increase in bioluminescence relative to cells treated with vehicle control (n = 4 per condition). **, p < 0.01; *, p < 0.05.
Since CXCR7 signaling through β-arrestin 2 may activate MAPK pathways, we investigated phosphorylation of ERK1/2 or JNK in response to incubation with CXCL12 and/or selected peptides [39]. For these studies, we used U343 glioma cells, which previously have been shown to activate MAPK pathways in a CXCR7-dependent manner [40]. All tested peptides added at 100 μM activated ERK1/2 with peptides 5 and 10 producing ≈ 25% more phosphorylation of ERK1/2 than 100 ng/ml full-length CXCL12 (Fig 9A, B). Combined treatment with peptide 5, 8, or 9 and CXCL12 also produced moderate (≈ 30%) increases in normalized phosphorylation of ERK1/2 above that observed for peptide alone. By comparison, only peptide 9 activated JNK above basal levels (Fig 9C, D). Similar to results with ERK1/2, both peptides 5 and 8 in combination with CXCL12 stimulated phosphorylation of JNK to a greater extent than either peptide or CXCL12 used singly. These data show that diverse N-terminal amino acids may activate CXCR7 signaling to ERK1/2 and to a lesser extent JNK, and selected peptide sequences modestly potentiate effects of CXCL12 on MAPK pathways.
Figure 9. Peptide regulation of CXCR7 signaling to MAPK pathways.
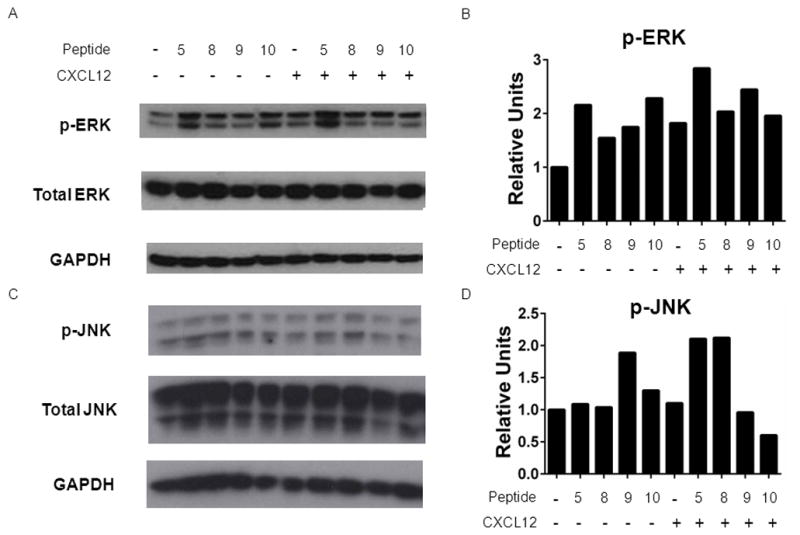
A, C) U343 cells were incubated with 100 ng/ml CXCL12, 100 μM listed peptide, or both for 20 minutes. Total cell lysates were analyzed by Western blot for phosphorylated ERK1/2 or JNK. Membranes were stripped and re-probed for total ERK1/2 or JNK and GAPDH as controls for loading. B, D) Graph shows normalized data from two experiments for phosphorylated ERK1/2 (B) or JNK (D) relative to total ERK1/2 (B) or JNK (D) and GAPDH.
4. Discussion
There is increasing interest in identifying allosteric modulators of seven transmembrane receptors as novel therapeutic agents [41, 42]. Allosteric modulators act at sites separate from the orthosteric binding site for ligands, preserving endogenous regulation of receptor signaling and dynamics. Particularly for sub-families of receptors that share a common ligand and/or critically regulate normal physiology, allosteric modulators potentially improve selectivity and safety of drugs. Allosteric modulators may have unique modes of action that control only selective functions of a receptor, expanding the range of therapeutic applications. In addition, allosteric modulators are important research tools for analyzing complexities of seven-transmembrane receptor biology.
