

Abstract
Objectives
This study was aimed to assess the diversity of the meconium microbiome and determine if the bacterial community is affected by maternal diabetes status.
Methods
The first intestinal discharge (meconium) was collected from 23 newborns stratified by maternal diabetes status: 4 mothers had pre-gestational type 2 diabetes mellitus (DM) including one mother with dizygotic twins, 5 developed gestational diabetes mellitus (GDM) and 13 had no diabetes. The meconium microbiome was profiled using multi-barcode 16S rRNA sequencing followed by taxonomic assignment and diversity analysis.
Results
All meconium samples were not sterile and contained diversified microbiota. Compared with adult feces, the meconium showed a lower species diversity, higher sample-to-sample variation, and enrichment of Proteobacteria and reduction of Bacteroidetes. Among the meconium samples, the taxonomy analyses suggested that the overall bacterial content significantly differed by maternal diabetes status, with the microbiome of the DM group showing higher alpha-diversity than that of no-diabetes or GDM groups. No global difference was found between babies delivered vaginally versus via Cesarean-section. Regression analysis showed that the most robust predictor for the meconium microbiota composition was the maternal diabetes status that preceded pregnancy. Specifically, Bacteroidetes (phyla) and Parabacteriodes (genus) were enriched in the meconium in the DM group compared to the no-diabetes group.
Conclusions
Our study provides evidence that meconium contains diversified microbiota and is not affected by the mode of delivery. It also suggests that the meconium microbiome of infants born to mothers with DM is enriched for the same bacterial taxa as those reported in the fecal microbiome of adult DM patients.
Introduction
Prenatal diabetes, which includes both pre-gestational Type 1 and Type 2 diabetes, and gestational diabetes that develops during pregnancy have been associated with an increased risk of obstetric and neonatal complications [1]–[3]. Both pre-gestational and gestational diabetes have also been associated with major birth defects [4] and congenital anomalies of the offspring [5], [6]. Moreover, gestational diabetes has been linked to the risk of childhood obesity [7], [8], which has both immediate and long-term implications on human health.
Human microbiome studies have demonstrated dynamic changes in bacterial composition in the gut during pregnancy and childhood development [9]–[11]. Moreover, the presence of pathogenic species, or absence of beneficial species, in early childhood has been suggested to play a key role in the initiation of preterm birth [12], development of asthma or eczema [13], [14], allergy [15], autism [16] or other immunological deficiency [17], [18].
Historically, the fetus, as well as the intrauterine environment, has been considered sterile, with the initial microbial exposure taking place at birth vaginally or via C-section through contacting maternal vaginal or skin microbiota, respectively [9], [11], [19], [20]. However, accumulating evidence suggests the presence of different microbes in amniotic fluid [21], [22], umbilical cord blood [23], meconium [24], [25], and placental [26] and fetal membranes [27]. Studies in mice have demonstrated the transmission of labeled bacterial strains from a mother to fetus during pregnancy [25]. Taken together, these results suggest that mother-to-baby efflux of commensal microbes may occur prior to birth.
However, despite the growing recognition that commensal microbes may contribute fundamentally to infant and childhood development and immunity [14], [15], [17], [18], [28], [29], only a few studies have determined the microbial composition of the first intestinal discharge, or meconium, in premature [30]–[32] and in term neonates [24], [25] and linked its bacterial content to maternal eczema and infant mucus congestion during the first year of life [24]. Therefore, the main objectives of this study were to further characterize the composition of the meconium and assess whether maternal diabetes status, prior to or during pregnancy, affects bacterial composition of the newborn's first stool.
Methods
Subjects
This study was approved by the Mount Sinai Institutional Review Board. Pregnant women before their second trimester were recruited during their regular prenatal visits at a prenatal obstetrics and gynecological (OB/GYN) clinic at Mount Sinai Medical Center and provided a written informed consent for themselves and their prospective infants. The exclusion criteria included: 1) any antibiotic treatment during pregnancy; women who eventually underwent C-section and received an immediate dose of Kefzol (cefazolin) <30 mins prior to C-section as a standard of care were retained in the study, or 2) obstetric risks, such as HIV positivity, significant congenital anomalies, neurological dysfunction, fetal chromosomal anomalies, or inborn errors in metabolism. Clinical characteristics of the infant included sex, birth weight (BW), birth length, time of sampling (hours after birth), neonatal complications, gestational age, and delivery method. Clinical variables of the mother included age, body mass index (BMI) at 1st and 3rd trimester, glucose level (1 hour glucose challenge test, or GCT, completed at 24–28 weeks), medications during pregnancy, maternal smoking, and diabetes status. A subclinical group included 4 no-diabetes mothers with glucose levels higher than the cut-off point of 130 ng/dL, who did not meet the criteria for gestational diabetes at the diagnostic 3-hour glucose tolerance test (GTT). Such women are known to carry additional obstetric risk, such as fetal macrosomia and other morbidities [33], [34]. Seven adult fecal samples used for the comparison purposes in the present study were collected for an unrelated study from healthy individuals with no diabetes and no antibiotic treatments for at least 6 months, who consented for their samples to be used for other research.
Sample collection
The neonate meconium from 23 enrolled infants was passed ranging between 2 hours and 48 hours after birth ( Table 1 ). The meconium was transferred from a diaper to sterile 15 ml Falcon tubes using a sterile tongue depressor by the research staff and stored at −80°C until processing. During the sample collection and processing, the mother did not handle the meconium in order to avoid possible contaminations.
