

Abstract
Purpose of review
This review summarizes recent progress in the development of myostatin inhibitors for the treatment of muscle wasting disorders. It also focuses on findings in myostatin biology that may have implications for the development of antimyostatin therapies.
Recent findings
There has been progress in evaluating antimyostatin therapies in animal models of muscle wasting disorders. Some programs have progressed into clinical development with initial results showing positive impact on muscle volume.
In normal mice myostatin deficiency results in enlarged muscles with increased total force but decreased specific force (total force/total mass). An increase in myofibrillar protein synthesis without concomitant satellite cell proliferation and fusion leads to muscle hypertrophy with unchanged myonuclear number. A specific force reduction is not observed when atrophied muscle, the predominant therapeutic target of myostatin inhibitor therapy, is made myostatindeficient.
Myostatin has been shown to be expressed by a number of tumor cell lines in mice and man.
Summary
Myostatin inhibition remains a promising therapeutic strategy for a range of muscle wasting disorders.
Keywords: ActRIIB, cancer cachexia, muscle function, muscle hypertrophy, muscle wasting, muscular dystrophy, myostatin, satellite cell
INTRODUCTION
Cachexia is a wasting syndrome exhibited by many cancer patients particularly at advanced stages of disease. It is characterized by the loss of skeletal muscle mass (with or without fat loss), despite adequate nutritional intake. Cancer cachexia is associated with diminished quality of life, functional performance and decreased survival. Cancer-related muscle loss is an independent predictor of poor outcome linked to increased immobility and mortality, and has also been associated with intolerance to chemotherapy. There has been increasing interest in therapeutic interventions that prevent, delay or treat cancer-related muscle wasting, in hopes of improving outcomes for the cancer patient. Some recent reviews and key articles in this area are available [1–16].
There are many commonalities at the molecular level in the pathways in skeletal muscle that result in atrophy, whether it is in the context of cancer cachexia or other noncancer muscle wasting situations. The mechanisms regulating skeletal muscle mass have recently been reviewed [17–21]. Myostatin has emerged as an intriguing therapeutic target [22]. Myostatin, a member of the TGFβ superfamily of growth factors, is a highly conserved negative regulator of skeletal muscle mass that is upregulated in many conditions of muscle wasting. Various induced or natural conditions leading to myostatin deficiency result in increased muscle mass and strength in normal animals and have been shown to treat or prevent a range of muscle wasting conditions.
Box 1.
no caption available
The myostatin signaling pathway and its role in regulating skeletal muscle has been recently reviewed [23,24]. At the molecular level, myostatin binds to and activates the activin receptor IIB (ActRIIB)/Alk 4/5 complex (Fig. 1). Although ActRIIB/Alk4/5 is broadly expressed, myostatin is produced and exhibits its effects primarily on skeletal muscle.
FIGURE 1.
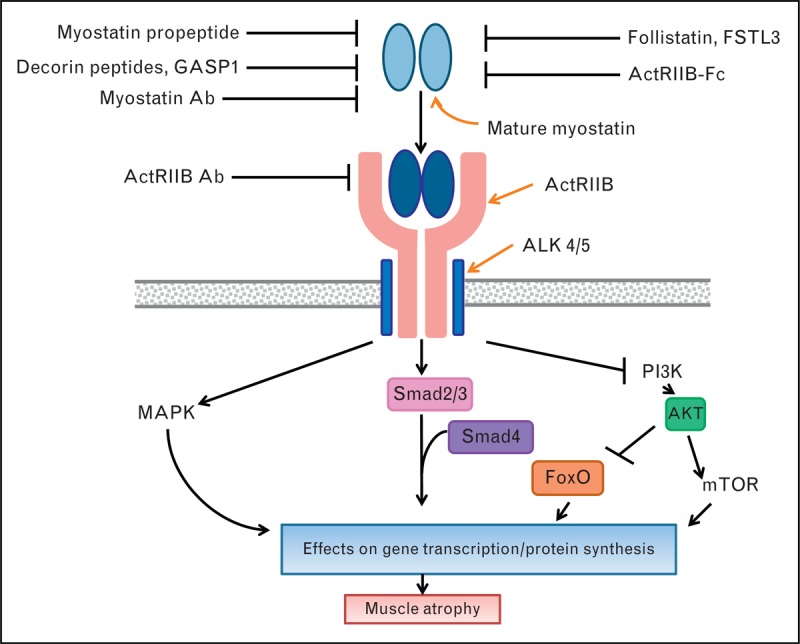
Summary of therapeutic invention points in the myostatin signaling pathway. Myostatin binds to its receptor complex ActRIIB/Alk 4 or 5 on skeletal muscle resulting in activation of the Smad 2/3, mitogen-activated protein kinase and inhibition of the PI3K intracellular signaling pathways that together result in gene transcriptional changes and effects on protein synthesis that ultimately give rise to muscle atrophy. Myostatin pathway inhibitors act extracellularly by either binding myostatin directly (Fstl3, Follistatin, myostatin antibody, GASP1, myostatin propeptide, decorin peptides, ActRIIB-Fc) or by binding its receptor complex (ActRIIB antibody) in order to block myostatin engaging its receptor complex and activating downstream signaling. Some of the inhibitors are naturally occurring (myostatin propeptide, Gasp1, follistatin, Fstl3) whereas others are engineered (myostatin antibody, ActRIIB antibody, ActRIIB-Fc). ---I represent inhibitory activities. → represent activating activities. Ab = antibody.