We previously identified two allosteric peptide modulators of CXCR4 using yeast to screen a large 17-mer library of peptides with semi-randomized amino acids at positions 1–4 and amino acids 5–17 from wild-type CXCL12. Two peptides with sequences RSVM and ASLW in positions 1–4 served as lead molecules to further analyze effects on CXCR4 signaling and investigate regulation of CXCR7, the second receptor for CXCL12. Using recruitment of the cytosolic adapter protein β-arrestin 2 as a reporter for CXCR4 signaling, peptides RSVM and ASLW stimulated substantially less signaling than full-length CXCL12 or even a 17-mer peptide with all wild-type amino acids. The RSVM and ASLW peptides also did not activate CXCR4-dependent signaling to ERK1/2, unlike full-length CXCL12. These results are consistent with prior studies showing that N-terminal amino acids critically determine CXCL12-CXCR4 signaling activity, while C-terminal truncations of the chemokine markedly reduce potency for CXCR4 [25–27].
Remarkably, we discovered multiple peptides that regulate CXCR7 signaling and function without using a library screening strategy. Including the wild-type 17-mer and peptides RSVM and ASLW, we found that all 11 peptides tested activated recruitment of β-arrestin 2 to CXCR7. These peptides contained a mixture of positive, neutral, polar, and/or non-polar amino acids at positions 1–4. Four of the peptides (peptides 3, ASLW; 5, RAVM; 8, GSLW; and 9, AALW) drove interaction of CXCR7 and β-arrestin 2 at least as effectively as the wild-type 17-mer peptide. The luciferase complementation signal from these peptides also was comparable to full-length CXCL12, although we used approximately 1000-fold higher concentrations of peptides than CXCL12. Nevertheless, we note that we used these same concentrations of peptides and CXCL12 for CXCR4-β-arrestin 2 assays in which peptides all were comparatively much less effective. Relative to CXCR4, these results establish that CXCR7 signaling to β-arrestin 2 is much less dependent upon specific N-terminal amino acids and full-length chemokine.
While all tested peptides promoted recruitment of CXCR7 with β-arrestin 2, data from CXCL12 binding and internalization assays indicate that some peptides functioned allosterically to drive this signaling pathway. In particular, peptide 8 (GSLW) only modestly blocked CXCL12 binding and had no effect on CXCR7-dependent internalization of this chemokine. The GSLW peptide, as well as peptide 9 (AALW), also potentiated CXCR7 interaction with β-arrestin 2 and modestly increased activation of ERK1/2 when combined with full-length CXCL12. These peptides establish essential proof-of-concept for identifying allosteric modulators of CXCR7 that dissociate inhibition of CXCL12 scavenging from activation of the β-arrestin 2 signaling pathway.
Previous studies have identified small molecule and peptide allosteric antagonists of CXCR4 in addition to the RSVM and ASLW peptide allosteric agonists we described previously [43, 44]. By comparison, very little is known about allosteric regulation of CXCR7. AMD3100, a clinically-approved small molecule inhibitor of CXCR4, acts allosterically to enhance binding of CXCL12 to CXCR7 and modestly activate CXCR7 signaling through β-arrestin 2 [22, 45]. However, allosteric modulation of CXCR7 requires relatively high concentrations of AMD3100, limiting utility of this molecule as a chemical probe for CXCR7 especially in small animal models. Another peptidomimetic antagonist of CXCR4, TC14012, also has been shown to stimulate recruitment of CXCR7 to β-arrestin 2, although studies have not determined if this compound acts at orthosteric or allosteric sites on CXCR7 [22, 46]. Since currently available small molecules targeting CXCR7 block both CXCL12 binding and internalization while driving recruitment of β-arrestin 2, peptides such as GSLW and AALW serve as probes to investigate distinct aspects of CXCR7 function.