Table 1. Clinical information of the neonates and mothers.
| Infant | Mother | ||||||||||||
| IDs | Sex | BW1 (gram) | Length (cm) | Complication & Treatment | TOS2 (hrs) | GADAYs3 (days) | Delivery Status | Type of C-section | BMI14 | BMI25 | Maternal Age | Glucose Level | Diabetic Status |
| BM62 | F | 1645 | 52 | NICU (dextrose, fat emulsion, multivitamins) | 24 | 259 | cesarean | Emergency, Induced labor | 42.45 | 40.78 | 29 | 106 | HC6 |
| BM110 | F | 3450 | 51 | 6 | 275 | cesarean | Emergency, Arrest of dilation | 30.11 | 34.96 | 24 | 93 | HC | |
| BM172 | F | 3690 | 52.5 | 43 | 287 | vaginal | 23.17 | 27.56 | 34 | 96 | HC | ||
| BM124 | M | 4060 | 53 | 2 | 291 | cesarean | Emergency, Arrest of dilation | 27.04 | 33.12 | 23 | 120 | HC | |
| BM158 | F | 3505 | 51 | n/a | 283 | vaginal | 19.38 | 23.58 | 21 | 75 | HC | ||
| BM38 | F | 3370 | 48 | 20 | 272 | vaginal | 20.67 | 21.77 | 30 | 117 | HC | ||
| BM188 | F | 3410 | 48.5 | 27 | 285 | vaginal | 19.00 | 26.30 | 17 | 81 | HC | ||
| BM50 | F | 2500 | 48 | 25 | 279 | vaginal | 30.00 | 37.19 | 24 | 90 | HC | ||
| BM177 | F | 3246 | 51 | 9 | 280 | vaginal | 26.94 | 32.90 | 20 | 139 | HC | ||
| BM3 | M | 3485 | 53 | 5 | 274 | vaginal | 25.70 | 30.30 | 26 | 168 | Subclinical | ||
| BM180 | F | 3040 | 49 | 12 | 255 | cesarean | Elective, Induced labor | 30.90 | 34.40 | 39 | 141 | Subclinical | |
| BM52 | M | 2660 | 49 | 15 | 251 | vaginal | 26.08 | 28.96 | 23 | 164 | Subclinical | ||
| BM276 | F | 2875 | 49.5 | 8 | 291 | vaginal | n/a | 44.07 | 39 | 165 | Subclinical | ||
| BM68 | M | 2630 | 49 | NICU (bacitracin ointment, nystatin, phenobarbital, rantidine, simethicone, dextrose, ferrous sulfate, multivitamins) | 48 | 250 | cesarean | Emergency, Arrest of descent | 22.90 | 23.63 | 27 | 285 | GDM7 |
| BM126 | F | 3410 | 52 | Jaundice | 4 | 280 | vaginal | 35.02 | 40.96 | 28 | 151 | GDM | |
| BM9 | M | 3240 | 56 | 4 | 266 | vaginal | 21.17 | 34.92 | 37 | 92 | GDM | ||
| BM256 | F | 3800 | 51 | 9 | 282 | vaginal | n/a | 31.82 | 31 | 131 | GDM | ||
| BM257 | F | 4000 | 52 | 16 | 266 | cesarean | Emergency, Arrest of dilation | n/a | 39.45 | 42 | 182 | GDM | |
| BM25 | F | 2770 | 48 | 2 | 291 | cesarean | Elective, Induced labor | 27.04 | 33.12 | 23 | 120 | DM8 | |
| BM181 | M | 3120 | 50.5 | 45 | 249 | vaginal | DM | ||||||
| BM155A | M | 3125 | 50 | 24 | 233 | cesarean | Emergency, Fetus stress | n/a | 27.5 | 41 | n/a | DM | |
| BM155B | M | 1750 | 40.5 | NICU (palivizumab, lidocaine-prilocaine cream, ferrous sulfate, dextrose, artificial tears, multivitamins) | 24 | 233 | cesarean | Emergency, Fetus stress | n/a | 27.5 | 41 | n/a | DM |
| BM140 | M | 2940 | 50 | NICU (lidocaine-prilocaine cream, lidocaine injection, artificial tears) | 40 | 270 | cesarean | Emergency, Induced labor | 23.84 | 25.95 | 23 | n/a | DM |
BW = body weight;
TOS = Time of sample collection;
GADAYs = gestational age in days;
BMI1 = body mass index(BMI) in 1st trimester;
BMI2 = body mass index(BMI) in 3rd trimester;
HC = Healthy control;
GDM = gestational diabetes;
DM = Type 2 diabetes.
Fecal DNA extraction and 16S ribosomal RNA (rRNA) amplification
Total meconium DNA was extracted using Qiagen stool kit (Qiagen, CA). Subsequent amplification of bacterial 16S rRNA v3–v4 region using barcoded PCR primers (Table S1 in File S1) was performed as previously described [35]. A 16-mer error-correcting Golay barcode was added to the reverse primer. A composite bar-coded sample for sequencing was created by combining equimolar amounts of amplicons from the 23 individual samples.
