

Abstract
Photodynamic therapy is an emerging treatment modality that is under intensive preclinical and clinical investigations for many types of disease including cancer. Despite the promise, there is a lack of a reliable drug delivery vehicle that can transport photosensitizers (PSs) to tumors in a site-specific manner. Previous efforts have been focused on polymer- or liposome-based nanocarriers, which are usually associated with a suboptimal PS loading rate and a large particle size. We report herein that a RGD4C-modified ferritin (RFRT), a protein-based nanoparticle, can serve as a safe and efficient PS vehicle. Zinc hexadecafluorophthalocyanine (ZnF16Pc), a potent PS with a high 1O2 quantum yield but poor water solubility, can be encapsulated into RFRTs with a loading rate as high as ~60 wt % (i.e., 1.5 mg of ZnF16Pc can be loaded on 1 mg of RFRTs), which far exceeds those reported previously. Despite the high loading, the ZnF16Pc-loaded RFRTs (P-RFRTs) show an overall particle size of 18.6 ± 2.6 nm, which is significantly smaller than other PS–nanocarrier conjugates. When tested on U87MG subcutaneous tumor models, P-RFRTs showed a high tumor accumulation rate (tumor-to-normal tissue ratio of 26.82 ±4.07 at 24 h), a good tumor inhibition rate (83.64% on day 12), as well as minimal toxicity to the skin and other major organs. This technology can be extended to deliver other metal-containing PSs and holds great clinical translation potential.
Keywords: photodynamic therapy, photosensitizer, targeted delivery, ferritin, nanoparticle
Photodynamic therapy (PDT) is an emerging treatment modality that has shown promise for many types of disease, including cancer.1,2 PDT consists of three components: a photosensitizer (PS), light, and oxygen. PSs are usually pharmacologically inactive in the dark. When light at a specific wavelength is applied, PSs are activated, producing reactive oxygen species (ROS) such as 1O2, which are cytotoxic and capable of killing nearby cells.3 Due to limited light penetration, PDT was first used in the clinic to treat superficial conditions, such as lupus vulgaris and skin cancer.4 This limitation has changed due to the establishment of methods that can deliver light to certain internal organs.5,6 Taking PDT for prostate cancer as an example, ultrasound imaging is used to guide the insertion of a needle and subsequently a laser fiber into the prostate.4 Once the optic fiber is in position, laser light will be applied to illuminate the tissue and to elicit PDT. The advances in light delivery have expanded the applications of PDT. For instance, porfimer sodium-based PDT has been approved for the treatment of esophageal cancer in the United States, early- and late-stage lung cancer in The Netherlands, and bladder cancer in Canada.7,8 PDT is also found to be effective in treating recurrence prostate tumors after irradiation, and the related application is in clinical trials.4
On the other hand, selective delivery of PSs to tumors remains a problem. In the clinic, PS delivery is achieved through a passive approach, by controlling the time interval between the PS injection and light irradiation.9 This lack of a targeting mechanism causes poor tumor selectivity, leading to a tumor/normal tissue accumulation ratio that is typically less than 2.10 As a result, PDT is often associated with off-target damage to the normal organs (e.g., the skin) and surrounding tissues. Patients undergoing PDT are required to stay away from sunlight, or even room light to avoid phototoxicity, a side effect that can last for 1–2 months.11 Efforts have been made to improve the tumor selectivity of PSs, for instance, by coupling them with a tumor-targeting ligand such as an antibody.4,12,13 However, issues such as low loading capacity, reduced phototoxicity, and heterogeneous expression of antigens throughout the tumor mass were found, and clinical translation of these technologies has not been seen.4 Alternatively, a PS can be loaded, via hydrophobic–hydrophobic interactions, into polymer- or liposome-based nanoparticles.14,15 This approach, however, is usually associated with a relatively low loading rate (less than 10 wt %16) and a large particle size (around or larger than 100 nm15), both factors are detrimental to the PS delivery.
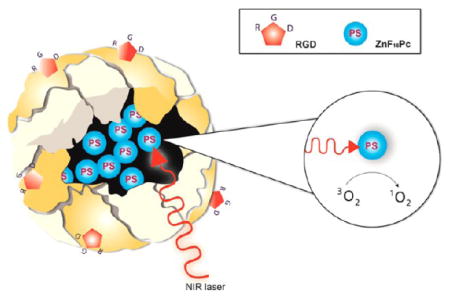
We herein report that surface-modified ferritin (FRT), a protein-based nanoparticle, can serve as an efficient PS delivery vehicle. In particular, we found that Cys-Asp-Cys-Arg-Gly-Asp-Cys-Phe-Cys (RGD4C)-modified FRTs (RFRTs) can encapsulate a large amount of zinc hexadecafluorophthalocyanine (ZnF16Pc), a potent but rather hydrophobic PS (λmax = 671 nm; ΦΔ = 0.85 in tetrahydrofuran17), and selectively deliver it to tumors to induce efficient PDT against cancer (Figure 1a). FRT is a major iron storage protein found in most living organisms including human beings. Each FRT nanocage is composed of 24 subunits, which self-assemble to form a cage-like nanostructure, with external and internal diameters of 12 and 8 nm, respectively.18,19 Our previous imaging studies showed that RFRTs can efficiently home to tumors through interactions with integrin αvβ3 overexpressed on tumor vasculatures and tumor cells.20 We and others have also demonstrated that non-iron metals (e.g., Cu, Pt, and Gd) or metal-containing compounds can be loaded into FRT or its derivatives by association with the metal binding sites at the interiors of the nanocages.20–23 Harnessing this characteristic to load metal-containing PSs and evaluating in vivo the PDT efficacy, however, has to our knowledge never been reported. FRT and its derivatives are safe and nonimmunogenic materials. Unlike conventional PS carriers, RFRTs were found to afford an extremely high ZnF16Pc loading rate (as much as ~60 wt %) and an ultrasmall post-loading size (less than 20 nm). These advantages lead to improved pharmacokinetics and treatment efficiency. Our in vivo studies with ZnF16Pc-loaded RFRTs found a high tumor accumulation rate (tumor-to-normal tissue ratio of 26.82 ±4.07 at 24 h), a good tumor inhibition rate (83.64% on day 12), as well as minimal toxicity to the skin and other normal tissues. All these features make FRT and its derivatives an attractive new type of PS carriers with great promise for selective PDT against cancer.
