

Abstract
Hepatitis B virus (HBV) is a DNA virus with complex replication, and high replication and mutation rates, leading to a heterogeneous viral population. The population is comprised of genomes that are closely related, but not identical; hence, HBV is considered a viral quasispecies. Quasispecies variability may be somewhat limited by the high degree of overlapping between the HBV coding regions, which is especially important in the P and S gene overlapping regions, but is less significant in the X and preCore/Core genes. Despite this restriction, several clinically and pathologically relevant variants have been characterized along the viral genome. Next-generation sequencing (NGS) approaches enable high-throughput analysis of thousands of clonally amplified regions and are powerful tools for characterizing genetic diversity in viral strains. In the present review, we update the information regarding HBV variability and present a summary of the various NGS approaches available for research in this virus. In addition, we provide an analysis of the clinical implications of HBV variants and their study by NGS.
Keywords: Hepatitis B virus, Next generation sequencing, Quasispecies, Linkage analysis, Gene overlapping
Core tip: We provide an update of hepatitis B virus (HBV) virology, focusing on its complex replication cycle which generates high genetic variability, which led HBV infection to evolve as viral quasispecies, complex distributions of variant populations that are closely related but not identical. We also discuss the clinical and virological implications of this population structure and the application of different next-generation sequencing approaches, which enable analysis of thousands of clonally amplified regions, to study these heterogeneous viral populations.
HEPATITIS B VIRUS INFECTION HAS SINGULAR VIROLOGY: IS IT A “PSEUDORETROVIRUS”?
Human hepatitis B virus (HBV; Hepadnaviridae family) causes acute and chronic infection in humans and chimpanzees. HBV infection is distributed throughout the world, and it is estimated that around 2000 million people worldwide have been in contact with this pathogenic agent. Despite successful vaccination programs and effective antiviral therapies, there are over 350 million carriers of HBV surface antigen (HBsAg). Some 150 million of them have active infection and a high risk of progression to cirrhosis or hepatocellular carcinoma (HCC). HBV-infected individuals have a 30-fold higher risk of developing HCC than the remainder of the population, and it is estimated that 53% of liver cancers worldwide are associated with HBV[1].
HBV consists of a spherical particle with a diameter of 42 nm. The outer envelope is comprised of a phospholipid/protein layer and the inner shell contains core antigen (HBcAg) with the viral genome. Based on differences in the nucleotide sequence of its genome, HBV virus is currently classified into 10 genotypes (A-J) and multiple subgenotypes, with characteristic ethnic/geographic distributions[2-4]. The viral genome is 3.2 kb in length, and within the double-shelled particle, the genome adopts the conformation of a circular, partially double-stranded DNA molecule (rcDNA), with a complete non-covalently closed minus strand and an incomplete plus strand.
The HBV genome has four highly overlapping open reading frames (ORFs) - S, P, C and X - that encode seven proteins. The S ORF encodes three different-sized surface antigens whose common region contains the main antigenic loop, the “a” epitope (amino acids 124-147). The a epitope is one of the main targets for neutralizing anti-HBs antibodies. The S ORF is completely overlapped by the P ORF, which encodes a polyprotein with different domains: the reverse transcriptase (RT), the polymerase, RNase H, and the terminal protein (TP). The catalytic center of RT is located at the C domain and is defined by four amino acids mapped at positions 203-206: tyrosine, methionine, aspartate, and aspartate (203YMDD206). The YMDD motif is highly conserved among viral polymerases/reverse transcriptases. The 3’ region of C ORF is overlapped by P ORF and contains two start (ATG) codons. The region between these codons is called the preCore region, while the remaining region from the second ATG is known as the Core gene. The two proteins translated from these two ATG start codons are the viral capsid (HBcAg) and the e antigen (HBeAg); the latter is a soluble antigen that acts as an immunomodulator. Finally, the 5’ end of the X ORF is overlapped by the P ORF, and the 3’ end is overlapped by part of the preCore. The X ORF encodes the x antigen (HBxAg), which is a multifunctional transactivator protein. Furthermore, the HBV genome contains various elements that regulate replication. One important element is enhancer II (ENHII), located in the X gene sequence, which modulates the promoters of transcription of viral mRNA.
Viral cycle: a DNA virus with a retrotranscription step, similar to a retrovirus
The HBV population is highly heterogeneous and is comprised of genomes that are closely related, but not identical; hence, it is considered a viral quasispecies, a term commonly associated with RNA viruses. The complex viral replication cycle of HBV may be the cause of this quasispecies nature.
HBV replication occurs exclusively in hepatocytes, but the specific and highly effective viral determinants that direct HBV into hepatocytes are still unknown. It has been hypothesized that HBV hepatotropism is mediated through specific binding of the myristoylated N-terminal preS1 domain of the large HBV surface protein to a hepatocyte-specific receptor that has not been completely defined[5]. After interacting with the cellular receptor, HBV particles enter the liver cell[6] and release the outer envelope. HBcAg from the inner shell directs the particle to the nucleus, where the rcDNA genome is repaired by host and viral polymerases to a fully double-stranded covalently closed circular genome (cccDNA). This molecule remains in the nucleus of the infected hepatocyte as a minichromosome until the cell dies[7]. cccDNA also acts as a template for synthesis of five different viral mRNAs: preCore mRNA, pre-S2 mRNA, X mRNA, S1 mRNA, and the pregenomic RNA (pgRNA). The largest of these, pgRNA, is 3.5 kb in length and contains a redundant fragment of the preCore region at both ends. pgRNA yields the polymerase and core transcripts. In addition, the 5’ and 3’ ends of pgRNA adopt a hairpin stem-loop structure, named epsilon. The epsilon signal at the 5’ end directly interacts with the polymerase and constitutes the first step in initiation of reverse transcription. The interaction induces recruitment of HBcAg monomers around the polymerase/pgRNA complex. These particles can be considered true previrions, with the HBV genome in RNA conformation. Within these particles, pgRNA serves as a template for reverse transcription to the minus strand of HBV DNA, which acts as a template for synthesis of the HBV DNA plus strand by the HBV polymerase. The plus strand is incomplete and yields a new particle with a new rcDNA genome[8].
Thus, the HBV viral cycle includes a transcription and translation step, but also a reverse transcription step similar to what occurs in retroviruses, mediated by the different activities of HBV polymerase[2,3]. One potential explanation for the quasispecies nature of the virus occurs at the retrotranscription step: the viral enzyme lacks proofreading activity and the new genomes are highly prone to accumulate errors[9]. This fact, in addition to the extremely high viral replication rate results in a remarkably heterogeneous viral population, referred to as a quasispecies[10]. In the section Variability only due to poor fidelity of the viral polymerase? we discuss further potential sources of the virus quasispecies formation.
Nonetheless, quasispecies variability may be somewhat limited by the high degree of overlap between the HBV coding regions (67%)[11]. This restriction would not be very important in the Core region because it is not overlapped and contains variable epitopic domains that play a central role in the immune response against the virus[12,13]. In contrast, the viral reverse transcriptase is highly conserved, due to its important functions and overlapping with the S ORF[3]. Positions at or near the initiation site for synthesis of the first viral strand are also highly conserved, and are homologous over 67 nucleotides to the U5 region; a region comparable to retroviral long terminal repeats[14].
The Core is the most suitable region to study the natural evolution of the viral genome, keeping in mind that the immune system is the main driver of evolutionary pressure[15]. Certain specific mutations observed in viral variants may have implications in the pathogenesis of viral disease. For example a G to A substitution at nucleotide 1896 yields a de novo stop codon in the preCore region that is associated with HBeAg seroconversion and a high risk of fulminant hepatic failure in acute infection[16]. In conclusion, depending on the HBV region analyzed, different features of the quasispecies will be examined.
Singular HBV virology: viral genome integration and nuclear reinfection of nascent viral capsids
One peculiarity of hepadnaviruses is that their genome is often found to be inserted in the hepatocyte genome[17-19], a fact that could indicate a possible common origin with retroviruses. These integrations can induce chromosome changes, genome instability, or changes in the expression of human genes[20] and have been found throughout the different chromosomal sites in the host genome [e.g., near the telomerase reverse transcriptase gene (tert) and fibronectin gene (fn1)][19,21-24]. Integrated HBV DNA sequences and episomal HBV genomes have been found in 85%-90% of HBV-related cases of HCC[19,21]. Recent studies using various genetic approaches and microarray technology have reported that HBV-related HCC displays higher rates of chromosomal abnormalities than HCC due to other risk factors, and even generates chimeric oncogenic proteins[25,26]. Ding et al[19] analyzed 286 unique integration sites from 40 pairs of HBV-related HCC and tumor-adjacent tissues by massive sequencing on the Illumina platform [see next-generation sequencing (NGS) methods in later sections], and seven HBV integrations per individual were detected. However, the relationship between HBV integration and HCC has not been completely elucidated.
Another peculiarity of viral replication is nuclear reinfection. The nascent capsids have the same structure and conformation as those that originally infected the hepatocytes and released the outer envelope, and this similarity enables nuclear reinfection. In this way, the new capsids can increase the amount and diversity of nuclear cccDNA during viral replication[27]. This leads to an increase in the intracellular and intranuclear viral quasispecies.
Based on its singular virology, HBV can be considered a non-enveloped RNA virus at the intracellular level and an enveloped DNA virus at the extracellular level. In addition, it has “pseudoretroviral” behavior consisting of an ability to integrate into the host cell genome, and a highly variable genome that leads to a quasispecies nature. In the next sections, the variability of the HBV genome will be discussed in the light of data obtained by recently developed massive sequencing technologies.
TOOLS FOR VIRAL QUASISPECIES STUDY: NGS TECHNOLOGIES AS A NEW STRATEGY FOR A DEEP ANALYSIS
Study of HBV quasispecies evolution together with disease progression is important to clarify the pathogenesis of this condition and improve the available treatment strategies[28]. This type of study can be performed by standard methods or by new, more powerful approaches.
Standard approaches to describe genomic variability
For the past 40 years, the most widely used method for sequencing DNA has been Sanger sequencing, which is based on a single sequencing reaction with fluorescent dideoxynucleotides, which act as chain terminators[29-31]. This produces a mixture of DNA fragments of different sizes, all ending with one of four terminators, each of which is marked with a different fluorochrome that can be detected when the fragments are separated by size in capillary electrophoresis-based sequencers. To obtain the maximum amount of product to be analyzed, polymerase chain reaction (PCR) is performed to amplify the region of interest. Analysis of a complex genomic sample (e.g., a viral quasispecies) by Sanger sequencing yields the most common nucleotide present in the quasispecies, known as the consensus sequence, but individual sequences are not obtained. Thus, analysis of the HBV RT domain C (putative active center) covering codons 202 to 206 can yield positions with different nucleotides (A/GGC202 TAT203 A/GTG204 GAT205 GAT206), but it is uncertain whether the changes are present in the same sequence or in two different sequences, for example GGC202.TAT203.ATG204.GAT205.GAT206 or AGC202.TAT203.GTG204.GAT205.GAT206. Minor nucleotide changes in the same position can only be detected as “inner small peaks” when their frequency is higher than 20% (e.g., a clear G peak with a small A peak inside in codon 202). Therefore, Sanger sequencing of a complex genomic mixture presents two main limitations: low sensitivity for minor variant detection and inability to perform haplotype analysis.
In addition to such direct sequencing methods, some indirect molecular techniques, mainly focussed on detection of specific variants, have also been extensively used. The most common is reverse hybridization in Line Probe Assays format that can detect specific variants present as at least 5% of the viral quasispecies[32,33]. Other, less commonly used methods for detecting specific variants include restriction fragment length polymorphism (RFLP) analysis[34-36], 5’-nuclease assays[35], melting point analysis[37], mass spectrometry[38], DNA chip technology[39] and real-time PCR using mutation-specific primers[40]. Although these methods can differentiate between population mixtures[41], they do not allow simultaneous detection of different substitutions in the same viral genome. Therefore, complex variants with two or more amino acid changes, such as those related to entecavir resistance or those causing a fitness increase during lamivudine treatment, cannot be properly studied by these indirect methods. Furthermore, they cannot be used to properly describe the composition of a quasispecies.
The traditional method used to analyze a heterogeneous genomic population, such as a viral quasispecies, is cloning and sequencing. This is a costly, labor-intensive process that requires multiple, complex experimental steps, and provides limited resolution regarding the mutation spectrum and frequencies. Analysis of a quasispecies by cloning requires a large number of clones (> 100) which significantly hampers its use.
NGS
The genome sequencing scenario is now changing, mainly because of the development of NGS techniques[42]. NGS approaches enable high-throughput analysis of thousands of clonally amplified regions and are powerful tools for characterizing genetic diversity in viral strains[43].
The most widespread platforms available are the 454 FLX (Roche)[44], the Solexa genome analyzer (Illumina)[45,46], the SOLiD system (Applied Biosystems)[47], and recently, the Ion Torrent system (Life Technologies)[48]. These platforms apply different experimental strategies (enzymological, chemical) and complex engineering (high-resolution optics, hardware, and software), and provide significant time saving and a minimal requirement for associated equipment in comparison to cloning. NGS technologies seek amplification of single strands of DNA with specific sequences (added by chemical binding or by specific PCR primers) and perform sequencing reactions on the amplified strands. The presence of specific sequences enables selection of PCR-amplified fragments without requiring a bacterial cloning step. The yield of sequence reads and total bases per instrument run is significantly higher than that produced in a capillary sequencer run: from several hundred thousand reads (454 FLX and Ion Torrent) to tens of millions of reads (Illumina and SOLiD)[44].
FLX-454 pyrosequencing platform (Roche): The 454 technology is based on ultra-deep pyrosequencing (UDPS), a DNA sequencing method that relies on chemiluminescent detection of pyrophosphate release during polymerase-mediated dNTPS incorporation. The pyrophosphate is converted into ATP, which allows generation of light by the enzyme luciferase in amounts proportional to the amount of ATP. UDPS is a fast method that can detect and quantify small viral subpopulations (present in < 5%)[43,49]; specifically, a recent study reported detection of HBV subpopulations accounting for 2% of the total viral population[50].
The 454 Sequencing System supports analysis of samples from a wide variety of starting materials, including genomic DNA, PCR products, artificial bacterial chromosomes, and cDNA. The system relies on fixing nebulized and adapter-ligated DNA fragments to small DNA-capture beads in a water-in-oil emulsion. The emulsion is prepared to achieve oil drops with a diameter that accomodates only one bead and a single DNA molecule. The oil drops act as microreactors for clonal PCR amplification. DNA-bound beads are placed on a PicoTiterPlate (PTP), together with a fiber optic chip and a mix of enzymes such as DNA polymerase, ATP sulfurylase, and luciferase. The PTP is then placed in the GS FLX system for sequencing. The sequencing process is monitored by the light flashes released once a nucleotide is incorporated. However, the calibrated base calling cannot properly interpret long stretches (> 6) of the same nucleotide and for this reason, homopolymeric regions (defined as repeats of ≥ 4 identical bases) are prone to artifactual base insertions and deletions. In contrast, as each incorporation step is nucleotide-specific, substitution errors are rarely encountered in Roche/454 sequence reads.
The raw reads are processed by the 454 analytical software and then screened by various quality filters to remove poor-quality sequences. Fragments > 400 nucleotides can be sequenced (currently around 700 nucleotides), allowing haplotypic analysis that would theoretically be useful even for phylogenetic studies. UDPS is currently the only platform for quantitative analysis of long clonal sequences that allows detection of particular mutations as well as combinations of mutations in the same sequence. It is, therefore, highly useful as a high-throughput, massive sequencing method to describe quasispecies.
Solexa (llumina): The single molecule amplification step of the Illumina Genome Analyzer starts with an Illumina-specific adapter library. This step automatically takes place on the oligo-derivatized surface of a flow cell, which is an eight-channel sealed glass microfabricated device that allows a special process, “bridge amplification”, of fragments on its surface. This process uses DNA polymerase to produce multiple DNA clusters, each representing the single molecule that initiated the cluster amplification. Bridge amplification generates multiple copies of a specific DNA molecule on an “oligo-decorated” solid support. Each cluster contains approximately 1 million copies of the original fragment.
The Illumina system utilizes a sequencing-by-synthesis approach in which all four nucleotides and the DNA polymerase are simultaneously added to flow cell channels for incorporation into the oligo-primed cluster fragments[46]. The nucleotides carry a unique base fluorescent label in which the 3’-OH group is chemically blocked. The DNA chains are then extended by one nucleotide, and an image is taken. Subsequently, the 3′ blocking group is chemically removed to prepare each strand for the next incorporation by DNA polymerase. This series of steps continues for a specific number of cycles, permitting discrete read lengths of 50-250 nucleotides. However, this approach is not suitable for haplotypic analysis or quasispecies studies.
SOLiD (Applied Biosystems/Life Technologies): The SOLiD platform, which is similar to that of other NGS methods, uses an adapter-ligated fragment library and an emulsion PCR (similar to 454 technology)[47]. However, it is based on the principle of “two-base encoding” and uses a DNA ligase as the enzyme. The system has random 8-mer fluorescent probes with all 16 combinations of di-bases at the 3’ ends. Once one of these fluorescent probes hybridizes over the sequence, the probe is linked to the region of interest by ligase. Each ligation step is followed by fluorescence detection and removal of the ligated 8-mer. This method sequences short fragments of 35-50 nucleotides and does not allow haplotypic studies.
Ion torrent: ion semiconductor sequencing (Life Technologies): This platform is similar to the FLX 454 method, but has a semiconductor-based detection system[48]. The sequencing system is based on detection of hydrogen ions (protons) released during DNA polymerization, instead of the image capture used in other sequencing systems. A microwell containing a DNA template is flooded with a single type of nucleotide. If the nucleotide introduced is complementary to the leading template nucleotide, it will be incorporated into the growing complementary strand. This phenomenon releases a hydrogen ion that triggers a hypersensitive ion sensor, whose signal indicates that a reaction has occurred. If homopolymer repeats are present in the template sequence, multiple nucleotides will be incorporated in a single cycle. This leads to a corresponding number of released hydrogen molecules and a proportionately higher electronic signal. In this sense, this method can be considered more accurate than FLX-454 for homopolymeric sequences. Currently, fragments up 200 nt can be sequenced, with an approximation to haplotypic analysis. However, the sequence length is too short for phylogenetic studies, and the method is not useful to describe quasispecies.
NGS applications and limitations
NGS techniques have been used to study some quasispecies, such as human immunodeficiency virus (HIV)[51,52], HBV[53-59], and hepatitis C virus (HCV)[46,60]. The 454 platform has been successful in analysis of HIV quasispecies, exhibiting high sensitivity for detecting treatment-resistant variants[51,61-63], in describing HCV heterogeneity[46,60], and more recently, in HBV quasisepecies studies[53-59]. UDPS has been used in studies to quantify HBV minority variants carrying resistant mutations[55,58], and to detect defective variants[53-58]. In addition, the UDPS NGS approach has enabled dynamic study of clonal evolution in cancer cells, detecting somatic mutations in rare subclones at a rate of 1 in 5000 copies[49].
Several factors should be remembered in relation to NGS reads. First, the length of an NGS sequence read is shorter than that of Sanger sequencing, and second, each NGS platform has a unique error model that differs from the error model established for capillary sequence reads. In consequence, complex, accurate computational algorithms are required to process the raw data from massive parallel sequencing (background subtraction, base calling, and quality assessment), and they should be specific for each platform. Bioinformatic filtering has been the main method developed to validate variants carrying substitutions, but it has not been completely optimized for variants with insertions and deletions, which are common in HBV, especially in the X ORF and basic Core promoter, mapped at the preCore region[64-68]. Therefore, study of insertion and deletion variants still requires classical cloning analysis, and that is why they have been little investigated.
