

Abstract
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy. High-resolution copy number analysis of 125 tumors of which 24 were subjected to whole-exome sequencing identified 135 homozygous deletions and 994 somatic gene mutations with predicted functional consequences. We identified new recurrent alterations in 6 genes (ARID1A, RPS6KA3, NFE2L2, IRF2, CDH8 and PROKR2) not previously described in HCC. Functional analyses demonstrated tumor suppressor properties for IRF2 whose inactivation, exclusively found in hepatitis B virus related tumors, leads to impaired TP53 function. Alternatively, inactivation of proteins involved in chromatin remodeling was frequent and predominant in alcohol related tumors. Moreover, activation of the oxidative stress metabolism and inactivation of RPS6KA3 were new pathways associated with WNT/β-catenin activation, thereby suggesting a cooperative effect in tumorigenesis. This study shows the dramatic somatic genetic diversity in HCC, it reveals interactions between oncogene and tumor suppressor gene mutations markedly related to specific risk factors.
Keywords: Carcinoma, Hepatocellular; genetics; DNA Copy Number Variations; Humans; Interferon Regulatory Factor-2; genetics; Liver Neoplasms; genetics; Mutation; Signal Transduction; genetics
Hepatocellular carcinoma is the 3rd cause of cancer-related mortality worldwide usually associated to specific risk factors: hepatitis B or C infection, high alcohol intake, hemochromatosis or nonalcoholic fatty liver disease (NAFLD) caused by obesity and insulin resistance[1,2]. More than 90% of HCC arise in the context of chronic hepatitis and cirrhosis and it becomes an increasing health problem. To increase our knowledge on the carcinogenesis mechanisms involved in HCC development, we analyzed the whole-exome sequence of 24 human tumors mainly related to high alcohol intake, the principal HCC risk factor in France (Supplementary Table 1). These samples were included in a larger series of 125 HCC tumors related to various risk factors to identify chromosome alterations and validate the recurrent mutated genes (Supplementary Table 1).
By exome-sequencing 24 HCC paired with their non-tumor liver tissues, we obtained a 73-fold mean sequence coverage of targeted exonic regions with 76% of loci covered at >25-fold (Supplementary Fig. 1). To search for somatic mutations, we identified nucleotide variants in all tumors without mutation in their corresponding non-tumor DNA by filtering the data according to an experimental flowchart applied to all the samples (Supplementary Fig. 2). Then, we selected the mutations to keep variants highly predicted to impair the function of the corresponding coded proteins. Finally, in the 24-paired HCC, we identified 994 mutations that were further verified using Sanger sequencing as somatic events altering 906 different genes (Supplementary Table 2). The number of somatic mutations with predicted functional consequences per tumor was highly variable, ranging from 5 to 121 events/sample (Fig. 1a). As usual in solid tumors[3], most of the variants led to missense (74%) by far more represented than small insertion/deletion (14%) and nonsense and splicing site modifications (12%).
Figure 1.