Figure 1.

(a) Schematic illustration of the formation and working mechanism of P-RFRTs. (b) Photographs of P-RFRTs and free ZnF16Pc in PBS under difierent conditions. AFM images of (c) RFRTs and (d) P-RFRTs. Scale bars, 100 nm.
RESULTS AND DISCUSSION
The drug loading was achieved by adding ZnF16Pc in DMSO into a RFRT solution in 0.01 M PBS (pH 7.4) and, after that, incubating at room temperature for 45 min. The raw products were subjected to purification through a NAP-5 column to remove the unloaded ZnF16Pc. The starting concentrations of ZnF16Pc and RFRT were tuned, and the loading capacity was investigated (Supporting Information, Table 1). We found that 1 mg of RFRTs can load up to 1.5 mg of ZnF16Pc, yielding a loading rate as high as 60 wt %. For stability considerations, however, we used a formulation with a loading rate of 41.2 wt % for the current investigations. The absorption spectra of P-RFRTs and free ZnF16Pc are shown in Figure S1. The sizes of the nanoparticles were studied by atomic force microscopy (AFM) analysis (Figure 1c,d). We found overall comparable sizes before and after the ZnF16Pc loading (18.3 ± 4.1 nm for RFRTs and 18.6 ± 2.6 nm for P-RFRTs).
Despite the heavy loading, the resulting P-RFRTs are highly stable in PBS. A photograph of P-RFRTs in PBS is shown in Figure 1b. The solution was stable for more than a week without visible precipitation. In comparison, free ZnF16Pc at the same concentration quickly precipitated out due to its poor solubility. We also investigated the stability of P-RFRTs at pH = 2. Within 10 min, a large amount of blue precipitation had formed at the bottom of the vial (Figure 1b). It is known that FRT nanocages are disassembled at pH 2.0. This pH-induced unloading suggests that the cargo was mostly internalized into hollow cores of the nanocages and was released upon particle decomposition.
The generation of 1O2 was studied using a singlet oxygen sensor green (SOSG) reagent (Invitrogen). SOSG is essentially a dye that is fluorescently quenched in its intact form but, upon activation in response to 1O2, produces an increase of fluorescence signals at 525 nm. We incubated P-RFRTs at difierent concentrations with the SOSG reagent and irradiated the samples with a 671 nm laser. The relative increase of readings at 525 nm was recorded 1 min later. As a comparison, ZnF16Pc was dispersed in PBS containing 1% tween and subjected to the analyses at the same conditions (Figure S2). We found no significant difierence between the results from the two groups, suggesting that ZnF16Pc is not quenched in the nanocarriers. We also examined the post-irradiated P-RFRTs under AFM. Instead of finding ~20 nm nanoparticles, we observed clusters of debris across the scope (Figure S3). The particle destruction was attributed to the 1O2 generated during the irradiation. This agrees with the above observation that ZnF16Pc was unloaded upon particle decomposition.
We then studied if the drug loading had affected the particles’ ability to interact with integrin αvβ3. To facilitate the tracking of particles, P-RFRTs were labeled with ZW800, a near-infrared dye molecule (ex/em = 780/800 nm).24 The coupling ratio was controlled so that on average one ZW800 was coupled to one RFRT. The in vitro studies were performed with U87MG human glioblastoma cells, which are known to express a high level of integrin αvβ3.20 As shown in Figure 2a, P-RFRTs demonstrated time-dependent internalization by U87MG cells. This internalization was blocked when free c(RGDyK) (30×, relative to protein concentration; it is noted that there are 24 RGD4C moieties on the surface of each RFRT nanoparticle) was coincubated (Figure 2a and Figure S4). The results suggest that, despite the heavy loading, P-RFRTs kept the targeting specificity toward integrin αvβ3.
Figure 2.

(a) Uptake of P-RFRTs (50 μg ZnF16Pc/mL) by U87MG cells at difierent time points. The uptake can be efficiently inhibited if c(RGDyK) (30×) was coincubated. The P-RFRTs were labeled with ZW800 (ex/em = 780/800 nm). Scale bars, 50 μm. (b) Cell viability studies with P-RFRTs on U87MG cells. Elevated cytotoxicity (red fluorescence) was found with increased incubation time in the presence of light irradiation (671 nm, 0.1 W/cm2 for 200 s). Without irradiation, no significant cell death was found. Red, ethidium homodimer-1 (ex/em = 528/617 nm), which stains dead cells. Scale bars, 50 μm.