Key functions of CXCR7 in diseases including cancer growth and metastasis, autoimmunity, and stroke make this receptor an active target for drug discovery and development. CXCR7 also regulates functions of CXCR4 in these and other diseases through processes such as scavenging CXCL12 and forming receptor heterodimers that alter downstream signaling dynamics [2, 47]. Therapy with an inhibitor of CXCR7 could not only block CXCR7 functions but also alter CXCR4-dependent pathways, potentially producing unexpected and toxic effects particularly when used chronically. The peptides identified in this study provide structural information for designing allosteric modulators of CXCR7 as potential new therapeutic agents to target pathologic functions of the receptor while maintaining functions essential for normal physiology. Ultimately, allosteric agonists or antagonists may advance understanding of CXCR7 under physiologic and pathologic conditions and lead to novel drugs targeting this receptor.
Acknowledgments
This work was supported by United States National Institutes of Health National Cancer Institute Grants R01CA136553, R01CA136829, R01CA142750, and P50CA093990 (GDL) and Allergy and Infectious Diseases Grant R01AI082295 (EL). Research also was supported by NIH grant 1S10RR28819-1. The authors thank ChemoCentryx for providing U343 cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yan X, Cai S, Xiong X, Sun W, Dai X, Chen S, et al. Chemokine receptor CXCR7 mediates human endothelial progenitor cells survival, angiogenesis, but not proliferation. J Cell Biochem. 2012;113:1437–46. doi: 10.1002/jcb.24015. [DOI] [PubMed] [Google Scholar]
- 2.Sierro F, Biben C, Martinez-Munoz L, Mellado M, Rashohoff R, Li M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A. 2007;104:14759–64. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez-Alcaniz J, Haege S, Mueller W, Pla R, Mackay F, Schulz S, et al. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron. 2011;69:77–90. doi: 10.1016/j.neuron.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Li G, Stanco A, Long J, Crawford D, Potter G, et al. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron. 2011;69:61–76. doi: 10.1016/j.neuron.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393:591–4. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 6.Li M, Chang C, Lathia J, Wang L, Pacenta H, Cotleur A, et al. Chemokine receptor CXCR4 signaling modulates the growth factor-induced cell cycle of self-renewing and multipotent neural progenitor cells. Glia. 2011;59:108–18. doi: 10.1002/glia.21080. [DOI] [PubMed] [Google Scholar]
- 7.Moll N, Ransohoff R. CXCL12 and CXCR4 in bone marrow physiology. Expert Rev Hematol. 2010;3:315–22. doi: 10.1586/ehm.10.16. [DOI] [PubMed] [Google Scholar]
- 8.Decaillot F, Kazmi M, Lin Y, Ray-Saha S, Sakmar T, Sachdev P. CXCR7/CXCR4 heterodimer constitutively recruits {beta}-arrestin to enhance cell migration. J Biol Chem. 2011;286:32188–97. doi: 10.1074/jbc.M111.277038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimizu N, Soda Y, Kanbe K, Liu H, Mukai R, Kitamura T, et al. A putative G protein-coupled receptor, RDC1, is a novel coreceptor for human and simian immunodeficiency viruses. J Virol. 2000;74:619–26. doi: 10.1128/jvi.74.2.619-626.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duda D, Kozin S, Kirkpatrick N, Xu L, Fukumura D, Jain R. CXCL12 (SDF1alpha) - CXCR4/CXCR7 Pathway Inhibition: An Emerging Sensitizer for Anti-Cancer Therapies? Clin Cancer Res. 2011;17:2074–80. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakondi B, Shimada I, Peterson B, Spees J. SDF-1α secreted by human CD133-derived multipotent stromal cells promotes neural progenitor cell survival through CXCR7. Stem Cells Dev. 2011;20:1021–9. doi: 10.1089/scd.2010.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Huang J, Li Y, Yang G. Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr Drug Targets. 2012;13:166–72. doi: 10.2174/138945012799201603. [DOI] [PubMed] [Google Scholar]