In addition, UDPS is associated with sequencing errors in homopolymeric regions, although some of the variants identified have been true variants previously reported by cloning[69,70]. Currently, it is uncertain how natural insertion/deletion variants can be differentiated from variants resulting from errors in UDPS processing; therefore, the computational analysis of UDPS reads must be improved. One possible approach could include systematic analysis of forward and reverse reads in duplicate by NGS and in parallel with cloning. Another strategy could be targeted searching for sequences with known deletions and insertions, previously detected by classical clonal analysis[70], or selection of insertions or deletions that cause relevant changes, such as de novo stop codons[69,70].
The importance of investigating insertions and deletions in the HBV quasispecies is underscored with the G2091 deletion, described by Schories et al[69] and recently detected by Ramírez et al[70]. This deletion produces three de novo stop codons: (wild type, 2086TGG.GGG.GAA.TTG.ATG.ACT.CTA.GCT, and the deleted variant, 2086TGG.GGG.AAT.TGA.TGA.CTC.TAG.CT), resulting in an HBV “capsid-defective genome”. The presence of potentially defective genomes in the HBV quasispecies has been recently reported in the S ORF[56,58], in the HBcAg start codon, and even in the HBxAg stop codon in studies using massive UDPS[56].
Another factor that must be taken into account in NGS data analysis is the possibility of recombination events occurring during PCR amplification[71] by different mechanisms; for example, short incomplete amplicons can act as primers for different sequences present in the quasispecies, or crossing of amplified sequences can occur, similar to chromosome recombination events. These phenomena may be a potential limitation of UDPS, but they can also occur in classical cloning and any other technique that includes a PCR amplification step. This confounding factor questions the reliability of haplotype analysis of the quasispecies, which requires minimization of recombination events. Thus, the true impact of these events should be carefully evaluated in the future by UDPS analysis of in vitro mixtures from different clonal sequences. It would be particularly interesting to know the impact of recombination in order to derive conclusions from linkage analysis of different relevant nucleotide substitutions reported in the HBV polymerase and preCore region by classical cloning[72] and UDPS[56-58].
Lastly, the limit of detection of the UDPS approach has not been established, and differences between studies from different groups must be taken into account when viewing the results.
QUASISPECIES NATURE OF HBV
The term quasispecies was coined to refer to RNA viruses that genetically evolve as complex distributions of variants that are closely related, but not identical[73]. In quasispecies infection, the genome is not precisely defined, and an average of different variants is used as the consensus genome. Study of quasispecies dynamics is important to understand the adaptability, pathogenic power, and persistence of viruses, and to design strategies to prevent and treat the diseases they cause. A viral quasispecies is seen as a swarm of variants in a host, among which variants carrying a biological advantage during replication are selected. When changes occur in the environmental conditions of the virus, the quasispecies structure responds by rebalancing its composition. The predominant sequence (master sequence) may even shift by selection of a variant that is better adapted to the new environment, in the classic Darwinian process of survival of the fittest[57].
Variability only due to poor fidelity of the viral polymerase?
The hepadnavirus family replicates by a reverse transcriptase that lacks proofreading activity, and this fact seems to justify their high mutation rate (3.2 × 10-5 - 7.9 × 10-5 nucleotide substitutions/replicative cycle); 100 times higher than other DNA viruses[74]. The high mutation rate, intense replicative activity (1012 viral particles/d), and small genome size of HBV, results in approximately 1010-11 point mutations being produced per day in individuals with active replication.
In addition, certain host factors are reported to be associated with hypermutational activity. One example is the innate antiviral defense mechanism that mediates host enzymes belonging to the APOBEC3 cytidine deaminase family, which cause extensive deamination of cytidine (C) to uridine (U) bases in negative-stranded DNA[75]. This activity results in a guanine (G) to adenine (A) hypermutation in positive-stranded DNA[76]. APOBEC-mediated G to A hypermutation was initially reported in HIV, but it has also been described in HBV[77-79], other retroviruses, and retrotransposons[76]. In fact, several clinically relevant HBV variants result from G to A mutations, such as rtA181T (GCN to ACN) and rtM204I (ATG to ATA), interpreted as RT resistance mutations, and the G1896A and G1899A main preCore variants (see next sections).
Thus, viral and host factors yield an HBV quasispecies provided with plasticity and rapid adaptation to the changing environmental conditions caused by the immune response or antiviral treatments[80,81]. In this line, nascent HBV capsids can contain new mutations produced during reverse transcription or APOBEC activity; therefore, the genomes may differ from those that previously infected the nucleus. This reinfection process generates a kind of intracellular quasispecies, in addition to the circulating one, thereby increasing the complexity of HBV. Another factor potentially responsible for causing variability is the fact that the same liver cell can be infected by multiple virions simultaneously or sequentially during its lifetime.
The versatility of the quasispecies structure may include mechanisms to tolerate variants with changes involving long fragments of the HBV genome, such as deletions reported by genome splicing[73,82,83]. This tolerance might yield defective genomes that can be maintained by transcomplementation mechanisms. For instance, HBsAg-defective variants might be enveloped by functional surface proteins encoded by other variants present in the quasispecies[58,84] (see next sections). For this reason, viral genomes that cannot self-replicate sufficiently may be detected in the viral quasispecies population[56,58,85]. Such defective genomes have been isolated from liver tissue of infected patients[86], and the proteins they encode, which are known as hepatitis B spliced proteins, appear to contribute to persistent replication in patients with HCC[86]. This suggests that defective genomes may have modulatory roles and provides evidence of regulatory mechanisms directly associated with the HBV quasispecies structure.
These data seem to indicate that the quasispecies nature of HBV can further increase the encoding capacity of the virus, which is somewhat limited by the high overlapping of its genome. ORF overlapping represents a strategy to restrict the viral genome size and maximize its coding capacity[3,87-89]. An indication of gene overlapping may be the presence of unusually strong constraints at third codon positions[90-92]. Hepadnaviruses are representative of this situation, and overlapping must be systematically taken into account when HBV viral infection is investigated at the virological and clinical level.
As was discussed above, it is generally assumed that the main source of quasispecies variability is the viral polymerase lack of error correction. However, certain characteristics of the target sequences themselves, such as the presence of homopolymeric regions, may be associated with an increased risk of errors by polymerase “sliding”. This phenomenon would result in errors by deletion or insertion, giving rise to altered reading frame variants and resulting in potentially deficient genomes[69,70]. The presence of these variants in the circulating viral quasispecies would provide evidence of complementary mechanisms whose effect on infection is unknown.
In this line, it cannot be excluded that truncated proteins encoded by these variants may have some type of regulatory function. For example, lengthening the life of infected cells by changing cell apoptosis (carcinogenesis) has been reported in association with truncated HBx proteins[93-96]. To explain the existence of these truncated versions of HBx, it has been hypothesized that transcomplementation could be achieved by using functional protein encoded by competent variants. This process would potentially modulate viral encapsidation and replication, with direct effects on the clinical evolution of the infection.
Furthermore, due to the extreme overlapping of HBV coding regions, a nucleotide substitution in one ORF can also induce changes in another ORF, such as the rtA181T change in HBV polymerase, which results in a stop codon in the surface ORF (Figure 1) and yields truncated versions of all envelope proteins[58,84]. P ORF mutations (e.g., rtA181T/sW172Stop) do not seem critical to the viral envelope, perhaps because of the large excess of HBsAg production during viral replication. However, the same process could alter the viral cycle by affecting components produced in more limited amounts such as Core or polymerase proteins, whose defective variants have been recently observed by massive sequencing[56,58].
Figure 1.
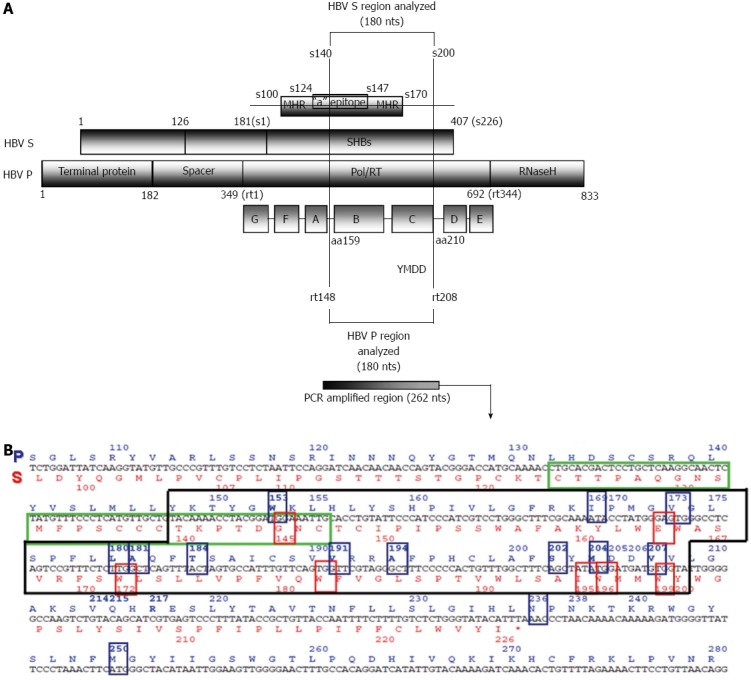
Overlapping region of the polymerase and surface genes (A) and its corresponding sequence (B). Translation into amino acids is depicted in blue in the P open reading frame (ORF) (above) and red in the S ORF (below). Most of the main codons related to nucleos(t)ide resistance (framed in blue), and the overlapping codons in the S ORF that may give rise to immune escape or stop codons (framed in red) are located within the fragment analyzed in a previous study by our group[58].
In summary, HBV quasispecies dynamics involves host factors (APOBEC3) and viral factors (lack proofreading of polymerase, high mutation rate, intense replicative activity, errors in homopolymeric regions, small genome size, and ORF overlapping). HBV variability makes the quasispecies highly versatile and allows defective particles to be present in the viral population. These particles may drive complementary regulatory mechanisms, which remain to be completely defined.
Parameters to study quasispecies complexity
The complexity of the HBV viral population may theoretically be associated with differing courses of infection, the probability of response to antiviral therapy, or seroconversion to antiHBe in HBeAg-positive chronic infection[28]. Shanon entropy is a useful parameter to study quasispecies complexity as related to the patients’ clinical evolution[46,97]. Several groups have studied the HBV quasispecies using classical clonal methods and Shannon entropy to determine its implications on the outcome of infection and the effects of antiviral therapy[33,72,98]. Liu et al[98] analyzed a group of entecavir-treated patients and found that responders presented a less complex quasispecies than the group of partial responders, whereas partial and non-responders showed similar patterns of complexity. A recent report based on a cloning method has shown that viral diversity after lamivudine treatment was higher in HBeAg-seroconverters than in non-seroconverters[72]. Nonetheless, these studies have the limitation of a low number of clones analyzed.
Nishijima et al[59] applied UDPS to study quasispecies complexity in a group of chronic hepatitis B patients, comparing the viral genome sequences determined in liver tissue with those in serum. Although the results cannot be compared with those of the cloning studies, the authors found no significant differences in the viral population between liver and serum from the same individual. In addition, they found no significant differences in viral complexity at the HBV DNA level, or according to age or degree of fibrosis. Nishijima et al[59] did not compare quasispecies diversity in relation to nucleos(t)ide analog (NUC) response or HBeAg seroconversion, but they observed similar complexity between naïve and NUC-treated cases. A preliminary study by our group using UDPS and Shannon entropy calculation indicated a decrease in quasispecies complexity at the HBV preCore/Core region after NUC treatment failure, likely due to selection of specific NUC-resistant variants. In contrast, diversity mainly increased during the natural evolution of the virus, probably because of immune system evolutive pressure. In this sense, it should be remembered that the main HBV epitopic regions are located in the preCore/Core region[99].
CLINICALLY SIGNIFICANT HBV GENOMIC VARIANTS: MASSIVE SEQUENCING FOR A DETAILED DEPICTION OF THE SITUATION
Several clinically and pathologically relevant variants have been characterized along the viral genome, despite the restriction resulting from the high degree of HBV genome overlapping. This restriction is especially important in the P and S gene overlapping regions, but is less significant in the regions corresponding to the X and preCore/Core genes. Therefore, the X and preCore/Core genes may be the most suitable for studying quasispecies variability, even though they contain the major enhancer of the viral genome (ENH II). The following sections will discuss the major variants of each of the HBV genome regions (P, S, X, preCore and Core) and their clinical and pathological implications, which are summarized in Table 1. Most of the reported data were obtained by direct sequencing or short clonal studies, which means that the variants were present in significant percentages. However, recent results obtained by massive sequencing are also included to provide a more detailed picture.
Table 1.
Main amino acid variants described in the hepatitis B virus genome and its effect
| ORF | Amino acid variant | Effect |
| P | A181T and M204 I/V (compensatory: L80I/V, T128N , R153Q, V173L L180M, A200V1, V207I1) | LMV resistance |
| N236T (compensatory: A181 T/V/S. Low sensitivity: V84M, S85A, L217R, I233V) | ADV resistance | |
| I169T, T184S, S202C/I/G, M250I | ETV resistance2 | |
| A194T1 | TDF resistance | |
| S | G145R W156Stop, W163Stop, W172Stop, W196Stop | Immune therapy failure |
| D144A, P142S, K141E, Q129H, I/T126N, T131I, M133L | HBsAg structural alterations | |
| X | I130M, V131I, F132Y | Contribution to HCC development |
| C | ||
| preCore | A1762T and G1764A | HBeAg negative forms |
| Core | G1896A and G1899A | Disease progression |
| P5H/L/T, D32N/H, C/E43K, cP50A/H/Y, E83D, I97F/L, L100I, A131G/N/P, S181H/P and C/Q182K/stop | HCC development | |
| Regions 50-69 and 74-84 | Immune scape variants |
Antiviral treatment resistances not completely defined[58];
Linked to lamivudine (LMV) signature (L180M + M204V). ORF: Open reading frames; ADV: Adefovir; ETV: Entecavir; TDF: Tenofovir; HBsAg: Hepatitis B virus (HBV) surface antigen; HBeAg: HBV e antigen; HCC: Hepatocellular carcinoma.
P gene variability and its implications in antiviral treatment
The polymerase gene (P ORF, nucleotides 2307-1623), the largest HBV gene, encodes the 90-kDa viral polymerase protein, a multifunctional enzyme involved in DNA synthesis and pgRNA retrotranscription (RT domain), with additional priming [TP domain and RNAse (RH) domain] functions. The main genomic variants of this region have been reported in the proper viral polymerase RT domain, which has both DNA polymerase and retrotranscriptase activities (retrotranscribes pgRNA to the minus DNA strand and synthesizes the incomplete plus strand). RT activity is located between the nonfunctional spacer region (Spc) and the RH domain. The TP domain, which acts as a primer for synthesis of the negative DNA strand is located at the N-terminal of the P ORF. The Spc region, located next to TP, is dispensable for enzyme function and therefore, easily tolerates mutations. The Spc region overlaps the preS region of the envelope gene and accumulates important mutations, such as long deletions[3,100,101]. The crystal structure of HBV polymerase has not yet been reported, but the 3D structure of HBV RT has been modeled, based on the crystal structure of HIV-1 RT[2,102]. The model shows the common right-handed configuration of both polymerases and identifies seven different domains (A-G). The catalytic center of RT activity (nucleotides 736-747) corresponds to the YMDD sequence, identical to what is observed in HIV-1[103]. The YMDD motif contains two of the three essential D (aspartate) residues of the polymerase. Attending to the specific nomenclature of the RT region, position 348 of the polymerase gene corresponds to the first amino acid of RT; hence, the catalytic motif is located at positions 203-206 (Y203M204D205D206)[32]. The RT region is highly conserved among retroviruses and hepadnaviruses[14]. In this putative structure, the YMDD main catalytic motif is identified in the C domain, located in the palm region of the right-handed RT structure[104,105].
As in HIV, therapy for HBV infection is currently based on the use of NUCs [lamivudine (LMV), adefovir (ADV), emtricitabin (EMT), telbivudin (LdT), entecavir (ETV), and tenofovir (TDF)], which are HBV RT inhibitors. Among them ETV and TDF are usually recommended as the first-line treatment option because of their high potency and low resistance rates. NUCs and host nucleotides, the natural polymerase substrates, bind at the YMDD motif. NUCs act as competitive inhibitors by blocking elongation of new HBV DNA strand. The central role of HBV polymerase in the viral replication cycle seems to justify the stability of its AA sequence relative to the remaining HBV protein products. However, non-synonymous nucleotide changes that result in amino acid substitutions have been reported in relation to resistance to NUC therapies. The presence of these drugs induces selection of HBV variants carrying amino acid substitutions in the RT domain. These mutations may cause structural changes in the polymerase, resulting in a decrease of drug affinity and antiviral activity. Thus, NUCs efficiently inhibit wild-type HBV variants present in the viral quasispecies, whereas variants carrying resistant mutations can maintain their replicative activity. Under NUC treatment, the percentage of resistant variants in the quasispecies may increase and ultimately be selected as the major variant, thereby causing treatment failure, manifested as viral breakthrough (VBK).
Because they can confer resistance to oral antiviral treatment, mutated strains are of great interest clinically. The intensity of viral resistance is related to the type of drug and the viral variant. The most commonly reported variants, rtM204V and rtM204I, are changes in the YMDD motif to YVDD or YIDD (located in RT domain C and analogous to the M184V/I LMV resistance mutation of the HIV-1 RT) (Table 1). Both these variants show low affinity for LMV, making them highly resistant to the drug and easily selected during treatment (70% of patients show resistance after 5 years of LMV therapy). These variants are also resistant to other nucleoside therapies such ETV and LdT, but with lower resistance rates than with LMV (e.g., 17% after 2 years of LdT treatment)[106].
The fitness of viruses with rtM204V and rtM204I variants is markedly reduced in comparison to wild-type[107], but they alone can replicate under LMV therapy[108,109]. Long-term LMV therapy increases the probability of new variants emerging, which will restore the replication capacity of the mutant and worsen the outcome of infection[110]. The most common of these additional, potentially compensatory mutations is rtL180M[107], which is often detected together with rtM204V and less often with rtM204I. Other mutations, such as rtV173L and rtL80I/V, do not alter the sensitivity of HBV to LMV, but instead, enhance its replication efficiency[111]. The rtL80I/V mutation (located in the RT A domain) is associated with severe disease in HBV genotype C patients[112]. rtV173L (combined with rtL180M and rtM204V) is the second most commonly detected compensatory mutant (19% of cases showing LMV resistance)[109]. Selection of compensatory mutants also occurs with the rtA200V/rtM204I combination[113] and with the variants rtT128N or rtR153Q in combination with the rtL180M/M204V polymerase mutations[114] (Table 1).
The compensatory effect of additional variants has been explained by molecular interactions between the various substitutions that provide HBV polymerase with a more efficient catalytic structure[102]. The rtA181T variant has been associated with LMV resistance in less than 1% of cases and has also shown resistance to ADV treatment[84]. The rare rtA181S mutation, which is similar to rtA181T, presents cross-resistance to LMV and ADV treatment in combination with M204I[115]. Emergence of resistance to ETV is uncommon in treatment-naïve patients (< 1% over 6 years), but in those with previous LMV failure, it increases dramatically to 40% after 4 years[106,116]. In fact, ETV resistance seems to be associated with the concomitant presence of LMV resistant variants and mutations in other RT codons (169, 184, 202 or 250, with at least four different substitutions in the same sequence), which confer decreased susceptibility to both LMV and ETV[117] in vitro, explaining the high genetic barrier of ETV.