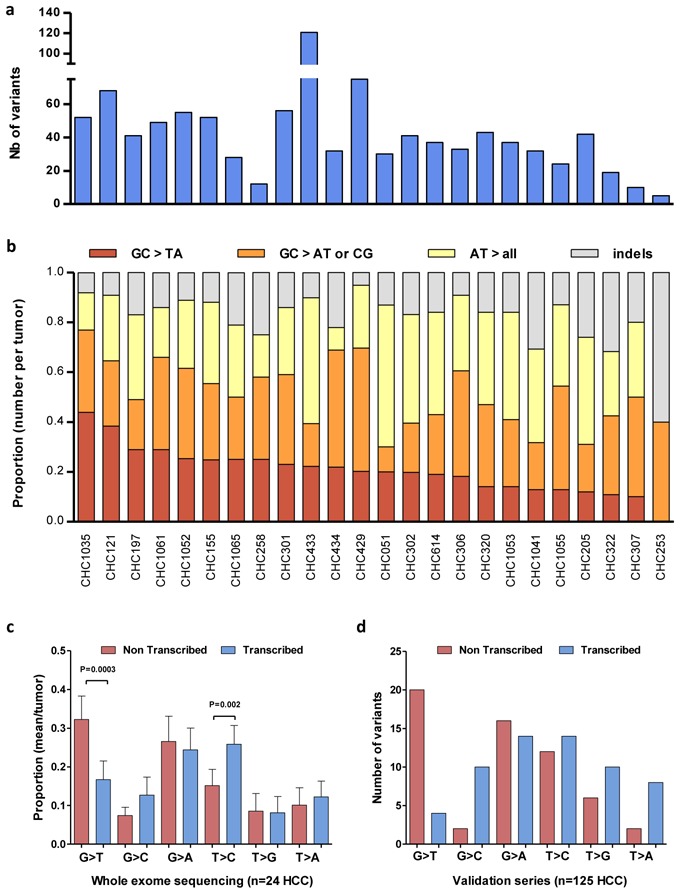
Comparison of mutation profiles in HCC. a, Number of gene mutations in each HCC. b, Frequency of indels and nucleotide substitutions in each HCC. c–d, Fraction of nucleotide substitutions on the transcribed (blue) and the non transcribed (red) strand from the list of somatic mutations identified the exome sequencing of 24 HCC (means with 95% IC, c) and the complete list of gene mutations found in a series of validation of 125 HCC (absolute number of variants per HCC is indicated, d). P-values refer to significant (P=0.0003 and 0.002) elevation in G-to-T transversion on the non-transcribed strand and T-to-C transition on the transcribed strand, respectively.
Analysis of the mutation spectrum could be indicative of specific mutagenesis mechanisms occurring in tumor cells. Interestingly, we found an over-representation of nucleotide transversion and particularly of G:C>T:A changes (Fig. 1b). This spectrum of mutations strongly differs from that usually observed in other solid tumors where C:G>T:A transitions are the most abundant[3]. Moreover, in HCC, G>T transversions were significantly enriched in the non-transcribed DNA strand (Fig. 1c), which is less efficiently repaired after genotoxic injury[4,5]. Then, we searched for association with clinical features and we found that G:C>T:A changes were significantly most frequent in HCC developed on non-cirrhotic livers (P=0.01) and in well-differentiated tumors (P=0.01). These results strongly suggest that exposure to genotoxic agents could contribute to hepatocarcinogenesis independently of the cirrhotic ground. The best characterized genotoxic inducer involved in HCC is exposure to Aflatoxin B1 in combination with HBV infection in sub-tropical areas associated with G>T transversion at codon 249 on TP53[6,7]. However, in the present series of patients living in France, the causative genotoxic agent remains to be determined by epidemiological/toxicological studies focusing on non-cirrhotic HCC patients.
Next, SNP array analysis of 125 HCC revealed frequent chromosome gains (1q, 5, 6p, 7, 8q, 17q and 20) and losses (1p, 4q, 6q, 8p, 13q, 16, 17p and 21) as usually described in HCC[8] (Supplementary Fig. 3a). Hyperploid DNA content (ploidy ≥ 3) was found in 20% of the cases and it was associated with poor tumor differentiation (Edmondson III-IV, P=0.03). We also evaluated the level of chromosomal instability in each tumor by calculating the “Fraction of Aberrant Arms” (FAA, i.e. the proportion of chromosome arms altered on more than 40% of their length). Highly rearranged copy-number profiles were associated with clinical and pathological features of aggressive tumors: HCC developed in non-cirrhotic liver (P=0.04), with HBV infection (P=0.01), large size (0.004), poorly differentiated (Edmondson III–IV, P=0.04) and with high serum alpha-fetoprotein (P=0.03). Focal chromosome amplifications (size < 0.3 Mb and copy number > ploidy+2) were identified in 32% of the tumors. We observed only one region, at 11q13.3, amplified in 2 cases (Supplementary Table 3); this region included CCND1 and FGF19, two genes previously found amplified in HCC[9]. Homozygous deletions were more frequent (40% of the tumors), usually focal (mean size = 0.2 Mb) and significantly associated with aggressiveness of the tumors and with poor survival (Supplementary Fig. 3b and Supplementary Table 4). A total of 12 regions were recurrently altered by homozygous deletion, the most frequent were located at the CDKN2A/B (6.4%), AXIN1 (3.2%) and IRF2 (3.2%) loci (Supplementary Fig. 4).