16S rRNA sequencing, taxonomy assignment and diversity computation
The pooled 16S rRNA PCR amplicons were sequenced on the Pacbio RS system. Two 45-minute movies were collected on each SMRT cell. Analysis was performed only on circular consensus (CCS) reads (CCS reads are generated when ≥3 full pass subreads are present). Final CCS reads were filtered by the sequencing quality score and read length before being exported to FASTA format. To test the sequencing accuracy and reproducibility, three healthy adult stool samples were processed twice and sequenced on two separated SMRT cells. The E.coli strain BL21 was sequenced to test the classification accuracy. Filtered high quality CCS reads were further processed by QIIME 1.5.0 [36]. The alpha-diversity was calculated using the Shannon Index as metric and represented the mean species diversity within each sample. The beta diversity was estimated using both weighted and unweighted UniFrac distance matrices and represented the measurement of compositional dissimilarity among samples.
Data Analysis
Wilcoxon-Mann-Whitney test was performed at various OTU (operational taxa unit) levels to compare between the adult feces and meconium with regard to and regardless of maternal diabetes status. P-values were adjusted for false-discovery rate (FDR) using the Benjamini and Hochberg method [37]. Non-parametric multiple dimensional scaling (nMDS) and Principal Coordinate Analysis (PCoA) were used to visualize the dissimilarities of the overall microbiome. The PerMANOVA test [38], [39], with the maximum number of permutations = 999, was performed using the [Adonis] function of the R package vegan 2.0–5 [40] to compare the overall microbiome differences between the meconium microbiota by maternal diabetes status or type of delivery. We also performed the univariate regression analysis using the [lm] function in R.
The dependent variables included the unweighted principle components (PC): PC1, PC2 and PC3, representing the dominant taxa Bacteroidetes, Firmicutes and Proteobacteria at the phyla level and the unweighted UniFrac distance matrices representing the overall microbiome profile. The predictor included newborn sex, time of sampling, gestational age, delivery method, maternal BMI at 1st trimester and maternal diabetes status.
Results
Clinical information
Twenty-three newborns were enrolled in this study ( Table 1 ). Five of the babies were born to mothers with established DM, 5 to mothers with GDM, and 13 to mothers with subclinical diabetes (n = 4) or no diabetes (n = 9). One of the mothers with DM had dizygotic twins. None of the study participants reported being smokers. Ten babies were delivered by C-section with the type of C-sections (emergency or elective), as well as additional participant characteristics, listed in Table 1 . No significant difference was observed for TOS, BW and length among DM, GDM, and no-diabetes groups (p-value >0.1 by t-test). Missing data resulted from the fact that some mothers only consented to certain components of the study.
Validation of the sequencing accuracy and reproducibility
In this study, on average, a single chip generated ∼55,000 post-filtered reads (mean mapped CCS read accuracy about 94%), of which ∼33,000 reads were longer than 2 kilobases (Figure S1A in File S1). Figure S1B in File S1 demonstrates that the mean CCS read length was 443 base pairs (bp, ranging between 400 and 500 bp). We tested reproducibility of our 16S rRNA sequencing technique by comparing duplicate samples. Taxonomy assignment showed that, the correlation between the duplicates was within the range of 99.5% to 99.9% at any taxonomy level (Figure S1C in File S1). When we examined the bacterial composition of the control E.coli strain BL21, 99.5% of the CCS reads were correctly assigned at the phyla through family level and 97.1% at the genus level, suggesting a high accuracy and low misclassification rate (Figure S1D in File S1).
Bacterial Composition of the Meconium and Comparison to the Adult and Infant Microbiome
After filtering by quality and length, we obtained, on average, 3,300 CCS reads per sample (range 1,464–6,641). Our results indicated that all 23 meconium samples were not sterile. At the phylum level, all four major phyla, Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria, were represented accounting for about 99% of the microbial content ( Figure 1A ). However, the average taxa abundance of the newborn meconium was very different from that of the adult stool samples ( Figure 1B ). Specifically, when compared with the adult feces ( Figure 2 ), the meconium from the neonates born to no-diabetes (healthy and preclinical DM) mothers had a significantly higher proportion of Proteobacteria (71.0% in the meconium vs. 3.1% in the adult stool), and a lower proportion of Bacteroidetes (2.5% in the meconium vs. 42.8% in the adult stool), Firmicutes (20.0% in the meconium vs. 44.7% in the adult stool) and Verrucomicrobia (0.06% in the meconium vs. 3.2% in the adult stool) (FDR-adjusted p<0.05). At the family level, Comamonadaceae and Enterobacteriaceae were significantly enriched in the meconium samples compared to the adult stool (FDR-adjusted p<0.05). Further analyses showed a significant reduction in Bacteroidaceae, Lachnospiraceae, Ruminococcaceae and Veillonellaceae (FDR-adjusted p<0.05) in the meconium. The four most abundant genera in the meconium included Comamonas, Escherichia/Shigella, Klebsiella and Proteus. Similar to the no-diabetes group, the meconium from DM and GDM groups also showed enrichment of Proteobacteria and reduction of Bacteroidetes and Firmicutes, in comparison with the adult stool samples.
Figure 1. Comparison of the microbiota communities of neonate meconium to adult stool samples.

