

Abstract
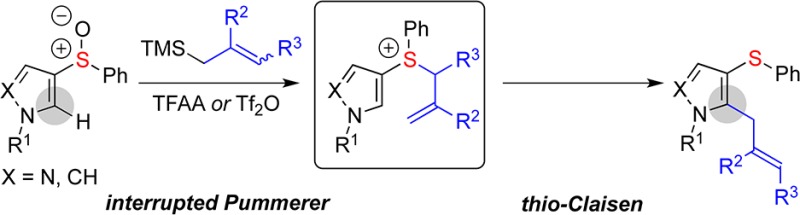
Arylsulfinyl groups direct the metal-free, regiospecific, nucleophilic ortho-allylation of pyrroles and pyrazoles. Mechanistic studies support the intermediacy of allylsulfonium salts that undergo facile thio-Claisen rearrangement onto the heterocyclic ring, giving products of coupling. The strategy has been adapted to allow regiospecific propargylation of the heterocyclic substrates.
Functionalized aromatic and heteroaromatic systems form the cores of many pharmaceuticals, agrochemicals, and functional materials. Specifically, substituted pyrroles1a−1c and pyrazoles1d−1g with their broad spectrum of biological activities find application in many fungicides, insecticides, herbicides, statins (Lipitor), anti-inflammation (Celebrex),2 and antitumor drugs (Figure 1), thus highlighting the importance of accessing elaborated systems. Functionalization of these heterocycles heavily relies on the electrophilic nature of the systems (exploited in Friedel–Crafts3 type reactions or C–H activation4 processes using transition-metal catalysts) and can be problematic due to regioselectivity issues. Alternative approaches involving the de novo synthesis of the functionalized heterocycles,5 using elaborated building blocks, are therefore often adopted. Although direct metalation6 with stoichiometric reagents and cross-couplings7 of heteroaryl derivatives are known, the use of activating substituents to facilitate nucleophilic substitution is an underexploited approach. In recent years, Pummerer-type8 coupling reactions utilizing sulfoxide substituents have begun to emerge for the nucleophilic alkylation of heteroaromatic9 systems. Oshima and Yorimitsu10a−10d as well as Maulide10e−10f have recently explored interrupted Pummerer reactions in approaches to benzofurans and in α-arylation reactions, while we have reported the allylation11a and propargylation11b of aromatic systems.
Figure 1.

Selected biologically active pyrroles and pyrazoles.
Herein we report a nucleophilic ortho-allylation of pyrrole and pyrazole sulfoxides, such as 1, that proceeds by a heterocycle accelerated, interrupted Pummerer/thio-Claisen12 rearrangement sequence involving allylsulfonium salts 4 (Scheme 1). The procedure is general, metal-free, and regiospecific with regard to both coupling partners.
Scheme 1. Nucleophilic ortho-Allylation (X = N, CH).

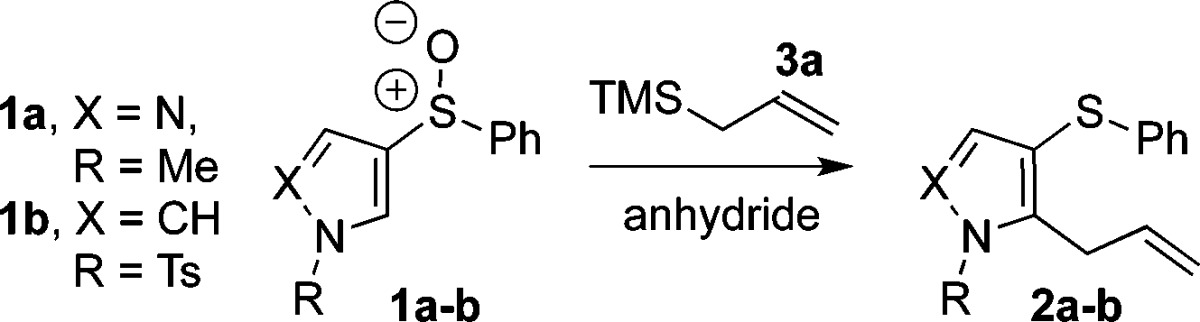
We began investigating the ortho-allylation reaction of pyrazole sulfoxide 1a with allyltrimethylsilane 3a (Table 1). Using our previously established conditions11a with TFAA (trifluoroacetic anhydride) as the electrophilic activating agent in MeCN (entry 1), the allylation product 2a was observed as a minor component. Changing the solvent to CH2Cl2 improved the reaction slightly (entries 2–3), while heating was not helpful. The use of Tf2O (trifluoromethanesulfonic anhydride) significantly enhanced the reaction to give 2a in 53% yield (entry 7).13 In contrast, when investigating the reaction of pyrrole sulfoxide 1b, TFAA appears to be the better activating agent (entry 9) and employing MeCN as solvent (entry 10) gave 2b in an excellent yield of 98%.13
Table 1. Optimization of the ortho-Allylation.