The NUC ADV is active against HBV wild-type virus and variants resistant to LMV, EMT, and LdT. Emergence of ADV-resistant variants is less frequent than with LMV therapy (29% at 5 years)[118]. ADV resistance has been associated with the A181T/V and rtN236T variants, and less frequently, with rtI233V[119-121]. Interestingly, variant A181T has also shown LMV resistance according to in vivo and in vitro evidence; thus, rtA181T is cross resistant to LMV and ADV[84]. Other amino acid changes have been occasionally linked to resistance or low sensitivity to ADV, such as rtV84M and rtS85A in the A domain and rtL217R; a natural polymorphism in the D domain observed in subgenotype A2 HBV strains[2,120,122]. In addition to the main resistant variant, other minor variants are selected after LMV or ADV failure[123], and some of them seem to be associated with the viral genotype (e.g., rtS85F, rtL91I, and C2456G associated with LMV resistance in HBV genotype D, or rtI53V, rtW153R and rtF221Y associated with ADV resistance in HBV genotype A). Of note, it seems that the main LMV resistant variants, M204V and M204I, are also associated with viral genotype (rtM204I is not often detected in genotype A)[123].
TDF treatment, which is extensively used in HIV-1 infection, is also highly active against wild-type and LMV-resistant HBV polymerase variants[122,124,125]. TDF is associated with high sustained viral response (SVR) rates and a low rate of resistances: 0% after 6 years of therapy[106]. However, the rtA194T variant observed in some HIV/HBV coinfected patients has been related with TDF resistance, and shows a reduced in vitro replication rate in combination with the rtL180M and rtM204V LMV variants[110]. Use of LMV, ETV, LdT, and ADV has been largely replaced by the new potent NUCs, ETV and TDF[106]. However, extensive application of LMV and the use of ADV as rescue therapy for LMV failure over many years has resulted in a considerable percentage of chronic HBV patients in whom resistance to these NUCs has developed, and a quasispecies enriched in LMV-resistant variants can be expected. These variants can limit the response to new-generation NUCs. This is the case of the rtL180M + rtM204V combination (known as the LMV signature) in ETV therapy and rtN236T in TDF treatment.
For all these reasons, early detection of RT resistant variants by highly sensitive methods, even minor components of the HBV quasispecies in treatment-naïve patients, could be highly useful for therapy purposes. Detection of minor variants can easily be performed by conventional techniques (reverse hybridization or clonal sequencing) or by NGS methods[53-58]. An essential consideration to bear in mind is the S and P gene overlapping, which can lead to reciprocal consequences when there is a nucleotide substitution in either ORF. For example, the rtA181T and rtM204I polymerase variants also produce a stop codon in the S gene (sW172stop and sW196stop, respectively) (Figure 1). Therefore, antiviral treatment pressure may cause selection of viral genomes that are potentially defective for envelope proteins[58,84]. Another relevant variant in this regard is rtW153Q, which leads to the sG145R variant in the S ORF, associated with failure of immunotherapy.
Complex variants with two or more amino acid changes, such as those conferring ETV resistance or causing a fitness increase during LMV treatment, cannot be properly studied by indirect methods (LiPa, RFLP, 5’-nuclease assays, melting points, mass spectrometry, DNA chip technology, or real-time PCR), because they do not allow simultaneous detection of different substitutions in the same viral genome. Identification of these substitutions requires clonal techniques, such as classical clonal sequencing or the recent NGS methods. Specifically, UDPS enables simultaneous analysis of thousand of clonally amplified long fragments (700 nucleotides), and deeper and more sensitive detection of minor populations in complex mixtures; therefore, it may be the most suitable method for viral quasispecies studies.
As was mentioned above, the limit of detection of UDPS remains to be resolved. To date, different studies have achieved mixed results. Ijaz et al[126] reported a lower quantitation limit of 2% for minor HBV populations. More recently, Mello et al[50] reported values of 4%-17% for LMV-resistant variants[50]. Our group recently reported a cut-off value of 0.03% that enabled detection of extremely low percentages (0.04%-0.09%) of RT variants in treatment-naïve patients. In another recent UDPS study we detected RT variants in treatment-naïve samples at values of 0.1%-0.55%[56]. In the study by Nishijima et al[59], in which drug-resistant mutants were investigated in chronic-naïve cases by Illumina, frequencies of 0.3%-30% were reported. These three recent studies[56,58,59] as well as the previously reported ones[53,54] have all shown that resistant RT variants are present at baseline in treatment-naïve chronically HBV-infected individuals. This suggests that a reservoir of RT variants may exist, which would be prone to selection by the effect of antiviral therapies. Furthermore, LMV-resistant variants (LMV signature) linked to specific mutations responsible for ETV resistance have been simultaneously detected with 454 UDPS[58,127] (Figure 2). Moreover, Margeridon-Thermet et al[55] reported low-level persistence of LMV-resistant variants even 1 year after LMV treatment discontinuation using UDPS with a sensitivity level of 0.5%.
Figure 2.
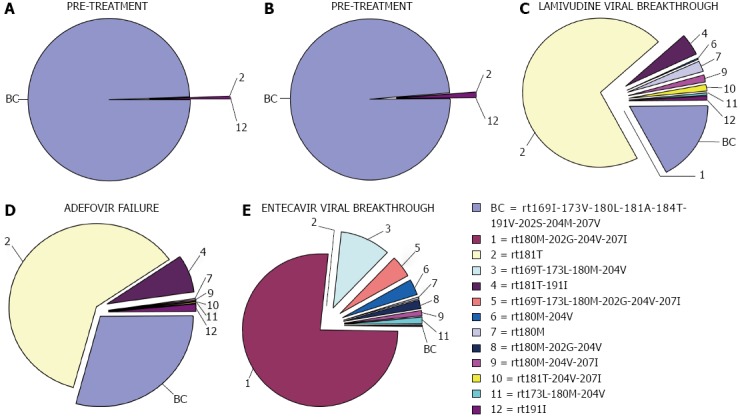
Changes in percentages of reverse transcriptase variants during follow-up of a patient included in one of our studies. Reproduced from Rodriguez-Frías et al[58]. BC: Baseline combination.
Nevertheless, the clinical significance of these minor drug-resistant mutations remains uncertain. Nishijima et al[59] concluded that pre-existing drug-resistant mutants (at naïve status), such as low-abundance mutant clones, may provide the opportunity to develop drug resistance against NUCs through selection of dominant mutations. However, a predictive cut-off value for baseline percentages to define preferential selection after NUC treatment has not been reported. Along this line, we found[56,58] that after VBK on LMV treatment, the variants selected were not the ones most frequently detected at baseline, suggesting that the low percentages observed at baseline (≤ 1%) do not determine the variant selected at VBK. Interestingly, we detected a small population of ETV-resistant variants[58] after LMV treatment in a patient who developed ETV resistance. See examples of UDPS haplotypic study in Figure 2. Additional sequential studies with a larger number of cases must be performed to define a cut-off value for the baseline percentage of resistant RT variants that can predict drug resistance. Hence, massive sequencing has opened the door to a more profound knowledge of the dynamic behavior of the HBV quasispecies that may clarify the role of minor variants in the HBV RT region on the outcome of infection[55,58].
S gene variability and its implications in immunoprophylaxis
As was discussed in the HBV virology section, the surface ORF (nucleotides 2848-835) is completely overlapped by the P gene (Figure 1). S ORF has three in-frame start codons encoding the three types of surface antigens present on the outer envelope: small (SHBS), middle (MHBs), and large (LHBs, 39 kDa). SHBS, which is common to all three, contains the main antigenic loop, also called the “a” determinant (codons 124-147 within the major hydrophilic region, which covers codons 100-170).
The antigenic loop activates the primary response of the neutralizing antibodies in HBV infection. In the so-called preS region, located in front of the SHBS start (positions 2950 and 3125 of the HBV genome), there is a highly variable sequence that is used to distinguish four major HBV serotypes (adr, adw, ayr, and ayw). HBsAg can self-assemble without containing the HBV genome, and adopt non-infective structural spheres or filamentous forms, which represent nearly the entire population of HBV-related particles (> 99.9%)[128]. The preS region overlaps the dispensable spacer domain of the P protein, which allows high heterogeneity. In contrast, the SHBS region (shared by all S-derived proteins) overlaps the essential RT domain of the P protein, which strongly restricts its variability. Deletions are the most relevant variants in the preS region[129]; being the main cause of the different genome lengths of the HBV genotypes and affecting the balance between the various types of HBsAg proteins[130]. Thus, it can be speculated that a partial or complete HBsAg defect may decrease virion assembly and secretion and lead to a parallel increase in the amount of cccDNA by enhancement of the capsid recycling pathway. An increase in preS deletions has been reported in HBeAg-positive chronic cases (35% in the sixth decade of infection), and has been related to a poor outcome (detected in 60% of HCC patients)[130,131]. The study of Kao et al[100] has reported a significantly higher rate of preS deletions in HCC patients than in chronic carriers, mainly clustered (> 70%) in the C-terminal preS1 and N-terminal pre-S2 sequences. Interestingly, all these deletions encompassed T and B cell epitopes, and functional mapping showed that they affect the viral secretion site.
In addition, preS deletions can induce oxidative DNA damage and genomic instability; upregulation of certain genes, such as cyclooxygenase-2 and cyclin A, induces cell cycle progression and hepatocyte proliferation - a phenomenon linked to a high risk of developing HCC[130]. It must be kept in mind that the S region encoding HBsAg is completely overlapped with the HBV RT region. A mutation in RT codon 153, selected as compensatory after lengthy LMV therapy, causes an sG145R change in the S region (Figure 1), which is strongly associated with immunotherapy failure[111] (Table 1). Other relevant examples are the rtA181T or rtM204I RT variants, which result in stop codons in envelope proteins (sW172stop and sW196stop, Figure 1). In the shared S/P region, the main variants have been described in the antigenic determinant “a” (located the major hydrophilic region, amino acids 100-170). These variants (mainly sG145R) were first reported in an Italian boy, son of an HBsAg/HBeAg-positive mother, who had HBsAg and anti-HBs in serum despite receiving both active (vaccine) and passive [hepatitis B immune globulin (HBIG)] immunization[132]. The variant was not detected in his mother[129,132], which suggests that sR145 presumably arose by immune selection pressure in the infant after HBV vaccination (vaccine escape variant), selected from a very small population in the mother’s HBV quasispecies. Most hepatitis B vaccines contain the major surface protein, SHBS, which induces an immune response against the ‘‘a” determinant and constitutes an evolutive factor for variant selection.
Other minor substitutions in the S gene include sG145R (again, a G to A mutation in nucleotide 587), followed by sD144A, sP142S, sK141E, sQ129H, sI/T126N/A, sT131I, and sM133L (Table 1), all of which strongly affect the HBsAg structure[114,133,134]. The sR145 main variant seems to alter the projecting loop (aa 139-147) of the “a” determinant, inhibiting recognition of induced neutralizing antibody[135]. This variant can horizontally infect[136] and replicate for several years[137], but at lower rate than the wild-type sG145 variant, probably because of a decrease in virion stability[138]. However, the presence of anti-HBs (hepatitis B immunoglobulin prophylaxis or vaccine-induced), which would block the sG145 strain, may allow selection of sR145 when it is present in the quasispecies as a minor variant. This mechanism would ultimately establish infection with the predominant presence of this variant in a clear manifestation of the adaptation capability of quasispecies structures. Such immune selection from the quasispecies would explain the strong association of this and other “a” determinant variants with HBV vaccination failure.
Variants observed in low percentages under immunoprophylaxis escape are detected in low prevalence in practically all the clinical stages of HBV infection[133,139]. Longitudinal studies have reported their accumulation during the course of chronic infection[140,141] as the major cause (70% of cases) of the paradoxical coexistence of HBsAg and anti-HBs[142]. Variant sG145R in conjunction with other S mutations located in the HLA I T cell epitope have been observed in fulminant HBV cases[143]. However, unlike sG145R, other variants appearing after vaccination often rapidly revert to the strain seen in the mother[144].
Vaccination at birth is an ideal situation for escape variant selection, similar to administration of high-titer anti-HBs preparations to prevent graft infection in liver transplantation[145-147]. A screening program for school-age children in Taiwan found a 0.7% prevalence of “a” determinant mutants[148]. Interestingly, the percentage of HBsAg mutants increased from 8% to 25% over 10 years after introduction of a universal vaccination program from 1984 to 1994, but remained stable (23%) in 1999. This study clearly suggests a role for HBV vaccine in selecting HBsAg mutations. In a study in the United States, only 0.8% of vaccinated children born to HBeAg-positive mothers were infected with sG145R[149]. The prevalence of this variant in North Americans and in Europeans seems to be low[150]. However, some recent studies have reported relevant new data about this type of variant. Perinatal transmission of HBV has not been fully controlled despite adequate immunoprophylaxis in infants in Thailand, with escape mutants in the “a” determinant region (residues 144 and 145) being observed in 14% of infected infants[151]. Shahmoradi et al[152] reported HBV-DNA activity in 28% of children born to HBsAg-positive mothers, and 62% of these cases carried envelope variants, mainly (77%) the sG145R variant. In fact, these variants seem to be present as minor populations in 9% of HBV carriers who have not been exposed to HBV vaccination or HBIG prophylaxis[153].
HBIG therapy is used to prevent recurrent HBV infection after liver transplantation (LT) for end-stage HBV liver disease. However, in some LT patients who become HBsAg- and HBV DNA-positive on HBIG therapy, emergence of mutations in putative neutralizing epitopes such as sG145R, similar to what occurs in vaccine failure, has been described[154-156]. A recent report found that 50% of reinfected LT recipients had mutations in the “a” determinant region and flanking sequences; a fact suggesting that quasispecies formation contributes to HBV reinfection following LT[156]. Therefore, HBIG-associated variants, like HBV vaccine-related variants, would arise from a pre-existing, but extremely minor population. Confirmation of this hypothesis would require application of ultrasensitive NGS massive sequencing methods, as has been used for other HBV regions[53-59].
The longer the duration of HBIG therapy, the greater likelihood that “a” determinant variants will arise. Discontinuation of HBIG often leads to reversion to the pretransplant sequence[140,155], thus providing further evidence of HBV quasispecies dynamics and reinforcing the idea that these variants are less replication-efficient than the wild-type strain. HBIG-treated liver transplant patients infected with “a” determinant escape variants in positions 144 or 145 showed a poorer clinical outcome than those infected with other variants or wild-type viruses[157]. In order to decrease selection of these variants, HBIG plus LMV combination therapy is used. This strategy has drastically reduced recurrence rates from 35% to less than 10%[158]. However, it should be remembered that there is a relationship between these variants and those in the overlapping polymerase region that arise during NUC therapy; especially the polymerase variant rtR153Q, which partially restores the replication efficiency of LMV-resistant variants such as rtM204V, and is associated with the main “a” determinant sG145R variant[114]. Therefore, patients with pre-LT polymerase variants may have a high risk of reinfection despite HBIG therapy[159], and patients with “a” determinant variants due to HBIG therapy may develop polymerase variants, associated with high viral replication and a poor outcome[160].
Currently, there are no available studies in which the S ORF is analyzed by NGS techniques. However, because of the complete overlapping between the S and P ORFs, some NGS studies mainly focussing on P ORF variability have reported interesting findings about the HBV quasispecies in S ORF[53,58]. Our group performed massive UDPS sequencing of samples from patients sequentially treated with LMV and ADV (Figure 2), and found a high frequency of rtA181T[58]; the substitution that causes the sW172stop stop codon in S ORF. The consequence of this change is that two-thirds of all HBV particles lacked 50 amino acids in HBsAg (in all types of envelope proteins) including several essential codons (hot spots sS174 and sL175, sV177, sQ181, sW191, sL192, and sI195 placed in the Core-Surface interphase and residues sV184, sL186, sS187, and sW190 from the S-S interaction interphase)[161]. Therefore, it is suggested that HBsAg carrying this stop codon may not be completely functional. Moreover, despite this drastic alteration, samples with a high prevalence of this defective variant showed considerable replication, which suggests that the quasispecies has some type of recourse to compensate for this theoretical handicap. That mechanism could be transcomplementation of the defective S protein genomes for enveloping with complete S proteins encoded by other quasispecies members. As has been suggested by Villet et al[84], existence of such a mechanism may be a requirement for emergence and maintenance of this incomplete variant. Hence, this may be an example of cooperation within the quasispecies[162], as a relevant property that fits in with the idea that this population structure has remarkable plasticity. In our UDPS study[58], other surface positions showed de novo stop codons at frequencies of 0.13% to 0.17% (sW156stop, sW163stop, sW165stop, and sW191stop); of note, sW156 is involved in HBV infectivity[163]. These de novo stop codons accounted for around 1.5% of the viral population in our study[58] and between 1% and 2.8% in the study of Solmone et al[53]; both using UDPS. Therefore, UDPS has brought to light what seems to be a systematic phenomenon in the HBV quasispecies: the presence of defective genomes that participate in HBV virology. The pathological consequence of this phenomenon must be defined in further studies.
X gene variability and its implications in HCC
The X gene (nucleotides 1374-1838) encodes the HBV X protein (HBx). HBxAg is a 154-amino-acid protein with an N-terminal negative regulatory domain and a C-terminal transactivation/coactivation domain that plays a key role in control of cell proliferation, viability, and transformation[164-166]. This protein has been detected in both the cytoplasm and nuclei of infected hepatocytes[167-169]. HBx is a regulatory protein that is not packaged in virions during assembly, but is expressed in the new host cell to allow epigenetic control of HBV transcription from cccDNA[7,170]. In contrast to the other HBV genes, but similar to retroviral oncogenes, Miller et al[14] reported that HBx shows codon usage preference (third nucleotide in degenerated codons), which is more related to the behavior of eukaryotic genes than viral genes and suggests that HBxAg is of eukaryotic origin. However, the X ORF lacks homology to host protein[167].
The high conservation of X gene in mammalian hepadnavirus genomes strongly suggests that HBx is essential to the viral life cycle. It has been reported that HBx is required to initiate and maintain HBV replication, making HBx the key regulator of the natural infection process[170]. It is believed that HBx contributes to HBV oncogenicity[96,167,171-173] and it is reported to transform SV40-immortalized murine hepatocytes, induce cell cycle progression within the regenerating liver, and cause or accelerate liver cancer in transgenic mouse models[174-177]. HBx expression affects several cellular functions in transfected cells, such as cytoplasmic calcium regulation, cell signaling, transcription, cell proliferation, DNA repair and apoptosis[169,177-180].
To develop its functions, HBx interacts with many cellular partners, such as nuclear proteins involved in regulation of transcription and transcription factors[167,179]. Furthermore, HBx interacts and cooperates with cAMP response element binding (CREB)-binding protein/p300 to modify chromatin dynamics of target genes and to synergistically enhance CREB activity[181].
HBx stimulates HBV replication 5-10-fold; likely by enhancing transcription of pgRNA[167] by activating the proteasome[182]. In this sense, HBx regions 61-69 and 105-140 seem to be essential for viral replication and expression. Paradoxically to its antiapoptotic capacity related to inhibition of tumor suppressor genes, the HBx 68-104 region is associated with mitochondrial membrane alterations that promote cell death[172]. Multiple evidence has related HBV infection with inhibition of the innate antiviral immune response, such as inhibition of the Toll-like receptor response[183]. HBx directly binds to interferon promoter stimulator-1 factor and inhibits activation of interferon β[184], thereby inhibiting the innate antiviral immune response; a pathway in common with other viruses, and even inhibiting signals through the mitochondrial proteins[185]. These and other data clearly indicate that HBx protein has a key role in HBV infection.
In liver tumor tissue, the X gene is often integrated into the genome of infected hepatocytes while retaining its functionality, especially antiapoptotic capability, mediated by inhibition of p53 transcriptional activation[96,186] or adenosylmethionine[187]. HBx stimulates methyltransferases leading to hypermethylation, which is associated with chromosomal instability[96]. HBx also activates cell proliferation by repression of tumor suppressor genes, such as melanoma inhibitory activity 2[188], or by increasing β-catenin expression[189]. HBx activity over nuclear factor κB has been associated with antitumor therapy failure[187,190-192]. HBx does not directly bind to DNA; it acts through the activation process of various transduction signal cascades in cis[172]. It seems that HBx binds to the cccDNA HBV minichromosome histone-like transcriptional complex, thus modifying epigenetic regulation of this essential structure[7].