Many approaches are being taken both preclinically and clinically to inhibit the myostatin signaling pathway (Fig. 1). The majority of these approaches acts extracellularly to block myostatin engaging with the ActRIIB/Alk4/5 receptor complex, either by binding directly to myostatin itself or by binding to components of this receptor complex. Due to the fact that multiple ligands signal through, and therefore bind, ActRIIB apart from myostatin (including activin A, gdf11, bmp9) [25–27] the approaches that target the ActRIIB receptor or use ActRIIB as a soluble decoy receptor may not specifically block myostatin action. Similarly, the naturally occurring myostatin binding proteins follistatin and Fstl3 are known to bind a number of growth factors in addition to myostatin [28,29]. The added risk/benefit of these multitargeted approaches is under investigation (see below).
PROGRESS IN VALIDATION OF MYOSTATIN AS A TARGET FOR MUSCLE WASTING DISORDERS
Preclinical results
Two recent studies, performed in mouse models of cancer cachexia, have examined the effects of myostatin inhibitors on physical performance and muscle function, building on previous data that showed positive effects on muscle mass [30,31]. Mice with Lewis Lung carcinoma treated with ActRIIB-Fc (Fig. 1), a soluble myostatin receptor that binds myostatin, activin and other ligands, showed increases in body weight and muscle weights with grip strength significantly increased and resting time significantly decreased by treatment [32▪]. A myostatin antibody in the same model was able to completely abrogate the tumor-induced reduction in total muscle force in various limb and diaphragm muscles [33▪]. The results of these recent studies are encouraging as the value of myostatin inhibitors to cancer patients exhibiting muscle wasting is ultimately to affect functional performance through increased muscle function.
Aside from models of cancer cachexia, most recently published preclinical activity with myostatin inhibitors has focused on developing therapies in the area of rare or orphan diseases, in which symptoms are devastating to patients and few if any significant treatment options are available. Testing of myostatin inhibitors in animal models of muscular dystrophy [34] has shown generally positive effects on muscle mass but inconsistent effects on muscle function and histopathology [reviewed in [35▪]]. ActRIIB-Fc or ActRIIB shRNA given to mdx mice, a well used but not ideal model of human muscular dystrophy [36,37▪▪], produced increases in muscle mass and total force but specific force was unchanged [38,39▪,40]. In contrast, a recent study reported an increase in specific force of the soleus muscle in mdx mice after long-term exposure to a myostatin propeptide [41]. Studies with myostatin inhibitors have not shown any improvement on eccentric contraction-induced force drop, a key measure of myofiber structural integrity [40,42,43]. Therefore, there is increasing evidence that myostatin inhibitors can improve muscle function in the mdx mouse through an increase in muscle mass and total force but do not consistently improve the underlying weakness of dystrophic muscle. There has been hope that myostatin inhibitors might attenuate the muscle fibrosis that is a hallmark of muscular dystrophy, given myostatin's role in inducing dystrophic muscle fibroblast proliferation [44▪] and the observation of decreased connective tissue in myostatin null mice [45▪]. Although earlier observations in mdx mice [34] and more recent observations in the golden retriever muscular dystrophy model [GRMD [46]], showed improvement in fibrosis with myostatin antibody or myostatin propeptide treatment, respectively, no improvement on muscle histopathology was seen after ActRIIB-Fc treatment of mdx mice [40,42]. It has been suggested that the degree of muscle disease at the time of treatment may influence outcome [43]. Human muscular dystrophy disorders display paradoxical muscle wasting and selective hypertrophy of skeletal muscles, leading to imbalance, contractures and postural instabilities [37▪▪]. When the muscle hypertrophic myostatin heterozygote whippet [47] was crossed with the GRMD dog, selective muscle hypertrophy seen in the GRMD dog was exaggerated resulting in more pronounced postural instability and worsened clinical scores, cautioning that further hypertrophy of already selective hypertrophic muscle in muscular dystrophy may not be beneficial [37▪▪]. Dysferlin null mice, a model of dysferlin-deficiency muscular dystrophy [48], expressing the myostatin inhibitor follistatin, demonstrated a transient increase in muscle mass followed by decreased muscle mass and function and increased muscle fibrosis [Lee et al. MDA meeting, San Diego, 2013].
There is excitement regarding disease-modifying therapies currently in clinical development for muscular dystrophy based on exon skipping methods, which overcome the underlying genetic defect of the dystrophin gene and improve specific muscle force without effects on muscle mass [reviewed in [49,50]]. Myostatin inhibitors are currently being investigated preclinically as possible adjunct therapy with these molecules [39▪,42,51–53].
The recently described increase in axon number together with delay in age-related neural degeneration in myostatin null mice have added support to the investigation of myostatin inhibitors for the treatment of severe neuromuscular disorders [54▪,55]. However, SOD1 null mice, a model of amyotrophic lateral sclerosis, did not exhibit any improvements in survival (despite improvements in muscle mass) when exposed to myostatin inhibitors [56]. In another report, crossing of SMN null mice, a model of Spinal Muscular Atrophy, with myostatin null mice did not lead to increases in muscle mass or effects on survival [57], consistent with results using myostatin inhibitors from Sumner et al. [58] but inconsistent with the positive effects reported by Rose et al. [59]. In contrast to the above reports, treatment of the myotubularin-deficient mouse, a model of X-linked myotubular myopathy, with ActRIIB-Fc did lead to transient increases in muscle mass and strength and a 17% increase in survival [60▪▪].