| entry | HetAr | solvent | anhyd. | conditions | yield (%) |
|---|---|---|---|---|---|
| 1 | 1a | MeCN | TFAA | –40 °C to rt; 2 h | <5 |
| 2 | 1a | CH2Cl2 | TFAA | –78 °C to rt; 2 h | 14 |
| 3 | 1a | CH2Cl2 | TFAA | –78 °C to rt; 18 h | 24 |
| 4 | 1a | DCE | TFAA | –78 to 60 °C 2 h | 13 |
| 5 | 1a | DCE | Tf2O | –78 to 60 °C; 2 h | 18 |
| 6 | 1a | CH2Cl2 | Tf2O | –78 °C to rt; 2 h | 47 |
| 7 | 1a | CH2Cl2 | Tf2O | -78 °C to rt; 18 h | 53 |
| 8 | 1b | CH2Cl2 | Tf2O | –78 °C to rt; 18 h | 14a |
| 9 | 1b | CH2Cl2 | TFAA | –78 °C to rt; 18 h | 41a |
| 10 | 1b | MeCN | TFAA | -40 °C to rt; 18 h | 98a |
| 11 | 1b | MeCN | TFAA | –40 °C to rt; 2 h | 95a |
| 12 | 1b | MeCN | TFAA | –40 °C; 2 h | 92a |
Yields determined by 1H NMR.13
Having identified optimized conditions for the allylation of both pyrazoles and pyrroles, we next investigated the substrate scope. Pleasingly, the ortho-allylation of pyrrole was not restricted with regard to the position of the sulfoxide moiety; both 1b and its regioisomer 1c (entries 1 and 6; Table 2) underwent allylation in high yields. The reaction is also tolerant to various allylsilanes, allowing high yielding allylation when using functionalized silanes 3b–c (entries 2–3) and the extended allylsilanes 3d–e (entries 4–5). Interestingly, the reaction is stereoconvergent with regard to alkene geometry, since both silanes 3d and 3e give products of allylation favoring the E-isomer (entries 4–5). Although we mainly explored the reactivity with tosyl-protected pyrrole, the unprotected N–H pyrrole 1d also successfully underwent allylation (entry 7). Analogously, phenylsulfinyl-pyrazoles 1a, 1e, 1f, and 1g were successfully allylated under these conditions in good yields (entries 8–10 and 13), showing more efficient reactivity when ortho-allylation at the 4-position of pyrazole is possible.14 A variety of commonly used protecting groups are tolerated in the allylation of pyrazoles (entries 9, 10, and 13). Functionalized silanes 3b and 3c can also be used with phenylsulfinyl-pyrazoles to give products of allylation 2k and 2l (entries 11–12).
Table 2. Scope of ortho-Allylationa.