A. Classification of the microbiota at the Phylum level. Samples BM62-BM140 are neonate meconium and 7 samples are from healthy adults. The total reads for each sample are normalized to 2000. Neonate meconium samples are grouped by maternal clinical status: Healthy, subclinical, GDM (gestational diabetes) and DM (type 2 diabetes). B. Average taxa abundances between the neonate meconium and adult stool samples. HC group combined healthy and subclinical samples. C. Alpha rare plot shows the average microbiota diversity of the neonate meconium grouped by maternal status (HC, GDM and DM) and adult stool samples.
Figure 2. Comparison of the OTUs at the Phylum and Family level between healthy adult stool samples and the meconium samples from neonates born to mothers with no diabetes.
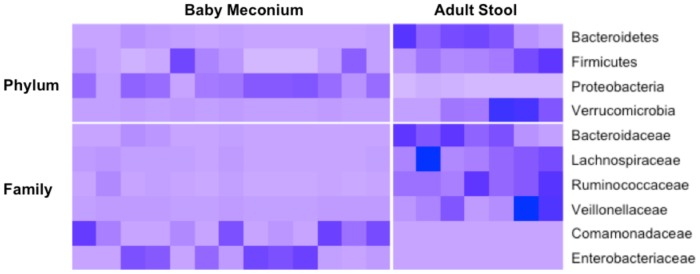
The meconium samples included both HC and subclinical groups. Only OTUs shown significance (p values <0.05 by WMW-test and adjusted by BH method) between the two groups are presented.
As the composition of the first stool is likely to change in response to environmental exposures, we compared the meconium microbiome content to the reported microbiome profiles from early infancy [9], [19], [20], [41]. Our data showed that, although at the phylum level, the meconium is more similar to that of infants than to adults, at the genus level, the dominant taxa found in the early infant stool from vaginal deliveries (Lactobacillus, Prevotella) or from C-sections (Acinetobacter, Propionibacterineae, Micrococcineae, Corynebacterineae) [9], [11], [19], [41] were not prevalent (<1%) in over 90% of the meconium samples.
Diversity of the Meconium Microbiota
The average microbiota diversity within the meconium microbiome was lower than that of adult feces (3.1 vs. 8.5, p = 1.7e-4, Figure 1C ). Within meconium samples, the DM group showed higher alpha-diversity (within-sample bacterial diversity) than that from the no-diabetes or GDM groups (6.2 vs. 3.1 and 2.9; FDR-adjusted p = 1e-3 and 0.08, respectively). Meconium samples of babies born to mothers with no diabetes or GDM had a higher variability in beta-diversity (between-sample bacterial diversity) than the DM group (p = 0.006 and 0.0017 by F-test, respectively) or the adult samples (p = 3e-5 and 2.3e-4 by F- test, respectively) (Figure S2A in File S1). The DM group showed a lower variability than the adult samples.
Meconium Microbiome and Maternal Diabetes Status
The nMDS analysis using unweighted UniFrac distance matrices showed a considerable separation of the overall microbiota by maternal diabetes status ( Figure 3 ). The PerMANOVA test detected that the DM and GDM groups were each significantly different from the no-diabetes group (p = 0.004, 0.022, respectively); however no significant difference was observed between the DM and GDM groups (p = 0.16). Consistent with the PerMANOVA results, weighted and unweighted UniFrac PCoA showed a separation of the DM from no-diabetes samples on the PC1 vs. PC2 plot (Figure S2B in File S1), mostly driven by the enrichment of Bacteroidetes and reduction of Proteobacteria in the DM meconium samples (Figure S3 in File S1). Notably, the 4 subjects with subclinical DM (subjects BM3, BM180, BM52 and BM276) showed no separation from other no-diabetes samples (Figure S3 in File S1). Furthermore, we compared the abundance of the OTUs among these three groups. Figure 4 depicts all OTUs that showed significant difference (p<0.05) at the phylum, class, family and genus level between DM and no-diabetes controls. Specifically, several OTUs, such as Bacteroidetes (phyla), Lachnospiraceae (family) and Parabacteriodes (genus) that were enriched in adult Type 2 diabetes patients [42], were also significantly enriched in the meconium samples of babies born to DM mothers compared to the no-diabetes group. No significance was achieved when p-values were adjusted for multiple hypothesis testing.
Figure 3. Comparison of meconium microbiota from neonates born to HC (healthy), DM (type 2 diabetes) and GDM (gestational diabetes) mothers using nMDS ordination.

The unweighted UniFrac distance matrices generated from QIIME were visualized in nMDS plot. Lines connect individual communities to the centroid values for each group. The ellipses were drawn to represent the class standard errors.
Figure 4. List of OTUs that showed significant difference between neonates from mothers with different diabetes states.

HC(healthy) group was combined with both healthy and subclinical groups. *unadjusted p-value from WMW-test between HC and DM(type 2 diabetes) group; **unadjusted p-value from WMW-test between HC and GDM(gestational diabetes) group.
Relationship between the Meconium Microbiome and Other Maternal and Newborn Characteristics
Previous work has shown the association of the infant microbiome with the delivery mode [9], [20]. In our study, the PerMANOVA test showed no significant difference between the two delivery methods in neonates born to no-diabetes mothers (p = 0.26). The weighted and unweighted PCoA plot also showed no separation (Figure S2C in File S1). Moreover, we found no significant differences in taxa between the two delivery modes at each OTU level. Despite the small sample size, we further investigated the impact of the type of C-section (i.e., emergency or elective) on newborn's gut microbiota and found no significant differences between the microbiome profiles of the 8 emergency and 2 elective C-sections.