The multiple functions of HBx may indicate that nucleotide substitutions in the X gene would have important consequences in HBV infection. Several specific substitutions have been reported in the region where X and ENH II overlap (T1485, C1479, A1613, T1653, T1689, A1753, T1766, A1768, and A1776)[82,193-195] and some of them seem to be related with the viral genotype[196]. These variants, which are found alone or in combination, can change the regulatory function of basal core promotor (BCP) motifs, thereby decreasing HBeAg expression and facilitating HBeAg seroconversion[197-199]. Furthermore, some of these variants, particularly the T1762/A1764 double mutation, are strongly associated with cirrhosis and HCC[200,201]. Other variants, such as those detected in preCore region, may also show this relationship and there may even be a link with HBeAg status[202,203]. In one study, 40% of HCC patients presented six or more of these mutations, while they were present in only 2.7% of non-HCC patients, indicating a predictive value of these variants for HCC similar to that of α-fetoprotein[201]. In another, more recent study performed in India, the A1753 mutation was detected in 35% of cases of HBV-related liver cirrhosis[204].
Multiple X gene variants directly affect the HBx amino acid sequence; these include xI130M, xV131I, and xF132Y, which are associated with the main BCP variants (Table 1). Mutations in amino acids 5, 130, and 131 may contribute to HCC development and could be useful to predict clinical outcome in patients with chronic HBV infection[205]. Some HBx amino acid variants are typically located in the region between TATA boxes TA2 and TA3 of the BCP sequence (overlapping with the X gene). Our group observed that the most common variants are 8-nucleotide deletions-some of these (unpublished) deletions in the quasispecies, obtained by UDPS are shown in Figure 3 - which result in frame shifting and often create a de novo stop codon at position 134, resulting in a 20-amino-acid truncation of the C terminus of the HBx protein[56]. This could cause a huge change in the proapoptotic functionality of this region[96] or in its transactivator role[206]. X gene deletions are commonly found in DNA integrated in liver tumor tissue[192] and are strongly associated with the development of HCC by multiple mechanisms, such as deregulation of the centromeric protein A[93], regulation of miRNA[94], and loss of the proapoptotic effect[96].
Figure 3.

Quasispecies of the X gene obtained by unpublished result. The four TA-like boxes are highlighted in red and the eight nucleotides detected are indicated in green.
It is likely that the highly complex scenario of the quasispecies in this extremely relevant region of the HBV genome can be clarified by applying massive sequencing. However, no UDPS studies in this line have been reported to date, probably because of the technical difficulties associated with the presence of deletions. In a recent UDPS study by our group[56], mainly focused on the preCore region but including the last eight HBX codons (positions 1814-1838), we found that around 3% of sequences carried the TAG amber stop codon instead of the major TAA codon. Even more interesting was the significant percentage of sequences (0.2%) in which a T1836C substitution changed the ochre stop codon TAA to CAA (Q). After this substitution, the HBx protein would be translated to the next in-frame stop codon TAG at position 1992, inducing expression of 51 additional amino acids at the C terminus, which could potentially alter HBx functionality. Thus, this preliminary UDPS study revealed a minor HBV population with highly relevant HBx changes (AA deletions or insertions) whose significance in the HBV quasispecies and in the outcome of infection requires a more systematic and extensive UDPS study of this region.
Variability in the preCore/Core regions
The Core gene (positions 1814-2548) has two in-frame start codons encoding two proteins: the component of the viral capsid or Core (HBc, start codon at 1901), and the preCore protein, the precursor of HBeAg (start codon at position 1814). HBeAg protein is secreted by infected hepatocytes and seems to have an immunomodulatory function, establishing immune tolerance and predisposing infants of HBV-infected mothers to develop persistent infection. HBeAg promotes a Th2 response that leads to suppression of the host immune response against the virus, preventing clearance and allowing viral persistence[207]. HBeAg-mediated IL-1 activation may also increase hepatocyte proliferation, inducing anti-apoptotic genes and promoting hepatocarcinogenesis[207].
In clinical practice, HBeAg expression differentiates between chronic HBV carriers as HBeAg-positive and HBeAg-negative; the latter are usually positive for the corresponding antibody (anti-HBe). The anti-HBe seroconversion event is a crucial turning point in the natural history of chronic hepatitis B. Anti-HBe seroconversion is considered a favorable prognostic factor in the disease, and sustained virological response in HBeAg-positive cases is a treatment endpoint[106]. The factors involved in HBeAg seroconversion have been extensively studied. However, seroconversion is not always followed by establishment of HBV inactive carrier status. Some patients show elevated transaminases and active viral replication after seroconversion to anti-HBe, indicating that the disease continues to be active in the absence of HBeAg[208]. In addition, significant differences in treatment response have been seen between HBeAg-positive and -negative status. In particular, HBeAg-negative patients present response rates 10%-20% higher than HBeAg-positive ones; even with the last generation antivirals ETV and TDF[106]. This phenomenon may be associated with different levels of viral replication between patients positive and negative to HBeAg[106]. However, other mechanisms associated with viral characteristics may also be implicated in response rates, such as baseline quasispecies composition[72,98] and differing sensitivities to antivirals of certain HBV variants[57,59]. In fact, as described below, certain preCore and BCP variants that prevent or significantly decrease HBeAg expression (HBeAg-negative variants) may promote the process of HBeAg seroconversion. In many HBV cases from the Mediterranean area, the disease remains active after seroconversion, and HBeAg-negative preCore or BCP variants have been detected in these patients[209,210]. Once these variants become detectable, they perpetuate the infection beyond seroconversion[211]. Nonetheless, recent evidence seems to suggest that some HBeAg-negative variants may be more senstitive to antivirals[56,57,59].
Variants in the preCore region (between start codons 1814 and 1901) are clinically important because they abolish HBeAg expression. The most common is G1896A (interestingly another G to A substitution), which leads to premature termination of the preCore protein (the HBeAg precursor) and creates a de novo in-frame stop codon at codon 28 (TGG to TAG) in the preCore region (pcW28stop, Table 1). This change is located in a region of four G residues that are prone to G to A mutations[129]. G1896A abrogates HBeAg synthesis, does not disturb HBcAg production[212], and has been detected in virtually all clinical stages of HBV infection, from extremely severe to the most benign forms[210-214]. Although G1896A-containing strains are independently transmissible[214], HBeAg-negative HBV carriers do not develop chronic infection, but acute or fulminant hepatitis can occur[204,215,216]. This mutation shows a strong association with viral genotype, and is commonly found in Mediterranean and oriental anti-HBe-positive chronic HBV patients who show a high prevalence of genotypes D and B, respectively.
In contrast, the mutation is notably less prevalent in chronic HBV patients from Western Europe or North America, where genotype A is more prevalent[217]. This genotype association is clearly related to the overlapping of the preCore and the encapsidation signal sequences of ε. In this essential pgRNA secondary structure, positions 1896 and 1858 are paired in the lower stem of the ε stem loop secondary structure[218,219] (Figure 4). The G1896A substitution results in stabilization of the ε structure in genotypes B, D, E, G and H, and in C strains in which there is a T in the paired position, 1858. In contrast, in genotype A and F strains, there is a C in position 1858, and the G1896A substitution would result in a loss of thermodynamic stability of ε, producing a decrease in encapsidation and viral replication[209,220,221]. This observation explains the frequent detection of G1896A in genotype D strains, but rarely in genotype A strains[222]. The G1899A substitution (again, G to A) produces a pcG29D change, is frequently associated with liver cirrhosis, and is usually detected together with G1896A[222].
Figure 4.
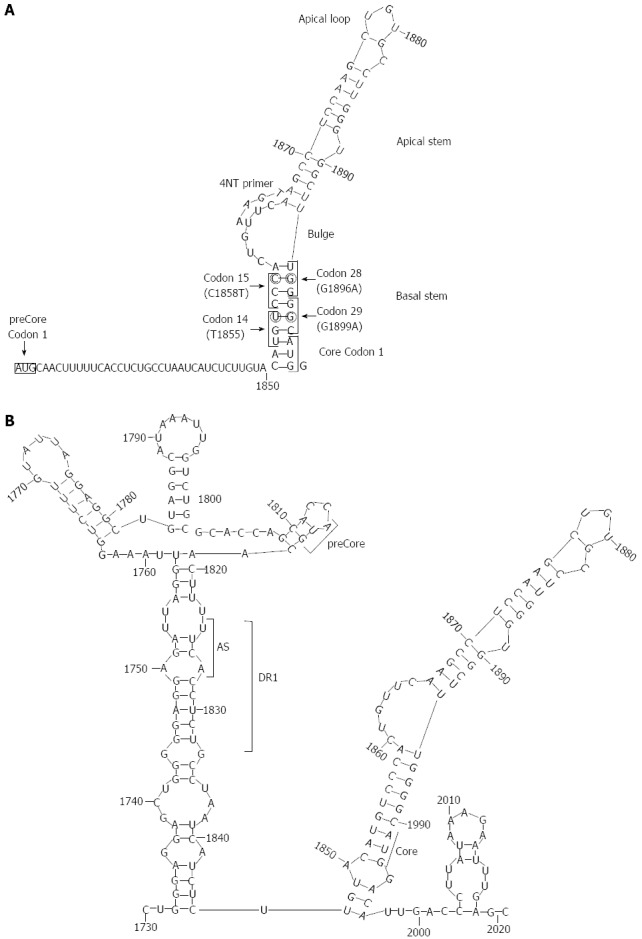
Secondary structures adopted at the 5’ and 3’ ends of hepatitis B virus pgRNA reported by our group. A: Stem-loop structure of the 5’ with main structural motifs (loops and stems), main codons (1, 14, 15, 28, and 29 of preCore, and 1 of Core gene), and the location of 4-nt primer annealing in the 5’; B: Stem-loop structure of the 3’. The acceptor site (AS), direct repeat region 1 (DR1), and preCore and Core start codons are indicated in the figure. Reproduced from Homs et al[56].
A lack of HBeAg expression can also be the result of mutations in any of the three nucleotides of the preCore start codon. Although they are less common than G1896A, these mutations represent a large percentage of preCore defective variants[210]. Mutations in the preCore start codon do not disturb the encapsidation process, because they are located outside of the canonical ε signal sequence[219] and are not restricted by genotype. preCore frameshift mutations due to insertions or deletions are uncommon events occurring outside essential regions, such as ε or DR1 (e.g., deletion in nt 1836-9)[129]. In contrast, the rare G1862T variant (pcV17F) affects the ε structure bulge and disrupts viral replication, but not HBeAg expression.
The G1862T variant is found in the African A1 subgenotype, is associated with lower viremia than European A1[223], and is more common in HBeAg-positive than -negative patients (37% vs 11%)[224]. In addition to the G1896A change, three other preCore stop codons have been observed (positions 1817, 1874 and 1897), all of which disrupt the secondary pgRNA structure (as deduced by the secondary structure predicted by magnetic resonance study)[219]. The 1817, 1874 and 1897 changes need compensatory changes to stabilize the pgRNA structure, a fact that seems justify their low-frequency detection.
In HBeAg-positive patients (immunotolerance phase), defective preCore variants are rarely found as minority populations by conventional direct Sanger sequencing or even by clonal analysis. However, these variants emerge following activation of hepatitis, often associated with anti-HBe seroconversion. In fact, the G1896A preCore mutation and the double A1762T/G1764A BCP mutation are associated with HBeAg seroconversion[197,225]. These variants accumulate along seroconversion and are the predominant viral population in anti-HBe-positive patients[226]. Whether HBeAg-defective preCore variants are selected from the viral quasispecies in persistent infection is still uncertain. However, the host immune system seems to be the main cause of emergence of preCore variants.
Hypermutations in the B and T epitopes of the HBcAg sequence have been observed in the presence of preCore mutations (HBeAg-negative)[209], allowing escape of these variants. Some preCore variants, such as G1899A, have been clearly associated with HCC progression in HBeAg-positive patients, while G1896A plays a similar role in HBeAg-negative cases[202]. Therefore, early detection of preCore variants seems to have predictive value for spontaneous or interferon-induced seroconversion[220,227].
Our group investigated the variability of the preCore region and its relationship with ε structure functionality using UDPS at a sensitivity of 0.03%[56]. Minor populations of G1896A, G1899A variants, and preCore start codon variants were observed, but these mutations showed thermodynamic and structural restrictions in the encapsidation signal. Minor percentages of preCore variants were also seen in HBeAg-positive patients, but not in HBeAg-negative cases[56]. The study included a small number of patients, however, and did not explain the presence of a minor population of preCore variants in the HBV quasispecies. Hence, NGS studies including a large number of patients are needed to elucidate the significance of these findings.
The C gene also encodes HBcAg, which forms the HBV nucleocapsid. This protein is highly immunogenic, whereas the envelope proteins have low immunogenicity. Thus, HBV-infected individuals develop an early, intense anti-HBc humoral response that remains after clinical recovery. Multiple epitopic regions in nearly the entire HBc sequence have been described in relation with B cell and T helper epitope response[228,229]. Among them, the region flanked by codons 74 to 84 (B74-84) located at the tip of the spike structures of the capsid and included in the so-called major immunodominant region should be highlighted. This epitope is shared by HBc and HBeAg, hence it constitutes the major epitope for B cells[13,230]. Other regions stimulate CD4+ Th cells, the best known being the immunodominant epitope flanked by amino acids 50 and 69 (TH50-69). Variability in these two regions has been related with immune response escape, disease persistence, and interferon therapy failure[57,231]. Furthermore, amino acid variants in the C gene, specifically cE83D and cP50A/H/Y (Table 1), are associated with HCC[232].
In the report by Kim et al[232], mutations in the C region were not randomly distributed, but were mainly clustered in the major histocompatibility complex class II restricted region and significantly related to HCC. These included the main preCore variants G1896A (pcW28stop) and G1899A (pcG29D), and certain C variants (Table 1), such as cP5H/L/T, cD32N/H, cC/E43K, cP50A/H/Y, cE83D, cI97F/L, cL100I, cA131G/N/P, cS181H/P and cC/Q182K/stop. In a recent study performed in India, single nucleotide substitutions such as C1914G were found in 32% of HCC cases[204].
In addition to amino acid substitutions, deletions leading to truncated HBc proteins have been described, such as the single G deletion in position 2090 in a group of six Gs[69,70]. It has been reported that these C-defective genomes are unable to replicate, but they are always present in the HBV quasispecies, regardless of the region studied (S or C)[69,70]. Hence, their presence might indicate that there are active transcomplementation mechanisms in HBV infection that tolerate them. The natural tendency to maintain these defective particles may be because they confer a benefit for viral evolution[69,70], although they seem to be poorly represented in the baseline HBV quasispecies, making them difficult to detect by conventional sequencing technologies, even clonal analysis. In contrast to what was previously thought, UDPS study has brought to light the fact that these defective variants can replicate and form minor populations in the HBV quasispecies[56,58]. Study of these variants by massive sequencing may help to establish whether their presence is only occasional and a mere side effect of the quasispecies structure, or whether they are more prevalent and have functions affecting the outcome of infection.
Significant changes in the immunodominant epitopes of the viral capsid have been observed by direct Sanger sequencing, mainly under the effect of antiviral treatment with the immunomodulator interferon and even with NUCs[231]. Similar results were obtained in a preliminary study using massive sequencing[57]. However, it should be noted that HBeAg shares many epitopes of the HBc protein, and an association between the presence of preCore HBeAg-negative variants and mutations in Core epitopes has been reported[209]. This relationship shows that in the absence of HBeAg, immune pressure stimulates selection of some Core variants and provides further evidence that the immune system is an evolutionary factor acting on the HBV quasispecies[57,209].
The baseline presence of HBV RT variants resistant to antiviral therapy has been reported in multiple studies by conventional direct sequencing and cloning[233], and recently by NGS methods[53-56,58,234]. However, study of the preCore region, which could help to understand the phenomenon of anti-HBe seroconversion and its association with SVR rates, has only been reported by our group[56,57]. Highly sensitive methods are required to detect minor variants in certain environmental situations, and evaluate the evolution of these variants within the quasispecies. We found strong selection of a minor member of the quasispecies (around 1%) in the absence of antiviral therapy in an HBeAg-negative case; possibly as a result of immune pressure. In this case, variants carrying amino acid substitutions were found in a motif commonly recognized by T helper cells (D64VTN67 instead of E64LMT67, located in the main TH 50-69 epitope). To date, only one preliminary study has analyzed epitope variability in the C region of the viral capsid by massive sequencing[99]. The results seem to confirm the dynamics of variant selection in the major epitopes of the C region in the absence of treatment, as well as some degree of simplification or decreased quasispecies diversity after antiviral treatment, as evaluated by Shannon entropy[70]. Study of the variability of immunodominant epitopes might permit a redefinition of these epitopic motifs (e.g., the B74-84 epitope could include positions 71, 72 and 87 or putative new immune-stimulating positions, such as 40, 41, 92 and 93).
The BCP region in preCore/Core controls expression of the preCore mRNA (precursor of HBeAg) and pregenomic RNA. BCP is located between positions 1742 and 1842 and overlaps with the 3’ end of the X gene and the 5’ end of precore/Core. BCP contains 4 TATA boxes (TA1-TA4) and includes a fragment of the 3’ terminal region of ENHII, which is involved in controlling S and X gene expression[210]. Despite its essential nature, clinically relevant BCP variants have been detected. The most common of these changes is the double mutation A1762T/G1764A associated with significant decreases in HBeAg levels and in viral activity[64,235-241], and with severe liver disease[199-201,205].
The liver-specific activity of ENHII is regulated by multiple liver-enriched transcription factors, specifically nuclear receptor hepatocyte nuclear factor (HNF) 4 or 1, which span the TATA box-like sequence of the pre-C promoter. A functional synergism between some nuclear receptors (including HNF4) upregulates the liver-specific activity of ENHII[242]. The effect of BCP variants at ENHII is associated with modification of specific sites recognized by hepatic factors. For example, the HNF1-binding site created by the A1762T/G1764A double mutation is imperfect for a response to HNF1, which results in suppression of preCore mRNA synthesis and increases in pregenomic RNA. Both these actions seem to explain the decrease in HBeAg (promoting seroconversion) and the increase in viral activity in the presence of the A1762T/G1764A mutation. In addition, this double mutation is associated with HBx substitutions in amino acids 130 and 131, which may contribute to HCC development[205]. Other minor mutations in the BCP region, such as C1740, C1753, and T/A1768, have been associated with severe liver damage[200,210,243-245] and even fulminant hepatitis[85,246]. The presence of BCP mutations in inactive carriers is significantly lower than in patients with active chronic infection and those with persistently elevated ALT levels[210]. Both BCP and preCore mutations (e.g., A1762T/G1764A and G1896A stop codon) are present in most cases of HBV reactivation in immunosuppressed anti-HBc-positive individuals[247,248]. Early, sensitive detection of BCP and/or preCore mutant populations in the HBV quasispecies could reveal potential predictors of seroconversion to anti-HBe or of evolution of the disease[82,190,211,232]. Fortunately, quantification of HBV variants is currently possible by massive sequencing techniques[70].
Due to the overlapping of BCP and X, BCP changes can also cause changes in the 3’ region of the X gene[205]. Some such changes have been reported in the region immediately preceding the precore start codon (positions -5, -3 and -2 from 1814, the preCore start). This region corresponds to Kozak sequences, and associated nucleotide changes have been found with effects similar to those related to BCP mutations[249]. BCP variants between positions 1763 and 1779, which cause X gene deletions and decrease HBeAg expression have been associated with reactivation after chemotherapy and with disease severity[64,65]. However the effect of these variants on viral replication varies depending on the specific deletion (e.g., deletion affecting TA3 1768-1775 causes a decrease in pregenomic and preCore RNA with a concomitant decrease in viral activity)[66-68]. Many BCP deletions produce a frame shift that results in a 20-amino-acid truncation of the HBx protein (amino acid 154 passes to 134) and partial loss of the C-terminal region, which contains the HBx transactivating activity. All these mutations have enormous clinical-pathological interest and have been related to interferon failure[250].