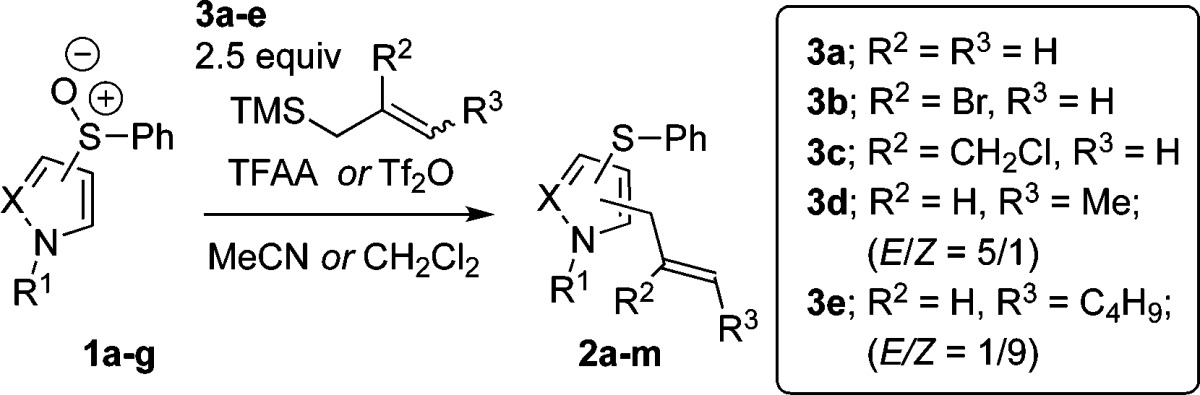
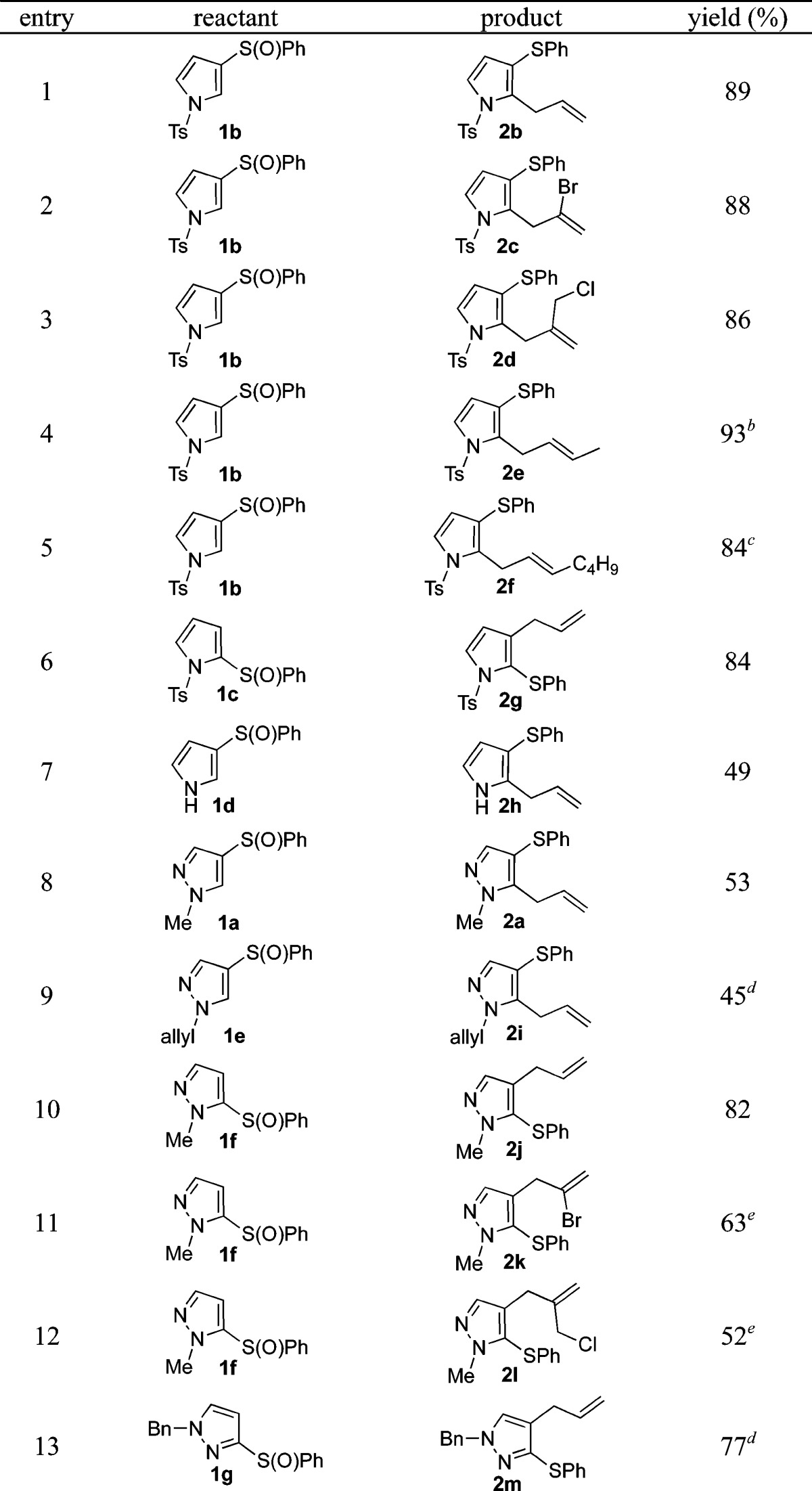
Conditions: entries 1–6: TFAA (2.5 equiv), MeCN, −40 °C to rt, 18 h; entry 7: TFAA (2.5 equiv), CH2Cl2, −40 to 0 °C, 2 h; entries 8–13: Tf2O (2.5 equiv), CH2Cl2, −78 °C to rt, 18 h.
Crotyl-TMS (E/Z, 5/1), giving mixture (E/Z, 11/1).
Hept-2-en-1-yltrimethylsilane (E/Z, 1/9), giving mixture (E/Z, 17/1).
100 °C MW, 2 h.
60 °C MW, 2 h.
From a mechanistic perspective, the activation of sulfoxides with TFAA or Tf2O15 allows for two plausible mechanisms. In a vinylogous Pummerer reaction,9bN-lone pair donation with concomitant triflate expulsion could form an extended thionium ion, allowing direct nucleophilic attack at the heterocycle followed by rearomatization.16a Alternatively, the activated sulfoxide could undergo an interrupted Pummerer reaction11 followed by a thio-Claisen rearrangement and rearomatization. The former pathway could in theory lead to regioisomeric products of allylation, whereas the process described shows complete ortho-selectivity. Moreover, the use of the extended alkenylsilanes 3d and 3e (entries 4 and 5) selectively produced linear alkylation products from double-allylic inversion. This strongly suggests a pathway proceeding through a sigmatropic rearrangement of allylsulfonium salt 4. Alternatively, allylsulfonium salt 4, from an interrupted Pummerer reaction, could participate in intermolecular Friedel–Crafts type alkylations in which 4 acts as an electrophilic alkylating agent.16a The lack of crossover products from the reaction of 1b when 1 equiv of 3-(p-tolylthio)-1-tosyl-pyrrole 5b was present (Scheme 2A) demonstrates the intramolecularity of the alkylation and rules out an intermolecular Friedel–Crafts-type process. The experiment resulted in complete allylation of the starting sulfoxide 1b and recovery of 5b. Similar results were obtained from an analogous reaction of sulfoxide 1h in the presence of sulfide 5a.
Scheme 2. Competition Experiments.

TFAA (2.5 equiv), MeCN, −40 °C to rt, 18 h. b Tf2O (2.5 equiv), CH2Cl2, −78 to 60 °C MW, 2 h.