To draw a more comprehensive picture of the HCC-altered gene landscape, we further screened for the mutations present in 13 genes expressed in liver tissues and modified by homozygous deletion and/or mutation in at least 3 different tumors (Supplementary Table 2 and Fig. 2). We also screened 3 other genes well known as recurrently altered in hepatocellular adenoma (IL6ST[10], HNF1A[11]) or in rare cases of HCC (KRAS[12]). Screening 125 HCC revealed four genes (CTNNB1, TP53, ARID1A and AXIN1) altered in more than 10% of the cases, whereas the others were less frequently mutated. We identified a total of 118 somatic point mutations in 125 tumors (Supplementary Table 5). The spectrum of mutations demonstrated a distribution similar to that observed in the exome-sequencing with an over-representation of G>T on the non-transcribed DNA strand (Fig. 1d). Thus, we confirmed a “genotoxic signature” in this entire series of HCC related to various etiologies.
Figure 2.
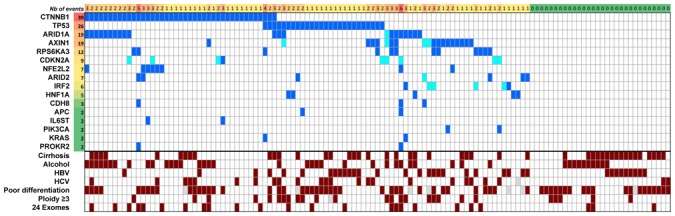
Overview of mutations and major associated clinical features. The heatmap displays genes (row) and tumors (columns) with or without mutations (dark blue) or homozygous deletion (clear blue). The total number of cases (resp. genes) in which each gene (resp. case) contained a mutation is shown. The number of events is indicated. Potential occurrence with major clinical and pathological features and whole-exome sequenced cases are pointed (dark red).
In the exome-sequencing and in the tumor validation set, the WNT/s-catenin pathway was the most frequently altered by either activating mutation of s-catenin (32.8%), inactivation of AXIN1 (15.2%) or APC (1.6%) (Fig. 3a). CTNNB1, AXIN1 and APC gene alterations were mutually exclusive (only one HCC was mutated for both CTNNB1 and AXIN1). Their mutation spectra were classical excepted for 4 rare CTNNB1 mutations identified in exon 6 to 8 (Fig. 3b and Supplementary Table 5). CTNNB1 mutations define a homogenous sub-type of HCC not related to HBV infection (P=0.001) and with a specific transcriptomic signature (G5-6 subclasses as defined in Boyault et al[13], P<10−9 Fig. 2). In contrast, AXIN1 and APC mutations occurred in HCC related to various etiologies including HBV infection. In exome-sequencing, unique mutations of FZR1, CSNK1E and CDC16 genes related to the s-catenin pathway were also identified but not further evaluated.
Figure 3.
