

Abstract
Accumulating evidence has implicated Helicobacter pylori (H. pylori) infection in extragastrointestinal diseases, including obesity, type 2 diabetes mellitus, cardiovascular disease, and liver disease. Recently, there has been a special focus on H. pylori infection as a risk factor for the development of nonalcoholic fatty liver disease (NAFLD). NAFLD is currently considered to be the most common liver disorder in western countries, and is rapidly becoming a serious threat to public health. The mechanisms of pathogenesis underlying NAFLD remain unclear at present and therapeutic options are limited. The growing awareness of the role of H. pylori in NAFLD is thus important to aid the development of novel intervention and prevention strategies, because the eradication of H. pylori is easy and much less expensive than long-term treatment of the other risk factors. H. pylori infection is involved in the pathogenesis of insulin resistance (IR), which is closely linked with NAFLD. It provides a new insight into the pathogenesis of NAFLD. This review probes the possible relationship between H. pylori and NAFLD, from the perspective of the potential mechanism of how H. pylori infection brings about IR and other aspects concerning this correlation.
Keywords: Helicobacter pylori, Nonalcoholic fatty liver disease, Insulin resistance, Inflammation, Cytokines
Core tip: A growing body of evidence suggests that Helicobacter pylori (H. pylori) infection is linked with nonalcoholic fatty liver disease (NAFLD). There are some potential pathogenic mediators and mechanisms involved in this progress, including fetuin-A, tumor necrosis factor-α and adiponectin. Long-term H. pylori infection may cause insulin resistance and inflammation, contributing to NAFLD. H. pylori toxins in the portal circulation arising from the gastroduodenal area may be another intriguing point, which might be related to the increased intestinal permeability in patients with NAFLD. It is hoped that eradication of H. pylori will provide a new treatment strategy for NAFLD.
INTRODUCTION
Helicobacter pylori (H. pylori) is a Gram-negative and microaerophilic bacterium[1]. The incidence of H. pylori infection in adults is particularly high in developing countries compared with developed countries[2].
H. pylori colonizes the stomach in childhood and persists throughout life, causing diseases mainly in adults, including chronic gastritis, peptic ulcer disease, gastric mucosa-associated lymphoid tissue lymphoma, and gastric cancer[3,4]. This persistent infection elicits a chronic inflammatory and immune response[5], inducing both local and remote lesions. The interaction of the host with H. pylori can have profound systemic effects[6]. A growing body of evidence has implicated H. pylori infection in extragastrointestinal diseases such as cardiovascular, liver and biliary diseases[7-9]. The possible causative role of H. pylori infection in these diseases is intriguing. The contribution of H. pylori to the development of hepatic encephalopathy and hyperammonemia has been revealed[10], and the possible correlation of H. pylori with other liver diseases has attracted a lot of attention[8,11]. Recent reports have emerged on the relationship between H. pylori infection and nonalcoholic fatty liver disease (NAFLD).
In recent years, there has been increased appreciation of the significance of NAFLD, which is currently considered to be the most common liver disorder in western countries, affecting up to 25%-30% of individuals[12-14]. NAFLD encompasses a range of related disorders[15]. The earliest stage is simple steatosis. It can progress to nonalcoholic steatohepatitis (NASH) with the cardinal features including hepatocyte injury, and inflammation with or without fibrosis[16-18]. NASH, in turn, may progress to cirrhosis and ultimately liver cancer in some patients[19]. Consequently, NAFLD is rapidly becoming a serious threat to public health. However, the mechanisms of the pathogenesis underlying NAFLD are complex and remain unknown at present[20,21].
NAFLD is now regarded as the liver manifestation of the metabolic syndrome (MetS)[22]. It is strongly associated with obesity, diabetes, cardiovascular disease (CVD) and dyslipidemia[23], because they spring from a “common soil”, namely, insulin resistance (IR)[24]. A growing body of experimental evidence suggests that NAFLD and IR are closely related[25,26].
H. pylori infection is implicated in the pathogenesis of IR[27], which is important in the development of NAFLD, therefore, investigating the impact of H. pylori infection as a risk factor for IR might have implications in understanding its effect on NAFLD. It is hoped that the eradication of H. pylori can provide a novel strategy for the treatment of NAFLD. This review focuses on the possible relationship between H. pylori and NAFLD; mainly from the perspective of the potential mechanism of how H. pylori infection brings about IR and other aspects concerning this correlation (Figure 1).
Figure 1.
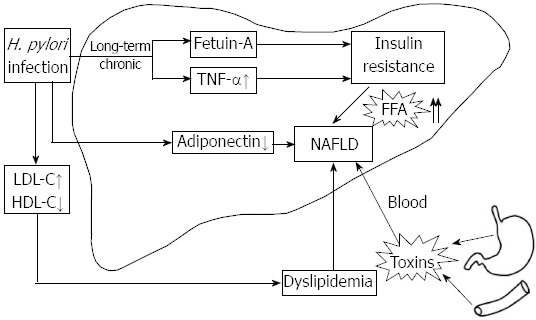
Potential mechanism of how Helicobacter pylori contribute to nonalcoholic fatty liver disease. Insulin resistance (IR) may be an important link between Helicobacter pylori (H. pylori) infection and nonalcoholic fatty liver disease (NAFLD). IR favors accumulation of free fatty acids (FFAs) in the liver. H. pylori-induced IR may be mediated through fetuin-A. Tumor necrosis factor (TNF)-α plays a central role in the response to inflammation elicited by long-term H. pylori infection. The decrease in adiponectin is implicated in H. pylori-induced NAFLD. H. pylori toxins arising from the gastrointestinal area may cause liver damage. H. pylori may be associated with the altered lipid profile, leading to dyslipidemia, which is involved in the pathogenesis of NAFLD.