TREATMENT RESPONSE: VIRAL VARIANTS AND GENOTYPE
BCP and precore variants
As was discussed above, HBeAg-negative variants result from changes in the preCore and/or BCP regions, and are observed in all clinical forms of HBV infection (from asymptomatic to severe) and in all the viral genotypes. However, their distribution and prevalence show important differences[210,246]. PreCore mutations are more common in genotype D[209,251] and BCP mutations are more frequent in genotype A[210]. In Asia, where HBV infection is widespread, genotype C is associated with a higher prevalence of BCP mutations, whereas genotype B shows more preCore mutations[252,253]. The poorer prognosis of genotype C is probably associated with the prevalence of BCP mutations[68,240,254]. During seroconversion in genotype C, BCP variants are selected before preCore variants, whereas in genotype B the opposite occurs[253]. BCP and preCore mutations are more prevalent in severe forms of infection[68,240,240,255], in which BCP deletions are particularly important[254]. Although the impact of HBV genomic features during seroconversion is unknown, the viral quasispecies structure and its qualitative and quantitative composition is likely associated with this essential phenomenon in natural infection[28] and in the response to antiviral therapy[98]. In this sense, it has been reported that the evolutionary pattern of the viral reverse transcriptase region differs between responders and partial responders during the initial phase of ETV treatment[98].
Treatment of chronic HBV infection and its relationship with variability: is the RT region the only one involved?
Chronic HBV infection is now primarily treated with NUCs, and these treatments are associated with a risk of selecting resistant RT variants that cause treatment failure. Currently, ETV or TDF are universally recommended as first-line therapy[106]. Both treatments present a small risk of resistance development (< 1% for ETV and 0% for TDF after 6 years of treatment). In contrast, LMV has been associated with a high risk of emerging resistance (> 80% at 5 years of treatment). LMV has been widely used, and a large number of LMV-treated patients have developed treatment-resistant variants. The percentage of RT variants increases after LMV relative to their baseline proportion. We looked into this situation by UDPS study and found the following: the LMV resistant variant L180M + M204V was undetectable in pre-LMV samples, was selected during LMV therapy, remained in significant percentages after ADV treatment, and was re-selected after ETV treatment[58]. A recent UDPS study by Margeridon-Thermet et al[55] also reported a high percentage of resistant RT variants after LMV treatment.
In general, HBeAg-negative patients (with preCore variants) showing viral activity after seroconversion present higher rates of SVR to NUCs than HBeAg-positive patients[106]. However, the response rates to interferon treatment differ: HBeAg-negative patients present lower response rates than HBeAg-positive ones[256,257]. These differences seem to be related to viral genotype, because significantly higher SVR rates have been found for genotype A, in which preCore variants are strongly restricted[258], than for the remaining genotypes, especially genotype D. The reason for this genotype-dependent response is not clear, but the diversity of the viral quasispecies may play a role, as has been described for HBeAg seroconversion and treatment response[98].
In chronic LMV-treated HBV patients, major defective precore mutations occasionally revert to wild-type forms, with reappearance of HBeAg[238,259]. This seems to suggest that HBeAg-negative variants are more sensitive to antiviral treatment than HBeAg-positive ones[238,260]. In vitro studies indicate that defective preCore mutations restore the replication efficacy of LMV-resistant mutants[261]. In one UDPS study, we found that the percentage of HBeAg-negative G1896A preCore variants in the quasispecies of HBeAg-positive cases (present as minor populations and undetectable by conventional sequencing) decreased after a period of treatment with LMV[56,57]. This was also seen in BCP variants associated with low HBeAg expression in a preliminary study[99]. These data suggest that these variants are more sensitive to LMV treatment than HBeAg-positive variants, and could explain the seroreversion phenomenon. In a recent study using quantitative real-time PCR, Nishijima et al[59] reported that liver tissue of NUC-treated HBeAg-negative cases showed low levels of the G1896A preCore mutant (0.0%-1.1%). The authors also reported a significant reduction in the percentage of this mutant following treatment in 13 of 14 cases (92.9%). These results reinforce the hypothesis that the G1896A preCore mutant is highly sensitive to NUCs. A decline in the percentage of HBeAg-negative variants (preCore and/or BCP mutations) and selection of HBeAg-positive variants (without preCore or BCP mutations) after antiviral treatment[56,57,99] seems to agree with the lower SVR rates in the presence of HBeAg than in the absence of this antigen[106].
Interestingly, preCore mutations reappear after prolonged therapy, with return to HBeAg-negative status (seroconversion)[238]. It has also been reported that HBeAg-negative preCore and BCP variants compensate for the decreased replicative capacity of resistant RT variants[261]. This observation, and the reappearance of HBeAg-negative variants after therapy continuation seem to be contradictory and suggest that additional factors such quasispecies complexity or adaptation to the host immune system would be involved. The application of NGS to analyze quasispecies dynamics in cases with seroreversion after anti-HBe loss might elucidate this phenomenon.
CONCLUSION
In conclusion, HBV infection remains a major health problem worldwide, despite the continuing advances in treatment and development of effective vaccines. The high incidence of HBV infection is mainly due to the virological characteristics of this pathogen. HBV shows high variability that characterizes it as a quasispecies in a manner similar to other infectious agents such as HCV and HIV, but with a much higher prevalence than these. Detailed study of the viral population structure is essential to combat these infections successfully, and this type of study is now possible with NGS methods. These techniques have contributed important data over recent years, and they are expected to undergo spectacular development in the medium term. NGS promises to be a powerful resource that will help the scientific community in the management of HBV and other infections, as well as other key problems related to human health, such as genetic and oncologic disease.
ACKNOWLEDGMENTS
The authors thank Celine Cavallo for English language support and helpful editing suggestions. This study was financed by a grant (PI 12/1893) of the Instituto Carlos III (Spanish Government).
Footnotes
Supported by Instituto de Salud Carlos III, grant PI12/1893, co-financed by the European Regional Development Fund (ERDF) and Instituto Carlos III of the Spanish Ministry of Health and Consumer Affairs
P- Reviewers: Scherer A, Shimizu Y S- Editor: Zhai HH L- Editor: Kerr C E- Editor: Wang CH
References
- 1.Lupberger J, Hildt E. Hepatitis B virus-induced oncogenesis. World J Gastroenterol. 2007;13:74–81. doi: 10.3748/wjg.v13.i1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez-Frias F, Jardi R, Schaper M, Buti M, Ferrer-Costa C, Tabernero D, Homs M, Esteban R. Adefovir for chronic hepatitis B treatment: identification of virological markers linked to therapy response. Antivir Ther. 2008;13:991–999. [PubMed] [Google Scholar]
- 3.Kay A, Zoulim F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007;127:164–176. doi: 10.1016/j.virusres.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 4.Tong W, He J, Sun L, He S, Qi Q. Hepatitis B virus with a proposed genotype I was found in Sichuan Province, China. J Med Virol. 2012;84:866–870. doi: 10.1002/jmv.23279. [DOI] [PubMed] [Google Scholar]
- 5.Schieck A, Schulze A, Gähler C, Müller T, Haberkorn U, Alexandrov A, Urban S, Mier W. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology. 2013;58:43–53. doi: 10.1002/hep.26211. [DOI] [PubMed] [Google Scholar]
- 6.Penna A, Del Prete G, Cavalli A, Bertoletti A, D’Elios MM, Sorrentino R, D’Amato M, Boni C, Pilli M, Fiaccadori F, et al. Predominant T-helper 1 cytokine profile of hepatitis B virus nucleocapsid-specific T cells in acute self-limited hepatitis B. Hepatology. 1997;25:1022–1027. doi: 10.1002/hep.510250438. [DOI] [PubMed] [Google Scholar]
- 7.Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA. 2009;106:19975–19979. doi: 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang GH, Seeger C. Novel mechanism for reverse transcription in hepatitis B viruses. J Virol. 1993;67:6507–6512. doi: 10.1128/jvi.67.11.6507-6512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murray JM, Purcell RH, Wieland SF. The half-life of hepatitis B virions. Hepatology. 2006;44:1117–1121. doi: 10.1002/hep.21364. [DOI] [PubMed] [Google Scholar]
- 10.Hollinger FB. Hepatitis B virus genetic diversity and its impact on diagnostic assays. J Viral Hepat. 2007;14 Suppl 1:11–15. doi: 10.1111/j.1365-2893.2007.00910.x. [DOI] [PubMed] [Google Scholar]
- 11.Kidd-Ljunggren K, Miyakawa Y, Kidd AH. Genetic variability in hepatitis B viruses. J Gen Virol. 2002;83:1267–1280. doi: 10.1099/0022-1317-83-6-1267. [DOI] [PubMed] [Google Scholar]
- 12.Torre F, Cramp M, Owsianka A, Dornan E, Marsden H, Carman W, Williams R, Naoumov NV. Direct evidence that naturally occurring mutations within hepatitis B core epitope alter CD4+ T-cell reactivity. J Med Virol. 2004;72:370–376. doi: 10.1002/jmv.20016. [DOI] [PubMed] [Google Scholar]
- 13.Belnap DM, Watts NR, Conway JF, Cheng N, Stahl SJ, Wingfield PT, Steven AC. Diversity of core antigen epitopes of hepatitis B virus. Proc Natl Acad Sci USA. 2003;100:10884–10889. doi: 10.1073/pnas.1834404100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller RH, Robinson WS. Common evolutionary origin of hepatitis B virus and retroviruses. Proc Natl Acad Sci USA. 1986;83:2531–2535. doi: 10.1073/pnas.83.8.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Klundert MA, Cremer J, Kootstra NA, Boot HJ, Zaaijer HL. Comparison of the hepatitis B virus core, surface and polymerase gene substitution rates in chronically infected patients. J Viral Hepat. 2012;19:e34–e40. doi: 10.1111/j.1365-2893.2011.01506.x. [DOI] [PubMed] [Google Scholar]
- 16.Omata M, Ehata T, Yokosuka O, Hosoda K, Ohto M. Mutations in the precore region of hepatitis B virus DNA in patients with fulminant and severe hepatitis. N Engl J Med. 1991;324:1699–1704. doi: 10.1056/NEJM199106133242404. [DOI] [PubMed] [Google Scholar]
- 17.Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012;22:593–601. doi: 10.1101/gr.133926.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding D, Lou X, Hua D, Yu W, Li L, Wang J, Gao F, Zhao N, Ren G, Li L, et al. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing-based approach. PLoS Genet. 2012;8:e1003065. doi: 10.1371/journal.pgen.1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007;252:157–170. doi: 10.1016/j.canlet.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Murakami Y, Saigo K, Takashima H, Minami M, Okanoue T, Bréchot C, Paterlini-Bréchot P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut. 2005;54:1162–1168. doi: 10.1136/gut.2004.054452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horikawa I, Barrett JC. cis-Activation of the human telomerase gene (hTERT) by the hepatitis B virus genome. J Natl Cancer Inst. 2001;93:1171–1173. doi: 10.1093/jnci/93.15.1171. [DOI] [PubMed] [Google Scholar]
- 23.Ferber MJ, Montoya DP, Yu C, Aderca I, McGee A, Thorland EC, Nagorney DM, Gostout BS, Burgart LJ, Boix L, et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22:3813–3820. doi: 10.1038/sj.onc.1206528. [DOI] [PubMed] [Google Scholar]
- 24.Tokino T, Matsubara K. Chromosomal sites for hepatitis B virus integration in human hepatocellular carcinoma. J Virol. 1991;65:6761–6764. doi: 10.1128/jvi.65.12.6761-6764.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchio A, Pineau P, Meddeb M, Terris B, Tiollais P, Bernheim A, Dejean A. Distinct chromosomal abnormality pattern in primary liver cancer of non-B, non-C patients. Oncogene. 2000;19:3733–3738. doi: 10.1038/sj.onc.1203713. [DOI] [PubMed] [Google Scholar]
- 26.Garcia M, de Thé H, Tiollais P, Samarut J, Dejean A. A hepatitis B virus pre-S-retinoic acid receptor beta chimera transforms erythrocytic progenitor cells in vitro. Proc Natl Acad Sci USA. 1993;90:89–93. doi: 10.1073/pnas.90.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, Trepo C, Marcellin P, Goodman Z, Delaney WE, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 28.Lim SG, Cheng Y, Guindon S, Seet BL, Lee LY, Hu P, Wasser S, Peter FJ, Tan T, Goode M, et al. Viral quasi-species evolution during hepatitis Be antigen seroconversion. Gastroenterology. 2007;133:951–958. doi: 10.1053/j.gastro.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 30.Smith LM, Fung S, Hunkapiller MW, Hunkapiller TJ, Hood LE. The synthesis of oligonucleotides containing an aliphatic amino group at the 5’ terminus: synthesis of fluorescent DNA primers for use in DNA sequence analysis. Nucleic Acids Res. 1985;13:2399–2412. doi: 10.1093/nar/13.7.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith LM, Sanders JZ, Kaiser RJ, Hughes P, Dodd C, Connell CR, Heiner C, Kent SB, Hood LE. Fluorescence detection in automated DNA sequence analysis. Nature. 1986;321:674–679. doi: 10.1038/321674a0. [DOI] [PubMed] [Google Scholar]
- 32.Stuyver L, De Gendt S, Van Geyt C, Zoulim F, Fried M, Schinazi RF, Rossau R. A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J Gen Virol. 2000;81:67–74. doi: 10.1099/0022-1317-81-1-67. [DOI] [PubMed] [Google Scholar]
- 33.Cheng Y, Guindon S, Rodrigo A, Wee LY, Inoue M, Thompson AJ, Locarnini S, Lim SG. Cumulative viral evolutionary changes in chronic hepatitis B virus infection precedes hepatitis B e antigen seroconversion. Gut. 2013;62:1347–1355. doi: 10.1136/gutjnl-2012-302408. [DOI] [PubMed] [Google Scholar]
- 34.Allen MI, Gauthier J, DesLauriers M, Bourne EJ, Carrick KM, Baldanti F, Ross LL, Lutz MW, Condreay LD. Two sensitive PCR-based methods for detection of hepatitis B virus variants associated with reduced susceptibility to lamivudine. J Clin Microbiol. 1999;37:3338–3347. doi: 10.1128/jcm.37.10.3338-3347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chayama K, Suzuki Y, Kobayashi M, Kobayashi M, Tsubota A, Hashimoto M, Miyano Y, Koike H, Kobayashi M, Koida I, et al. Emergence and takeover of YMDD motif mutant hepatitis B virus during long-term lamivudine therapy and re-takeover by wild type after cessation of therapy. Hepatology. 1998;27:1711–1716. doi: 10.1002/hep.510270634. [DOI] [PubMed] [Google Scholar]
- 36.Jardi R, Buti M, Rodriguez-Frias F, Cotrina M, Costa X, Pascual C, Esteban R, Guardia J. Rapid detection of lamivudine-resistant hepatitis B virus polymerase gene variants. J Virol Methods. 1999;83:181–187. doi: 10.1016/s0166-0934(99)00125-1. [DOI] [PubMed] [Google Scholar]
- 37.Cane PA, Cook P, Ratcliffe D, Mutimer D, Pillay D. Use of real-time PCR and fluorimetry to detect lamivudine resistance-associated mutations in hepatitis B virus. Antimicrob Agents Chemother. 1999;43:1600–1608. doi: 10.1128/aac.43.7.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HS, Han KH, Ahn SH, Kim EO, Chang HY, Moon MS, Chung HJ, Yoo W, Kim SO, Hong SP. Evaluation of methods for monitoring drug resistance in chronic hepatitis B patients during lamivudine therapy based on mass spectrometry and reverse hybridization. Antivir Ther. 2005;10:441–449. [PubMed] [Google Scholar]
- 39.Tran N, Berne R, Chann R, Gauthier M, Martin D, Armand MA, Ollivet A, Teo CG, Ijaz S, Flichman D, et al. European multicenter evaluation of high-density DNA probe arrays for detection of hepatitis B virus resistance mutations and identification of genotypes. J Clin Microbiol. 2006;44:2792–2800. doi: 10.1128/JCM.00295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang RS, Zhang H, Zhu YF, Han B, Yang ZJ. Detection of YMDD mutants using universal template real-time PCR. World J Gastroenterol. 2006;12:1308–1311. doi: 10.3748/wjg.v12.i8.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malmström S, Hannoun C, Lindh M. Mutation analysis of lamivudine resistant hepatitis B virus strains by TaqMan PCR. J Virol Methods. 2007;143:147–152. doi: 10.1016/j.jviromet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 43.Astrovskaya I, Tork B, Mangul S, Westbrooks K, Măndoiu I, Balfe P, Zelikovsky A. Inferring viral quasispecies spectra from 454 pyrosequencing reads. BMC Bioinformatics. 2011;12 Suppl 6:S1. doi: 10.1186/1471-2105-12-S6-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bentley SD, Parkhill J. Comparative genomic structure of prokaryotes. Annu Rev Genet. 2004;38:771–792. doi: 10.1146/annurev.genet.38.072902.094318. [DOI] [PubMed] [Google Scholar]
- 46.Nasu A, Marusawa H, Ueda Y, Nishijima N, Takahashi K, Osaki Y, Yamashita Y, Inokuma T, Tamada T, Fujiwara T, et al. Genetic heterogeneity of hepatitis C virus in association with antiviral therapy determined by ultra-deep sequencing. PLoS One. 2011;6:e24907. doi: 10.1371/journal.pone.0024907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 48.Merriman B, Rothberg JM. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis. 2012;33:3397–3417. doi: 10.1002/elps.201200424. [DOI] [PubMed] [Google Scholar]