- 13.Karin N. The multiple faces of CXCL12 (SDF-1α) in the regulation of immunity during health and disease. J Leukoc Biol. 2010;88:463–74. doi: 10.1189/jlb.0909602. [DOI] [PubMed] [Google Scholar]
- 14.Tang Y, Zhu W, Cheng M, Chen L, Zhang J, Sun T, et al. Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing CXCR4 expression. Circ Res. 2009;104:1209–16. doi: 10.1161/CIRCRESAHA.109.197723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teicher B, Fricker S. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 16.Balabanian K, Lagane B, Infantino S, Chow K, Harriague J, Moepps B, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–6. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- 17.Burns J, Summers B, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–13. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boldajipour B, Mahabaleshwar S, Kardash E, Reichman-Fried M, Blaser H, Minina S, et al. Control of chemokine-guided cell migration by ligand sequestration. Cell. 2008;132:463–73. doi: 10.1016/j.cell.2007.12.034. [DOI] [PubMed] [Google Scholar]
- 19.Luker K, Steele J, Mihalko L, Luker G. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene. 2010;29:4599–610. doi: 10.1038/onc.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajagopal S, Kim J, Ahn S, Craig S, Lam C, Gerard N, et al. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628–32. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray P, Mihalko L, Coggins N, Moudgil P, Ehrlich A, Luker K, et al. Carboxy-terminus of CXCR7 regulates receptor localization and function. Int J Biochem Cell Biol. 2012;44:669–78. doi: 10.1016/j.biocel.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luker K, Gupta M, Steele J, Foerster B, Luker G. Imaging ligand-dependent activation of CXCR7. Neoplasia. 2009;11:1022–35. doi: 10.1593/neo.09724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witmans M, Maussang D, Sirci F, Scholten D, Canals M, Mujic-Delic A, et al. Synthesis, modeling and functional activity of substituted styrene-amides as small-molecule CXCR7 agonists. Eur J Med Chem. 2012;51:184–92. doi: 10.1016/j.ejmech.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 24.Zabel B, Wang Y, Lewen S, Berahovich R, Penfold M, Zhang P, et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol. 2009;183:3204–11. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- 25.Cho S, Xu M, Roboz J, Lu M, Mascarenhas J, Hoffman R. The effect of CXCL12 processing on CD34+ cell migratioin in myeloproliferative neoplasms. Cancer Res. 2010;70:3402–10. doi: 10.1158/0008-5472.CAN-09-3977. [DOI] [PubMed] [Google Scholar]
- 26.Heveker N, Montes M, Germeroth L, Amara A, Trautmann A, Alizon M, et al. Dissociation of the signalling and antiviral properties of SDF-1-derived small peptides. Curr Biol. 1998;8:369–76. doi: 10.1016/s0960-9822(98)70155-1. [DOI] [PubMed] [Google Scholar]
- 27.Loetscher P, Gong J, Dewald B, Baggiolini M, Clark-Lewis I. N-terminal peptides of stromal cell-derived factor-1 with CXC chemokine receptor 4 agonist and antagonist activities. J Biol Chem. 1998;278:22279–83. doi: 10.1074/jbc.273.35.22279. [DOI] [PubMed] [Google Scholar]
- 28.Struyf S, Noppen S, Loos T, Mortier A, Gouwy M, Verbeke H, et al. Citrullination of CXCL12 differentially reduces CXCR4 and CXCR7 binding with loss of inflammatory and anti-HIV activity via CXCR4. J Immunol. 2009;182:666–74. doi: 10.4049/jimmunol.182.1.666. [DOI] [PubMed] [Google Scholar]
- 29.Sachpatzidis A, Benton B, Manfredi J, Wang H, Hamilton A, HGD, et al. Identification of allosteric peptide agonists of CXCR4. J Biol Chem. 2003;278:896–907. doi: 10.1074/jbc.M204667200. [DOI] [PubMed] [Google Scholar]
- 30.Coggins N, Trakimas D, Chang S, Ehrlich A, Ray P, Luker K, et al. Systems level analysis of integrated CXCR4 and CXCR7 signaling outputs to β-arrestin2. BMC Comput Biol. 2013 submitted. [Google Scholar]