Other animal models of muscle wasting have been used to determine if inhibition of myostatin has therapeutic potential in treating a range of muscle wasting conditions. Positive results have been reported in models of chronic kidney disease, disuse atrophy and age and hypogonadism-induced muscle loss [61,62▪,63]. Overexpression of the myostatin interacting protein GASP1 [64] has been shown to induce muscular hypertrophy in mice but has not yet been tested in models of muscle wasting. Identification of myostatin-blocking decorin peptides are at an even earlier stage of preclinical development [65]. There is increasing preclinical evidence to suggest that inhibition of myostatin may also have metabolic benefits. Myostatin deficiency or myostatin inhibition in mice has been shown to result in decreased fat mass and increased insulin sensitivity raising the therapeutic potential of myostatin inhibition in obesity and insulin resistance associated with obesity [reviewed in [23]].
Clinical results
Some myostatin inhibitors have progressed into clinical development as summarized in Table 1.
Table 1. Summary of clinical development of myostatin inhibitors for treatment of muscle wasting associated with cancer and other disorders.
| Name | Type of inhibitor | Sponsor | Condition | Patient population | Phase of development | Current status | Outcome | CT identifier/Ref. |
| LY2495655 | Myostatin antibody | Lilly | – | Healthy volunteers | Phase 1 SAD | Completed | Well tolerated, increased TMV | [66▪▪] |
| – | Healthy Japanese volunteers | Phase 1; SAD, MAD | Completed | NCT01341470 | ||||
| Muscular atrophy | Hip arthroplasty | Phase 2 | Actively recruiting | NCT01369511 | ||||
| Muscle weakness | Older weak fallers | Phase 2 | Active, not recruiting | NCT01604408 | ||||
| Advanced cancer | Cancer patients | Phase 1 | Active, not recruiting | Well tolerated, increased muscle volume (interim report) | [66▪▪]; NCT01524224 | |||
| Advanced cancer | Pancreatic cancer | Phase 2 | Recruiting | NCT01505530 | ||||
| MYO-029 | Myostatin antibody | Wyeth | – | Healthy volunteers | Phase 1 | Completed | NCT00563810 | |
| Adult muscular dystrophy | BMD; Facioscapulohumeral; muscular dystrophy; Limb-Girdle muscular dystrophy | Phase 1/2 | Terminated | Increase in LBM; no effects on strength or function; skin hypersensitivity at highest doses | [70▪▪]; NCT00104078 | |||
| ACE-031/Ramatercept | ActRIIb-Fc | Acceleron/Shire | Muscle loss | Healthy postmenopausal women | Phase Ia (SAD) | Completed | Generally well tolerated, increased LBM and TMV | [70▪▪]; NCT00755638 |
| Muscle atrophy | Healthy postmenopausal women | Phase Ib (MAD) | Terminated | Common AE: nosebleed. Increased LBM, TMV | Borgstein et al. 2010, WMS, Japan; NCT00952887 | |||
| DMD | DMD boys | Phase 2; MAD | Terminated | Reversible telangiectasia and nosebleed; increased LBM, attenuated TMV and 6MWD | [71▪▪]; NCT01239758; NCT01099761 | |||
| PF-06252616 | Myostatin antibody | Pfizer | – | Healthy volunteers | Phase I; SAD, MAD | Recruiting | NCT01616277 | |
| BYM338 | ActRIIB antibody | Novartis | Muscle wasting | Healthy volunteers | Phase 1 SAD | Well tolerated, increase in TMV | D. Rook, Intl conference on Sarcopenia research, Orlando, Dec, 2012 | |
| sIBM | sIBM | Phase 2; Single dose | Completed | Well tolerated, increase in TMV, LBM, quadriceps strength and 6MWD | Amato et al. MDA meeting San Diego, 2013; NCT01423110 | |||
| COPD | COPD patients with cachexia | Phase 2 | Recruiting | NCT01669174 | ||||
| Skeletal muscle | Sarcopenic adults | Phase 2 | Recruiting | NCT01601600 | ||||
| Cachexia | Cancer cachexia (lung or pancreas) | Phase 2 | Recruiting | NCT01433263 | ||||
| REGN1033/SAR391786 | Myostatin antibody | Regeneron/Sanofi | Rehabilitation postorthopedic surgery | Healthy volunteers | Phase 1 SAD, MAD | Active, not recruiting | NCT01507402 NCT01720576 | |
| FS344 | Follistatin-AAV gene therapy | Nationwide Children's Hospital/Milo Biotech | BMD and SIBM | BMD and SIBM | Phase 1 | Enrolling by invitation | NCT01519349 | |
| AMG-745 | Myostatin peptibody | Amgen | Age-associated muscle loss | Age-associated muscle loss | Phase 2 | Withdrawn prior to enrolment | NCT00975104 |
BMD, Becker muscular dystrophy; COPD, chronic obstructive pulmonary disease; DMD, Duchenne muscular dystrophy; CT identifier, clinical trial identifier at ClinicalTrials.gov; LBM, lean body mass ; MAD, multiple ascending dose; SAD, single ascending dose; sIBM, sporadic inclusion body myositis; TMV, thigh muscle volume; 6MWD, six minute walk distance.
LY2495655 is a myostatin antibody that is currently in clinical development for muscle wasting associated with cancer and other disorders (see Table 1). Results of a study in healthy volunteers demonstrated the drug to be well tolerated and led to an increase in thigh muscle volume (TMV) [66▪▪]. Interim results of a Phase 1 safety study of LY2495655 in advanced cancer patients without chemotherapy reported that a maximum tolerated dose was not reached and increased muscle volume with concomitant increases in hand grip strength and other functional measures were observed; however, a clear dose–response was not observed, ascribed to small sample size and patient heterogeneity [66▪▪]. A Phase 2 trial of LY2495655 in patients with locally advanced/inoperable or metastatic pancreatic cancer receiving standard of care chemotherapy is ongoing with overall survival as the primary endpoint of the trial (Table 1).