The relative t1/2 values as determined by individual reactions for 1b/1c/1f/1a/diphenylsulfoxide11a are 1500:1500:1020:3.2:1 (±10%), respectively.16b In addition, competition experiments reacting pyrrole sulfoxide 1b in the presence of 1c and analogously, pyrazole sulfoxide 1a in the presence of 1f, with limiting allylTMS, showed a preference for the consumption of sulfoxides 1b and 1a, suggesting steric factors control the formation of 4 (Scheme 2B).17 Furthermore, competitions between 1a and 1i, and 1b and 1i, with limiting allylTMS indicated a preference for consumption of the heterocyclic substrates (Scheme 2C). These results, in conjunction with the relative reaction rates, suggest that rearrangement onto the heterocyclic systems is accelerated: the overall allylation process to form 2a and 2b is much faster comparatively to the allylation of diphenyl sulfoxide, and the intramolecular competition shows complete selectivity for reaction on the heterocyclic ring.
A proposed mechanism for the ortho-allylation of pyrrole and pyrazole sulfoxides involves an interrupted Pummerer reaction of the activated sulfoxide 6, to form allylsulfonium salt 4, which can then undergo a thio-Claisen rearrangement followed by rearomatization to give the product 2 (Scheme 3).18
Scheme 3. Mechanism of ortho-Allylation (X = N, CH).

The products of ortho-allylation are rich in synthetic potential, as both the allyl and organosulfanyl10d,11a,19 groups can be utilized as handles for further manipulation. Preliminary studies show how the process can be exploited in a tandem nucleophilic ortho-allylation/electrophilic substitution sequence in the formation of 7a, or as part of an approach to more complex heterocycles such as 7b (Scheme 4).
Scheme 4. Ortho-Allylation and Product Manipulation.

Furthermore, under the conditions developed here, propargylsilanes in conjunction with pyrazole and pyrrole sulfoxides give the respective products of nucleophilic ortho-propargylation 8a and 8b in high yields (Scheme 5).
Scheme 5. Ortho-Propargylation of Pyrroles and Pyrazoles (2,6-DTBP = 2,6-Di-tert-butylpyridine).

In summary, pyrrole and pyrazole sulfoxides undergo ortho-allylation by a heterocycle-accelerated interrupted Pummerer/thio-Claisen rearrangement sequence. The operationally simple and metal-free process allows the addition of allylic and propargylic nucleophiles with C–H substitution and shows complete regiospecificity with regard to both coupling partners.
Acknowledgments
We thank Merck Sharp & Dohme (CASE award to A.J.E.) and the EPSRC (CPhD award to A.J.E.) for financial support and Dr. Jason E. Imbriglio for additional discussions during an industrial placement at Merck, Rahway, NJ, USA.
Supporting Information Available
Optimization table, additional mechanistic studies, experimental procedures, characterization data, 1H and 13C spectra, and X-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Teixeira C.; Barbault F.; Rebehmed J.; Liu K.; Xie L.; Lu H.; Jiang S.; Fan b.; Maurel F. Bioorg. Med. Chem. 2008, 16, 3039. [DOI] [PubMed] [Google Scholar]; b Biava M.; Porretta G. C.; Deidda D.; Pompei R.; Tafic A.; Manettic F. Bioorg. Med. Chem. 2004, 12, 1453. [DOI] [PubMed] [Google Scholar]; c Boger D. L.; Boyce C. W.; Labroli M. A.; Sehon C. A.; Jin Q. J. Am. Chem. Soc. 1999, 121, 54. [Google Scholar]; d Daidone G.; Maggio B.; Plescia S.; Raffa D.; Musiu C.; Milia C.; Perra G.; Marongiu M. E. Eur. J. Med. Chem. 1998, 33, 375. [Google Scholar]; e Li Y.; Zhang H. Q.; Liu J.; Yang X. P.; Liu Z. J. J. Agric. Food Chem. 2006, 54, 3636. [DOI] [PubMed] [Google Scholar]; f Chen L.; Ou X. M.; Mao C. H.; Shang J.; Huang R. Q.; Bi F. C.; Wang Q. M. Bioorg. Med. Chem. 2007, 15, 3678. [DOI] [PubMed] [Google Scholar]; g Gamage S. A.; Spicer J. A.; Rewcastle G. W.; Milton J.; Sohal S.; Dangerfield W.; Mistry P.; Vicker N.; Charlton P. A.; Denny W. A. J. Med. Chem. 2002, 45, 740. [DOI] [PubMed] [Google Scholar]; h Wu J.; Shi Q.; Chen Z.; He M.; Jin L.; Hu D. Molecules 2012, 17, 5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipitor (1st) and Celebrex were in the 50 top selling drugs in 2011: http://www.pharmacytimes.com/publications/issue/2012/July2012/Top-200-Drugs-of-2011.