The particles’ phototoxicity was also studied with U87MG cells. Briefly, we incubated cells with P-RFRTs in the dark and at difierent time points and irradiated them with a 671 nm laser (0.1 W/cm2, 200 s). The cell viability was studied 120 min post-irradiation by ethidium homodimer-1 staining (Invitrogen), which marked dead cells (Figure 2b). Increased red fluorescence was found to be correlated with elevated incubation time and was suppressed when free c(RGDyK) (30×) was coincubated. On the other hand, if no laser was applied, the red fluorescence remained at a background level. Similar observations were made from MTT assays, where we found a marginal drop in cell viability when there was no light irradiation and concentration-dependent cell death when there was (Figure S5). These observations suggest that cytotoxicity only occurs when both ZnF16Pc and light are present, which is the hallmark of PDT-induced phototoxicity.
We then studied the tumor selectivity of P-RFRT in subcutaneous (s.c.) U87MG tumor models. We injected ZW800-labeled P-RFRTs (5 mg RFRTs/kg) intravenously (i.v.) and acquired fluorescence images on a Maestro scanner using an “orange” filter (640 to 820 nm) at difierent time points. Tumor-to-normal tissue (T/N) ratios were evaluated to be 3.82 ±0.56, 14.47 ±1.69, and 26.82 ± 4.07 at 1, 4, and 24 h time points, suggesting good tumor accumulation (Figure 3a and Figure S6). When c(RGDyK) (30×) was injected prior to the P-RFRT injection, the tumor uptake was significantly diminished. This conforms that tumor accumulation was mainly mediated by RGD–integrin interaction. Post-mortem ex vivo imaging was performed with tumors and other major organs (Figure 3a). In addition to accumulation in the tumors, we also found a high level of fluorescence activity in the liver, which is common for a nanoparticle-based drug formulation. The uptake in other organs was much lower. We also examined the particle distribution in tumors by immunofluorescence staining using an anti-integrin β3 antibody (Figure 3b). As expected, positive β3 staining was found on both tumor vasculature and tumor cells.25,26 The ZW800 signals overlapped well with the positive β3 staining, conforming that the tumor retention was mainly caused by RGD–integrin interactions.
Figure 3.

(a) In vivo and ex vivo fluorescence imaging results. P-RFRTs were i.v. injected, and images were taken at 1, 4, and 24 h. In the control group, c(RGDyK) (30×) was injected to block the tumor homing. The organs were arranged in the following order: (1) tumor; (2) heart; (3) liver; (4) spleen; (5) skin; (6) lung; (7) kidneys; (8) intestine; (9) muscle; (10) brain. (b) Immunofluorescence microscopic imaging results. Integrin β3 is upregulated on both tumor vasculature and tumor cells. The distribution of P-RFRTs (ZW800, ex/em = 780/800 nm) was well correlated with the positive integrin β3 staining (Cy5, ex/em = 650/670 nm), indicating that the tumor homing was mostly mediated by RGD–integrin interactions. In the control group, where c(RGDyK) was preinjected, minimal ZW800 signals were found in tumors. Scale bars, 50 μm. (c) Tumor growth curve. The animals were divided into four groups. Group 1: P-RFRTs, with irradiation. Group 2: P-RFRTs, without irradiation. Group 3: ZnF16Pc, with irradiation. Group 4: PBS, without irradiation. Significant tumor suppression was found in group 1 (P < 0.05). On day 12, a TIR of 83.64 ± 2.52% was found. (d) Weight growth curve. No significant weight drop was found with animals injected with P-RFRTs, with or without irradiation. (e) Caspase 3 staining with tumor tissues. High level of apoptosis was found in tumors from group 1 but not in the other control groups. Red, caspase 3 (Cy5, ex/em = 650/670 nm); blue, DAPI. Scale bars, 50 μm. (f) H&E staining with tumor tissues. Densely packed neoplastic cells were found in the controls. In the treatment group, markedly disturbed tumor architecture was observed. Scale bars, 10 μm.
We then evaluated the treatment efficacy of P-RFRTs on a s.c. U87MG tumor model. We i.v. injected P-RFRTs (1.5 mg ZnF16Pc/kg) into the animals and illuminated (671 nm) the tumor surface over a 1 cm diameter beam spot (0.3 W/cm2, 15 min) 24 h after the injection (n = 5). The three control groups are (1) P-RFRTs (1.5 mg ZnF16Pc/kg, without irradiation); (2) free ZnF16Pc (1.5 mg ZnF16Pc/kg, with irradiation); (3) PBS (without irradiation). Tumor growth was similar in all the control groups but was significantly suppressed in the treatment group (Figure 3c). On day 12, we observed an average tumor inhibition rate (TIR) of 83.64% from the treatment group and found no impact on animals’ body weights (Figure 3d).
After therapy, we sacrificed the mice and dissected the tumors for histology studies. The apoptosis level in the tumors was examined by caspase 3 staining. We found positive staining with the samples from the treatment group but not with those from the controls (Figure 3e). Also, H&E staining demonstrates densely packed neoplastic cells in the controls but markedly disturbed tumor architecture in the treatment group (Figure 3f). These observations agree with the therapy results. To evaluate the size effects, we also performed caspase 3 staining with the skin and H&E staining with other normal organ tissues (e.g., the skin, heart, liver, spleen, lung, kidneys, intestine, muscle, and brain). No abnormalities were observed (Figure 4a,b), conforming that P-RFRT has minimal off-target damage.
Figure 4.