BYM-338 is an antibody directed to ActRIIB that is currently in Phase 2 for the treatment of cachexia in patients with stage IV nonsmall cell lung cancer or Stage III/IV adenocarcinoma of the pancreas. The primary endpoint of the trial is TMV at 8 weeks as measured by MRI. BYM-338 is also in Phase 2 trials for other muscle wasting disorders (Table 1). Single infusions of BYM-334 in healthy volunteers were reported to be well tolerated and resulted in an increase in TMV (D. Rook; International conference on sarcopenia research, Orlando, December 2012).
The correlation of increases in muscle volume to clinically meaningful functional outcomes for patients treated with myostatin inhibitors still awaits validation. Interestingly, myostatin protein levels were found to be upregulated in muscle biopsies taken from early stage gastric cancer patients even before significant weight loss (>10%) had occurred, leading to the suggestion of early intervention to prevent cancer cachexia [67▪].
Clinical development of myostatin inhibitors for the treatment of muscular dystrophy has made recent progress [49,68]. Phase 1/2 results of MYO-029, a myostatin antibody, failed to show effects on muscle strength or function in adult Becker, limb-girdle and facioscapulohumeral muscular dystrophy patients [69]. PF-06252616 is a myostatin antibody currently in Phase 1 testing in healthy volunteers; it was recently given orphan drug designation by the European Medical Agency (EMA) for treatment of Duchenne muscular dystrophy (DMD). ACE-031, a human ActRIIB-Fc, in single and multiple ascending studies in healthy volunteers showed significant increases in lean body mass (LBM) and TMV [70▪▪] [Borgstein et al. World Muscle Society, Japan, 2010]. ACE-031 was awarded orphan status and accelerated review by the Food and Drug Administration (FDA) for muscular dystrophy in 2010. Results from a Phase 2 study with ACE-031 in DMD boys showed an increase in LBM and attenuation of declines in TMV and six minute walk distance (6MWD) [71▪▪]. However, the observation of reversible nosebleeds and skin telangiectasias in the healthy volunteer MAD study as well as in the Phase 2 muscular dystrophy study [71▪▪] has led to the termination of these trials. The underlying mechanism behind these adverse events is not understood although the genetic associations of mutations in the ALK1 type I receptor with hereditary haemorrhagic telangiectasias type 2 [72], and the elucidation of the Alk1/ActRIIB complex as the main signaling pathway for BMP9 [27,73] warrant further investigation into this particular ligand.
Skeletal muscle weakness is associated with a large array of conditions that involve muscle wasting ranging from age-related atrophy, termed sarcopenia, to the wasting associated with immobility, termed disuse atrophy (reviewed in [18,22]). Several clinical trials are in progress that seek to prove the concept that myostatin inhibitors may be therapeutically beneficial and provide meaningful benefit to these wasting states. Trials are ongoing in chronic obstructive pulmonary disease (COPD) patients, in rehabilitation postorthopedic surgery/hip replacement subjects and in the sarcopenic adult and older weak fallers (Table 1). In a Phase 2 trial in sporadic inclusion body myositis (sIBM), a rare autoimmune disorder [74], BYM-338 was able to increase LBM, TMV and improve quadriceps strength and 6MWD (Amato et al. Annual MDA conference, San Diego, 2013; Table 1).
EMERGING MYOSTATIN PATHWAY BIOLOGY WITH IMPLICATIONS FOR THERAPEUTIC TARGETING: ROLE OF SATELLITE CELLS IN HYPERTROPHY OF SKELETAL MUSCLE INDUCED BY MYOSTATIN DEFICIENCY
In general muscles enlarged beyond normal size, or ‘supersized’ as a result of myostatin deficiency have increased total force but reduced specific force [75–78]. In myostatin null mice, the increase in total force does not match the increase in muscle mass [79]. Analysis of the contractility of single fibers from MSTN null mice demonstrated that the specific force deficits were at the level of the muscle myofiber [80,81▪▪]. Historically, it has been thought that a major function of myostatin was to maintain muscle satellite cell quiescence and that the relief of this inhibitory influence led to satellite proliferation and fusion to existing myofibers resulting in hypertrophy, akin to the mechanisms of muscle enlargement after exercise [81▪▪,82]. However, recent evidence demonstrates that myostatin exerts its effects directly on the myofiber with little effect on satellite cell activity [83▪]. The number of myonuclei in muscle is unchanged between normal and myostatin null mice resulting in larger ratios of cytoplasm volume : nuclei or myonuclear domains (MNDs) in null mice [81▪▪,84]. Qaisar et al. [81▪▪] suggested that there is a threshold size of MND under which the fiber is able to maintain the myofibrillar contractile apparatus and hence specific force. However, when a specific MND threshold is reached the fiber is unable to maintain the specific force. Interestingly, food restriction of the MSTN–/– mouse, by reducing muscle fiber size, restored the MND to normal with a corresponding normalization of force generation capacity of the muscle [85]. Lee et al. [86▪▪] demonstrated that muscle hypertrophy still occurred in animals with satellite cell deficiencies treated with myostatin inhibitors and in mice with a myofiber specific ablation of the myostatin receptor ActRIIB, confirming that the myofiber itself is the target of myostatin action. Myostatin deficiency leads to an increase in myofibrillar protein synthesis although a decrease in protein degradation may also be at play [80,87▪,88▪]. These findings suggest that myostatin inhibition in mice leading to ‘supersized’ muscles occurs with minimal satellite cell activation and can lead to a MND size in which there may not be concomitant increases in function. It remains to be determined if this same mechanism is active in man but with the increased attention and speculation around misuse of myostatin inhibitors this might prove to be a blessing in disguise [89,90]. As exemplified above, treating disorders with myostatin inhibitors in which muscle is atrophied and MND size is below normal has not been confounded by negative effects on specific force measures. These results also suggest that myostatin inhibitors would be effective in states of satellite cell dysfunction or depletion such as muscular dystrophy [91].