- 49.Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, Follows GA, Green AR, Futreal PA, Stratton MR. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci USA. 2008;105:13081–13086. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mello FC, Lago BV, Lewis-Ximenez LL, Fernandes CA, Gomes SA. Detection of mixed populations of wild-type and YMDD hepatitis B variants by pyrosequencing in acutely and chronically infected patients. BMC Microbiol. 2012;12:96. doi: 10.1186/1471-2180-12-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 2007;17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le T, Chiarella J, Simen BB, Hanczaruk B, Egholm M, Landry ML, Dieckhaus K, Rosen MI, Kozal MJ. Low-abundance HIV drug-resistant viral variants in treatment-experienced persons correlate with historical antiretroviral use. PLoS One. 2009;4:e6079. doi: 10.1371/journal.pone.0006079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solmone M, Vincenti D, Prosperi MC, Bruselles A, Ippolito G, Capobianchi MR. Use of massively parallel ultradeep pyrosequencing to characterize the genetic diversity of hepatitis B virus in drug-resistant and drug-naive patients and to detect minor variants in reverse transcriptase and hepatitis B S antigen. J Virol. 2009;83:1718–1726. doi: 10.1128/JVI.02011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Margeridon-Thermet S, Shulman NS, Ahmed A, Shahriar R, Liu T, Wang C, Holmes SP, Babrzadeh F, Gharizadeh B, Hanczaruk B, et al. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J Infect Dis. 2009;199:1275–1285. doi: 10.1086/597808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Margeridon-Thermet S, Svarovskaia ES, Babrzadeh F, Martin R, Liu TF, Pacold M, Reuman EC, Holmes SP, Borroto-Esoda K, Shafer RW. Low-level persistence of drug resistance mutations in hepatitis B virus-infected subjects with a past history of Lamivudine treatment. Antimicrob Agents Chemother. 2013;57:343–349. doi: 10.1128/AAC.01601-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Homs M, Buti M, Quer J, Jardí R, Schaper M, Tabernero D, Ortega I, Sanchez A, Esteban R, Rodriguez-Frias F. Ultra-deep pyrosequencing analysis of the hepatitis B virus preCore region and main catalytic motif of the viral polymerase in the same viral genome. Nucleic Acids Res. 2011;39:8457–8471. doi: 10.1093/nar/gkr451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Homs M, Buti M, Tabernero D, Quer J, Sanchez A, Corral N, Esteban R, Rodriguez-Frias F. Quasispecies dynamics in main core epitopes of hepatitis B virus by ultra-deep-pyrosequencing. World J Gastroenterol. 2012;18:6096–6105. doi: 10.3748/wjg.v18.i42.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodriguez-Frías F, Tabernero D, Quer J, Esteban JI, Ortega I, Domingo E, Cubero M, Camós S, Ferrer-Costa C, Sánchez A, et al. Ultra-deep pyrosequencing detects conserved genomic sites and quantifies linkage of drug-resistant amino acid changes in the hepatitis B virus genome. PLoS One. 2012;7:e37874. doi: 10.1371/journal.pone.0037874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishijima N, Marusawa H, Ueda Y, Takahashi K, Nasu A, Osaki Y, Kou T, Yazumi S, Fujiwara T, Tsuchiya S, et al. Dynamics of hepatitis B virus quasispecies in association with nucleos(t)ide analogue treatment determined by ultra-deep sequencing. PLoS One. 2012;7:e35052. doi: 10.1371/journal.pone.0035052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang GP, Sherrill-Mix SA, Chang KM, Quince C, Bushman FD. Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J Virol. 2010;84:6218–6228. doi: 10.1128/JVI.02271-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ji H, Massé N, Tyler S, Liang B, Li Y, Merks H, Graham M, Sandstrom P, Brooks J. HIV drug resistance surveillance using pooled pyrosequencing. PLoS One. 2010;5:e9263. doi: 10.1371/journal.pone.0009263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruselles A, Rozera G, Bartolini B, Prosperi M, Del Nonno F, Narciso P, Capobianchi MR, Abbate I. Use of massive parallel pyrosequencing for near full-length characterization of a unique HIV Type 1 BF recombinant associated with a fatal primary infection. AIDS Res Hum Retroviruses. 2009;25:937–942. doi: 10.1089/aid.2009.0083. [DOI] [PubMed] [Google Scholar]
- 63.Vandenbroucke I, Van Marck H, Mostmans W, Van Eygen V, Rondelez E, Thys K, Van Baelen K, Fransen K, Vaira D, Kabeya K, et al. HIV-1 V3 envelope deep sequencing for clinical plasma specimens failing in phenotypic tropism assays. AIDS Res Ther. 2010;7:4. doi: 10.1186/1742-6405-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moriyama K. Reduced antigen production by hepatitis B virus harbouring nucleotide deletions in the overlapping X gene and precore-core promoter. J Gen Virol. 1997;78(Pt 6):1479–1486. doi: 10.1099/0022-1317-78-6-1479. [DOI] [PubMed] [Google Scholar]
- 65.Yeo W, Zhong S, Chan PK, Ho WM, Wong HT, Chan AS, Johnson PJ. Sequence variations of precore/core and precore promoter regions of hepatitis B virus in patients with or without viral reactivation during cytotoxic chemotherapy. J Viral Hepat. 2000;7:448–458. doi: 10.1046/j.1365-2893.2000.00257.x. [DOI] [PubMed] [Google Scholar]
- 66.Kohno K, Nishizono A, Terao H, Hiraga M, Mifune K. Reduced transcription and progeny virus production of hepatitis B virus containing an 8-bp deletion in basic core promoter. J Med Virol. 2000;61:15–22. doi: 10.1002/(sici)1096-9071(200005)61:1<15::aid-jmv3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 67.Li KS, Yamashiro T, Sumie A, Terao H, Mifune K, Nishizono A. Hepatitis B virus harboring nucleotide deletions in the core promoter region and genotype B correlate with low viral replication activity in anti-HBe positive carriers. J Clin Virol. 2001;23:97–106. doi: 10.1016/s1386-6532(01)00212-8. [DOI] [PubMed] [Google Scholar]
- 68.Chen CH, Lee CM, Lu SN, Changchien CS, Eng HL, Huang CM, Wang JH, Hung CH, Hu TH. Clinical significance of hepatitis B virus (HBV) genotypes and precore and core promoter mutations affecting HBV e antigen expression in Taiwan. J Clin Microbiol. 2005;43:6000–6006. doi: 10.1128/JCM.43.12.6000-6006.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schories M, Peters T, Rasenack J. Isolation, characterization and biological significance of hepatitis B virus mutants from serum of a patient with immunologically negative HBV infection. J Hepatol. 2000;33:799–811. doi: 10.1016/s0168-8278(00)80313-x. [DOI] [PubMed] [Google Scholar]
- 70.Ramírez C, Gregori J, Buti M, Tabernero D, Camós S, Casillas R, Quer J, Esteban R, Homs M, Rodriguez-Frías F. A comparative study of ultra-deep pyrosequencing and cloning to quantitatively analyze the viral quasispecies using hepatitis B virus infection as a model. Antiviral Res. 2013;98:273–283. doi: 10.1016/j.antiviral.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 71.Görzer I, Guelly C, Trajanoski S, Puchhammer-Stöckl E. The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. J Virol Methods. 2010;169:248–252. doi: 10.1016/j.jviromet.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 72.Cheng Y, Guindon S, Rodrigo A, Lim SG. Increased viral quasispecies evolution in HBeAg seroconverter patients treated with oral nucleoside therapy. J Hepatol. 2013;58:217–224. doi: 10.1016/j.jhep.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 73.Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 74.Dandri M, Murray JM, Lutgehetmann M, Volz T, Lohse AW, Petersen J. Virion half-life in chronic hepatitis B infection is strongly correlated with levels of viremia. Hepatology. 2008;48:1079–1086. doi: 10.1002/hep.22469. [DOI] [PubMed] [Google Scholar]
- 75.Reuman EC, Margeridon-Thermet S, Caudill HB, Liu T, Borroto-Esoda K, Svarovskaia ES, Holmes SP, Shafer RW. A classification model for G-to-A hypermutation in hepatitis B virus ultra-deep pyrosequencing reads. Bioinformatics. 2010;26:2929–2932. doi: 10.1093/bioinformatics/btq570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol. 2006;80:1067–1076. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Noguchi C, Ishino H, Tsuge M, Fujimoto Y, Imamura M, Takahashi S, Chayama K. G to A hypermutation of hepatitis B virus. Hepatology. 2005;41:626–633. doi: 10.1002/hep.20580. [DOI] [PubMed] [Google Scholar]
- 78.Noguchi C, Imamura M, Tsuge M, Hiraga N, Mori N, Miki D, Kimura T, Takahashi S, Fujimoto Y, Ochi H, et al. G-to-A hypermutation in hepatitis B virus (HBV) and clinical course of patients with chronic HBV infection. J Infect Dis. 2009;199:1599–1607. doi: 10.1086/598951. [DOI] [PubMed] [Google Scholar]
- 79.Suspène R, Guétard D, Henry M, Sommer P, Wain-Hobson S, Vartanian JP. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci USA. 2005;102:8321–8326. doi: 10.1073/pnas.0408223102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carman W, Thomas H, Domingo E. Viral genetic variation: hepatitis B virus as a clinical example. Lancet. 1993;341:349–353. doi: 10.1016/0140-6736(93)90146-8. [DOI] [PubMed] [Google Scholar]
- 81.Osiowy C, Giles E, Tanaka Y, Mizokami M, Minuk GY. Molecular evolution of hepatitis B virus over 25 years. J Virol. 2006;80:10307–10314. doi: 10.1128/JVI.00996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Asim M, Malik A, Sarma MP, Polipalli SK, Begum N, Ahmad I, Khan LA, Husain SA, Akhtar N, Husain S, et al. Hepatitis B virus BCP, Precore/core, X gene mutations/genotypes and the risk of hepatocellular carcinoma in India. J Med Virol. 2010;82:1115–1125. doi: 10.1002/jmv.21774. [DOI] [PubMed] [Google Scholar]
- 83.Franco S, Giménez-Barcons M, Puig-Basagoiti F, Furcic I, Sánchez-Tapias JM, Rodés J, Sáiz JC. Characterization and evolution of NS5A quasispecies of hepatitis C virus genotype 1b in patients with different stages of liver disease. J Med Virol. 2003;71:195–204. doi: 10.1002/jmv.10470. [DOI] [PubMed] [Google Scholar]
- 84.Villet S, Pichoud C, Billioud G, Barraud L, Durantel S, Trépo C, Zoulim F. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J Hepatol. 2008;48:747–755. doi: 10.1016/j.jhep.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 85.Parekh S, Zoulim F, Ahn SH, Tsai A, Li J, Kawai S, Khan N, Trépo C, Wands J, Tong S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J Virol. 2003;77:6601–6612. doi: 10.1128/JVI.77.12.6601-6612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma ZM, Lin X, Wang YX, Tian XC, Xie YH, Wen YM. A double-spliced defective hepatitis B virus genome derived from hepatocellular carcinoma tissue enhanced replication of full-length virus. J Med Virol. 2009;81:230–237. doi: 10.1002/jmv.21393. [DOI] [PubMed] [Google Scholar]
- 87.Pavesi A. Origin and evolution of overlapping genes in the family Microviridae. J Gen Virol. 2006;87:1013–1017. doi: 10.1099/vir.0.81375-0. [DOI] [PubMed] [Google Scholar]
- 88.Krakauer DC. Stability and evolution of overlapping genes. Evolution. 2000;54:731–739. doi: 10.1111/j.0014-3820.2000.tb00075.x. [DOI] [PubMed] [Google Scholar]
- 89.Pavesi A, De Iaco B, Granero MI, Porati A. On the informational content of overlapping genes in prokaryotic and eukaryotic viruses. J Mol Evol. 1997;44:625–631. doi: 10.1007/PL00006185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pavesi A. Detection of signature sequences in overlapping genes and prediction of a novel overlapping gene in hepatitis G virus. J Mol Evol. 2000;50:284–295. doi: 10.1007/s002399910033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walewski JL, Keller TR, Stump DD, Branch AD. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA. 2001;7:710–721. doi: 10.1017/s1355838201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spiropoulou CF, Nichol ST. A small highly basic protein is encoded in overlapping frame within the P gene of vesicular stomatitis virus. J Virol. 1993;67:3103–3110. doi: 10.1128/jvi.67.6.3103-3110.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu L, Li Y, Zhang S, Yu D, Zhu M. Hepatitis B virus X protein mutant upregulates CENP-A expression in hepatoma cells. Oncol Rep. 2012;27:168–173. doi: 10.3892/or.2011.1478. [DOI] [PubMed] [Google Scholar]
- 94.Yip WK, Cheng AS, Zhu R, Lung RW, Tsang DP, Lau SS, Chen Y, Sung JG, Lai PB, Ng EK, et al. Carboxyl-terminal truncated HBx regulates a distinct microRNA transcription program in hepatocellular carcinoma development. PLoS One. 2011;6:e22888. doi: 10.1371/journal.pone.0022888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lizzano RA, Yang B, Clippinger AJ, Bouchard MJ. The C-terminal region of the hepatitis B virus X protein is essential for its stability and function. Virus Res. 2011;155:231–239. doi: 10.1016/j.virusres.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26 Suppl 1:144–152. doi: 10.1111/j.1440-1746.2010.06546.x. [DOI] [PubMed] [Google Scholar]
- 97.Cabot B, Martell M, Esteban JI, Sauleda S, Otero T, Esteban R, Guàrdia J, Gómez J. Nucleotide and amino acid complexity of hepatitis C virus quasispecies in serum and liver. J Virol. 2000;74:805–811. doi: 10.1128/jvi.74.2.805-811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu F, Chen L, Yu DM, Deng L, Chen R, Jiang Y, Chen L, Huang SY, Yu JL, Gong QM, et al. Evolutionary patterns of hepatitis B virus quasispecies under different selective pressures: correlation with antiviral efficacy. Gut. 2011;60:1269–1277. doi: 10.1136/gut.2010.226225. [DOI] [PubMed] [Google Scholar]
- 99.Homs M, Gregori J, Ramirez C, Buti M, Quer J, Camos S, Tabernero D, Esteban R, Rodriguez-Frias F. Ultra-deep pyrosequencing and Shannon Entropy to analyze the effect of natural evolution and treatment resistance on mutations in the hepatitis B virus quasispecies. Hepatology. 2012;56(S1):433A. [Google Scholar]
- 100.Kao JH, Liu CJ, Jow GM, Chen PJ, Chen DS, Chen BF. Fine mapping of hepatitis B virus pre-S deletion and its association with hepatocellular carcinoma. Liver Int. 2012;32:1373–1381. doi: 10.1111/j.1478-3231.2012.02826.x. [DOI] [PubMed] [Google Scholar]
- 101.Radziwill G, Tucker W, Schaller H. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J Virol. 1990;64:613–620. doi: 10.1128/jvi.64.2.613-620.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Das K, Xiong X, Yang H, Westland CE, Gibbs CS, Sarafianos SG, Arnold E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC) J Virol. 2001;75:4771–4779. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Poch O, Sauvaget I, Delarue M, Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989;8:3867–3874. doi: 10.1002/j.1460-2075.1989.tb08565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 105.Doublié S, Sawaya MR, Ellenberger T. An open and closed case for all polymerases. Structure. 1999;7:R31–R35. doi: 10.1016/S0969-2126(99)80017-3. [DOI] [PubMed] [Google Scholar]
- 106.European Association For The Study Of The Liver. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J Hepatol. 2012;57:167–185. doi: 10.1016/j.jhep.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 107.Ono SK, Kato N, Shiratori Y, Kato J, Goto T, Schinazi RF, Carrilho FJ, Omata M. The polymerase L528M mutation cooperates with nucleotide binding-site mutations, increasing hepatitis B virus replication and drug resistance. J Clin Invest. 2001;107:449–455. doi: 10.1172/JCI11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Allen MI, Deslauriers M, Andrews CW, Tipples GA, Walters KA, Tyrrell DL, Brown N, Condreay LD. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology. 1998;27:1670–1677. doi: 10.1002/hep.510270628. [DOI] [PubMed] [Google Scholar]
- 109.Westland CE, Yang H, Delaney WE, Wulfsohn M, Lama N, Gibbs CS, Miller MD, Fry J, Brosgart CL, Schiff ER, et al. Activity of adefovir dipivoxil against all patterns of lamivudine-resistant hepatitis B viruses in patients. J Viral Hepat. 2005;12:67–73. doi: 10.1111/j.1365-2893.2005.00578.x. [DOI] [PubMed] [Google Scholar]
- 110.Sheldon J, Camino N, Rodés B, Bartholomeusz A, Kuiper M, Tacke F, Núñez M, Mauss S, Lutz T, Klausen G, et al. Selection of hepatitis B virus polymerase mutations in HIV-coinfected patients treated with tenofovir. Antivir Ther. 2005;10:727–734. [PubMed] [Google Scholar]
- 111.Delaney WE, Yang H, Westland CE, Das K, Arnold E, Gibbs CS, Miller MD, Xiong S. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J Virol. 2003;77:11833–11841. doi: 10.1128/JVI.77.21.11833-11841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ogata N, Fujii K, Takigawa S, Nomoto M, Ichida T, Asakura H. Novel patterns of amino acid mutations in the hepatitis B virus polymerase in association with resistance to lamivudine therapy in japanese patients with chronic hepatitis B. J Med Virol. 1999;59:270–276. [PubMed] [Google Scholar]
- 113.Fu L, Cheng YC. Role of additional mutations outside the YMDD motif of hepatitis B virus polymerase in L(-)SddC (3TC) resistance. Biochem Pharmacol. 1998;55:1567–1572. doi: 10.1016/s0006-2952(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 114.Torresi J. The virological and clinical significance of mutations in the overlapping envelope and polymerase genes of hepatitis B virus. J Clin Virol. 2002;25:97–106. doi: 10.1016/s1386-6532(02)00049-5. [DOI] [PubMed] [Google Scholar]
- 115.Karatayli E, Karayalçin S, Karaaslan H, Kayhan H, Türkyilmaz AR, Sahin F, Yurdaydin C, Bozdayi AM. A novel mutation pattern emerging during lamivudine treatment shows cross-resistance to adefovir dipivoxil treatment. Antivir Ther. 2007;12:761–768. [PubMed] [Google Scholar]
- 116.Colonno RJ, Rose R, Baldick CJ, Levine S, Pokornowski K, Yu CF, Walsh A, Fang J, Hsu M, Mazzucco C, et al. Entecavir resistance is rare in nucleoside naïve patients with hepatitis B. Hepatology. 2006;44:1656–1665. doi: 10.1002/hep.21422. [DOI] [PubMed] [Google Scholar]
- 117.Tenney DJ, Levine SM, Rose RE, Walsh AW, Weinheimer SP, Discotto L, Plym M, Pokornowski K, Yu CF, Angus P, et al. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob Agents Chemother. 2004;48:3498–3507. doi: 10.1128/AAC.48.9.3498-3507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tillmann HL. Antiviral therapy and resistance with hepatitis B virus infection. World J Gastroenterol. 2007;13:125–140. doi: 10.3748/wjg.v13.i1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Angus P, Vaughan R, Xiong S, Yang H, Delaney W, Gibbs C, Brosgart C, Colledge D, Edwards R, Ayres A, et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology. 2003;125:292–297. doi: 10.1016/s0016-5085(03)00939-9. [DOI] [PubMed] [Google Scholar]
- 120.Schildgen O, Sirma H, Funk A, Olotu C, Wend UC, Hartmann H, Helm M, Rockstroh JK, Willems WR, Will H, et al. Variant of hepatitis B virus with primary resistance to adefovir. N Engl J Med. 2006;354:1807–1812. doi: 10.1056/NEJMoa051214. [DOI] [PubMed] [Google Scholar]
- 121.Chang TT, Lai CL. Hepatitis B virus with primary resistance to adefovir. N Engl J Med. 2006;355:322–323; author reply 323. doi: 10.1056/NEJMc066267. [DOI] [PubMed] [Google Scholar]