EXPERIMENTAL EVIDENCE LINKING
H. PYLORI WITH NAFLD
A novel finding in the study of Cindoruk et al[28] was the presence of 16S rDNA of H. pylori in the liver sample of a 44-year-old woman with NASH. They used polymerase chain reaction-based techniques to amplify 16S rDNA sequences of H. pylori. In that study, 27 of 75 patients with suspected liver disease were diagnosed with NASH. It turned out in one sample that 16S rDNA of H. pylori was detected. This observation suggested that H. pylori play a role in NAFLD.
H. pylori is mainly identified by 16S rDNA sequencing[8]. In bacteria, there are three types of rDNA that are readily identifiable by size: the 120-nucleotide (nt) 5S rDNA, the 1600-nt 16S rDNA, and the 3000-nt 23S rDNA[29]. Researchers have recently tended to use genetic criteria including virulence gene and 16S rDNA sequencing for distinguishing it from other curved Gram-negative rods[30].
In 2009 another study added credence to this finding, in which Pirouz et al[31] noticed that patients with various chronic liver diseases (CLDs) had a greater probability of positive H. pylori 16S rDNA compared with the control group. In the 46 liver biopsies from patients with CLD, they detected H. pylori DNA in five of 11 samples from patients who were diagnosed with NAFLD.
CLINICAL EVIDENCE LINKING H. PYLORI WITH NAFLD
Polyzos et al[32] showed that NAFLD patients had significantly higher anti-H. pylori IgG, insulin, homeostatic model of assessment insulin resistance (HOMA-IR), and tumor necrosis factor (TNF)-α, but less total and high-molecular-weight adiponectin compared to the control group. However, there were no significant differences in steatosis grade, fibrosis stage, lobular or portal inflammation, or ballooning, when NAFLD patients were divided into subgroups according to H. pylori IgG seropositivity or 13C-urea breath test positivity. This might be a clue that H. pylori infection is strongly linked to the pathogenesis of early-stage NAFLD, which is described as simple steatosis. Yet at the same time, it indicates that H. pylori infection may not contribute to the progression of NASH.
A randomized controlled single-blind study from Doğan et al[33] showed that fatty liver was significantly more frequent in H. pylori-positive patients. The severity of the fatty appearance assessed by ultrasonography was also higher in the H. pylori-positive group. A study conducted in Japan demonstrated that H. pylori infection was one of the independent risk factors for the development of NAFLD[7]. These studies had some limitations and further research is warranted with larger, longer-term studies to confirm their findings. It is hard to determine if H. pylori is responsible for the natural course of NAFLD, or if it is merely an incidental finding. If this association is confirmed, eradicating H. pylori infection may have certain therapeutic perspectives in NAFLD.
POTENTIAL PATHOGENETIC MEDIATORS AND MECHANISMS
IR: a possible bond linking H. pylori with NAFLD
IR is a key pathogenic factor in NASH[34]. It leads to hyperinsulinemia and favors accumulation of free fatty acids (FFAs) in the liver because of decreased mitochondrial β-oxidation, on account that insulin inhibits hepatic mitochondrial β-oxidation of fatty acids[35]. Moreover, IR predisposes the liver to oxidative stress by stimulating microsomal lipid peroxidases[23].
H. pylori infection is involved in diverse biological processes[36], comprising inflammation, metabolism and oncogenic transformation[31,37]. In view of its effect on metabolic variables, H. pylori is associated with IR.
H. pylori infection is implicated in the pathogenesis of obesity[38] and type 2 diabetes mellitus (T2DM)[39-41], which are closely related to MetS. IR is thought to be the underlying mechanism for MetS. However, so far, only limited clinical data directly suggest that H. pylori infection is involved in the development of NAFLD. Polyzos et al[32] showed that the contribution of H. pylori to NAFLD might be achieved indirectly through increasing IR, or directly, given that it can predict NAFLD independently from IR. Inspired by this hypothesis, studies investigating the relationship between H. pylori infection and IR would be of importance to infer the mechanism of how H. pylori induce NAFLD. A few studies have explored the possible link between H. pylori infection and IR. Besides, research on the influence of eradication therapy on IR and other metabolic parameters has been conducted. However, results are controversial, and whether H. pylori infection plays a role in IR remains to be determined. Assessed by HOMA-IR, existing data indicate a potential association between H. pylori infection and IR. HOMA-IR, which derives from fasting insulin and glucose levels[42], is the most common method for assessment of IR in clinical practice and epidemiological studies[43]. Using this method, a high HOMA-IR score denotes low insulin sensitivity. In 2011, Polyzos et al[27] performed a systematic review[27], summarizing the evidence for the association between H. pylori infection and quantitative indexes of IR. Summary data indicate a potential association between H. pylori infection and IR.
Eshraghian et al[44] showed that HOMA-IR score was significantly higher in the H. pylori-positive group compared with the negative group. They suggest that H. pylori infection is a risk factor for IR. However, Naja et al[45] have suggested no association of H. pylori infection with IR or MetS. They take the view that eradication of H. pylori infection to prevent IR or MetS is not warranted.