In the univariate regression analysis, we found that the maternal diabetes status was significantly associated with the abundance of Bacteroidetes (p = 0.029), PC2 (p = 0.015) and the overall microbiome profile (p = 0.038) (Table S2 in File S1). No other demographic or clinical characteristics were found to be associated with the meconium microbiome profile.
Discussion
We implemented 16S rRNA sequencing using the Pacbio RS system to explore the meconium microbiome of 23 newborns. Our findings showed that the meconium was not sterile in all of our study subjects and contained diverse microbiota communities, regardless of the demographic and clinical characteristics. A number of studies using culture-dependent or independent methods have established the presence of live bacteria in the fetus prior to birth [12], [21]–[26], [30]–[32], [43]. However, until the development of the next generation sequencing (NGS) technology, the detailed characterization of the microbiome at the taxa level was challenging. Moreover, while a number of studies have compared the bacterial composition of the adult stool with early infant stool [9], [11], [19], [20], [28], only a handful of small studies have been carried out in meconium [24], [25], [32]. Using a culture-independent NGS technology, we demonstrated that the meconium microbiota substantially differ from those in adult feces, with the overall microbiome diversity being significantly lower than that of the adult stool ( Figure 1 ). While at the phylum level the meconium microbiome was more similar to that of infants [9], [11], [19], [41] than adults, at the genus level, the dominant taxa found in the early infant stool were not prevalent in the meconium samples.
Among the meconium samples, we observed a significant increase in the diversity of various bacterial types if mothers had pre-gestational DM compared to those with no diabetes or GDM.
These results are also consistent with the recent findings that have suggested that certain microbiota components are more prevalent in adult individuals with DM [42], [44]. Specifically, at the OTU level, the Bacteroides, Parabacteroides and Lachnospiraceae, which have been reportedly prevalent in adult diabetes patients [29], [42], were enriched in the meconium of babies born to mothers with DM. Despite the enrichment of certain bacteria, a previous study have reported no difference in the mean OTU diversity within each sample between the adult DM cases and controls [45]. However, in our study, we observed that the meconium in the DM group showed higher alpha-diversity than that in the no-diabetes or GDM groups with a possible explanation being that the bacterial transmission to fetus may be more permissible under maternal diabetic conditions.
An additional illustration of the link between the meconium microbiota composition and maternal and infant health has been provided by Gosalbes et al., who reported that the meconium microbiota types dominated by lactic acid or enteric bacteria are differentially associated with maternal eczema and respiratory problems in infants [24]. Moreover, they have shown that some of the species that reach the fetal gastrointestinal tract prenatally can be present in the infant long into the first year, underlining the potential long-term clinical consequences of the initial microbiota content. We speculate that the bacterial diversity in the gut of a fetus may reflect pathophysiological processes occurring during pregnancy or represent a direct transmission of maternal intestinal bacteria. Prospective studies will be required to further clarify the path of the mother-to-baby efflux of commensal microbes during pregnancy, as well as the impact of the maternal microbiome on predisposition to adult-onset diseases.
Microbiome studies of early infancy have demonstrated a significant effect of the mode of delivery on the microbiome composition, suggesting the likely association of the infant gut bacteria with maternal vaginal or skin microbiome habitats [9], [20]. However, in our study, no significant differences in the overall microbiome composition by the mode of delivery were detected in babies born to mothers with and without diabetes. Furthermore, we observed that neither of the dominant OTUs reported previously [9] at the genus level in the stool of young infants delivered vaginally (Lactobacillus, Prevotella) or via C-section (Acinetobacter, Propionibacterineae, Micrococcineae, Corynebacterineae) were prevalent in the majority of the meconium samples. Instead, the most common OTUs at the genus level in the meconium included Comamonas, Escherichia/Shigella, Klebsiella and Proteus, which are predominantly aerobic or facultative anaerobic organisms. Comamonas was found in human appendix, a small pouch attached to the first portion of the large intestine [46] and Escherichia/Shigella, Klebsiella and Proteus occur naturally in the human gut [47], further supporting our hypothesis that the initial colonization in the human gut starts prior to birth. While our findings are consistent with a recent report that also showed no substantial overlap of the meconium microbiota with maternal habitats, especially in babies born via C-section [24], it contrasts previous analyses demonstrating that the microbiota recovered from multiple newborn sites closely resembles vaginal or skin microbiota, depending on the mode of delivery [9]. Emerging studies point toward pregnancy as the beginning of bacterial exposure for the developing fetus [23]–[25]. A recent study on the microbiome dynamics in the gut of pregnant women has shown that the bacterial composition evolves between the 1st and 3rd trimesters, with the maternal gut microbiome in the 3rd trimester being more similar to what we observed in the meconium, including the increased variability in beta-diversity, the compositional dissimilarity among samples, increasing abundance of Proteobacteria, and the reduction in alpha-diversity between samples [48]. Taken together, it further suggests that the maternal microbiota can be transferred to the fetus during pregnancy.
For this study, we used the Pacbio RS system. Compared with other NGS platforms, Pacbio RS has the advantage of a much shorter sequencing time (1 to 2 hours) and the much longer read length (up to 8,000 bases). However, the embedded relatively high random sequencing error rate was until recently considered a limiting factor for applying this technology to 16S-based taxonomy assignment and phylogenetic analysis. Our result showed that using CCS reads we were able to replicate an earlier experiment and correctly assign the OTUs to at least the genus level.