- 31.Luker K, Gupta M, Luker G. Imaging CXCR4 signaling with firefly luciferase complementation. Anal Chem. 2008;80:5565–73. doi: 10.1021/ac8005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luker K, Gupta M, Luker G. Bioluminescent CXCL12 fusion protein for cellular studies of CXCR4 and CXCR7. Biotechniques. 2009;47:625–32. doi: 10.2144/000113126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luker K, Mihalko L, Schmidt B, Lewin S, Ray P, Shcherbo D, et al. In vivo imaging of ligand receptor binding with Gaussia luciferase complementation. Nat Med. 2012;18:172–7. doi: 10.1038/nm.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salomonnson E, Stacer A, Ehrlich A, Luker K, Luker G. Imaging CXCL12-CXCR4 signaling in ovarian cancer. PLoS ONE. 2013;8:e51500. doi: 10.1371/journal.pone.0051500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho H, Kyoung K, Seo M, Kim Y, Bae Y, Jung J. Overexpression of CXCR4 increases migration and proliferation of human adipose tissue stromal cells. Stem Cells Dev. 2006;15:853–64. doi: 10.1089/scd.2006.15.853. [DOI] [PubMed] [Google Scholar]
- 36.Song J, Cavnar S, Walker A, Luker K, Gupta M, Tung Y, et al. Microfluidic endothelium for studying the intravascular adhesion of metastatic breast cancer cells. PLoS One. 2009;4:e5756. doi: 10.1371/journal.pone.0005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friand V, Haddad O, Papy-Garcia D, Hlawaty H, Vassy R, Hamma-Kourbali Y, et al. Glycosaminoglycan mimetics inhibit SDF-1/CXCL12-mediated migration and invasion of human hepatoma cells. Glycobiology. 2009;19:1511–24. doi: 10.1093/glycob/cwp130. [DOI] [PubMed] [Google Scholar]
- 38.Naumann U, Cameroni E, Pruenster M, Mahabaleshwar S, Raz E, Zerwes H, et al. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One. 2010;5:e9175. doi: 10.1371/journal.pone.0009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma W, Liu Y, Ellison N, Shen J. Induction of C-X-C chemokine receptor type 7 (CXCR7) switches stromal cell-derived factor-1 (SDF-1) signaling and phagocytic activity in macrophages linked to atherosclerosis. J Biol Chem. 2013;288:15481–94. doi: 10.1074/jbc.M112.445510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hattermann K, Held-Feindt J, Lucius R, Muerkoster S, Penfold M, Schall T, et al. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010;70:3299–308. doi: 10.1158/0008-5472.CAN-09-3642. [DOI] [PubMed] [Google Scholar]
- 41.Conn P, Christopoulos A, Lindsley C. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Martin B, Brenneman R, Luttrell L, Maudsley S. Allosteric modulators of g protein-coupled receptors: future therapeutics for complex physiological disorders. J Pharmacol Exp Ther. 2009;331:340–8. doi: 10.1124/jpet.109.156380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janz J, Ren Y, Looby R, Kazmi M, Sachdev P, Grunbeck A, et al. Direct interaction between an allosteric agonist pepducin and the chemokine receptor CXCR4. J Am Chem Soc. 2011;133:15878–81. doi: 10.1021/ja206661w. [DOI] [PubMed] [Google Scholar]
- 44.Mosi R, Anastassova V, Cox J, Darkes M, Idzan S, Labrecque J, et al. The molecular pharmacology of AMD11070: an orally bioavailable CXCR4 HIV entry inhibitor. Biochem Pharmacol. 2012;83:472–9. doi: 10.1016/j.bcp.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Kalatskaya I, Berchiche Y, Gravel S, Limberg B, Rosenbaum J, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75:1240–7. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 46.Gravel S, Malouf C, Boulais P, Berchiche Y, Oishi S, Fujii N, et al. The peptidomimetic CXCR4 antagonist TC14012 recruits beta-arrestin 2 to CXCR7: roles of receptor domains. J Biol Chem. 2010;285:37939–43. doi: 10.1074/jbc.C110.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luker K, Gupta M, Luker G. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009;23:823–34. doi: 10.1096/fj.08-116749. [DOI] [PMC free article] [PubMed] [Google Scholar]