(a) Caspase 3 staining with the skin tissues from animals treated with P-RFRTs. No obvious apoptosis was found, either with or without light irradiation. Red, caspase 3 (Cy5, ex/em = 650/670 nm); blue, DAPI. Scale bars, 50 μm. (b) H&E staining with normal tissues. No abnormalities were observed. Scale bar, 100 μm.
ZnF16Pc is a ZnPc analogue with a good 1O2 quantum yield17,27 but poor water solubility (Figure 1c).28 As shown in the therapy studies (Figure 3c), ZnF16Pc is not a satisfactory PS. As a matter of fact, poor stability is a problem shared by many PSs, most of which are porphyrin-like hydrophobic compounds.29 These PSs are easily aggregated in the blood, causing self-quenching and poor pharmacokinetics.30 There have been efforts to develop polymer-31–33 and liposome-based17,34 nanoparticle carriers for the delivery of PSs like ZnPc31 or ZnF16Pc.35 These approaches, however, are usually associated with a loading rate that is typically less than 10 wt %16 and an overall particle size that is around or above 100 nm.15 On the other hand, using protein nanocages as a PS vehicle has seldom been exploited and, to the best of our knowledge, has never been investigated in vivo. We showed in the current study that, despite the heavy loading, the size of P-RFRTs remains below 20 nm. This is much smaller than conventional nanoparticle–PS conjugates.16 Both the high loading rate and the small overall size contributed to the good pharmacokinetics observed.
Although the possibility of surface docking cannot be excluded, several observations seem to support the idea that ZnF16Pc is mostly loaded into the interiors of the RFRT nanocages: First, the nanoparticle size was almost unchanged after the ZnF16Pc loading, as shown by AFM. Second, ZnF16Pc is unloaded by the decomposition of the nanocarriers, either by reducing the pH or by inducing PDT. Third, P-RFRTs kept the targeting specificity against integrin αvβ3. This was conformed by both in vitro and in vivo imaging studies. Fourth, preincubating RFRTs with Cu(II) can significantly suppress the loading of ZnF16Pc (from 60 to 25 wt %, Table 1 in Supporting Information). This indicates that most ZnF16Pc is competing for the same binding sites as Cu(II). The latter, according to our previous studies, is mainly encapsulated into the interiors of RFRTs.20 However, the exact mechanism behind the heavy loading is still unclear at this stage. It is known that there are both three-fold and four-fold symmetric channels present on the FRT surface.36 The three-fold channel, with a potential gradient directing toward the cavity of the nanocages, serves as a pathway to transfer metal cations into the protein cages. The four-fold channel, on the other hand, has a potential gradient in the opposite direction and is believed to be used to expel species from the cavity.36 The fact that Cu can suppress the ZnF16Pc uptake suggests that ZnF16Pc is internalized via the three-fold channel as Cu does. However, the three-fold channel on intact FRTs has a size of 3–4 Å,36,37 which is considered to be small compared to the bulky ZnF16Pc. It is possible that the exposure to ZnF16Pc or DMSO (a trace amount of DMSO may have been introduced during the drug loading) leads to an enlarged channel size that facilitates the uptake. It is also plausible that the planar structure of ZnF16Pc makes it relatively easy to penetrate the protein shell. Further investigations are needed to fully elucidate the mechanism.
PDT can target either tumor cells or tumor vasculature to cause damage.3 In the former mechanism, PDT-in-duced 1O2 acts on tumor cell membrane or mitochondria to cause necrosis or apoptosis.38 In the second mechanism, PDT causes vascular collapse and embolization, terminating the supply of oxygen and nutrients to the tumor cells.7 In the current study, ZnF16Pc was delivered by RFRTs to both tumor vasculature and U87MG tumor cells through RGD–integrin interactions (Figure 3b). Hence, both mechanisms may have played a role in the tumor destruction. It should be pointed out, however, that U87MG is chosen in the current study for its high expression of integrin αvβ3, not because of its origin. There is no indication that PDT can be used for the treatment of brain tumors, to which the delivery of either the drug or light is very challenging. For proof-of-concept, the current study was performed with subcutaneous tumors. To better assess the efficacy of P-RFRT-based PDT, it is important to investigate the modality in orthotopic tumor models with better clinical relevance (e.g., orthotopic prostate tumor models).39
As aforementioned, a most critical issue of PDT is skin toxicity.40 We, however, found negligible skin accumulation with P-RFRTs; instead, many of them were accumulated in the liver. In the context of PDT, however, uptake by the liver is a minor concern given its deep location and, hence, limited accessibility by light. Indeed, histology studies conformed that the treatment caused little impact to the liver, as well as to other major organs (Figure 4b). In the current study, we injected ZnF16Pc at 1.5 mg/kg and irradiated tumors at a fluence rate of 0.3 W/cm2 for 15 min. Similar conditions were used in the previous studies.41 Given that no adverse effects were observed, it is possible to increase the dose to improve the treatment. It is also possible to improve the efficacy by adjusting the fluence and fluence rate.