Emerging linkage of myostatin with tumor biology
Perhaps not surprisingly skeletal muscle tumors, specifically rhabdomyosarcomas (RMS), the most common soft tissue tumor in children, are known to overexpress myostatin [92]. Blocking myostatin activity with a dominant negative form of ActRIIB resulted in decreased proliferation and promoted differentiation of a human RMS cell line suggesting that myostatin inhibition may be a valuable target for interventions for RMS [93]. More interestingly, Lokireddy et al. [94▪▪] for the first time reported that myostatin protein is expressed and secreted from the mouse adenocarcinoma cell line C26, and from several human cancer cell lines. A characterization of myostatin expression in primary tumors is warranted in light of these initial findings.
CONCLUSION
There are a number of intervention points being exploited to inhibit myostatin signaling in order to enhance muscle mass under the conditions of muscle atrophy. Many of these therapies have now progressed into early stage clinical trials. Results of several Phase 2 trials underway are awaited to determine if increased muscle volumes translate into muscle strength, performance and outcomes that are clinically meaningful to patients.
Acknowledgements
The authors wish to thank B. Utterback for technical assistance with Figure 1.
Conflicts of interest
The authors are currently full time employees of Eli Lilly and Company.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
Footnotes
Correspondence to Rosamund C. Smith, DPhil, Lilly Corporate Center, Indianapolis, IN 46285, USA. Tel: +1 317 277 5229; e-mail: smith_ros@lilly.com
REFERENCES
- 1. Baracos VE. Clinical trials of cancer cachexia therapy, now and hereafter. J Clin Onc 2013; 31:1257–1258 [DOI] [PubMed] [Google Scholar]
- 2. Das UN. Can cancer cachexia be prevented/treated? Nutrition 2012; 28:844–848 [DOI] [PubMed] [Google Scholar]
- 3. Di Sebastiano KM, Mourtzakis M. A critical evaluation of body composition modalities used to assess adipose and skeletal muscle tissue in cancer. Appl Physiol Nutr Metab 2012; 37:811–821 [DOI] [PubMed] [Google Scholar]
- 4. Fearon KCH, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling and metabolic pathways. Cell Metab 2012; 16:153–166 [DOI] [PubMed] [Google Scholar]
- 5. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013; 10:90–99 [DOI] [PubMed] [Google Scholar]
- 6. Lee S-J, Glass DJ. Treating cancer cachexia to treat cancer. Skeletal Muscle 2011; 1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Macciò A, Madeddu C, Mantovani G. Current pharmacotherapy options for cancer anorexia and cachexia. Expert Opin Pharmacother 2012; 13:2453–2472 [DOI] [PubMed] [Google Scholar]
- 8. Martin L, Birdsell L, MacDonald N, et al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J Clin Oncol 2013; 31:1539–1547 [DOI] [PubMed] [Google Scholar]
- 9. Murphy KT, Lynch GS. Editorial update on emerging drugs for cancer cachexia. Expert Opin Emerg Drugs 2012; 17:5–9 [DOI] [PubMed] [Google Scholar]
- 10. Pausch T, Hartwig W, Hinz U, et al. Cachexia but not obesity worsens the postoperative outcome after pancreatoduodenectomy in pancreatic cancer. Surgery 2012; 152:S81–S88 [DOI] [PubMed] [Google Scholar]
- 11. Shum AMY, Polly P. Cancer cachexia: molecular targets and pathways for diagnosis and drug intervention. Endocr Metab Immune Disord Drug Targets 2012; 12:247–259 [DOI] [PubMed] [Google Scholar]
- 12. Suzuki H, Asakawa A, Amitani H, et al. Cancer cachexia – pathophysiology and management. J Gastroenterol 2013; 48:574–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsai S. Importance of lean body mass in the oncologic patient. Nutr Clin Pract 2012; 27:593–598 [DOI] [PubMed] [Google Scholar]
- 14. Utech AE, Tadros EM, Hayes TG, Garcia JM. Predicting survival in cancer patients: the role of cachexia and hormonal, nutritional and inflammatory markers. J Cachexia Sarcopenia Muscle 2012; 3:245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vaughan VC, Martin P, Lewandowski PA. Cancer cachexia: impact, mechanisms and emerging treatments. J Cachexia Sarcopenia Muscle 2013; 4:95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fearon K, Strasser F, Anker SD, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011; 12:489–495 [DOI] [PubMed] [Google Scholar]
- 17. Banerjee A, Guttridge DC. Mechanisms for maintaining muscle. Curr Opin Support Palliat Care 2012; 6:451–456 [DOI] [PubMed] [Google Scholar]
- 18. Fanzani A, Conraads VM, Penna F, Martinet W. Molecular and cellular mechanisms of skeletal muscle atrophy: an update. J Cachexia Sarcopenia Muscle 2012; 3:163–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lenk K, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 2010; 1:9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakuma K, Yamaguchi A. Sarcopenia and cachexia: the adaptations of negative regulators of skeletal muscle mass. J Cachexia Sarcopenia Muscle 2012; 3:77–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schiaffino S, Dyar KA, Ciciliot S, et al. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 2013; 280:4294–4314 [DOI] [PubMed] [Google Scholar]