We must take into consideration that most of the studies that investigated the association between H. pylori and IR or MetS were small and did not adjust for potential confounders. Many of them did not control for the bacterial strain type or the host genetic factors. Due to the limited quantity and quality of these studies, more high-quality, multicenter, large-scale randomized controlled trials are required to clarify the association between H. pylori infection and IR development. A positive link between H. pylori infection and IR could have certain therapeutic prospects.
A peculiar intermediary: fetuin-A: H. pylori infection has been proposed in an attempt to elucidate the multifaceted aspects of the pathogenesis of IR. However, the pathogenetic link between H. pylori infection and IR is not fully understood as yet.
Among the various factors capable of inducing IR and subsequent IR syndrome, fetuin-A is peculiar because it is a glycoprotein that is produced exclusively in the liver and then secreted into the circulation in high concentrations[46]. Previous studies have shown that fetuin-A is closely related to IR[47-49], and it has been linked with impaired insulin sensitivity, glucose metabolism, and the onset of diabetes mellitus[50,51].
Recent studies have investigated whether the H. pylori-induced IR is mediated through fetuin-A. In the study of Kebapcilar et al[52], fetuin-A level significantly decreases in H. pylori-infected patients when compared to control subjects. Moreover, H. pylori eradication reduces the levels of proinflammatory cytokines such as migration inhibitory factor and high-sensitivity C-reactive protein (CRP), with a significant increase in fetuin-A. They regarded fetuin-A as a potential anti-inflammatory cytokine[53], on the basis of the theory that anti-inflammatory cytokines produced during inflammation tend to modulate the inflammatory reaction.
However, the findings of Manolakis et al[54] were the opposite. They noted that H. pylori-infected individuals showed higher levels of fetuin-A, insulin and HOMA-IR than controls. In addition, there was a positive correlation between fetuin-A and HOMA-IR. This has interesting therapeutic implications because it suggests that H. pylori eradication might decrease IR. This observation coincides with the results of Ou et al[55], who showed that fetuin-A is considered to be a key proinflammatory mediator that plays a pivotal role in inflammatory and immune diseases. They suggest that elevated fetuin-A level has clinical implications in NAFLD and impaired glucose tolerance, which are features of IR[55].
Fetuin-A is an endogenous inhibitor of insulin receptor tyrosine kinase in the liver and skeletal muscle[48]. Srinivas et al[48] have revealed that fetuin-A specifically inhibits insulin-stimulated insulin receptor autophosphorylation in vitro and in vivo, as well as exogenous substrate tyrosine phosphorylation. Moreover, they have demonstrated that fetuin-A influences insulin signaling by inhibiting insulin-induced tyrosine phosphorylation of insulin receptor substrate (IRS)-1[56] and insulin-dependent mitogenesis. The glucoregulatory effects derived from insulin are predominantly exerted in three tissues consisting of liver, muscle and fat[57]. Thus, when IR occurs, the liver can be the point of attack. Based on the theory that fetuin-A inhibits insulin signaling in hepatocytes, it is reasonable to assume that the elevated fetuin-A in patients with NAFLD may contribute to the deteriorated hepatic IR.
Although the mechanism underlying fetuin-A-mediated IR remains elusive, it is a novel concept that fetuin-A may represent a promising index for assessing the H. pylori-related contributions to IR and MetS. If this particular association is confirmed, fetuin-A could be a potential target for therapy of IR and IR-related disorders, including T2DM, CVD and NAFLD.
Chronic inflammation, cytokines and adipokines: Helicobacter spp. are strong inducers of proinflammatory cytokines[58]. Long-standing H. pylori infection induces inflammation by stimulating excessive release of proinflammatory cytokines and vasoactive substances, such as interleukin (IL)-6, IL-8, IL-1β and TNF-α[59-61]. H. pylori-positive individuals exhibit elevated levels of these proinflammatory cytokines[62].
A growing body of evidence supports that inflammation is involved in the pathogenesis of IR and IR-related disorders[63]. Festa et al[64] have suggested that low-grade inflammation is a risk factor for the development of T2DM[64]. Several studies indicate that chronic subclinical inflammation is associated with CVD[65].
Hotamisligil et al[66] and Feinstein et al[67] have demonstrated that TNF-α is able to induce IR. Therefore, we reckon that TNF-α may be a key mediator of both direct and indirect effects of H. pylori infection on NAFLD.
TNF-α interferes with insulin signaling, thereby favoring steatosis, and may play a proinflammatory role in the pathogenesis of NASH[68,69]. On the one hand, TNF-α promotes Ser phosphorylation of IRS-1[70], resulting in a net decrease in insulin-receptor-mediated signaling. On the other hand, TNF-α can inhibit the autophosphorylation of insulin receptor or tyrosyl phosphorylation of IRS-1[67]. In addition, TNF-α downregulates the expression of key genes in adipose cells such as GLUT4[71], resulting in decreased glucose transport[72]. Besides, TNF-α is capable of accelerating lipolysis, leading to an increase in FFAs, which can cause detrimental effects in hepatocytes, including oxidative stress[73], induction of endoplasmic reticulum stress[74] and subsequent expression of proinflammatory cytokines. TNF-α promotes and is activated by IR via activation of IKK-β[17] , which is a central coordinator of inflammatory responses through activation of nuclear factor (NF-κB)[75]. NF-κB is a proinflammatory “master switch” that regulates inflammatory mediators including CRP, plasminogen activator inhibitor, TNF-α, IL-6 and IL-1β[76].