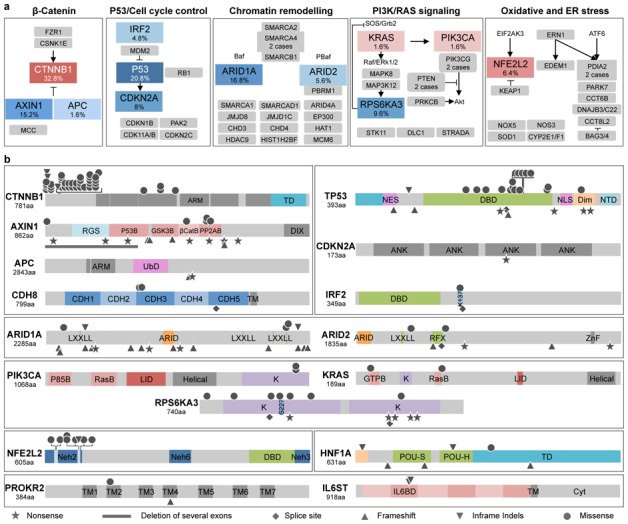
Altered pathways and somatic mutation spectra in 125 HCC. a, Major pathways commonly altered by somatic mutations or homozygous gene deletions. Alteration frequencies are expressed as a percentage mutation and/or homozygous deletion in the validation series of 125 (red or blue when activated or inactivated, respectively) or 24 exome-sequenced (grey) HCC; for unique gene mutation, no frequency is indicated. Arrows are positive interactions and lines are inhibitory interactions. b, Spectrum of somatic mutations identified in HCC. Inactivating and activating mutations are shown above and below the core protein, respectively. Functional domains are colored boxes. ANK: Ankyrin repeat, ARM: Armadillo repeat, βCatB: βCatenin binding Domain, CDH: Cadherin Domain, Cyt: Cytosolic Domain, DBD: DNA Binding domain, Dim: Dimerization Domain, GSK3B: GSK3 Binding Domain, GTPB: GTP binding Domain, IL6BD: IL6 Binding Domain, K: Kinase domain, LID: Lipid interaction domain, Neh: Nrf2-ECH Homology Domain, NES: Nuclear Export Signal, NLS: Nuclear Localization Signal, NTD: Negative Transactivation Domain, P53B: P53 Binding Domain, P85B: P85 Binding Domain, PP2AB: PP2A Binding Domain, RasB: Ras Binding Domain, RGS: regulator of G-protein signalling, TD: Transactivation domain, TM: Transmembrane domain, UbD: Ubiquitinylated domain.
TP53, was identified as the second pathway the most frequently altered in HCC. This manifested by TP53 inactivating mutations (20.8%) and CDKN2A homozygous deletion or mutations (8%) (Fig. 3a). TP53 alterations were usually exclusive from CTNNB1 mutations (P=0.0001), but not from AXIN1 and APC. In accordance with the well-known function of TP53 in the maintenance of chromosome stability, TP53 mutations were more frequent in HCC displaying a high number of chromosome rearrangements (P=0.003). IRF2 (interferon regulatory factor 2) inactivation was also identified in 6/125 HCC (4.8%), due to homozygous deletion, splicing site or missense mutations (Supplementary Table 5 and Supplementary Fig. 4). IRF2, a partner of the TP53 inhibitor MDM2, acts as a transcriptional regulator through its DNA-binding activity and through protein-protein interactions[14]. It has been reported to play a major role in cell growth regulation and immune response. Interestingly both splicing and missense mutant altered the K137 residue that is known to be SUMOylated in IRF2, thereby affecting its transcriptional activity[15]. All 6 tumors showed a biallelic alteration of IRF2 and were associated with HBV infection (P=0.0003). IRF2 mutations were also associated with hyperploidy and high chromosome instability (P=0.01). We searched for a putative tumor suppressor function of IRF2 in hepatocellular cell lines. Accordingly, we showed that siRNA-mediated silencing of IRF2 in 2 different cell lines (HepaRG and HepG2, wild type for TP53) significantly increased cell proliferation whereas IRF2 over-expression was responsible for dramatic apoptotic cell death (Fig. 4a–c and Supplementary Fig. 5a–c). In vivo, stable IRF2 extinction by shRNA in HepaRG HCC cell lines resulted in increased tumor growth using sub-cutaneous xenograft in CD1 nude mice (Fig. 4d and supplementary Fig. 5d). Looking for association in gene mutations, we showed that IRF2 and TP53 mutations were mutually exclusive, whereas tumors mutated for either IRF2 or TP53 mainly belonged to the same transcriptomic subclass G2[13]. Because IRF2 is known to bind MDM2[16], we hypothesized that the lack of IRF2 could impair P53 function. Accordingly, we showed that IRF2 silencing decreased P53 protein levels and P53 target genes expression in HepaRG (Fig. 4e). Moreover, a strong correlation between IRF2 and P53 protein expression levels was observed. Thus, our study demonstrated for the first time the role of IRF2 as a tumor suppressor in HBV-associated HCC and its function as a regulator of the P53 pathway.