We reported very recently that drug molecules like doxorubicin can be loaded onto RFRTs with high efficiency.23 It is envisioned that both types of therapeutics can be docked onto RFRTs and delivered to diseased areas simultaneously. PDT can then work in combination with chemotherapy to achieve synergistic therapeutic effects. Moreover, it is an interesting finding that, for ZnF16Pc-loaded RFRTs, the nanocage structure can be broken down upon exposure to photoirradiation. This suggests a means to control the cargo unloading. That is, for dually loaded FRTs, PDT can be used as a mechanism to facilitate the release of the other drug to the surroundings. The FRT nanocages can be fabricated for delivery to a difierent target. Currently, RGD4C was used in the design as a tumor-targeting ligand and was imparted onto the nanocage surface through genetic modification. It is anticipated that other targeting ligands can also be introduced, through either genetic modification or chemical conjugation, onto FRT nanocages, and the new derivatives can transport drugs to difierent targets. Also, difierent types of metal-containing PSs, such as Pd-bacteriopheophor-bide and motexafin lutetium, are expected to be loaded similarly. These possibilities and optimizations will be also exploited in our future investigations.
CONCLUSION
Overall, we demonstrated in this work that RFRT nanoparticles are safe and efficient carriers for ZnF16Pc. The resulting conjugates can home to tumors through RGD–integrin interactions and, with light irradiation, induce phototoxicity to tumors while leaving normal tissues unaffected. Boasting an extremely high PS loading rate and an ultrasmall particle size, this technology is expected to find widespread use in PDT and holds great potential in clinical translation.
MATERIALS AND METHODS
Preparation and Purification of RFRTs
Production and purification of RFRTs have been reported previously. We constructed the DNA plasmid of R-Fn by introducing the RGD4C peptide sequence to the N-terminus of Fn with restrictions sites, NcoI and XhoI, at the 5′- and 3′-ends, respectively. Primers were designed as follows: (+) 5′ ATA TAC CAT GGG CTG CGA CTG CCG CGG AGA CTG CTT CTG CGG AGG CGG AGG CAC CAC CGC GTC T 3′; (−) 5′ CCA GAC TCG AGT TAG CTC TCA TCA 3′. The double digested PCR product was ligated into NcoI/XhoI digested plasmid pRSF with T4 DNA ligase, and the ligation mixture was used to transform competent cells of E. coli XL1-Blue by standard procedures. The resulting pRSF/RFRT plasmids were screened by appropriate restriction digests, verified by DNA sequencing, and then used to transform the expression strain E. coli BL21(DE3). For expression, a 1 L LB-kanamycin (50 μg/mL) culture of E. coli BL21(DE3)/RFRT was grown at 37 °C until an OD600 of 0.8 was reached. For induction, 1 mM IPTG was added to the culture and the culture was heated at 37 °C for 4 h. After sonication, the cell lysate was centrifuged at 10 400 rpm (12 930g) for 30 min to remove the cell debris. The supernatant was heated at 60 °C for 10 min and centrifuged at 13 000 rpm for 30 min to remove the precipitates. 2-Mercaptoethanol (10 mM) was added to stabilize the product. The raw product was purified by HPLC using a Superose 6 size exclusion column. The concentration of RFRTs was determined by Bradford protein assay. The purified FRTs/RFRTs were stored at −80 °C. For ZW800 labeling, RFRTs were incubated with ZW800-NHS24 for 30 min and purified through a NAP-5 column to remove uncoupled dye molecules. A starting ratio of 2:1 (ZW800-NHS to RFRTs) was used. The coupling efficiency was assessed spectroscopically by comparing with a predetermined standard curve (by monitoring absorbance at 780 nm). It was determined that the final conjugates have on average one ZW800 per particle.
Loading ZnF16Pc into FRTs/RFRTs
The ZnF16Pc loading was achieved without breaking down the nanocages. Briefly, 10 μL of ZnF16Pc (5 mg/mL) in DMSO was dropwise added into 490 μL of RFRTs in PBS (0.5 mg/mL), and the mixture was gently shaken for ~45 min at room temperature. The raw products were then purified using a NAP-5 column to remove the unloaded ZnF16Pc. The ZnF16Pc content was determined spectroscopically by comparing with a standard absorption curve of ZnF16Pc. The protein concentration was determined by Bradford protein assay. The loading rate was expressed in weight percent (wt %).
In Vitro Assays
U87MG cells were cultured in DMEM medium containing 10% nonessential amino acids, 10% fetal bovine serum, 0.1 mg/mL streptomycin sulfate, and 100 U/mL penicillin (MediaTech, USA) at 37 °C in a humidified atmosphere with 5% CO2. For cell uptake studies, 105 U87MG cells were seeded onto each well of a four-chamber slide (Lab-teck) one day prior to the studies. ZW800-labeled P-RFRTs were added to the solution to reach a final concentration of 50 μg ZnF16Pc/mL. In the control group, 30× free c(RGDyK) was used to coincubate with P-RFRTs. At different time points, incubation was stopped. The cells were washed with PBS five times and fixed with 75% ethanol overnight at 4 °C. The slides were mounted with DAPI containing mounting medium (Vector Inc.) and imaged under an Olympus X71 fluorescence microscope. For PDT studies, the cells were exposed to a 671 nm laser at 0.1 W/cm2 for 200 s. The cell viability was determined by MTT assays using a gradient of P-RFRTs (ZnF16Pc concentrations of 3, 6.25, 12.5, 25, and 50 μg/mL). In control groups, either no irradiation was applied or free ZnF16Pc at the same dose was used. Live/dead assays were performed by following a protocol provided by the vendor.
Animal Models
Animal models were established by subcutaneous injection of 106 human glioblastoma U87MG onto the hind legs of 5–6 week athymic nude mice (Harlan). Animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of University of Georgia.