Lower adiponectin level was observed in H. pylori-positive patients with NAFLD in the study of Polyzos et al[32]. This finding gives us a new clue that adiponectin may play a part in the process of H. pylori-induced NAFLD. Adiponectin, the adipocyte-derived hormone, is implicated in the pathogenesis of IR and NASH. Low serum adiponectin levels in NAFLD patients are suggestive of advanced hepatic fibrosis[77]. Adiponectin exerts several anti-inflammatory effects[78], including inhibition of NF-κB activation[79] and suppression of macrophage function. Adiponectin and TNF-α are mutually antagonizing adipokines[80]. In contrast to TNF-α, adiponectin has an antilipogenic effect[81]. Thus, when adiponectin is decreased, its effects of controlling FFA entry and oxidation in the mitochondria are subsequently weakened, allowing FFAs to accumulate in the cytoplasm[82].
Lipid metabolism
Inflammation, IR and aberrant lipid metabolism may be interlinked components of the MetS[83,84]. Abnormalities of serum lipid concentrations are common in patients with NASH[23]. It is acknowledged that hypertriglyceridemia is involved in the pathogenesis of NASH. Each of the steps involved in hepatic lipid accumulation is altered in NAFLD, although to a different extent[85]. Satoh et al[86] have shown that H. pylori infection is a significant and independent risk factor for a modified lipid profile, including high low-density lipoprotein cholesterol (LDL-C) and low high-density lipoprotein cholesterol (HDL-C) in Japanese men, whereas these associations are not significant in women. Kebapcilar et al[52] demonstrated that H. pylori infection is significantly associated with lower HDL-C[52], but they showed that eradication of H. pylori had no effect on the lipid profile. Akbas et al[87] reported that there was no significant difference in serum HDL-C, LDL-C, or total cholesterol between H. pylori-seropositive and H. pylori-seronegative individuals, whereas serum triglyceride level was higher in the H. pylori-positive group.
Increased intestinal permeability, H. pylori toxins and cross-reactive antibody response
H. pylori is thought to have deleterious consequences on the hepatobiliary tract because the biliary epithelium can easily be colonized by bacteria from the duodenum[88]. The human gastrointestinal tract is an ecosystem integrated by microbiota. The mucosal epithelium of the small intestine is the barrier between the microbiota and gut lumen[89]. It is reported that increased intestinal permeability and small intestinal bacterial overgrowth (SIBO), may reflect qualitative and quantitative changes in the microbiota, leading to disruption of the intestinal barrier, subsequent bacterial translocation, and development of portal endotoxemia[90]. As a result, lipopolysaccharide, which is produced by Gram-negative bacteria, is increased in the portal circulation and accompanied by increased levels of endotoxin-mediated cytokines in the liver. Bacterial translocation occurs due to impaired barrier function[91], and bacterial constituents enhance hepatic inflammation and fibrosis[92]. Miele et al[93] found that in NAFLD patients, increased gut permeability and the prevalence of SIBO correlated with the severity of steatosis but not with the presence of NASH.
In light of the above considerations, we speculate that the liver may be damaged by H. pylori toxins and constituents circulating in the blood coming out from the gastroduodenal area. And it is probably linked with the increased intestinal permeability in patients with NAFLD.
Abenavoli et al[94] reported a case of a 36-year-old woman with diagnosis of celiac disease (CD), primary biliary cirrhosis (PBC) and H. pylori infection. They found that strict adherence to a gluten-free diet, associated with ursodeoxycholic acid administration and eradication of H. pylori infection, led to a marked histological and serological improvement of PBC. Helicobacter spp. are implicated in the pathogenesis of PBC, because microbial DNA is found in liver tissue and bacterial antibodies in the serum of patients with PBC[95,96], which is characterized by the presence of antimitochondrial antibodies directed predominantly against the E2 subunit of the pyruvate dehydrogenase complex[97]. They indicated that increased permeability to intraluminal antigens could induce an immune response against antigens sharing common epitopes to self-liver proteins and/or against cryptic antigens unmasked by the reaction with gliadin. Their study supports the pathogenetic role of increased intestinal permeability in the course of CD and H. pylori infection to induce PBC. However, this concept remains obscure. More studies are needed to clarify the reality of this association.
CONCLUSION AND OUTLOOK
NAFLD affects both adults and children who present with particular risk factors, including obesity, sedentary lifestyle and/or a predisposing genetic background[98]. In some individuals it can progress to cirrhosis, or even hepatocellular carcinoma. Treatment strategies ranging from simple lifestyle modifications to pharmacological agents and even invasive surgical procedures[99] have been investigated as potential treatments for NAFLD. However, at present, it is regrettable that there is no one ideal therapy suitable for all patients[100]. Thus, new treatment strategies are important to halt the progression of NAFLD.
IR assumes importance in the pathogenesis of NAFLD progression[101]. Recent studies, largely on the possible contribution of H. pylori to IR, provide new insights into the link between H. pylori and NAFLD. Eradication of H. pylori is easy and relatively inexpensive, therefore, the interest in exploring its involvement in extragastric diseases is of importance for public health. Thus, understanding the pathogenetic role of H. pylori in NAFLD is important for devising new specific management strategies.