Figure 4.

IRF2 is a new tumor suppressor gene in HCC that controls the P53 pathway. a, Effect of in vitro IRF2 silencing in HepaRG cell line: increased cell proliferation with IRF2 siRNA (siIRF2) when compared to control siRNA (siControl) in triplicate with regression analysis. Relative mRNA and protein expression were quantified by qRT-PCR and Western blot, respectively (n=3; mean ± SD). b, IRF2 overexpression in HepaRG cell line (pIRF2) induced dramatic cell death when compared to HepaRG transfected with an empty plasmid (pControl) in triplicate with regression analysis. Relative mRNA and protein expression (n=3; mean ± SD). c, Increased apoptosis in HepaRG cells overexpressing IRF2 compared to pControl: flow cytometry analysis shows a higher percentage of AnV+, IP+ cells with pIRF2 (n=3; mean ± SD). d. Stable IRF2 silencing by shRNAs (shIRF2(1) and shIRF2(2)) in HepaRG cell line enhanced tumor growth in a subcutaneous xenograft mouse model (n=5; mean tumor volume ± SD). Immunohistochemical analysis of IRF2 in xenograft tumor with shControl and shIRF2 HepaRG cell confirmed IRF2 silencing. e, Western-blot analysis comparing P53 (53 kDa), IRF2 (39 kDa) and βActin (42 kDa) expression levels between HepaRG cells transfected with siControl and siIRF2 (siIRF2(1) and siIRF2(2)) after 48h, 72h and 96h transfection. Western-blot analysis shows reduced expression of P53 following IRF2 silencing. Strong positive correlation between P53 and IRF2 expression levels was observed (linear regression analysis, Pearson correlation). f, P53 pathway target genes expression in HepaRG cells with silenced (blue) or enhanced (red) IRF2 expression, qRT-PCR data were normalised to the mean expression with siControl or pControl indicated by a line, respectively (n=3; mean ± SD).
Genes belonging to chromatin remodeling complexes were the third most frequently altered class in the exome screening and we validated ARID1A and ARID2 mutations in 16.8% and 5.6% of the cases in the tumor validation set, respectively. ARID1A and ARID2 are part of the SWI/SNF-related chromatin-remodeling complex that controls accessibility of the promoter regions by the transcriptional machinery and demonstrates tumor suppressor functions[17,18]. ARID1A belongs to the BAF complex and was recently identified inactivated in several tumors types including gastric, ovarian and bladder carcinoma but its involvement in HCC has not been reported yet[19–22]. Here, we identified a spectrum of mutations predicted to inactivate ARID1A function similar to that observed in other tumor types (Fig. 3b). ARID1A mutations were significantly more frequent in HCC related to alcohol intake (P=0.002) and showed a significant association with CTNNB1 mutations (P=0.05). In contrast, mutations in ARID2, that belongs to the PBAF complex, were less frequent but exclusive from ARID1A mutations. Overall ARID2 mutation frequency was similar to that previously observed in HCC[23] but we did not observe a significant relationship with HCV infection or any other risk factors. In the exome sequencing, we identified some mutations in additional genes participating to the chromatin remodeling (i.e. PBRM1, SMARCA1, SMARCA2, SMARCA4, SMARCAB1 and SMARCD1) (Fig. 3a). However, most of these mutations were not recurrent and were not exclusive from ARID1A and ARID2 alterations. The precise mutation frequency of the six genes mentioned above remains to be evaluated in a large series of HCC, but overall, more than 24.8% of HCC exhibited a mutation in at least one gene related to chromatin remodeling, thereby indicating that this pathway might be a major contributor to hepatocyte tumorigenesis.