- 22. Han HQ, Mitch WE. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr Opin Support Palliat Care 2011; 5:334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Argilés JM, Orpí M, Busquets S, et al. Myostatin: more than just a regulator of muscle mass. Drug Discov Today 2012; 17:702–709 [DOI] [PubMed] [Google Scholar]
- 24. Elkina Y, von Haehling S, Anker SD, Springer J. The role of myostatin in muscle wasting: an overview. J Cachexia Sarcopenia Muscle 2011; 2:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee S-J, Reed LA, Davies MV, et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors 2005. Proc Natl Acad Sci U S A 2005; 13:18117–18122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sako D, Grinberg AV, Liu J, et al. Characterization of the ligand binding functionality of the extracellular domain of the activin receptor type IIb. J Biol Chem 2010; 285:21037–21048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Townson SA, Martinez-Hackert E, Greppi C, et al. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J Biol Chem 2012; 287:27313–27325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hill JJ, Davies MV, Pearson AA, et al. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem 2002; 277:40735–40741 [DOI] [PubMed] [Google Scholar]
- 29. Sidis Y, Mukherjee A, Keutmann H, et al. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinol 2006; 147:3586–3597 [DOI] [PubMed] [Google Scholar]
- 30. Benny Klimek ME, Aydogdu T, Link MJ, et al. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010; 391:1548–1554 [DOI] [PubMed] [Google Scholar]
- 31. Zhou X, Wang JL, Lu J, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010; 142:531–543 [DOI] [PubMed] [Google Scholar]
- 32▪. Busquets S, Toledo M, Orpí M, et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012; 3:37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]; Improved muscle force as well as muscle mass when treating a mouse model of cancer cachexia with ActRIIB-Fc.
- 33▪. Murphy KT, Chee A, Gleeson BG, et al. Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am J Physiol Regul Integr Comp Physiol 2011; 301:R716–R726 [DOI] [PubMed] [Google Scholar]; Myostatin antibody treatment of a mouse model of cancer cachexia was able to enhance muscle mass and function.
- 34. Bogdanovich S, Krag TOB, Barton ER, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002; 420:418–421 [DOI] [PubMed] [Google Scholar]
- 35▪. Amthor H, Hoogaars WMH. Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Curr Gene Ther 2012; 12:245–259 [DOI] [PubMed] [Google Scholar]; Comprehensive review of preclinical and clinical data of myostatin pathway inhibitors as therapies for muscular dystrophies.
- 36. Chamberlain JS. Duchenne muscular dystrophy models show their age. Cell 2010; 143:1040–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37▪▪. Kornegay JN, Childers MK, Bogan DJ, et al. The paradox of muscle hypertrophy in muscular dystrophy. Phys Med Rehabil Clin N Am 2012; 23:149–172 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reviews the pros and cons of available animal models of muscular dystrophy and reports that crossing the myostatin heterozygous whippet dog with the golden retriever model of muscular dystrophy results in selective muscular hypertrophy and worsening clinical scores of the myo+/– dys–/– progeny.
- 38. Carlson CG, Bruemmer K, Sesti J, et al. Soluble activin receptor type IIB increases forward pulling tension in the mdx mouse. Muscle Nerve 2011; 43:694–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39▪. Dumonceaux J, Marie S, Beley C, et al. Combination of myostatin pathway interference and dystrophin rescue enhances titanic and specific force in dystrophic mdx mice. Mol Ther 2010; 18:881–887 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports the additive effects on muscle mass and specific force of myostatin inhibition and dystrophin rescue, respectively.
- 40. Pistilli EE, Bogdanovich S, Goncalves MD, et al. Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am J Physiol 2011; 178:1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morine KJ, Bish LT, Pendrak K, et al. Systemic myostatin inhibition via liver-targeted gene transfer in normal and dystrophic mice. PLoS One 2010; 5:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoogaars WMH, Mouisel E, Pasternack A, et al. Combined effect of AAV-U7-induced dystrophin exon skipping and soluble activin type IIB receptor in mdx mice. Hum Gene Ther 2012; 23:1269–1279 [DOI] [PubMed] [Google Scholar]
- 43. Murphy KT, Ryall JG, Snell SM, et al. Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am J Pathol 2010; 176:2425–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44▪. Li ZB, Zhang J, Wagner KR. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J Cell Sci 2012; 125:3957–3965 [DOI] [PubMed] [Google Scholar]; This article reports that mdx mice treated with ActRIIB-Fc have reduced muscle fibrosis.
- 45▪. Elashry MI, Collins-Hooper H, Vaiyapuri S, Patel K. Characterisation of connective tissue from the hypertrophic skeletal muscle of myostatin null mice. J Anat 2012; 220:603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports that the hypertrophic muscles from the myostatin null mouse have decreased connective tissue content.