The clinical data regarding H. pylori infection in NAFLD are limited, thus, it is premature to advocate intervention measures in patients with NAFLD. However, once this particular association is confirmed, it could drastically change our understanding of pathophysiology and treatment of NAFLD. Therefore, further studies are warranted to verify such associations before the strategy can be recommended in routine clinical practice.
Footnotes
Supported by The National Natural Science Foundation of China, No. 81230012; Zhejiang Provincial Laboratory Animal Science Technology Program of China, No. 2011C37088; the National Natural Science Foundation of China, No. 81300303
P- Reviewers: Abenavoli L, Celinski K S- Editor: Zhai HH L- Editor: A E- Editor: Wang CH
References
- 1.Dunn BE, Cohen H, Blaser MJ. Helicobacter pylori. Clin Microbiol Rev. 1997;10:720–741. doi: 10.1128/cmr.10.4.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu MS, Lee WJ, Wang HH, Huang SP, Lin JT. A case-control study of association of Helicobacter pylori infection with morbid obesity in Taiwan. Arch Intern Med. 2005;165:1552–1555. doi: 10.1001/archinte.165.13.1552. [DOI] [PubMed] [Google Scholar]
- 3.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banatvala N, Mayo K, Megraud F, Jennings R, Deeks JJ, Feldman RA. The cohort effect and Helicobacter pylori. J Infect Dis. 1993;168:219–221. doi: 10.1093/infdis/168.1.219. [DOI] [PubMed] [Google Scholar]
- 5.Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306–323. doi: 10.1053/j.gastro.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol. 2012;30:677–706. doi: 10.1146/annurev-immunol-020711-075008. [DOI] [PubMed] [Google Scholar]
- 7.Takuma Y. Helicobacter pylori infection and liver diseases. Gan To Kagaku Ryoho. 2011;38:362–364. [PubMed] [Google Scholar]
- 8.Pellicano R, Ménard A, Rizzetto M, Mégraud F. Helicobacter species and liver diseases: association or causation? Lancet Infect Dis. 2008;8:254–260. doi: 10.1016/S1473-3099(08)70066-5. [DOI] [PubMed] [Google Scholar]
- 9.Pellicano R, Fagoonee S, Rizzetto M, Ponzetto A. Helicobacter pylori and coronary heart disease: which directions for future studies? Crit Rev Microbiol. 2003;29:351–359. doi: 10.1080/713608015. [DOI] [PubMed] [Google Scholar]
- 10.Abdel-Hady H, Zaki A, Badra G, Lotfy M, Selmi C, Giorgini A, El-Sayed M, Badr R. Helicobacter pylori infection in hepatic encephalopathy: Relationship to plasma endotoxins and blood ammonia. Hepatol Res. 2007;37:1026–1033. doi: 10.1111/j.1872-034X.2007.00146.x. [DOI] [PubMed] [Google Scholar]
- 11.Esmat G, El-Bendary M, Zakarya S, Ela MA, Zalata K. Role of Helicobacter pylori in patients with HCV-related chronic hepatitis and cirrhosis with or without hepatocellular carcinoma: possible association with disease progression. J Viral Hepat. 2012;19:473–479. doi: 10.1111/j.1365-2893.2011.01567.x. [DOI] [PubMed] [Google Scholar]
- 12.de Alwis NM, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. J Hepatol. 2008;48 Suppl 1:S104–S112. doi: 10.1016/j.jhep.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Bhala N, Jouness RI, Bugianesi E. Epidemiology and natural history of patients with NAFLD. Curr Pharm Des. 2013;19:5169–5176. doi: 10.2174/13816128113199990336. [DOI] [PubMed] [Google Scholar]
- 14.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- 15.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 17.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 18.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 19.Day CP. Natural history of NAFLD: remarkably benign in the absence of cirrhosis. Gastroenterology. 2005;129:375–378. doi: 10.1053/j.gastro.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 20.Larrain S, Rinella ME. A myriad of pathways to NASH. Clin Liver Dis. 2012;16:525–548. doi: 10.1016/j.cld.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 21.McClain CJ, Mokshagundam SP, Barve SS, Song Z, Hill DB, Chen T, Deaciuc I. Mechanisms of non-alcoholic steatohepatitis. Alcohol. 2004;34:67–79. doi: 10.1016/j.alcohol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Yilmaz Y. NAFLD in the absence of metabolic syndrome: different epidemiology, pathogenetic mechanisms, risk factors for disease progression? Semin Liver Dis. 2012;32:14–21. doi: 10.1055/s-0032-1306422. [DOI] [PubMed] [Google Scholar]
- 23.Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis. 2001;21:27–41. doi: 10.1055/s-2001-12927. [DOI] [PubMed] [Google Scholar]
- 24.Stern MP. Diabetes and cardiovascular disease. The “common soil” hypothesis. Diabetes. 1995;44:369–374. doi: 10.2337/diab.44.4.369. [DOI] [PubMed] [Google Scholar]
- 25.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, Song XY, Diehl AM. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 27.Polyzos SA, Kountouras J, Zavos C, Deretzi G. The association between Helicobacter pylori infection and insulin resistance: a systematic review. Helicobacter. 2011;16:79–88. doi: 10.1111/j.1523-5378.2011.00822.x. [DOI] [PubMed] [Google Scholar]