This study's limitations include the fact that the sample size may not have allowed us to detect modest differences in bacterial distribution. Nevertheless, it was comparable to the size of a recent study with similar objectives and design [24] and allowed us to detect significant differences with regard to maternal diabetes status. Also, we did not have access to the matched maternal samples; thus, evidence connecting the fetal microbiome directly to the maternal microbiome is lacking. Future studies comparing bacterial composition of the matched maternal (vaginal, placental and fecal) and neonatal microbiome are warranted to determine the major source of the newborn microbiota. In addition, since the gut microbiota changes dramatically during pregnancy [48], a direct comparison between pregnant and non-pregnant women with DM will be important to explore the effect of pregnancy on bacterial content in the context of DM. Furthermore, numerous prenatal factors, such as chorioamnonitis or premature rupture of membranes, maternal prenatal smoking, maternal stress, and others , may contribute to the initial colonization of the gut microbiome. Women were also excluded from the study if they received antibiotic treatment over the course of pregnancy, precluding testing the relationship between maternal infection, diabetes, and meconium content. However, women undergoing C-section and receiving a prophylactic dose of an antibiotic as a standard of care were retained in the study, which could potentially affect the meconium microbiota, especially if antibiotics were given before cord clamping. The sample size of this study would not allow to systematically assess the effect of these and other factors on the meconium microbiome profile. Larger studies collecting extensive prenatal and neonatal data are underway to explore the role of other factors that have effects on the meconium microbiome. Also, health-related information, such as BMI, maternal smoking or glucose level, was not accessible on several enrolled subjects due to the lack of consent or ethical issues associated with administering GCT to individuals with established DM. Therefore, we were unable to establish the relationship between these traits and the meconium microbial composition. Moreover, the time of sampling widely varied among the newborns raising the possibility that environmental exposures may have influenced the bacterial content for those who passed their first stool at a later time. However, the time of sampling was not significantly different between the study groups; therefore, it is unlikely to systematically bias our results. Also, it is known that maternal diabetes can increase the risk of pre-term birth, due to an increase in both fetal and maternal indications for delivery. In our study, we did not see microbiota differences by gestational age among DM, GDM, and no-diabetes groups; however, future studies with a larger sample size will have to examine whether gestational age at delivery is associated with the meconium microbiota. In addition, we did not account for susceptibility loci predisposing to DM, which could also be transmitted from a mother to an infant and potentially affect the microbiome. Future studies will have to determine if host DM risk alleles are associated with a “diabetic” microbial profile.
In summary, our study provides further evidence that meconium contains diversified microbiota and suggests that the initial colonization of the gut flora may start prior to birth. Furthermore, the meconium microbiome of babies born to DM mothers is enriched for the bacterial OTUs observed in the fecal microbiome of adult DM patients. These findings can enhance our understanding of a non-genetic risk of transmission of DM, and help design novel preventive measures for adult onset diseases.
Supporting Information
File includes Tables S1 and S2, and Figures S1–S3. Table S1. Sequences of bar-coded primers for PCR to generate 16S sequencing amplicons. Table S2. Association between the microbiome composition and clinical data. P-values are shown. Figure S1. Description statistic of Pacbio RS 16S sequencing results. A. Histogram of the counts of reads and CCS (circular consensus sequencing) reads at different read length in three chips. This figure demonstrates that a single run of Pacbio RS for 2×45 minutes generated ∼55 k reads, of which ∼33 k reads were longer than 2 kb. B. Histogram to demonstrate the mean read length of CCS reads is 443 bp (range 400–500). C. Bar plots to compare the taxa classification and abundance of 3 stool samples and their repeated measurements at all 5 taxonomy levels. D. Bar plots to show the misclassification rate of E.coli strain at all 5 taxonomy levels. Figure S2. Beta diversity of the microbiome from neonate meconium samples. A. Variability in beta-diversity of the microbiome using unweighted or weighted Unifrac distances. B. Unifrac unweighted and weighted PCoA PC1 vs PC2 plot to show the overall similarity of the microbiome of neonates born to mothers with three different diabetic statuses. C. Unifrac unweighted and weighted PCoA plot to show the overall similarity of the microbiome from born via different delivery methods. Figure S3. Biplot of the principal coordinates analysis (PCoA) for stool bacteria comparing the healthy adult (Adult stool, red), neonates born to healthy mothers (HC, green), neonates born to mothers with gestational diabetes(GDM, yellow) and neonates born to mothers with type 2 diabetes(DM, blue). Significant separation can be observed between the DM and HC groups.
(PDF)
Acknowledgments
We thank Drs. Yasmin Hurd and Rhoda Sperling for critical reviewing of this manuscript. We also thank Ms. Stephanie Gampel, Ms. Nancy Huynh, Mr. Mordechai Grabie, Ms. Jenny Ly, Mr. Solomon Bienstock and Ms. Jackie Finik for helping with subject recruitment, clinical data collection, and reviewing the manuscript.
Funding Statement
Work was supported by a CCFA Career Development Award (JH) and grants from The Chemotherapy Foundation (SI), the National Cancer Institute UH3CA140233, R01CA159036, and R03CA159414 (ZP) and the National Institute of Mental Health K01MH080062-03 (YN). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Barker DJ (2004) The developmental origins of adult disease. J Am Coll Nutr 23: 588S–595S. [DOI] [PubMed] [Google Scholar]
- 2.Buchanan TA (1995) Pregnancy in preexisting diabetes. National Diabetes Data Group: Diabetes in America. pp. 719–733.