In Vivo Imaging
The imaging studies were performed when tumors reached a size between 350 and 500 mm3. We intravenously (i.v.) injected ZW800-labeled P-RFRTs (5 mg RFRT/mL) into mice (n = 3). For the control group, 30× c(RGDyK) was administrated 30 min prior to the R-RFRT injection (n = 3). Fluorescence images were acquired on a Maestro II imaging system using an orange filter (640 to 820 nm) at 1, 4, and 24 h time points. The images were unmixed using the Maestro software. The average signal (106 photons/cm2/s) for each region of interest (ROI) was measured. Tumor-to-normal tissue ratio (T/N) was determined and was expressed as mean ± SD. All mice were euthanized after the 24 h imaging. Tumors as well as major organs were collected and subjected to ex vivo imaging. After imaging, the tissues were snapfrozen in O.C.T. (Tissue-Tek) and stored in a −80 °C freezer.
Therapy Studies
For PDT studies, 20 mice bearing U87MG tumors were randomly divided into four groups. The treatment scheme is as follows: (1) P-RFRTs (1.5 mg ZnF16Pc/kg), with irradiation; (2) P-RFRTs (1.5 mg ZnF16Pc/kg), without irradiation; (3) free ZnF16Pc (1.5 mg ZnF16Pc/kg), with irradiation; (4) PBS, no irradiation. The photoirradiation was applied 24 h after the injection of P-RFRTs (671 nm laser, 0.3 W/cm2 for 15 min). The tumor sizes and body weights were inspected every 3 days. The tumor weight was estimated using the formula, tumor volume = length × (width)2/2, assuming a tumor density of 1 mg/mL. After therapy, major organs as well as tumors were collected and sectioned to 8 μm slices for caspase 3 and H&E staining.
Immunofluorescence Staining
The cryogenic slides were fixed with cold acetone for 30 min, washed by running water for 5 min, and blocked by 10% goat serum for 1 h. Anti-integrin β3 or anti-caspase 3 antibodies were incubated with the slides at 4 °C overnight. Cy5.5-labeled secondary antibody was then added and incubated for 1 h at 37 °C. After gently rinsing with PBS, the slides were mounted and ready for microscopic imaging.
H&E Staining
H&E staining was performed according to a protocol provided by the vendor (BBC Biochemical). Briefly, 8 μm cryogenic slides were prepared and fixed with 10% formalin for about 30 min at room temperature. After washing with running water for 5 min, the slides were treated with gradient concentrations of alcohol (100, 95, and 70%), each for 20 s. The hematoxylin staining was performed for about 3 min and washed with water for 1 min. The eosin staining was performed for about 1 min. The slides were washed, treated with xylene, and mounted with Canada balsam. The images were acquired on a Nikon Eclipse 90i microscope.
Supplementary Material
Acknowledgments
This work was supported by an NCI/NIH R00 grant (5R00CA153772, J.X.), a UGA startup grant (J.X.), U.S. National Science Foundation grants (ECCS0823849 and CBET 1139057, B.X.), NCI/NIH R01 grant (R01CA156775, B.F.), Georgia Cancer Coalition Distinguished Clinicians and Scientists Award (B.F.), and the Intramural Research Program of NIBIB, NIH. We thank the support from the Major State Basic Research Development Program of China (973 Program, 2013CB733802), the National Nature Science of Foundation of China (NSFC, 81101101, 51273165), and the Key Project of Chinese Ministry of Education (212149). We are grateful to Prof. J. Frangioni and Prof. H. Soo at Harvard Medical School for generously providing the ZW800 dye.
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information Available: Cellular uptake, MTT assay, and AFM results. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.Bessler NM. Verteporfin Therapy in Age-Related Macular Degeneration (VAM): An Open-Label Multicenter Photo-dynamic Therapy Study of 4,435 Patients. Retina. 2004;24:512–520. doi: 10.1097/00006982-200408000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Gross S, Gilead A, Scherz A, Neeman M, Salomon Y. Monitoring Photodynamic Therapy of Solid Tumors Online by Bold-Contrast MRI. Nat Med. 2003;9:1327–1331. doi: 10.1038/nm940. [DOI] [PubMed] [Google Scholar]
- 3.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, et al. Photodynamic Therapy of Cancer: An Update. Ca—Cancer J Clin. 2011;61:250–281. doi: 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore CM, Pendse D, Emberton M. Photodynamic Therapy for Prostate Cancer—A Review of Current Status and Future Promise. Nat Clin Pract Urol. 2009;6:18–30. doi: 10.1038/ncpuro1274. [DOI] [PubMed] [Google Scholar]