- 28.Cindoruk M, Cirak MY, Unal S, Karakan T, Erkan G, Engin D, Dumlu S, Turet S. Identification of Helicobacter species by 16S rDNA PCR and sequence analysis in human liver samples from patients with various etiologies of benign liver diseases. Eur J Gastroenterol Hepatol. 2008;20:33–36. doi: 10.1097/MEG.0b013e3282efa4f2. [DOI] [PubMed] [Google Scholar]
- 29.Battles JK, Williamson JC, Pike KM, Gorelick PL, Ward JM, Gonda MA. Diagnostic assay for Helicobacter hepaticus based on nucleotide sequence of its 16S rRNA gene. J Clin Microbiol. 1995;33:1344–1347. doi: 10.1128/jcm.33.5.1344-1347.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monstein H, Nikpour-Badr S, Jonasson J. Rapid molecular identification and subtyping of Helicobacter pylori by pyrosequencing of the 16S rDNA variable V1 and V3 regions. FEMS Microbiol Lett. 2001;199:103–107. doi: 10.1111/j.1574-6968.2001.tb10658.x. [DOI] [PubMed] [Google Scholar]
- 31.Pirouz T, Zounubi L, Keivani H, Rakhshani N, Hormazdi M. Detection of Helicobacter pylori in paraffin-embedded specimens from patients with chronic liver diseases, using the amplification method. Dig Dis Sci. 2009;54:1456–1459. doi: 10.1007/s10620-008-0522-5. [DOI] [PubMed] [Google Scholar]
- 32.Polyzos SA, Kountouras J, Papatheodorou A, Patsiaoura K, Katsiki E, Zafeiriadou E, Zavos C, Anastasiadou K, Terpos E. Helicobacter pylori infection in patients with nonalcoholic fatty liver disease. Metabolism. 2013;62:121–126. doi: 10.1016/j.metabol.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Doğan Z, Filik L, Ergül B, Sarikaya M, Akbal E. Association between Helicobacter pylori and liver-to-spleen ratio: a randomized-controlled single-blind study. Eur J Gastroenterol Hepatol. 2013;25:107–110. doi: 10.1097/MEG.0b013e3283590c10. [DOI] [PubMed] [Google Scholar]
- 34.Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, Karim R, Lin R, Samarasinghe D, Liddle C, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–379. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 35.Fong DG, Nehra V, Lindor KD, Buchman AL. Metabolic and nutritional considerations in nonalcoholic fatty liver. Hepatology. 2000;32:3–10. doi: 10.1053/jhep.2000.8978. [DOI] [PubMed] [Google Scholar]
- 36.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang FY, Chan AO, Rashid A, Wong DK, Cho CH, Yuen MF. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1β activation of nitric oxide production in gastric cancer cells. Cancer. 2012;118:4969–4980. doi: 10.1002/cncr.27519. [DOI] [PubMed] [Google Scholar]
- 38.Arslan E, Atilgan H, Yavaşoğlu I. The prevalence of Helicobacter pylori in obese subjects. Eur J Intern Med. 2009;20:695–697. doi: 10.1016/j.ejim.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Vaarala O, Yki-Järvinen H. Diabetes: Should we treat infection or inflammation to prevent T2DM? Nat Rev Endocrinol. 2012;8:323–325. doi: 10.1038/nrendo.2012.31. [DOI] [PubMed] [Google Scholar]
- 40.Jeon CY, Haan MN, Cheng C, Clayton ER, Mayeda ER, Miller JW, Aiello AE. Helicobacter pylori infection is associated with an increased rate of diabetes. Diabetes Care. 2012;35:520–525. doi: 10.2337/dc11-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eshraghian A, Pellicano R. Comment on: Jeon et al. Helicobacter pylori infection is associated with an increased rate of diabetes. Diabetes Care 2012; 35: 520-525. Diabetes Care. 2013;36:e20. doi: 10.2337/dc12-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kantartzis K, Machann J, Schick F, Fritsche A, Häring HU, Stefan N. The impact of liver fat vs visceral fat in determining categories of prediabetes. Diabetologia. 2010;53:882–889. doi: 10.1007/s00125-010-1663-6. [DOI] [PubMed] [Google Scholar]
- 43.Bonora E, Targher G, Alberiche M, Bonadonna RC, Saggiani F, Zenere MB, Monauni T, Muggeo M. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care. 2000;23:57–63. doi: 10.2337/diacare.23.1.57. [DOI] [PubMed] [Google Scholar]
- 44.Eshraghian A, Hashemi SA, Hamidian Jahromi A, Eshraghian H, Masoompour SM, Davarpanah MA, Eshraghian K, Taghavi SA. Helicobacter pylori infection as a risk factor for insulin resistance. Dig Dis Sci. 2009;54:1966–1970. doi: 10.1007/s10620-008-0557-7. [DOI] [PubMed] [Google Scholar]
- 45.Naja F, Nasreddine L, Hwalla N, Moghames P, Shoaib H, Fatfat M, Sibai A, Gali-Muhtasib H. Association of H. pylori infection with insulin resistance and metabolic syndrome among Lebanese adults. Helicobacter. 2012;17:444–451. doi: 10.1111/j.1523-5378.2012.00970.x. [DOI] [PubMed] [Google Scholar]
- 46.Denecke B, Gräber S, Schäfer C, Heiss A, Wöltje M, Jahnen-Dechent W. Tissue distribution and activity testing suggest a similar but not identical function of fetuin-B and fetuin-A. Biochem J. 2003;376:135–145. doi: 10.1042/BJ20030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279–1285. doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- 48.Srinivas PR, Wagner AS, Reddy LV, Deutsch DD, Leon MA, Goustin AS, Grunberger G. Serum alpha 2-HS-glycoprotein is an inhibitor of the human insulin receptor at the tyrosine kinase level. Mol Endocrinol. 1993;7:1445–1455. doi: 10.1210/mend.7.11.7906861. [DOI] [PubMed] [Google Scholar]