- 122.Schildgen O, Schewe CK, Vogel M, Däumer M, Kaiser R, Weitner L, Matz B, Rockstroh JK. Successful therapy of hepatitis B with tenofovir in HIV-infected patients failing previous adefovir and lamivudine treatment. AIDS. 2004;18:2325–2327. doi: 10.1097/00002030-200411190-00014. [DOI] [PubMed] [Google Scholar]
- 123.Mirandola S, Sebastiani G, Rossi C, Velo E, Erne EM, Vario A, Tempesta D, Romualdi C, Campagnolo D, Alberti A. Genotype-specific mutations in the polymerase gene of hepatitis B virus potentially associated with resistance to oral antiviral therapy. Antiviral Res. 2012;96:422–429. doi: 10.1016/j.antiviral.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 124.Núñez M, Pérez-Olmeda M, Díaz B, Ríos P, González-Lahoz J, Soriano V. Activity of tenofovir on hepatitis B virus replication in HIV-co-infected patients failing or partially responding to lamivudine. AIDS. 2002;16:2352–2354. doi: 10.1097/00002030-200211220-00023. [DOI] [PubMed] [Google Scholar]
- 125.Núñez M, Puoti M, Camino N, Soriano V. Treatment of chronic hepatitis B in the human immunodeficiency virus-infected patient: present and future. Clin Infect Dis. 2003;37:1678–1685. doi: 10.1086/379774. [DOI] [PubMed] [Google Scholar]
- 126.Ijaz S, Arnold C, Dervisevic S, Mechurova J, Tatman N, Tedder RS, Naoumov NV. Dynamics of lamivudine-resistant hepatitis B virus during adefovir monotherapy versus lamivudine plus adefovir combination therapy. J Med Virol. 2008;80:1160–1170. doi: 10.1002/jmv.21206. [DOI] [PubMed] [Google Scholar]
- 127.Ko SY, Oh HB, Park CW, Lee HC, Lee JE. Analysis of hepatitis B virus drug-resistant mutant haplotypes by ultra-deep pyrosequencing. Clin Microbiol Infect. 2012;18:E404–E411. doi: 10.1111/j.1469-0691.2012.03951.x. [DOI] [PubMed] [Google Scholar]
- 128.Patient R, Hourioux C, Roingeard P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell Microbiol. 2009;11:1561–1570. doi: 10.1111/j.1462-5822.2009.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Günther S, Fischer L, Pult I, Sterneck M, Will H. Naturally occurring variants of hepatitis B virus. Adv Virus Res. 1999;52:25–137. doi: 10.1016/s0065-3527(08)60298-5. [DOI] [PubMed] [Google Scholar]
- 130.Wang HC, Huang W, Lai MD, Su IJ. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006;97:683–688. doi: 10.1111/j.1349-7006.2006.00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang HC, Chang WT, Chang WW, Wu HC, Huang W, Lei HY, Lai MD, Fausto N, Su IJ. Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology. 2005;41:761–770. doi: 10.1002/hep.20615. [DOI] [PubMed] [Google Scholar]
- 132.Carman WF, Zanetti AR, Karayiannis P, Waters J, Manzillo G, Tanzi E, Zuckerman AJ, Thomas HC. Vaccine-induced escape mutant of hepatitis B virus. Lancet. 1990;336:325–329. doi: 10.1016/0140-6736(90)91874-a. [DOI] [PubMed] [Google Scholar]
- 133.Weber B. Genetic variability of the S gene of hepatitis B virus: clinical and diagnostic impact. J Clin Virol. 2005;32:102–112. doi: 10.1016/j.jcv.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 134.Zuckerman JN, Zuckerman AJ. Mutations of the surface protein of hepatitis B virus. Antiviral Res. 2003;60:75–78. doi: 10.1016/j.antiviral.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 135.Coleman PF. Detecting hepatitis B surface antigen mutants. Emerg Infect Dis. 2006;12:198–203. doi: 10.3201/eid1202.050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Oon CJ, Chen WN, Goo KS, Goh KT. Intra-familial evidence of horizontal transmission of hepatitis B virus surface antigen mutant G145R. J Infect. 2000;41:260–264. doi: 10.1053/jinf.2000.0751. [DOI] [PubMed] [Google Scholar]
- 137.Thakur V, Kazim SN, Guptan RC, Hasnain SE, Bartholomeusz A, Malhotra V, Sarin SK. Transmission of G145R mutant of HBV to an unrelated contact. J Med Virol. 2005;76:40–46. doi: 10.1002/jmv.20321. [DOI] [PubMed] [Google Scholar]
- 138.Kalinina T, Iwanski A, Will H, Sterneck M. Deficiency in virion secretion and decreased stability of the hepatitis B virus immune escape mutant G145R. Hepatology. 2003;38:1274–1281. doi: 10.1053/jhep.2003.50484. [DOI] [PubMed] [Google Scholar]
- 139.Hou J, Wang Z, Cheng J, Lin Y, Lau GK, Sun J, Zhou F, Waters J, Karayiannis P, Luo K. Prevalence of naturally occurring surface gene variants of hepatitis B virus in nonimmunized surface antigen-negative Chinese carriers. Hepatology. 2001;34:1027–1034. doi: 10.1053/jhep.2001.28708. [DOI] [PubMed] [Google Scholar]
- 140.Rodriguez-Frias F, Buti M, Jardi R, Vargas V, Quer J, Cotrina M, Martell M, Esteban R, Guardia J. Genetic alterations in the S gene of hepatitis B virus in patients with acute hepatitis B, chronic hepatitis B and hepatitis B liver cirrhosis before and after liver transplantation. Liver. 1999;19:177–182. doi: 10.1111/j.1478-3231.1999.tb00032.x. [DOI] [PubMed] [Google Scholar]
- 141.Avellón A, Echevarria JM. Frequency of hepatitis B virus ‘a’ determinant variants in unselected Spanish chronic carriers. J Med Virol. 2006;78:24–36. doi: 10.1002/jmv.20516. [DOI] [PubMed] [Google Scholar]
- 142.Lada O, Benhamou Y, Poynard T, Thibault V. Coexistence of hepatitis B surface antigen (HBs Ag) and anti-HBs antibodies in chronic hepatitis B virus carriers: influence of “a” determinant variants. J Virol. 2006;80:2968–2975. doi: 10.1128/JVI.80.6.2968-2975.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kalinina T, Riu A, Fischer L, Will H, Sterneck M. A dominant hepatitis B virus population defective in virus secretion because of several S-gene mutations from a patient with fulminant hepatitis. Hepatology. 2001;34:385–394. doi: 10.1053/jhep.2001.26516. [DOI] [PubMed] [Google Scholar]
- 144.Ogata N, Zanetti AR, Yu M, Miller RH, Purcell RH. Infectivity and pathogenicity in chimpanzees of a surface gene mutant of hepatitis B virus that emerged in a vaccinated infant. J Infect Dis. 1997;175:511–523. doi: 10.1093/infdis/175.3.511. [DOI] [PubMed] [Google Scholar]
- 145.Cariani E, Ravaggi A, Tanzi E, Romanò L, Fiordalisi G, Bellati G, Caccamo L, Galmarini D, Albertini A, Zanetti A. Emergence of hepatitis B virus S gene mutant in a liver transplant recipient. J Med Virol. 1995;47:410–415. doi: 10.1002/jmv.1890470419. [DOI] [PubMed] [Google Scholar]
- 146.Hawkins AE, Gilson RJ, Gilbert N, Wreghitt TG, Gray JJ, Ahlers-de Boer I, Tedder RS, Alexander GJ. Hepatitis B virus surface mutations associated with infection after liver transplantation. J Hepatol. 1996;24:8–14. doi: 10.1016/s0168-8278(96)80179-6. [DOI] [PubMed] [Google Scholar]
- 147.Carman WF. Molecular variants of hepatitis B virus. Clin Lab Med. 1996;16:407–428. [PubMed] [Google Scholar]
- 148.Hsu HY, Chang MH, Liaw SH, Ni YH, Chen HL. Changes of hepatitis B surface antigen variants in carrier children before and after universal vaccination in Taiwan. Hepatology. 1999;30:1312–1317. doi: 10.1002/hep.510300511. [DOI] [PubMed] [Google Scholar]
- 149.Nainan O, Stevens C, Taylo , R , Margolis H. Hepatitis B Virus (HBV) Antibody Resistant Mutants Among Mothers and Infants with Chronic HBV Infection. In: Rizzetto M, Purcell R, Gerin J, Verme G, editors. Viral Hepatitis and Liver Disease. Turin: Edizioni Minerva Medica; 1997. pp. 132–134. [Google Scholar]
- 150.Tabor E. Infections by hepatitis B surface antigen gene mutants in Europe and North America. J Med Virol. 2006;78 Suppl 1:S43–S47. doi: 10.1002/jmv.20606. [DOI] [PubMed] [Google Scholar]
- 151.Sa-Nguanmoo P, Tangkijvanich P, Tharmaphornpilas P, Rasdjarmrearnsook AO, Plianpanich S, Thawornsuk N, Theamboonlers A, Poovorawan Y. Molecular analysis of hepatitis B virus associated with vaccine failure in infants and mothers: a case-control study in Thailand. J Med Virol. 2012;84:1177–1185. doi: 10.1002/jmv.23260. [DOI] [PubMed] [Google Scholar]
- 152.Shahmoradi S, Yahyapour Y, Mahmoodi M, Alavian SM, Fazeli Z, Jazayeri SM. High prevalence of occult hepatitis B virus infection in children born to HBsAg-positive mothers despite prophylaxis with hepatitis B vaccination and HBIG. J Hepatol. 2012;57:515–521. doi: 10.1016/j.jhep.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 153.Komatsu H, Inui A, Sogo T, Konishi Y, Tateno A, Fujisawa T. Hepatitis B surface gene 145 mutant as a minor population in hepatitis B virus carriers. BMC Res Notes. 2012;5:22. doi: 10.1186/1756-0500-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Terrault NA, Zhou S, McCory RW, Pruett TL, Lake JR, Roberts JP, Ascher NL, Wright TL. Incidence and clinical consequences of surface and polymerase gene mutations in liver transplant recipients on hepatitis B immunoglobulin. Hepatology. 1998;28:555–561. doi: 10.1002/hep.510280237. [DOI] [PubMed] [Google Scholar]
- 155.Ghany MG, Ayola B, Villamil FG, Gish RG, Rojter S, Vierling JM, Lok AS. Hepatitis B virus S mutants in liver transplant recipients who were reinfected despite hepatitis B immune globulin prophylaxis. Hepatology. 1998;27:213–222. doi: 10.1002/hep.510270133. [DOI] [PubMed] [Google Scholar]
- 156.Shen ZY, Zheng WP, Deng YL, Song HL. Variations in the S and P regions of the hepatitis B virus genome under immunosuppression in vitro and in vivo. Viral Immunol. 2012;25:368–378. doi: 10.1089/vim.2012.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Protzer-Knolle U, Naumann U, Bartenschlager R, Berg T, Hopf U, Meyer zum Büschenfelde KH, Neuhaus P, Gerken G. Hepatitis B virus with antigenically altered hepatitis B surface antigen is selected by high-dose hepatitis B immune globulin after liver transplantation. Hepatology. 1998;27:254–263. doi: 10.1002/hep.510270138. [DOI] [PubMed] [Google Scholar]
- 158.Han SH, Ofman J, Holt C, King K, Kunder G, Chen P, Dawson S, Goldstein L, Yersiz H, Farmer DG, et al. An efficacy and cost-effectiveness analysis of combination hepatitis B immune globulin and lamivudine to prevent recurrent hepatitis B after orthotopic liver transplantation compared with hepatitis B immune globulin monotherapy. Liver Transpl. 2000;6:741–748. doi: 10.1053/jlts.2000.18702. [DOI] [PubMed] [Google Scholar]
- 159.Fontana RJ, Lok AS. Lamivudine treatment in patients with decompensated hepatitis B cirrhosis: for whom and when? J Hepatol. 2000;33:329–332. doi: 10.1016/s0168-8278(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 160.Bock CT, Tillmann HL, Torresi J, Klempnauer J, Locarnini S, Manns MP, Trautwein C. Selection of hepatitis B virus polymerase mutants with enhanced replication by lamivudine treatment after liver transplantation. Gastroenterology. 2002;122:264–273. doi: 10.1053/gast.2002.31015. [DOI] [PubMed] [Google Scholar]
- 161.van Hemert FJ, Zaaijer HL, Berkhout B, Lukashov VV. Mosaic amino acid conservation in 3D-structures of surface protein and polymerase of hepatitis B virus. Virology. 2008;370:362–372. doi: 10.1016/j.virol.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 162.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Salisse J, Sureau C. A function essential to viral entry underlies the hepatitis B virus “a” determinant. J Virol. 2009;83:9321–9328. doi: 10.1128/JVI.00678-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee SA, Mun HS, Kim H, Lee HK, Kim BJ, Hwang ES, Kook YH, Kim BJ. Naturally occurring hepatitis B virus X deletions and insertions among Korean chronic patients. J Med Virol. 2011;83:65–70. doi: 10.1002/jmv.21938. [DOI] [PubMed] [Google Scholar]
- 165.Sirma H, Giannini C, Poussin K, Paterlini P, Kremsdorf D, Bréchot C. Hepatitis B virus X mutants, present in hepatocellular carcinoma tissue abrogate both the antiproliferative and transactivation effects of HBx. Oncogene. 1999;18:4848–4859. doi: 10.1038/sj.onc.1202867. [DOI] [PubMed] [Google Scholar]
- 166.Tu H, Bonura C, Giannini C, Mouly H, Soussan P, Kew M, Paterlini-Bréchot P, Bréchot C, Kremsdorf D. Biological impact of natural COOH-terminal deletions of hepatitis B virus X protein in hepatocellular carcinoma tissues. Cancer Res. 2001;61:7803–7810. [PubMed] [Google Scholar]
- 167.Tang H, Oishi N, Kaneko S, Murakami S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. 2006;97:977–983. doi: 10.1111/j.1349-7006.2006.00299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Doria M, Klein N, Lucito R, Schneider RJ. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 1995;14:4747–4757. doi: 10.1002/j.1460-2075.1995.tb00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725–12734. doi: 10.1128/JVI.78.23.12725-12734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, Zoulim F, Hantz O, Protzer U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55:996–1003. doi: 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 171.Kremsdorf D, Soussan P, Paterlini-Brechot P, Brechot C. Hepatitis B virus-related hepatocellular carcinoma: paradigms for viral-related human carcinogenesis. Oncogene. 2006;25:3823–3833. doi: 10.1038/sj.onc.1209559. [DOI] [PubMed] [Google Scholar]
- 172.Koike K. Hepatitis B virus X gene is implicated in liver carcinogenesis. Cancer Lett. 2009;286:60–68. doi: 10.1016/j.canlet.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 173.Ng SA, Lee C. Hepatitis B virus X gene and hepatocarcinogenesis. J Gastroenterol. 2011;46:974–990. doi: 10.1007/s00535-011-0415-9. [DOI] [PubMed] [Google Scholar]
- 174.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 175.Madden CR, Finegold MJ, Slagle BL. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J Virol. 2001;75:3851–3858. doi: 10.1128/JVI.75.8.3851-3858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Slagle BL, Lee TH, Medina D, Finegold MJ, Butel JS. Increased sensitivity to the hepatocarcinogen diethylnitrosamine in transgenic mice carrying the hepatitis B virus X gene. Mol Carcinog. 1996;15:261–269. doi: 10.1002/(SICI)1098-2744(199604)15:4<261::AID-MC3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 177.Terradillos O, Billet O, Renard CA, Levy R, Molina T, Briand P, Buendia MA. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene. 1997;14:395–404. doi: 10.1038/sj.onc.1200850. [DOI] [PubMed] [Google Scholar]
- 178.Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376–2378. doi: 10.1126/science.294.5550.2376. [DOI] [PubMed] [Google Scholar]
- 179.Seeger C, Mason W, Zoulim F. Hepadnaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia (PA): Lippincott Williams and Wilkins; 2007. pp. 2977–3029. [Google Scholar]
- 180.Chirillo P, Pagano S, Natoli G, Puri PL, Burgio VL, Balsano C, Levrero M. The hepatitis B virus X gene induces p53-mediated programmed cell death. Proc Natl Acad Sci USA. 1997;94:8162–8167. doi: 10.1073/pnas.94.15.8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cougot D, Wu Y, Cairo S, Caramel J, Renard CA, Lévy L, Buendia MA, Neuveut C. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J Biol Chem. 2007;282:4277–4287. doi: 10.1074/jbc.M606774200. [DOI] [PubMed] [Google Scholar]
- 182.Zhang Z, Sun E, Ou JH, Liang TJ. Inhibition of cellular proteasome activities mediates HBX-independent hepatitis B virus replication in vivo. J Virol. 2010;84:9326–9331. doi: 10.1128/JVI.00579-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, Bucchi A, Sowa JP, Dittmer U, Yang D, et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology. 2009;49:1132–1140. doi: 10.1002/hep.22751. [DOI] [PubMed] [Google Scholar]
- 184.Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol. 2011;85:987–995. doi: 10.1128/JVI.01825-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z, Jia Y, Yuan Y, Guan K, Xu Y, et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol. 2010;185:1158–1168. doi: 10.4049/jimmunol.0903874. [DOI] [PubMed] [Google Scholar]
- 186.Knoll S, Fürst K, Thomas S, Villanueva Baselga S, Stoll A, Schaefer S, Pützer BM. Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC. Cell Cycle. 2011;10:3554–3565. doi: 10.4161/cc.10.20.17856. [DOI] [PubMed] [Google Scholar]
- 187.Liu Q, Chen J, Liu L, Zhang J, Wang D, Ma L, He Y, Liu Y, Liu Z, Wu J. The X protein of hepatitis B virus inhibits apoptosis in hepatoma cells through enhancing the methionine adenosyltransferase 2A gene expression and reducing S-adenosylmethionine production. J Biol Chem. 2011;286:17168–17180. doi: 10.1074/jbc.M110.167783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Xu Y, Yang Y, Cai Y, Liu F, Liu Y, Zhu Y, Wu J. The X protein of hepatitis B virus activates hepatoma cell proliferation through repressing melanoma inhibitory activity 2 gene. Biochem Biophys Res Commun. 2011;416:379–384. doi: 10.1016/j.bbrc.2011.11.046. [DOI] [PubMed] [Google Scholar]
- 189.Keng VW, Tschida BR, Bell JB, Largaespada DA. Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology. 2011;53:781–790. doi: 10.1002/hep.24091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ma NF, Lau SH, Hu L, Xie D, Wu J, Yang J, Wang Y, Wu MC, Fung J, Bai X, et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin Cancer Res. 2008;14:5061–5068. doi: 10.1158/1078-0432.CCR-07-5082. [DOI] [PubMed] [Google Scholar]
- 191.Zhang H, Shan CL, Li N, Zhang X, Zhang XZ, Xu FQ, Zhang S, Qiu LY, Ye LH, Zhang XD. Identification of a natural mutant of HBV X protein truncated 27 amino acids at the COOH terminal and its effect on liver cell proliferation. Acta Pharmacol Sin. 2008;29:473–480. doi: 10.1111/j.1745-7254.2008.00764.x. [DOI] [PubMed] [Google Scholar]
- 192.Liu XH, Lin J, Zhang SH, Zhang SM, Feitelson MA, Gao HJ, Zhu MH. COOH-terminal deletion of HBx gene is a frequent event in HBV-associated hepatocellular carcinoma. World J Gastroenterol. 2008;14:1346–1352. doi: 10.3748/wjg.14.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhu Y, Jin Y, Guo X, Bai X, Chen T, Wang J, Qian G, Groopman JD, Gu J, Li J, et al. Comparison study on the complete sequence of hepatitis B virus identifies new mutations in core gene associated with hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev. 2010;19:2623–2630. doi: 10.1158/1055-9965.EPI-10-0469. [DOI] [PubMed] [Google Scholar]