- 46. Bish LT, Sleeper MM, Forbes SC, et al. Long-term systemic myostatin inhibition via liver-targeted gene transfer in golden retriever muscular dystrophy. Hum Gene Ther 2011; 22:1499–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mosher DA, Quignon P, Bustamante CD, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet 2007; 3:779–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mariano A, Henning A, Han R. Dysferlin-deficient muscular dystrophy and innate immune activation. FEBS J 2013; 280:4165–4176 [DOI] [PubMed] [Google Scholar]
- 49. Malik V, Rodino-Klapac LR, Mendell JR. Emerging drugs for Duchenne muscular dystrophy. Expert Opin Emerg Drugs 2012; 17:261–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pichavant C, Aartsma-Rus A, Clemens PR, et al. Current status of pharmaceutical and genetic approaches to treat DMD. Mol Ther 2011; 19:830–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kang JK, Malerba A, Popplewell L, et al. Antisense-induced myostatin exon skipping leads to muscle hypertrophy in mice following octa-guanidine morpholino oligomer treatment. Mol Ther 2011; 19:159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kemaladewi DU, Hoogaars WMH, van Heiningen SH, et al. Dual exon skipping in myostatin and dystrophin for Duchenne muscular dystrophy. BMC Med Genomics 2011; 4:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Malerba A, Kang JK, McClorey G, et al. Dual myostatin and dystrophin exon skipping by morpholino nucleic acid oligomers conjugated to a cell-penetrating peptide is a promising therapeutic strategy for the treatment of Duchenne muscular dystrophy. Mol Ther Nucleic Acids 2012; 1:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54▪. Elashry MI, Otto A, Matsakas A, et al. Axon and muscle spindle hyperplasia in the myostatin null mouse. J Anat 2011; 218:173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors of this article report that there is an increased number of nerve fibers innervating the hypertrophic muscle skeletal muscle seen in the myostatin null mouse.
- 55. Gay S, Jublanc E, Bonnieu A, Bacou F. Myostatin deficiency is associated with an increase in number of total axons and motor axons innervating mouse tibialis anterior muscle. Muscle Nerve 2012; 45:698–704 [DOI] [PubMed] [Google Scholar]
- 56. Walsh FS, Rutkowski JL. Myostatin as a therapeutic target in amyotrophic lateral sclerosis. Neurochem Int 2012; 61:931–935 [DOI] [PubMed] [Google Scholar]
- 57. Rindt H, Buckley DM, Vale SM, et al. Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of spinal muscular atrophy. Neuromuscul Disord 2012; 22:277–285 [DOI] [PubMed] [Google Scholar]
- 58. Sumner CJ, Wee CD, Warsing LC, et al. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum Mol Genet 2009; 18:31245–33152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rose FF, Mattis VB, Rindt H, Lorson CL. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum Mol Genet 2009; 18:997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60▪▪. Lawlor MW, Read BP, Edelstein R, et al. Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am J Pathol 2011; 178:784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]; Treatment of the myotubularin-deficient mouse model of X-linked myotubular myopathy with ActRIIB-Fc resulted in improved survival and transient increases in muscle mass and strength.
- 61. Chiu C-S, Peekhaus N, Weber H, et al. Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. J Gerontol A Biol Sci Med Sci 2013; 68:1181–1192 [DOI] [PubMed] [Google Scholar]
- 62▪. Murphy KT, Cobani V, Ryall JG, et al. Acute antibody-directed myostatin inhibition attenuates disuse atrophy and weakness in mice. J Appl Physiol 2011; 110:1065–1072 [DOI] [PubMed] [Google Scholar]; Myostatin antibody treatment of a murine model of disuse atrophy demonstrated the ability to prevent loss of muscle mass and function.
- 63. Zhang L, Rajan V, Lin E, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 2011; 25:1653–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Monestier O, Brun C, Heu K, et al. Ubiquitous Gasp1 overexpression in mice leads mainly to a hypermuscular phenotype. BMC Genomics 2012; 13:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guiraud S, van Wittenberghe L, Georger C, et al. Identification of decorin derived peptides with a zinc dependent antimyostatin activity. Neuromuscul Disord 2012; 22:1057–1068 [DOI] [PubMed] [Google Scholar]
- 66▪▪. Jameson GS, von Hoff DD, Weiss GL, et al. Safety of the antimyostatin monoclonal antibody LY2495655 in healthy subjects and patients with advanced cancer. J Clin Oncol 2012; 30 (Suppl): [Google Scholar]; This abstract reports the ability of the myostatin antibody LY2495655 to increase TMV in healthy volunteers and in cancer patients.
- 67▪. Aversa Z, Bonetto A, Penna F, et al. Changes in myostatin signaling in nonweight-losing cancer patients. Ann Surg Oncol 2012; 19:1350–1356 [DOI] [PubMed] [Google Scholar]; This article reports that myostatin protein levels are elevated in muscles of early stage gastric cancer patients prior to significant weight loss.
- 68. Beytía MdlA, Vry J, Kirschner J. Drug treatment of Duchenne muscular dystrophy: available evidence and perspectives. Acta Myol 2012; 31:4–8 [PMC free article] [PubMed] [Google Scholar]
- 69. Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 2008; 63:561–571 [DOI] [PubMed] [Google Scholar]
- 70▪▪. Attie KM, Borgstein NG, Yang Y, et al. A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve 2012; 47:416–423 [DOI] [PubMed] [Google Scholar]; This article reports the positive effects on LBM and TMV of a single dose of ActRIIB-Fc in healthy postmenopausal women.
- 71▪▪. Campbell C, Escolar D, Mah J, et al. A Phase 2, randomized, placebo-controlled, multiple ascending-dose study of ACE-031, a soluble activin receptor Type IIB, in boys with Duchenne muscular dystrophy (DMD). Neurology 2012; 78:P04.088 [Google Scholar]; This abstract reports the positive efficacy results and adverse effects of ActRIIB-Fc in a Phase 2 trial of DMD.