- 49.Ishibashi A, Ikeda Y, Ohguro T, Kumon Y, Yamanaka S, Takata H, Inoue M, Suehiro T, Terada Y. Serum fetuin-A is an independent marker of insulin resistance in Japanese men. J Atheroscler Thromb. 2010;17:925–933. doi: 10.5551/jat.3830. [DOI] [PubMed] [Google Scholar]
- 50.Stefan N, Fritsche A, Weikert C, Boeing H, Joost HG, Häring HU, Schulze MB. Plasma fetuin-A levels and the risk of type 2 diabetes. Diabetes. 2008;57:2762–2767. doi: 10.2337/db08-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun Q, Cornelis MC, Manson JE, Hu FB. Plasma levels of fetuin-A and hepatic enzymes and risk of type 2 diabetes in women in the U.S. Diabetes. 2013;62:49–55. doi: 10.2337/db12-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kebapcilar L, Bilgir O, Cetinkaya E, Akyol M, Bilgir F, Bozkaya G. The effect of Helicobacter pylori eradication on macrophage migration inhibitory factor, C-reactive protein and fetuin-a levels. Clinics (Sao Paulo) 2010;65:799–802. doi: 10.1590/S1807-59322010000800011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H, Zhang M, Bianchi M, Sherry B, Sama A, Tracey KJ. Fetuin (alpha2-HS-glycoprotein) opsonizes cationic macrophagedeactivating molecules. Proc Natl Acad Sci USA. 1998;95:14429–14434. doi: 10.1073/pnas.95.24.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manolakis AC, Tiaka EK, Kapsoritakis AN, Georgoulias P, Tsiopoulos F, Valotassiou V, Potamianos SP. Increased fetuin A levels in Helicobacter pylori infection: a missing link between H. pylori and insulin resistance? Diabetologia. 2011;54:472–474. doi: 10.1007/s00125-010-1995-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ou HY, Yang YC, Wu HT, Wu JS, Lu FH, Chang CJ. Increased fetuin-A concentrations in impaired glucose tolerance with or without nonalcoholic fatty liver disease, but not impaired fasting glucose. J Clin Endocrinol Metab. 2012;97:4717–4723. doi: 10.1210/jc.2012-2414. [DOI] [PubMed] [Google Scholar]
- 56.Hennige AM, Staiger H, Wicke C, Machicao F, Fritsche A, Häring HU, Stefan N. Fetuin-A induces cytokine expression and suppresses adiponectin production. PLoS One. 2008;3:e1765. doi: 10.1371/journal.pone.0001765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moller DE, Flier JS. Insulin resistance--mechanisms, syndromes, and implications. N Engl J Med. 1991;325:938–948. doi: 10.1056/NEJM199109263251307. [DOI] [PubMed] [Google Scholar]
- 58.Straubinger RK, Greiter A, McDonough SP, Gerold A, Scanziani E, Soldati S, Dailidiene D, Dailide G, Berg DE, Simpson KW. Quantitative evaluation of inflammatory and immune responses in the early stages of chronic Helicobacter pylori infection. Infect Immun. 2003;71:2693–2703. doi: 10.1128/IAI.71.5.2693-2703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crabtree JE, Shallcross TM, Heatley RV, Wyatt JI. Mucosal tumour necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut. 1991;32:1473–1477. doi: 10.1136/gut.32.12.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Basso D, Plebani M, Kusters JG. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2010;15 Suppl 1:14–20. doi: 10.1111/j.1523-5378.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- 61.Suganuma M, Kuzuhara T, Yamaguchi K, Fujiki H. Carcinogenic role of tumor necrosis factor-alpha inducing protein of Helicobacter pylori in human stomach. J Biochem Mol Biol. 2006;39:1–8. doi: 10.5483/bmbrep.2006.39.1.001. [DOI] [PubMed] [Google Scholar]
- 62.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Festa A, D’Agostino R, Tracy RP, Haffner SM. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 2002;51:1131–1137. doi: 10.2337/diabetes.51.4.1131. [DOI] [PubMed] [Google Scholar]
- 65.Lagrand WK, Visser CA, Hermens WT, Niessen HW, Verheugt FW, Wolbink GJ, Hack CE. C-reactive protein as a cardiovascular risk factor: more than an epiphenomenon? Circulation. 1999;100:96–102. doi: 10.1161/01.cir.100.1.96. [DOI] [PubMed] [Google Scholar]
- 66.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 67.Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem. 1993;268:26055–26058. [PubMed] [Google Scholar]
- 68.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 69.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 70.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 71.Stephens JM, Lee J, Pilch PF. Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem. 1997;272:971–976. doi: 10.1074/jbc.272.2.971. [DOI] [PubMed] [Google Scholar]