- For a review on Friedel–Crafts reactions, see:Rueping M.; Nachtsheim B. J. Beilstein J. Org. Chem. 2010, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on C–H activation on pyrroles, see:Beck E. M.; Gaunt M. J. Top. Curr. Chem. 2010, 292, 85. [DOI] [PubMed] [Google Scholar]
- a Joule J. A.; Mills K.. Heterocyclic Chemistry, 5th ed.; John Wiley & Sons: 2010. [Google Scholar]; b Fustero S.; Sánchez-Roselló M.; Barrio P.; Simón-Fuentes A. Chem. Rev. 2011, 111, 6984. [DOI] [PubMed] [Google Scholar]; c Estévez V.; Villacampa M.; Menéndez J. C. Chem. Soc. Rev. 2010, 39, 4402. [DOI] [PubMed] [Google Scholar]
- For selected reviews on metalation, see:; a Kishbaugh T. L. S. Top. Heterocycl. Chem. 2012, 29, 1. [Google Scholar]; b Roy S.; Roy S.; Gribble G. W. Top. Heterocycl. Chem. 2012, 29, 155. [Google Scholar]; c Chinchilla R.; Nájera C.; Yus M. Chem. Rev. 2004, 104, 2667. [DOI] [PubMed] [Google Scholar]
- For selected reviews on cross-couplings, see:; a Schnürch M.; Flasik R.; Khan A. F.; Spina M.; Mihovilovic M. D.; Stanetty P. Eur. J. Org. Chem. 2006, 3283. [Google Scholar]; b Banwell M. G.; Goodwin T. E.; Ng S.; Smith J. A.; Wong D. J. Eur. J. Org. Chem. 2006, 3043. [Google Scholar]; c See ref (3)b.
- For selected reviews of the Pummerer reaction, see:; a Smith L. H. S.; Coote S. C.; Sneddon H. F.; Procter D. J. Angew. Chem., Int. Ed. 2010, 49, 5832. [DOI] [PubMed] [Google Scholar]; b Akai S.; Kita Y. Top. Curr. Chem. 2007, 274, 35. [Google Scholar]; c Feldman K. S. Tetrahedron 2006, 62, 5003. [Google Scholar]; d Bur S. K.; Padwa A. Chem. Rev. 2004, 104, 2401. [DOI] [PubMed] [Google Scholar]
- a Akai S.; Kawashita N.; Wada Y.; Satoh H.; Alinejad A. H.; Kakiguchi K.; Kuriwaki I.; Kita Y. Tetrahedron Lett. 2006, 47, 1881. [Google Scholar]; b Akai S.; Kawashita N.; Satoh H.; Wada Y.; Kakiguchi K.; Kuriwaki I.; Kita Y. Org. Lett. 2004, 6, 3793. [DOI] [PubMed] [Google Scholar]; c Akai S.; Morita N.; Iio K.; Nakamura Y.; Kita Y. Org. Lett. 2000, 2, 2279. [DOI] [PubMed] [Google Scholar]; d Feldman K. S.; Vidulova D. B. Org. Lett. 2004, 6, 1869. [DOI] [PubMed] [Google Scholar]; e Feldman K. S.; Skoumbourdis A. P. Org. Lett. 2005, 7, 929. [DOI] [PubMed] [Google Scholar]; f Feldman K. S.; Vidulova D. B.; Karatjas A. G. J. Org. Chem. 2005, 70, 6429. [DOI] [PubMed] [Google Scholar]; g Feldman K. S.; Karatjas A. G. Org. Lett. 2006, 8, 4137. [DOI] [PubMed] [Google Scholar]; h Feldman K. S.; Fodor M. D. J. Org. Chem. 2009, 74, 3449. [DOI] [PubMed] [Google Scholar]; i Padwa A.; Nara S.; Wang Q. Tetrahedron Lett. 2006, 47, 595. [Google Scholar]