Among the genes less frequently mutated in HCC, we identified for the first time in solid tumors, recurrent mutations in RPS6KA3 (9.6%), a gene located on chromosome X that encodes the ribosomal S6 protein kinase 2 (RSK2). RSK2 is a serine/threonine kinase of the RAS/MAPK signaling pathway directly phosphorylated and activated by ERK1/2. RSK2 exerts a feedback inhibition on the ERK1/2 pathway by phosphorylating and inhibiting SOS[24,25]. Half of RPS6KA3 mutations lead to premature stop codons or altered splicing sites, whereas 4 out of 7 missense mutations were located close to S227 and T557 phosphorylation sites required for RSK2 activation (Fig. 3b). Thus, RPS6KA3 somatic mutations were predicted to inactivate RSK2 function. Other components of the RAS/MAPK and PIK3 pathways were rarely mutated (Fig. 3a). Interestingly, in 11 cases out of 12, RPS6KA3 mutations were found in HCC developed without cirrhosis (P=0.05) and RPS6KA3 was frequently associated with AXIN1 mutations (P=0.02) suggesting cooperation between RPS6KA3 inactivation and WNT/s-catenin activation in tumorigenesis.
NFE2L2 coding for NRF2 a transcription factor crucial for cellular redox homeostasis, was mutated in 6.4% of HCC (Fig. 3a). All the mutations were located within the DLG and ETGE motifs, hotspots of somatic mutations previously identified in lung, esophageal, laryngeal and skin squamous cell carcinomas[26–28] (Fig. 3b). These mutations are known to inhibit KEAP1-mediated degradation of NRF2. Interestingly, 6 out of 8 NFE2L2 mutated HCC were also mutated for CTNNB1 (P=0.015) and another case was mutated for AXIN1. Moreover, an additional tumor demonstrating a KEAP1 inactivating mutation was also mutated for CTNNB1. Hence, these results identified for the first time in HCC the role of alteration in the oxidative stress pathway mainly in WNT/s-catenin activated tumors. Finally, other genes such as CDH8 and PROKR2 were rarely mutated in 2.4% and 1.6% of HCC, respectively.
In conclusion, we identified new oncogenes and tumor suppressor genes recurrently altered in HCC enlightening the major role of the SNF/SWI chromatin remodeling complexes and the new involvement of the interferon and the oxidative stress pathways in hepatocellular malignant proliferation and transformation.
Methods summary
All tumors and corresponding non-tumor liver tissues were frozen after surgical resection. These tumors were clinically and genetically characterized; they were previously included in genetic and phenotypic studies[13]. The study was approved by the local Ethics Committee (Paris Saint-Louis), informed consent was obtained in accordance with French legislation and the exome-sequencing project was approved by the national INSERM IRB committee in 2010. Exome-DNA sequencing was performed as described in the supplementary method. All mutations were validated by sequencing independent PCR product on both strands. In all cases the somatic origin of the mutation found in tumor was verified by sequencing the corresponding adjacent, normal liver sample. HepaRG cells were kindly provided by Fabien Zoulim (Centre de recherche en cancerologie de Lyon (CRCL) INSERM U-1052/CNRS UMR 5286), HuH7 were purchased from ATCC. In cell experiments, quantitative RT-PCR and proliferation assays were performed in triplicate in at least 2 different cell lines and with two different siRNAs.
Supplementary Material
Acknowledgments
We warmly thank Thomas Burguiere, Gilles Thomas, Robin Fahraeus and Coraline Mlynarczyk for their helpful participation to this work. We also thank Jean Saric, Christophe Laurent, Brigitte Le Bail, Anne Rullier, Antonio Sa Cunha (CHU Bordeaux) and Jeanne Tran Van Nhieu, Daniel Cherqui, Daniel Azoulay (CHU Henri Mondor, Créteil) for contributing to the tissue collection. This work was supported by the INCa with the ICGC project, the Ligue Nationale Contre le Cancer (“Cartes d’identité des tumeurs” program), the PAIR-CHC project NoFLIC (funded by INCa and Association pour la recherche contre le Cancer, ARC), the Réseau national CRB Foie and BioIntelligence. C.G., G.A., are supported by a fellowship from the INCa and ANRS respectively.