- 72.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 73.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 74.Ibrahim SH, Kohli R, Gores GJ. Mechanisms of lipotoxicity in NAFLD and clinical implications. J Pediatr Gastroenterol Nutr. 2011;53:131–140. doi: 10.1097/MPG.0b013e31822578db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 76.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Savvidou S, Hytiroglou P, Orfanou-Koumerkeridou H, Panderis A, Frantzoulis P, Goulis J. Low serum adiponectin levels are predictive of advanced hepatic fibrosis in patients with NAFLD. J Clin Gastroenterol. 2009;43:765–772. doi: 10.1097/MCG.0b013e31819e9048. [DOI] [PubMed] [Google Scholar]
- 78.Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, Pedersen AA, Kalthoff C, Tullin S, Sams A, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285:6153–6160. doi: 10.1074/jbc.M109.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 80.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Masaki T, Chiba S, Tatsukawa H, Yasuda T, Noguchi H, Seike M, Yoshimatsu H. Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology. 2004;40:177–184. doi: 10.1002/hep.20282. [DOI] [PubMed] [Google Scholar]
- 82.Capeau J. Insulin resistance and steatosis in humans. Diabetes Metab. 2008;34:649–657. doi: 10.1016/S1262-3636(08)74600-7. [DOI] [PubMed] [Google Scholar]
- 83.Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–1292. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- 84.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 85.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 86.Satoh H, Saijo Y, Yoshioka E, Tsutsui H. Helicobacter Pylori infection is a significant risk for modified lipid profile in Japanese male subjects. J Atheroscler Thromb. 2010;17:1041–1048. doi: 10.5551/jat.5157. [DOI] [PubMed] [Google Scholar]
- 87.Akbas HS, Basyigit S, Suleymanlar I, Kemaloglu D, Koc S, Davran F, Demir I, Suleymanlar G. The assessment of carotid intima media thickness and serum paraoxonase-1 activity in Helicobacter pylori positive subjects. Lipids Health Dis. 2010;9:92. doi: 10.1186/1476-511X-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Figura N, Franceschi F, Santucci A, Bernardini G, Gasbarrini G, Gasbarrini A. Extragastric manifestations of Helicobacter pylori infection. Helicobacter. 2010;15 Suppl 1:60–68. doi: 10.1111/j.1523-5378.2010.00778.x. [DOI] [PubMed] [Google Scholar]
- 89.Abenavoli L, Milic N, De Lorenzo A, Luzza F. A pathogenetic link between non-alcoholic fatty liver disease and celiac disease. Endocrine. 2013;43:65–67. doi: 10.1007/s12020-012-9731-y. [DOI] [PubMed] [Google Scholar]
- 90.Korponay-Szabó IR, Halttunen T, Szalai Z, Laurila K, Király R, Kovács JB, Fésüs L, Mäki M. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. 2004;53:641–648. doi: 10.1136/gut.2003.024836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sandler NG, Koh C, Roque A, Eccleston JL, Siegel RB, Demino M, Kleiner DE, Deeks SG, Liang TJ, Heller T, et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology. 2011;141:1220–1230, 1230.e1-3. doi: 10.1053/j.gastro.2011.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gäbele E, Dostert K, Hofmann C, Wiest R, Schölmerich J, Hellerbrand C, Obermeier F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol. 2011;55:1391–1399. doi: 10.1016/j.jhep.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 93.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 94.Abenavoli L, Arena V, Giancotti F, Vecchio FM, Abenavoli S. Celiac disease, primary biliary cirrhosis and helicobacter pylori infection: one link for three diseases. Int J Immunopathol Pharmacol. 2010;23:1261–1265. doi: 10.1177/039463201002300431. [DOI] [PubMed] [Google Scholar]
- 95.Nilsson I, Lindgren S, Eriksson S, Wadström T. Serum antibodies to Helicobacter hepaticus and Helicobacter pylori in patients with chronic liver disease. Gut. 2000;46:410–414. doi: 10.1136/gut.46.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shapira Y, Agmon-Levin N, Renaudineau Y, Porat-Katz BS, Barzilai O, Ram M, Youinou P, Shoenfeld Y. Serum markers of infections in patients with primary biliary cirrhosis: evidence of infection burden. Exp Mol Pathol. 2012;93:386–390. doi: 10.1016/j.yexmp.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 97.Neuberger J. Primary biliary cirrhosis. Lancet. 1997;350:875–879. doi: 10.1016/S0140-6736(97)05419-6. [DOI] [PubMed] [Google Scholar]
- 98.Alisi A, Feldstein AE, Villani A, Raponi M, Nobili V. Pediatric nonalcoholic fatty liver disease: a multidisciplinary approach. Nat Rev Gastroenterol Hepatol. 2012;9:152–161. doi: 10.1038/nrgastro.2011.273. [DOI] [PubMed] [Google Scholar]
- 99.Mathurin P, Gonzalez F, Kerdraon O, Leteurtre E, Arnalsteen L, Hollebecque A, Louvet A, Dharancy S, Cocq P, Jany T, et al. The evolution of severe steatosis after bariatric surgery is related to insulin resistance. Gastroenterology. 2006;130:1617–1624. doi: 10.1053/j.gastro.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 100.Kashi MR, Torres DM, Harrison SA. Current and emerging therapies in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:396–406. doi: 10.1055/s-0028-1091984. [DOI] [PubMed] [Google Scholar]
- 101.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]