- 5.Trachtenberg J, Weersink RA, Davidson SR, Haider MA, Bogaards A, Gertner MR, Evans A, Scherz A, Savard J, Chin JL, et al. Vascular-Targeted Photodynamic Therapy (Padoporfin, Wst09) for Recurrent Prostate Cancer after Failure of External Beam Radiotherapy: A Study of Escalating Light Doses. BJU Int. 2008;102:556–562. doi: 10.1111/j.1464-410X.2008.07753.x. [DOI] [PubMed] [Google Scholar]
- 6.Selman SH, Keck RW, Hampton JA. Transperineal Photodynamic Ablation of the Canine Prostate. J Urol. 1996;156:258–260. [PubMed] [Google Scholar]
- 7.Nishiyama N, Morimoto Y, Jang WD, Kataoka K. Design and Development of Dendrimer Photosensitizer-Incorporated Polymeric Micelles for Enhanced Photodynamic Therapy. Adv Drug Delivery Rev. 2009;61:327–338. doi: 10.1016/j.addr.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Fisher AM, Murphree AL, Gomer CJ. Clinical and Preclinical Photodynamic Therapy. Lasers Surg Med. 1995;17:2–31. doi: 10.1002/lsm.1900170103. [DOI] [PubMed] [Google Scholar]
- 9.Lovell JF, Liu TW, Chen J, Zheng G. Activatable Photosensitizers for Imaging and Therapy. Chem Rev. 2010;110:2839–2857. doi: 10.1021/cr900236h. [DOI] [PubMed] [Google Scholar]
- 10.Orenstein A, Kostenich G, Roitman L, Shechtman Y, Kopolovic Y, Ehrenberg B, Malik Z. A Comparative Study of Tissue Distribution and Photodynamic Therapy Selectivity of Chlorin E6, Photofrin II and Ala-Induced Proto-porphyrin IX in a Colon Carcinoma Model. Br J Cancer. 1996;73:937–944. doi: 10.1038/bjc.1996.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dougherty TJ, Kaufman JE, Goldfarb A, Weishaupt KR, Boyle D, Mittleman A. Photoradiation Therapy for the Treatment of Malignant Tumors. Cancer Res. 1978;38:2628–2635. [PubMed] [Google Scholar]
- 12.Renno RZ, Terada Y, Haddadin MJ, Michaud NA, Gragoudas ES, Miller JW. Selective Photodynamic Therapy by Targeted Verteporfin Delivery to Experimental Choroidal Neovascularization Mediated by a Homing Peptide to Vascular Endothelial Growth Factor Receptor-2. Arch Ophthalmol. 2004;122:1002–1011. doi: 10.1001/archopht.122.7.1002. [DOI] [PubMed] [Google Scholar]
- 13.Sharman WM, van Lier JE, Allen CM. Targeted Photodynamic Therapy via Receptor Mediated Delivery Systems. Adv Drug Delivery Rev. 2004;56:53–76. doi: 10.1016/j.addr.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Konan YN, Gurny R, Allemann E. State of the Art in the Delivery of Photosensitizers for Photodynamic Therapy. J Photochem Photobiol B. 2002;66:89–106. doi: 10.1016/s1011-1344(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 15.Bechet D, Couleaud P, Frochot C, Viriot ML, Guillemin F, Barberi-Heyob M. Nanoparticles as Vehicles for Delivery of Photodynamic Therapy Agents. Trends Biotechnol. 2008;26:612–621. doi: 10.1016/j.tibtech.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee DK, Fong LS, Zhang Y. Nanoparticles in Photodynamic Therapy: An Emerging Paradigm. Adv Drug Delivery Rev. 2008;60:1627–1637. doi: 10.1016/j.addr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Garcia AM, Alarcon E, Munoz M, Scaiano JC, Edwards AM, Lissi E. Photophysical Behaviour and Photodynamic Activity of Zinc Phthalocyanines Associated to Liposomes. Photochem Photobiol Sci. 2011;10:507–514. doi: 10.1039/c0pp00289e. [DOI] [PubMed] [Google Scholar]
- 18.Maham A, Tang Z, Wu H, Wang J, Lin Y. Protein-Based Nanomedicine Platforms for Drug Delivery. Small. 2009;5:1706–1721. doi: 10.1002/smll.200801602. [DOI] [PubMed] [Google Scholar]
- 19.Theil EC. Ferritin: Structure, Gene Regulation, and Cellular Function in Animals, Plants, and Microorganisms. Annu Rev Biochem. 1987;56:289–315. doi: 10.1146/annurev.bi.56.070187.001445. [DOI] [PubMed] [Google Scholar]
- 20.Lin X, Xie J, Niu G, Zhang F, Gao H, Yang M, Quan Q, Aronova MA, Zhang G, Lee S, et al. Chimeric Ferritin Nanocages for Multiple Function Loading and Multimodal Imaging. Nano Lett. 2011;11:814–819. doi: 10.1021/nl104141g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geninatti Crich S, Bussolati B, Tei L, Grange C, Esposito G, Lanzardo S, Camussi G, Aime S. Magnetic Resonance Visualization of Tumor Angiogenesis by Targeting Neural Cell Adhesion Molecules with the Highly Sensitive Gadolinium-Loaded Apoferritin Probe. Cancer Res. 2006;66:9196–9201. doi: 10.1158/0008-5472.CAN-06-1728. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z, Wang X, Diao H, Zhang J, Li H, Sun H, Guo Z. Encapsulation of Platinum Anticancer Drugs by Apoferritin. Chem Commun. 2007:3453–3455. doi: 10.1039/b705326f. [DOI] [PubMed] [Google Scholar]