Footnotes
List of Key genes/proteins: IRF2, RPS6KA3, NFE2L2, NRF2, CTNNB1, TP53, ARID1A, ARID2, CDKN2A, IL6ST, HNF1A, PROKR2, CDH8, KRAS, FGF19, KEAP1
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132 (7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International journal of cancer. Journal international du cancer. 2010;127 (12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 3.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446 (7132):153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denissenko MF, Pao A, Pfeifer GP, Tang M. Slow repair of bulky DNA adducts along the nontranscribed strand of the human p53 gene may explain the strand bias of transversion mutations in cancers. Oncogene. 1998;16 (10):1241–1247. doi: 10.1038/sj.onc.1201647. [DOI] [PubMed] [Google Scholar]
- 5.Hainaut P, Pfeifer GP. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22 (3):367–374. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 6.Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350 (6317):429–431. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- 7.Hsu IC, et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350 (6317):427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 8.Nault JC, Zucman-Rossi J. Genetics of hepatobiliary carcinogenesis. Seminars in liver disease. 2011;31 (2):173–187. doi: 10.1055/s-0031-1276646. [DOI] [PubMed] [Google Scholar]
- 9.Sawey ET, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer cell. 2011;19 (3):347–358. doi: 10.1016/j.ccr.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rebouissou S, et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumors. Nature. 2009;457 (7226):200–204. doi: 10.1038/nature07475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bluteau O, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nature genetics. 2002;32 (2):312–315. doi: 10.1038/ng1001. [DOI] [PubMed] [Google Scholar]
- 12.Weihrauch M, et al. Frequent k- ras -2 mutations and p16(INK4A)methylation in hepatocellular carcinomas in workers exposed to vinyl chloride. British journal of cancer. 2001;84 (7):982–989. doi: 10.1054/bjoc.2000.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyault S, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45 (1):42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 14.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annual review of immunology. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 15.Han KJ, Jiang L, Shu HB. Regulation of IRF2 transcriptional activity by its sumoylation. Biochemical and biophysical research communications. 2008;372 (4):772–778. doi: 10.1016/j.bbrc.2008.05.103. [DOI] [PubMed] [Google Scholar]
- 16.Pettersson S, Kelleher M, Pion E, Wallace M, Ball KL. Role of Mdm2 acid domain interactions in recognition and ubiquitination of the transcription factor IRF-2. The Biochemical journal. 2009;418 (3):575–585. doi: 10.1042/BJ20082087. [DOI] [PubMed] [Google Scholar]
- 17.Guan B, Wang TL, Shih Ie M. ARID1A, a Factor That Promotes Formation of SWI/SNF-Mediated Chromatin Remodeling, Is a Tumor Suppressor in Gynecologic Cancers. Cancer research. 2011;71 (21):6718–6727. doi: 10.1158/0008-5472.CAN-11-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nature reviews. Cancer. 2011;11 (7):481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 19.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330 (6001):228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiegand KC, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. The New England journal of medicine. 2010;363 (16):1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones S, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Human mutation. 2011 doi: 10.1002/humu.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nature genetics. 2011;43 (12):1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 23.Li M, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nature genetics. 2011;43 (9):828–829. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Douville E, Downward J. EGF induced SOS phosphorylation in PC12 cells involves P90 RSK-2. Oncogene. 1997;15 (4):373–383. doi: 10.1038/sj.onc.1201214. [DOI] [PubMed] [Google Scholar]
- 25.Schneider A, Mehmood T, Pannetier S, Hanauer A. Altered ERK/MAPK signaling in the hippocampus of the mrsk2_KO mouse model of Coffin-Lowry syndrome. Journal of neurochemistry. 2011;119 (3):447–459. doi: 10.1111/j.1471-4159.2011.07423.x. [DOI] [PubMed] [Google Scholar]
- 26.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475 (7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim YR, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. The Journal of pathology. 2010;220 (4):446–451. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
- 28.Shibata T, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105 (36):13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.