- 3. Dunne F, Brydon P, Smith K, Gee H (2003) Pregnancy in women with Type 2 diabetes: 12 years outcome data 1990–2002. Diabet Med 20: 734–738. [DOI] [PubMed] [Google Scholar]
- 4. Bell R, Glinianaia SV, Tennant PW, Bilous RW, Rankin J (2012) Peri-conception hyperglycaemia and nephropathy are associated with risk of congenital anomaly in women with pre-existing diabetes: a population-based cohort study. Diabetologia [DOI] [PubMed] [Google Scholar]
- 5. Fraser A, Nelson SM, Macdonald-Wallis C, Lawlor DA (2012) Associations of existing diabetes, gestational diabetes, and glycosuria with offspring IQ and educational attainment: the Avon Longitudinal Study of Parents and Children. Exp Diabetes Res 2012: 963735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ornoy A (2005) Growth and neurodevelopmental outcome of children born to mothers with pregestational and gestational diabetes. Pediatr Endocrinol Rev 3: 104–113. [PubMed] [Google Scholar]
- 7. Gillman MW, Rifas-Shiman S, Berkey CS, Field AE, Colditz GA (2003) Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics 111: e221–226. [DOI] [PubMed] [Google Scholar]
- 8. Oken E, Levitan EB, Gillman MW (2008) Maternal smoking during pregnancy and child overweight: systematic review and meta-analysis. Int J Obes (Lond) 32: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, et al. (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 107: 11971–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson CL, Versalovic J (2012) The human microbiome and its potential importance to pediatrics. Pediatrics 129: 950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO (2007) Development of the human infant intestinal microbiota. PLoS Biol 5: e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, et al. (2008) Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS One 3: e3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vaishampayan PA, Kuehl JV, Froula JL, Morgan JL, Ochman H, et al. (2010) Comparative metagenomics and population dynamics of the gut microbiota in mother and infant. Genome Biol Evol 2: 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hong PY, Lee BW, Aw M, Shek LPC, Yap GC, et al. (2010) Comparative Analysis of Fecal Microbiota in Infants with and without Eczema. PLoS One 5: e9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johansson MA, Sjogren YM, Persson JO, Nilsson C, Sverremark-Ekstrom E (2011) Early Colonization with a Group of Lactobacilli Decreases the Risk for Allergy at Five Years of Age Despite Allergic Heredity. PLoS One 6: e23031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L, Christophersen CT, Sorich MJ, Gerber JP, Angley MT, et al. (2011) Low Relative Abundances of the Mucolytic Bacterium Akkermansia muciniphila and Bifidobacterium spp. in Feces of Children with Autism. Applied and Environmental Microbiology 77: 6718–6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yap GC, Chee KK, Hong PY, Lay C, Satria CD, et al. (2011) Evaluation of stool microbiota signatures in two cohorts of Asian (Singapore and Indonesia) newborns at risk of atopy. Bmc Microbiology 11: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sjogren YM, Tomicic S, Lundberg A, Bottcher MF, Bjorksten B, et al. (2009) Influence of early gut microbiota on the maturation of childhood mucosal and systemic immune responses. Clinical and Experimental Allergy 39: 1842–1851. [DOI] [PubMed] [Google Scholar]
- 19. Guarino A, Wudy A, Basile F, Ruberto E, Buccigrossi V (2012) Composition and roles of intestinal microbiota in children. J Matern Fetal Neonatal Med 25 (Suppl 1) 63–66. [DOI] [PubMed] [Google Scholar]
- 20. Biasucci G, Rubini M, Riboni S, Morelli L, Bessi E, et al. (2010) Mode of delivery affects the bacterial community in the newborn gut. Early Hum Dev 86 (Suppl 1) 13–15. [DOI] [PubMed] [Google Scholar]
- 21. Hitti J, Riley DE, Krohn MA, Hillier SL, Agnew KJ, et al. (1997) Broad-spectrum bacterial rDNA polymerase chain reaction assay for detecting amniotic fluid infection among women in premature labor. Clin Infect Dis 24: 1228–1232. [DOI] [PubMed] [Google Scholar]
- 22. Oh KJ, Lee SE, Jung H, Kim G, Romero R, et al. (2010) Detection of ureaplasmas by the polymerase chain reaction in the amniotic fluid of patients with cervical insufficiency. J Perinat Med 38: 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jimenez E, Fernandez L, Marin ML, Martin R, Odriozola JM, et al. (2005) Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Current Microbiology 51: 270–274. [DOI] [PubMed] [Google Scholar]
- 24. Gosalbes MJ, Llop S, Valles Y, Moya A, Ballester F, et al. (2013) Meconium microbiota types dominated by lactic acid or enteric bacteria are differentially associated with maternal eczema and respiratory problems in infants. Clin Exp Allergy 43: 198–211. [DOI] [PubMed] [Google Scholar]
- 25. Jimenez E, Marin ML, Martin R, Odriozola JM, Olivares M, et al. (2008) Is meconium from healthy newborns actually sterile? Res Microbiol 159: 187–193. [DOI] [PubMed] [Google Scholar]
- 26. Satokari R, Gronroos T, Laitinen K, Salminen S, Isolauri E (2009) Bifidobacterium and Lactobacillus DNA in the human placenta. Lett Appl Microbiol 48: 8–12. [DOI] [PubMed] [Google Scholar]
- 27. Steel JH, Malatos S, Kennea N, Edwards AD, Miles L, et al. (2005) Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr Res 57: 404–411. [DOI] [PubMed] [Google Scholar]