- 72. Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996; 13:189–195 [DOI] [PubMed] [Google Scholar]
- 73. Van Meeteren LA, Thorikay M, Berggvist S, et al. Antihuman activin receptor-like kinase 1 (ALK1) antibody attenuates bone morphogenetic protein 9 (BMP9)-induced ALK1 signaling and interferes with endothelial cell sprouting. J Biol Chem 2012; 287:18551–18561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Breithaupt M, Schmidt J. Update on treatment of inclusion body myositis. Curr Rheumatol Rep 2013; 15:329. [DOI] [PubMed] [Google Scholar]
- 75. Amthor H, Macharia R, Navarrete R, et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci U S A 2007; 104:1835–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Giannesini B, Vilmen C, Amthor H, et al. Lack of myostatin impairs mechanical performance and ATP cost of contraction in exercising mouse gastrocnemius muscle in vivo. Am J Physiol Endocrinol Metab 2013; 305:E33–E40 [DOI] [PubMed] [Google Scholar]
- 77. Mendias CL, Marcin JE, Calerdon DR, Faulkner JA. Contractile properties of EDL and soleus muscles of myostatin-deficient mice. J Appl Physiol 2006; 101:898–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Personius KE, Jayaram A, Krull D, et al. Grip force, EDL contractile properties, and voluntary wheel running after postdevelopmental myostatin depletion in mice. J Appl Physiol 2010; 109:886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gentry BA, Ferreira JA, Phillips CL, Brown M. Hindlimb skeletal muscle function in myostatin deficient mice. Muscle Nerve 2011; 43:49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mendias CL, Kayupov E, Bradley JR, et al. Decreased specific force and power production of muscle fibers from myostatin-deficient mice are associated with a suppression of protein degradation. J Appl Physiol 2011; 111:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81▪▪. Qaisar R, Renaud G, Morine K, et al. Is functional hypertrophy and specific force coupled with the addition of myonuclei at the single muscle fiber level? FASEB J 2012; 26:1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]; Increased myofiber MND size in fast muscles of myostatin null mice correlate to decreases in muscle-specific force.
- 82. Pallafachina G, Blaauw B, Schiaffino S. Role of satellite cells in muscle growth and in maintenance of muscle mass. Nutr Metab Cardio Dis 2012; [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 83▪. Wang Q, McPherron AC. Myostatin inhibition induces muscle fibre hypertrophy prior to satellite cell activation. J Physiol 2012; 9:2151–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence that ActRIIB-Fc induces in mice a relatively small proliferation of satellite cells and this occurs after muscle hypertrophy can be detected leading the authors to suggest that myostatin inhibition in mice likely leads to muscular hypertrophy from direct effects on the myofiber rather than on the satellite cell itself.
- 84. Matsakas A, Macharia R, Otto A, et al. Exercise training attenuates the hypermuscular phenotype and restores skeletal muscle function in the myostatin null mouse. Exp Physiol 2012; 97:125–140 [DOI] [PubMed] [Google Scholar]
- 85. Matsakas A, Romanello V, Sartori R, et al. Food restriction reverses the hyper-muscular phenotype and force generation capacity deficit of the myostatin null mouse. Int J Sports Med 2013; 34:223–231 [DOI] [PubMed] [Google Scholar]
- 86▪▪. Lee S-J, Huynh TV, Lee Y-S, et al. Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proc Natl Acad Sci U S A 2012; 109:E2353–E2360 [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence that satellite cell proliferation is not required for skeletal muscle hypertrophy induced by Acvr2B deficiency or ActRIIB-Fc in mice.
- 87▪. Rodriguez J, Vernus B, Toubiana M, et al. Myostatin inactivation increases myotube size through regulation of translational initiation machinery. J Cell Biochem 2011; 112:3531–3542 [DOI] [PubMed] [Google Scholar]; This article reports that myofibers isolated from myostatin null mice are larger and have increased rates of protein synthesis.
- 88▪. Welle S, Mehta S, Burgess K. Effect of postdevelopmental myostatin depletion on myofibrillar protein metabolism. Am J Physiol Endocrinol Metab 2011; 300:E993–E1001 [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence that postnatal myostatin depletion results in an increase in total myofibrillar protein synthesis.
- 89. Schneider AJ, Fedoruk MN, Rupert JL. Human genetic variation: new challenges and opportunities for doping control. J Sports Sci 2012; 30:1117–1129 [DOI] [PubMed] [Google Scholar]
- 90. Van der Gronde T, de Hon O, Haisma HJ, Pieters T. Gene doping: an overview and current implications for athletes. Br J Sports Med 2013; 47:670–678 [DOI] [PubMed] [Google Scholar]
- 91. Sacco A, Mourkioti F, Tran R, et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010; 143:1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Langley B, Thomas M, McFarlane C, et al. Myostatin inhibits rhabdomyosarcoma cell proliferation through an Rb-independent pathway. Oncogene 2004; 23:524–534 [DOI] [PubMed] [Google Scholar]
- 93. Rossi S, Stoppani E, Puri PL, Fanzani A. Differentiation of human rhabdomyosarcoma RD cells is regulated by reciprocal, functional interactions between myostatin, p38 and extracellular regulated kinase signaling pathways. Eur J Cancer 2011; 47:1095–1105 [DOI] [PubMed] [Google Scholar]
- 94▪▪. Lokireddy S, Wijesoma IW, Bonala S, et al. Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem J 2012; 446:23–36 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; This article reports for the first time the expression of myostatin in a variety of mouse and human tumor cell lines and the ability of the conditioned media from C26 cells to induce muscle atrophy in vitro in a myostatin-dependent manner.