- 23.Zhen Z, Tang W, Chen H, Lin X, Todd T, Wang G, Cowger T, Chen X, Xie J. RGD-Modified Apoferritin Nanoparticles for Efficient Drug Delivery to Tumors. ACS Nano. 2013;7:4830–4837. doi: 10.1021/nn305791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi HS, Nasr K, Alyabyev S, Feith D, Lee JH, Kim SH, Ashitate Y, Hyun H, Patonay G, Strekowski L, et al. Synthesis and In Vivo Fate of Zwitterionic Near-Infrared Fluorophores. Angew Chem, Int Ed. 2011;50:6258–6263. doi: 10.1002/anie.201102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan M, Lu ZR. Integrin Targeted MR Imaging. Theranos-tics. 2011;1:83–101. doi: 10.7150/thno/v01p0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Yang Y, Cai W. Multimodality Imaging of Integrin αvβ3 Expression. Theranostics. 2011;1:135–148. doi: 10.7150/thno/v01p0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyle RW, Rousseau J, Kudrevich SV, Obochi MOK, van Lier JE. Hexadecafluorinated Zinc Phthalocyanine: Photodynamic Properties against the Emt-6 Tumour in Mice and Pharmacokinetics Using Zn-65 as a Radiotracer. Br J Cancer. 1996;73:49–53. doi: 10.1038/bjc.1996.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oda K, Ogura S, Okura I. Preparation of a Water-Soluble Fluorinated Zinc Phthalocyanine and Its Effect for Photo-dynamic Therapy. J Photochem Photobiol B. 2000;59:20–25. doi: 10.1016/s1011-1344(00)00129-9. [DOI] [PubMed] [Google Scholar]
- 29.Berg K, Selbo PK, Weyergang A, Dietze A, Prasmickaite L, Bonsted A, Engesaeter BO, Angell-Petersen E, Warloe T, Frandsen N, et al. Porphyrin-Related Photosensitizers for Cancer Imaging and Therapeutic Applications. J Microsc. 2005;218:133–147. doi: 10.1111/j.1365-2818.2005.01471.x. [DOI] [PubMed] [Google Scholar]
- 30.Dhami S, Phillips D. Comparison of the Photophysics of an Aggregating and Non-aggregating Aluminium Phtha-locyanine System Incorporated into Unilamellar Vesicles. J Photochem Photobiol A. 1996;100:77–84. [Google Scholar]
- 31.Conte C, Ungaro F, Maglio G, Tirino P, Siracusano G, Sciortino MT, Leone N, Palma G, Barbieri A, Arra C, et al. Biodegradable Core–Shell Nanoassemblies for the Delivery of Docetaxel and Zn(II)-Phthalocyanine Inspired by Combination Therapy for Cancer. J Controlled Release. 2013;167:40–52. doi: 10.1016/j.jconrel.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 32.van Nostrum CF. Polymeric Micelles To Deliver Photosensitizers for Photodynamic Therapy. Adv Drug Delivery Rev. 2004;56:9–16. doi: 10.1016/j.addr.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 33.Konan YN, Cerny R, Favet J, Berton M, Gurny R, Allemann E. Preparation and Characterization of Sterile Sub-200 nm meso-Tetra(4-hydroxylphenyl) Porphyrin-Loaded Nanoparticles for Photodynamic Therapy. Eur J Pharm Biopharm. 2003;55:115–124. doi: 10.1016/s0939-6411(02)00128-5. [DOI] [PubMed] [Google Scholar]
- 34.Ali MF. Topical Delivery and Photodynamic Evaluation of a Multivesicular Liposomal Rose Bengal. Lasers Med Sci. 2011;26:267–275. doi: 10.1007/s10103-010-0859-9. [DOI] [PubMed] [Google Scholar]
- 35.Allemann E, Brasseur N, Benrezzak O, Rousseau J, Kudrevich SV, Boyle RW, Leroux JC, Gurny R, van Lier JE. PEG-Coated Poly(lactic acid) Nanoparticles for the Delivery of Hexadecafluoro Zinc Phthalocyanine to Emt-6 Mouse Mammary Tumours. J Pharm Pharmacol. 1995;47:382–387. doi: 10.1111/j.2042-7158.1995.tb05815.x. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi T, Kuyucak S. Functional Properties of Threefold and Fourfold Channels in Ferritin Deduced from Electrostatic Calculations. Biophys J. 2003;84:2256–2263. doi: 10.1016/S0006-3495(03)75031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan F, Zhang Y, Yuan HK, Gregas MK, Vo-Dinh T. Apoferritin Protein Cages: A Novel Drug Nanocarrier for Photodynamic Therapy. Chem Commun. 2008:4579–4581. doi: 10.1039/b810949d. [DOI] [PubMed] [Google Scholar]
- 38.Fabris C, Valduga G, Miotto G, Borsetto L, Jori G, Garbisa S, Reddi E. Photosensitization with Zinc(II) Phthalocyanine as a Switch in the Decision between Apoptosis and Necrosis. Cancer Res. 2001;61:7495–7500. [PubMed] [Google Scholar]
- 39.Kosharskyy B, Solban N, Chang SK, Rizvi I, Chang Y, Hasan T. A Mechanism-Based Combination Therapy Reduces Local Tumor Growth and Metastasis in an Orthotopic Model of Prostate Cancer. Cancer Res. 2006;66:10953–10958. doi: 10.1158/0008-5472.CAN-06-1793. [DOI] [PubMed] [Google Scholar]
- 40.Baas P, van Mansom I, van Tinteren H, Stewart FA, van Zandwijk N. Effect of N-Acetylcysteine on Photofrin-Induced Skin Photosensitivity in Patients. Lasers Surg Med. 1995;16:359–367. doi: 10.1002/lsm.1900160407. [DOI] [PubMed] [Google Scholar]
- 41.Boyle RW, Rousseau J, Kudrevich SV, Obochi M, van Lier JE. Hexadecafluorinated Zinc Phthalocyanine: Photodynamic Properties against the Emt-6 Tumour in Mice and Pharmacokinetics Using 65zn as a Radiotracer. Br J Cancer. 1996;73:49–53. doi: 10.1038/bjc.1996.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.