- 28. Valles Y, Gosalbes MJ, de Vries LE, Abellan JJ, Francino MP (2012) Metagenomics and development of the gut microbiota in infants. Clin Microbiol Infect 18 (Suppl 4) 21–26. [DOI] [PubMed] [Google Scholar]
- 29. Wu X, Ma C, Han L, Nawaz M, Gao F, et al. (2010) Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr Microbiol 61: 69–78. [DOI] [PubMed] [Google Scholar]
- 30. Madan JC, Salari RC, Saxena D, Davidson L, O'Toole GA, et al. (2012) Gut microbial colonisation in premature neonates predicts neonatal sepsis. Arch Dis Child Fetal Neonatal Ed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mshvildadze M, Neu J, Shuster J, Theriaque D, Li N, et al. (2010) Intestinal Microbial Ecology in Premature Infants Assessed with Non-Culture-Based Techniques. Journal of Pediatrics 156: 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moles L, Gomez M, Heilig H, Bustos G, Fuentes S, et al. (2013) Bacterial Diversity in Meconium of Preterm Neonates and Evolution of Their Fecal Microbiota during the First Month of Life. PLoS One 8: e66986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langer O, Brustman L, Anyaegbunam A, Mazze R (1987) The significance of one abnormal glucose tolerance test value on adverse outcome in pregnancy. Am J Obstet Gynecol 157: 758–763. [DOI] [PubMed] [Google Scholar]
- 34. Lindsay MK, Graves W, Klein L (1989) The relationship of one abnormal glucose tolerance test value and pregnancy complications. Obstet Gynecol 73: 103–106. [PubMed] [Google Scholar]
- 35. Ahn J, Yang L, Paster BJ, Ganly I, Morris L, et al. (2011) Oral microbiome profiles: 16S rRNA pyrosequencing and microarray assay comparison. PLoS One 6: e22788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological 57: 289–300. [Google Scholar]
- 38. Price LB, Liu CM, Johnson KE, Aziz M, Lau MK, et al. (2010) The effects of circumcision on the penis microbiome. PLoS One 5: e8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, et al. (2012) Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 28: 2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oksanen J (2007) Multivariate analysis of ecological communities in R: vegan tutorial. R package version 2-0.
- 41. Sellitto M, Bai G, Serena G, Fricke WF, Sturgeon C, et al. (2012) Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS One 7: e33387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qin J, Li Y, Cai Z, Li S, Zhu J, et al. (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490: 55–60. [DOI] [PubMed] [Google Scholar]
- 43. Tsuji H, Oozeer R, Matsuda K, Matsuki T, Ohta T, et al. (2012) Molecular monitoring of the development of intestinal microbiota in Japanese infants. Benef Microbes 3: 113–125. [DOI] [PubMed] [Google Scholar]
- 44. Jeon CY, Haan MN, Cheng C, Clayton ER, Mayeda ER, et al. (2012) Helicobacter pylori infection is associated with an increased rate of diabetes. Diabetes Care 35: 520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, et al. (2010) Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 5: e9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma YF, Zhang Y, Zhang JY, Chen DW, Zhu Y, et al. (2009) The complete genome of Comamonas testosteroni reveals its genetic adaptations to changing environments. Appl Environ Microbiol 75: 6812–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, et al. (2009) Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5: e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, et al. (2012) Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150: 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File includes Tables S1 and S2, and Figures S1–S3. Table S1. Sequences of bar-coded primers for PCR to generate 16S sequencing amplicons. Table S2. Association between the microbiome composition and clinical data. P-values are shown. Figure S1. Description statistic of Pacbio RS 16S sequencing results. A. Histogram of the counts of reads and CCS (circular consensus sequencing) reads at different read length in three chips. This figure demonstrates that a single run of Pacbio RS for 2×45 minutes generated ∼55 k reads, of which ∼33 k reads were longer than 2 kb. B. Histogram to demonstrate the mean read length of CCS reads is 443 bp (range 400–500). C. Bar plots to compare the taxa classification and abundance of 3 stool samples and their repeated measurements at all 5 taxonomy levels. D. Bar plots to show the misclassification rate of E.coli strain at all 5 taxonomy levels. Figure S2. Beta diversity of the microbiome from neonate meconium samples. A. Variability in beta-diversity of the microbiome using unweighted or weighted Unifrac distances. B. Unifrac unweighted and weighted PCoA PC1 vs PC2 plot to show the overall similarity of the microbiome of neonates born to mothers with three different diabetic statuses. C. Unifrac unweighted and weighted PCoA plot to show the overall similarity of the microbiome from born via different delivery methods. Figure S3. Biplot of the principal coordinates analysis (PCoA) for stool bacteria comparing the healthy adult (Adult stool, red), neonates born to healthy mothers (HC, green), neonates born to mothers with gestational diabetes(GDM, yellow) and neonates born to mothers with type 2 diabetes(DM, blue). Significant separation can be observed between the DM and HC groups.
(PDF)