- 194.Cho HC, Kim YJ, Choi MS, Lee JH, Koh KC, Yoo BC, Paik SW. The Seroconversion Rate of Hepatitis A Virus Vaccination among Patients with Hepatitis B Virus-Related Chronic Liver Disease in Korea. Gut Liver. 2011;5:217–220. doi: 10.5009/gnl.2011.5.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kim S, Wang H, Ryu WS. Incorporation of eukaryotic translation initiation factor eIF4E into viral nucleocapsids via interaction with hepatitis B virus polymerase. J Virol. 2010;84:52–58. doi: 10.1128/JVI.01232-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yan CH, Zhao CY, Ding H, Peng YQ, Jin PY, Yan L, Zhuang H, Li T. Hepatitis B virus basal core promoter mutations A1762T/G1764A are associated with genotype C and a low serum HBsAg level in chronically-infected HBeAg-positive Chinese patients. Antiviral Res. 2012;96:108–114. doi: 10.1016/j.antiviral.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 197.Okamoto H, Yotsumoto S, Akahane Y, Yamanaka T, Miyazaki Y, Sugai Y, Tsuda F, Tanaka T, Miyakawa Y, Mayumi M. Hepatitis B viruses with precore region defects prevail in persistently infected hosts along with seroconversion to the antibody against e antigen. J Virol. 1990;64:1298–1303. doi: 10.1128/jvi.64.3.1298-1303.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Okamoto H, Tsuda F, Akahane Y, Sugai Y, Yoshiba M, Moriyama K, Tanaka T, Miyakawa Y, Mayumi M. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J Virol. 1994;68:8102–8110. doi: 10.1128/jvi.68.12.8102-8110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang D, Ma S, Zhang X, Zhao H, Ding H, Zeng C. Prevalent HBV point mutations and mutation combinations at BCP/preC region and their association with liver disease progression. BMC Infect Dis. 2010;10:271. doi: 10.1186/1471-2334-10-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kitab B, Essaid El Feydi A, Afifi R, Trepo C, Benazzouz M, Essamri W, Zoulim F, Chemin I, Alj HS, Ezzikouri S, et al. Variability in the precore and core promoter regions of HBV strains in Morocco: characterization and impact on liver disease progression. PLoS One. 2012;7:e42891. doi: 10.1371/journal.pone.0042891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jang JW, Chun JY, Park YM, Shin SK, Yoo W, Kim SO, Hong SP. Mutational complex genotype of the hepatitis B virus X /precore regions as a novel predictive marker for hepatocellular carcinoma. Cancer Sci. 2012;103:296–304. doi: 10.1111/j.1349-7006.2011.02170.x. [DOI] [PubMed] [Google Scholar]
- 202.Zheng JX, Zeng Z, Zheng YY, Yin SJ, Zhang DY, Yu YY, Wang F. Role of hepatitis B virus base core and precore/core promoter mutations on hepatocellular carcinoma in untreated older genotype C Chinese patients. J Viral Hepat. 2011;18:e423–e431. doi: 10.1111/j.1365-2893.2011.01458.x. [DOI] [PubMed] [Google Scholar]
- 203.Xiao L, Zhou B, Gao H, Ma S, Yang G, Xu M, Abbott WG, Chen J, Sun J, Wang Z, et al. Hepatitis B virus genotype B with G1896A and A1762T/G1764A mutations is associated with hepatitis B related acute-on-chronic liver failure. J Med Virol. 2011;83:1544–1550. doi: 10.1002/jmv.22159. [DOI] [PubMed] [Google Scholar]
- 204.Malik A, Singhal DK, Albanyan A, Husain SA, Kar P. Hepatitis B virus gene mutations in liver diseases: a report from New Delhi. PLoS One. 2012;7:e39028. doi: 10.1371/journal.pone.0039028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lee JH, Han KH, Lee JM, Park JH, Kim HS. Impact of hepatitis B virus (HBV) x gene mutations on hepatocellular carcinoma development in chronic HBV infection. Clin Vaccine Immunol. 2011;18:914–921. doi: 10.1128/CVI.00474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhu YZ, Zhu R, Fan J, Pan Q, Li H, Chen Q, Zhu HG. Hepatitis B virus X protein induces hypermethylation of p16(INK4A) promoter via DNA methyltransferases in the early stage of HBV-associated hepatocarcinogenesis. J Viral Hepat. 2010;17:98–107. doi: 10.1111/j.1365-2893.2009.01156.x. [DOI] [PubMed] [Google Scholar]
- 207.Yang CY, Kuo TH, Ting LP. Human hepatitis B viral e antigen interacts with cellular interleukin-1 receptor accessory protein and triggers interleukin-1 response. J Biol Chem. 2006;281:34525–34536. doi: 10.1074/jbc.M510981200. [DOI] [PubMed] [Google Scholar]
- 208.Fattovich G, Bortolotti F, Donato F. Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J Hepatol. 2008;48:335–352. doi: 10.1016/j.jhep.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 209.Rodriguez-Frias F, Buti M, Jardi R, Cotrina M, Viladomiu L, Esteban R, Guardia J. Hepatitis B virus infection: precore mutants and its relation to viral genotypes and core mutations. Hepatology. 1995;22:1641–1647. doi: 10.1002/hep.1840220605. [DOI] [PubMed] [Google Scholar]
- 210.Jardi R, Rodriguez F, Buti M, Costa X, Valdes A, Allende H, Schaper M, Galimany R, Esteban R, Guardia J. Mutations in the basic core promoter region of hepatitis B virus. Relationship with precore variants and HBV genotypes in a Spanish population of HBV carriers. J Hepatol. 2004;40:507–514. doi: 10.1016/j.jhep.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 211.Nie H, Evans AA, London WT, Block TM, Ren XD. Quantitative dynamics of hepatitis B basal core promoter and precore mutants before and after HBeAg seroconversion. J Hepatol. 2012;56:795–802. doi: 10.1016/j.jhep.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, Thomas HC. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;2:588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 213.Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clin Liver Dis. 2004;8:321–352, vi. doi: 10.1016/j.cld.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 214.Mphahlele MJ, Shattock AG, Boner W, Quinn J, McCormick PA, Carman WF. Transmission of a homogenous hepatitis B virus population of A1896-containing strains leading to mild resolving acute hepatitis and seroconversion to hepatitis B e antigen antibodies in an adult. Hepatology. 1997;26:743–746. doi: 10.1002/hep.510260329. [DOI] [PubMed] [Google Scholar]
- 215.Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen-negative chronic hepatitis B. Hepatology. 2001;34:617–624. doi: 10.1053/jhep.2001.27834. [DOI] [PubMed] [Google Scholar]
- 216.Ozasa A, Tanaka Y, Orito E, Sugiyama M, Kang JH, Hige S, Kuramitsu T, Suzuki K, Tanaka E, Okada S, et al. Influence of genotypes and precore mutations on fulminant or chronic outcome of acute hepatitis B virus infection. Hepatology. 2006;44:326–334. doi: 10.1002/hep.21249. [DOI] [PubMed] [Google Scholar]
- 217.Ngui SL, Hallet R, Teo CG. Natural and iatrogenic variation in hepatitis B virus. Rev Med Virol. 1999;9:183–209. doi: 10.1002/(sici)1099-1654(199907/09)9:3<183::aid-rmv248>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 218.Junker-Niepmann M, Bartenschlager R, Schaller H. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990;9:3389–3396. doi: 10.1002/j.1460-2075.1990.tb07540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kidd AH, Kidd-Ljunggren K. A revised secondary structure model for the 3’-end of hepatitis B virus pregenomic RNA. Nucleic Acids Res. 1996;24:3295–3301. doi: 10.1093/nar/24.17.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lok AS, Akarca U, Greene S. Mutations in the pre-core region of hepatitis B virus serve to enhance the stability of the secondary structure of the pre-genome encapsidation signal. Proc Natl Acad Sci USA. 1994;91:4077–4081. doi: 10.1073/pnas.91.9.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Tong SP, Li JS, Vitvitski L, Trépo C. Replication capacities of natural and artificial precore stop codon mutants of hepatitis B virus: relevance of pregenome encapsidation signal. Virology. 1992;191:237–245. doi: 10.1016/0042-6822(92)90185-r. [DOI] [PubMed] [Google Scholar]
- 222.Song BC, Cui XJ, Kim HU, Cho YK. Sequential accumulation of the basal core promoter and the precore mutations in the progression of hepatitis B virus-related chronic liver disease. Intervirology. 2006;49:266–273. doi: 10.1159/000093456. [DOI] [PubMed] [Google Scholar]
- 223.Guarnieri M, Kim KH, Bang G, Li J, Zhou Y, Tang X, Wands J, Tong S. Point mutations upstream of hepatitis B virus core gene affect DNA replication at the step of core protein expression. J Virol. 2006;80:587–595. doi: 10.1128/JVI.80.2.587-595.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Chauhan R, Kazim SN, Bhattacharjee J, Sakhuja P, Sarin SK. Basal core promoter, precore region mutations of HBV and their association with e antigen, genotype, and severity of liver disease in patients with chronic hepatitis B in India. J Med Virol. 2006;78:1047–1054. doi: 10.1002/jmv.20661. [DOI] [PubMed] [Google Scholar]
- 225.Kramvis A, Kew MC. The core promoter of hepatitis B virus. J Viral Hepat. 1999;6:415–427. doi: 10.1046/j.1365-2893.1999.00189.x. [DOI] [PubMed] [Google Scholar]
- 226.Hamasaki K, Nakata K, Nagayama Y, Ohtsuru A, Daikoku M, Taniguchi K, Tsutsumi T, Sato Y, Kato Y, Nagataki S. Changes in the prevalence of HBeAg-negative mutant hepatitis B virus during the course of chronic hepatitis B. Hepatology. 1994;20:8–14. doi: 10.1016/0270-9139(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 227.Chu CJ, Hussain M, Lok AS. Hepatitis B virus genotype B is associated with earlier HBeAg seroconversion compared with hepatitis B virus genotype C. Gastroenterology. 2002;122:1756–1762. doi: 10.1053/gast.2002.33588. [DOI] [PubMed] [Google Scholar]
- 228.Chan HL, Tsui SK, Tse CH, Ng EY, Au TC, Yuen L, Bartholomeusz A, Leung KS, Lee KH, Locarnini S, et al. Epidemiological and virological characteristics of 2 subgroups of hepatitis B virus genotype C. J Infect Dis. 2005;191:2022–2032. doi: 10.1086/430324. [DOI] [PubMed] [Google Scholar]
- 229.Pumpens P, Grens E, Nassal M. Molecular epidemiology and immunology of hepatitis B virus infection - an update. Intervirology. 2002;45:218–232. doi: 10.1159/000067915. [DOI] [PubMed] [Google Scholar]
- 230.Salfeld J, Pfaff E, Noah M, Schaller H. Antigenic determinants and functional domains in core antigen and e antigen from hepatitis B virus. J Virol. 1989;63:798–808. doi: 10.1128/jvi.63.2.798-808.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Homs M, Jardi R, Buti M, Schaper M, Tabernero D, Fernandez-Fernandez P, Quer J, Esteban R, Rodriguez-Frias F. HBV core region variability: effect of antiviral treatments on main epitopic regions. Antivir Ther. 2011;16:37–49. doi: 10.3851/IMP1701. [DOI] [PubMed] [Google Scholar]
- 232.Kim DW, Lee SA, Hwang ES, Kook YH, Kim BJ. Naturally occurring precore/core region mutations of hepatitis B virus genotype C related to hepatocellular carcinoma. PLoS One. 2012;7:e47372. doi: 10.1371/journal.pone.0047372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Villet S, Billioud G, Pichoud C, Lucifora J, Hantz O, Sureau C, Dény P, Zoulim F. In vitro characterization of viral fitness of therapy-resistant hepatitis B variants. Gastroenterology. 2009;136:168–176.e2. doi: 10.1053/j.gastro.2008.09.068. [DOI] [PubMed] [Google Scholar]
- 234.Rodriguez C, Chevaliez S, Bensadoun P, Pawlotsky JM. Characterization of the dynamics of hepatitis B virus resistance to adefovir by ultra-deep pyrosequencing. Hepatology. 2013;58:890–901. doi: 10.1002/hep.26383. [DOI] [PubMed] [Google Scholar]
- 235.Lin CL, Liao LY, Liu CJ, Chen PJ, Lai MY, Kao JH, Chen DS. Hepatitis B genotypes and precore/basal core promoter mutants in HBeAg-negative chronic hepatitis B. J Gastroenterol. 2002;37:283–287. doi: 10.1007/s005350200036. [DOI] [PubMed] [Google Scholar]
- 236.Moriyama K, Okamoto H, Tsuda F, Mayumi M. Reduced precore transcription and enhanced core-pregenome transcription of hepatitis B virus DNA after replacement of the precore-core promoter with sequences associated with e antigen-seronegative persistent infections. Virology. 1996;226:269–280. doi: 10.1006/viro.1996.0655. [DOI] [PubMed] [Google Scholar]
- 237.Scaglioni PP, Melegari M, Wands JR. Biologic properties of hepatitis B viral genomes with mutations in the precore promoter and precore open reading frame. Virology. 1997;233:374–381. doi: 10.1006/viro.1997.8594. [DOI] [PubMed] [Google Scholar]
- 238.Kuwahara R, Kumashiro R, Murashima S, Ogata K, Tanaka K, Hisamochi A, Hino T, Ide T, Tanaka E, Koga Y, et al. Genetic heterogeneity of the precore and the core promoter region of genotype C hepatitis B virus during lamivudine therapy. J Med Virol. 2004;72:26–34. doi: 10.1002/jmv.10558. [DOI] [PubMed] [Google Scholar]
- 239.Honda A, Yokosuka O, Ehata T, Tagawa M, Imazeki F, Saisho H. Detection of mutations in the enhancer 2/core promoter region of hepatitis B virus in patients with chronic hepatitis B virus infection: comparison with mutations in precore and core regions in relation to clinical status. J Med Virol. 1999;57:337–344. [PubMed] [Google Scholar]
- 240.Nakashima H, Furusyo N, Kubo N, Kashiwagi K, Etoh Y, Kashiwagi S, Hayashi J. Double point mutation in the core promoter region of hepatitis B virus (HBV) genotype C may be related to liver deterioration in patients with chronic HBV infection. J Gastroenterol Hepatol. 2004;19:541–550. doi: 10.1111/j.1440-1746.2003.03318.x. [DOI] [PubMed] [Google Scholar]
- 241.Kawabe N, Hashimoto S, Harata M, Nitta Y, Murao M, Nakano T, Shimazaki H, Arima Y, Komura N, Kobayashi K, et al. The loss of HBeAg without precore mutation results in lower HBV DNA levels and ALT levels in chronic hepatitis B virus infection. J Gastroenterol. 2009;44:751–756. doi: 10.1007/s00535-009-0061-7. [DOI] [PubMed] [Google Scholar]
- 242.Cai YN, Zhou Q, Kong YY, Li M, Viollet B, Xie YH, Wang Y. LRH-1/hB1F and HNF1 synergistically up-regulate hepatitis B virus gene transcription and DNA replication. Cell Res. 2003;13:451–458. doi: 10.1038/sj.cr.7290187. [DOI] [PubMed] [Google Scholar]
- 243.Yoo BC, Park JW, Kim HJ, Lee DH, Cha YJ, Park SM. Precore and core promoter mutations of hepatitis B virus and hepatitis B e antigen-negative chronic hepatitis B in Korea. J Hepatol. 2003;38:98–103. doi: 10.1016/s0168-8278(02)00349-5. [DOI] [PubMed] [Google Scholar]
- 244.Wang Y, Wei L, Jiang D, Cong X, Fei R, Chen H, Xiao J, Wang Y. In vitro resistance to interferon-alpha of hepatitis B virus with basic core promoter double mutation. Antiviral Res. 2007;75:139–145. doi: 10.1016/j.antiviral.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 245.Zhong S, Chan JY, Yeo W, Tam JS, Johnson PJ. Frequent integration of precore/core mutants of hepatitis B virus in human hepatocellular carcinoma tissues. J Viral Hepat. 2000;7:115–123. doi: 10.1046/j.1365-2893.2000.00209.x. [DOI] [PubMed] [Google Scholar]
- 246.Watanabe K, Takahashi T, Takahashi S, Okoshi S, Ichida T, Aoyagi Y. Comparative study of genotype B and C hepatitis B virus-induced chronic hepatitis in relation to the basic core promoter and precore mutations. J Gastroenterol Hepatol. 2005;20:441–449. doi: 10.1111/j.1440-1746.2004.03572.x. [DOI] [PubMed] [Google Scholar]
- 247.Park YN, Han KH, Kim KS, Chung JP, Kim S, Park C. Cytoplasmic expression of hepatitis B core antigen in chronic hepatitis B virus infection: role of precore stop mutants. Liver. 1999;19:199–205. doi: 10.1111/j.1478-3231.1999.tb00036.x. [DOI] [PubMed] [Google Scholar]
- 248.Sanchez MJ, Buti M, Homs M, Palacios A, Rodriguez-Frias F, Esteban R. Successful use of entecavir for a severe case of reactivation of hepatitis B virus following polychemotherapy containing rituximab. J Hepatol. 2009;51:1091–1096. doi: 10.1016/j.jhep.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 249.Ahn SH, Kramvis A, Kawai S, Spangenberg HC, Li J, Kimbi G, Kew M, Wands J, Tong S. Sequence variation upstream of precore translation initiation codon reduces hepatitis B virus e antigen production. Gastroenterology. 2003;125:1370–1378. doi: 10.1016/j.gastro.2003.07.016. [DOI] [PubMed] [Google Scholar]
- 250.Fernández M, Quiroga JA, Carreño V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J Gen Virol. 2003;84:2073–2082. doi: 10.1099/vir.0.18966-0. [DOI] [PubMed] [Google Scholar]
- 251.Rodriguez-Frias F, Jardi R, Buti M, Schaper M, Hermosilla E, Valdes A, Allende H, Martell M, Esteban R, Guardia J. Hepatitis B virus genotypes and G1896A precore mutation in 486 Spanish patients with acute and chronic HBV infection. J Viral Hepat. 2006;13:343–350. doi: 10.1111/j.1365-2893.2005.00691.x. [DOI] [PubMed] [Google Scholar]
- 252.Huang YH, Wu JC, Chang TT, Sheen IJ, Huo TI, Lee PC, Su CW, Lee SD. Association of core promoter/precore mutations and viral load in e antigen-negative chronic hepatitis B patients. J Viral Hepat. 2006;13:336–342. doi: 10.1111/j.1365-2893.2005.00688.x. [DOI] [PubMed] [Google Scholar]
- 253.Chen CH, Lee CM, Hung CH, Hu TH, Wang JH, Wang JC, Lu SN, Changchien CS. Clinical significance and evolution of core promoter and precore mutations in HBeAg-positive patients with HBV genotype B and C: a longitudinal study. Liver Int. 2007;27:806–815. doi: 10.1111/j.1478-3231.2007.01505.x. [DOI] [PubMed] [Google Scholar]
- 254.Chen WN, Oon CJ. Mutations and deletions in core promoter and precore stop codon in relation to viral replication and liver damage in Singaporean hepatitis B virus carriers. Eur J Clin Invest. 2000;30:787–792. doi: 10.1046/j.1365-2362.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 255.Chen CH, Hung CH, Lee CM, Hu TH, Wang JH, Wang JC, Lu SN, Changchien CS. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology. 2007;133:1466–1474. doi: 10.1053/j.gastro.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 256.Manesis EK, Hadziyannis SJ. Interferon alpha treatment and retreatment of hepatitis B e antigen-negative chronic hepatitis B. Gastroenterology. 2001;121:101–109. doi: 10.1053/gast.2001.25524. [DOI] [PubMed] [Google Scholar]
- 257.Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology. 2001;34:1225–1241. doi: 10.1053/jhep.2001.29401. [DOI] [PubMed] [Google Scholar]
- 258.Li JS, Tong SP, Wen YM, Vitvitski L, Zhang Q, Trépo C. Hepatitis B virus genotype A rarely circulates as an HBe-minus mutant: possible contribution of a single nucleotide in the precore region. J Virol. 1993;67:5402–5410. doi: 10.1128/jvi.67.9.5402-5410.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Cho SW, Hahm KB, Kim JH. Reversion from precore/core promoter mutants to wild-type hepatitis B virus during the course of lamivudine therapy. Hepatology. 2000;32:1163–1169. doi: 10.1053/jhep.2000.19618. [DOI] [PubMed] [Google Scholar]
- 260.Yuen MF, Sablon E, Libbrecht E, Van De Velde H, Wong DK, Fung J, Wong BC, Lai CL. Significance of viral load, core promoter/precore mutations and specific sequences of polymerase gene in HBV-infected patients on 3-year lamivudine treatment. Antivir Ther. 2006;11:779–786. [PubMed] [Google Scholar]
- 261.Tacke F, Gehrke C, Luedde T, Heim A, Manns MP, Trautwein C. Basal core promoter and precore mutations in the hepatitis B virus genome enhance replication efficacy of Lamivudine-resistant mutants. J Virol. 2004;78:8524–8535. doi: 10.1128/JVI.78.16.8524-8535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]