- a Yoshida S.; Yorimitsu H.; Oshima K. Org. Lett. 2009, 11, 2185. [DOI] [PubMed] [Google Scholar]; b Kobatake T.; Fujino D.; Yoshida S.; Yorimitsu H.; Oshima K. J. Am. Chem. Soc. 2010, 132, 11838. [DOI] [PubMed] [Google Scholar]; c Kobatake T.; Yoshida S.; Yorimitsu H.; Oshima K. Angew. Chem., Int. Ed. 2010, 49, 2340. [DOI] [PubMed] [Google Scholar]; d Ookubo Y.; Wakamiya A.; Yorimitsu H.; Osuka A. Chem.—Eur. J. 2012, 18, 12690. [DOI] [PubMed] [Google Scholar]; e Huang X.; Maulide N. J. Am. Chem. Soc. 2011, 133, 8510. [DOI] [PubMed] [Google Scholar]; f Huang X.; Patil M.; Farès C.; Thiel W.; Maulide N. J. Am. Chem. Soc. 2013, 135, 7312. [DOI] [PubMed] [Google Scholar]
- a Eberhart A. J.; Imbriglio J. E.; Procter D. J. Org. Lett. 2011, 13, 5882. [DOI] [PubMed] [Google Scholar]; b Eberhart A. J.; Procter D. J. Angew. Chem., Int. Ed. 2013, 52, 4008. [DOI] [PubMed] [Google Scholar]
- For thio-Claisen rearrangements of sulfonium ylides and sulfonium salts formed by alternative approaches, see:; a Huang X.; Klimczyk S.; Maulide N. Synthesis 2012, 44, 175. [Google Scholar]; b Harvey N. J.; Viehe H. G. J. Chem. Soc., Chem. Commun. 1995, 22, 2345. [Google Scholar]; c Boyarskikh V.; Nyong A.; Rainier J. D. Angew. Chem., Int. Ed. 2008, 47, 5374. [DOI] [PubMed] [Google Scholar]; d Furukawa N.; Shima H.; Ogawa S. Heteroat. Chem. 1995, 6, 559. [Google Scholar]; e Shima H.; Furukawa N. Tetrahedron 1995, 51, 12239. [Google Scholar]
- See Supporting Information for complete optimization table.
- This is in agreement with the typical relative reactivities of the positions on pyrazoles; see ref (3).
- The activation of sulfoxides with TFAA or Tf2O is well documented; see ref (8).
- See Supporting Information for (a) a schematic of alternative reaction pathways and (b) a table of relative reaction t1/2.
- Formation of allylsulfonium salt from diphenyl sulfoxide is fast; see ref (11).
- Preliminary studies show that similar products can be obtained from the corresponding sulfides using allyl iodide and silver tetrafluoroborate. However, complex mixtures of multiple allylation products as well as regioisomers are obtained.
- a For a review on transition metal catalyzed C–C bond formation reactions, see:Dubbaka S. R.; Vogel P. Angew. Chem., Int. Ed. 2005, 44, 7674. [DOI] [PubMed] [Google Scholar]; b For recent examples, see:Kanemura S.; Kondoh A.; Yorimitsu H.; Oshima K. Synthesis 2008, 2659. [Google Scholar]; c Hooper J. F.; Chaplin A. B.; González-Rodríguez C.; Thompson A. L.; Weller A. S.; Willis M. C. J. Am. Chem. Soc. 2012, 134, 2906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.