

Abstract
Background: MicroRNAs (miRNAs) are small noncoding RNAs which play crucial role in response to the adverse biotic and abiotic stress conditions at the post transcriptional level. The functions of the miRNAs are generally based on complementarity to their target region. Results: We used the online tool psRNA Target for the identification of submergence responsive miRNA using the gene expression profile related to the submergence condition. We wrote a perl script for the prediction of miRNA target gene. The position based feature of the script increases the overall specificity of the program. Our perl script performed well on the genomic data of Oryza sativa and produced significant results with their positions. These results were analyzed on the basis of complementarity and the statistical scores are used to find out the most probable binding regions. These predicted binding regions are aligned with their respective miRNAs to find out the consensus sequence. We scored the alignment using a position dependent, mismatch penalty system. We also identified the rate of conservation of bases at a particular position for all the predicted binding regions and it was found that all the predicted binding regions maintain above 70% rate of conservation of bases. Conclusion: Our approach provides a novel framework for screening the genome of Oryza sativa. It can be broadly applied to identify complementarity specific miRNA targets computationally by doing a little modification of the script depending on the type of the miRNA.
Background
Flood is a common phenomenon during monsoon season for rice cultivation in the rainfed lowlands of Northeast India. Flash flood occurs at various stages of crop growth. The regulation of gene expression in response to environmental cues is an important factor in plant's survival and adaptability. Most rice varieties are intolerant to flash flood but some have the ability to survive under flash flood. Transcriptome data have shown that the expression levels of a number of genes are altered in the cells of submerged plants. These genes are involved in a broad spectrum of biochemical, cellular and physiological processes, including glycolysis, energy metabolism, lipid metabolism, signal transduction, DNA transcription, protein biosynthesis and digestion, cell components and photosynthesis [1–4]. A few characters were identified that play a key role in submergence tolerance in rice, the most critical are: (a) maintenance of high carbohydrate concentration, (b) optimum rates of alcoholic fermentation (c) energy conservation by maintaining low elongation growth rates during submergence. The discovery of RNA interference (RNAi) has transformed our understanding of gene regulation, mechanisms of heterochromatin formation, and transposons control [5]. The term RNAi has come to encompass an increasingly broad family of related pathways, in which small RNAs from 20–30 nucleotides in length serve as guides to target recognition and regulation. In the canonical RNAi pathway, small RNAs are generated from double-stranded precursors by a ribonuclease enzyme termed, Dicer. Small RNAs act in complex with a second defining component of RNAi-related pathways, the argonaute (AGO) proteins, together forming the RNAinduced silencing complex (RISC) [6]. MicroRNAs (miRNAs) play significant role in response to the adverse biotic and abiotic stress conditions at the post-transcriptional level [7]. They are small, non-coding, single stranded RNAs that are abundantly found in prokaryotic and eukaryotic cells and can trigger translational repression or gene silencing by binding to complementary sequences on target mRNA transcripts. In the recent years, miRNAs have been reported to control a variety of biological processes, such as plant development, differentiation, signal transduction or stress responses [8–10]. Thus identification of miRNA targets is an essential step in understanding the miRNA functions.
A given miRNA may have multiple different mRNA targets, and a given target might similarly be targeted by multiple miRNAs. By coordinating the expression of multiple genes, miRNAs are responsible for guiding the embryonic development, immune and related inflammatory responses, as well as the cellular growth and proliferation. So, with the help of bioinformatics tools it is now possible to determine all the possible targets within the whole genome for a particular miRNA and thereby their cellular responses. The function of a miRNA closely resembles its structure, which can be determined by the bioinformatics program with the use of certain notions of secondary structure because miRNA genes are more conserved in secondary structure than their primary sequences [11]. Of late several miRNA target prediction tools have been developed but they are unable to discriminate the true miRNA targets from non targets. If gene expression profiles of transgenic lines with increased miRNA expression are available, it is possible to do high-throughput and more accurate screening of targets [12]. When the under-expressed genes are extracted, putative targets can be defined and the sets that overlapped with the computationally predicted ones can be targeted. Unfortunately, this kind of high-throughput expression profile dataset is difficult to generate due to the high cost and the labor-intensive experimental process. In this paper, we propose a novel approach for screening miRNA targets. Our approach is based on complementarity and statistical scores. We used a perl script for the prediction of miRNA target gene. At first we looked for which genes are expressed under submerged condition. Then the properties of these genes were analyzed and used as query sequence to find out the submergence responsive miRNAs. The script was written in such a way that it keeps the first eight to nine bases of the miRNA sequence constant to find out the most probable target within the genome of Oryza sativa.
Methodology
Identification of submergence responsive genes:
Genes delivering resistance to rice during the submergence were identified from GRAMENE database. These genes are: ABA 8'- hydroxylase1 (ABA8ox1), Reduced ADH activity (RAD), Submergence tolerance1 (SUB 1), Submergence tolerance2 (SUB 2), Oryza sativa cation transport protein (osCTP). Gene RAD & SUB 2 are not sequenced till now, so only three genes were used for our study. The nucleotide sequences of these genes were downloaded in FASTA format from NCBI database. Accession numbers of the genes are given in the Table 2 (see supplementary material). The comparative sequence statistics of the genes was done with the help of CLC MAIN WORKBENCH 6.8.2. The genomic data of Oryza sativa was downloaded from GRAMENE database.
Identification of Submergence responsive miRNA:
Submergence responsive miRNA were detected using the server psRNA target: A plant small RNA target analysis (http://plantgrn.noble.org/psRNATarget/?function=2). Where target transcript was submitted in FASTA format and searched against the database miRBase (Release 19, August 2012). The parameters used for the search are given in the Table 1 (see supplementary material). Selected miRNA sequences were downloaded in FASTA format from miRBase database. With the help of a perl script the miRNA sequences were converted to DNA format and then to their complementary sequences. Outputs of the program were saved in FASTA format. Which are then used a query sequence to find out the binding region within the genome of Oryza sativa.
Prediction of miRNA binding region within the genome of O.sativa:
A perl script was written to find out the binding region of miRNA (complementarity based) keeping the first eight to ten bases of miRNA constant. The code of the program was written in such a way that it extracts the target sequence and also gives the position of the genome where the miRNA binds. The perl script gives a number of outputs, but the most probable region was selected based on the statistical scores and complementarity values. The regions having mismatch within the range of 0-5 nt with their respective miRNA were selected for further analysis.
Creation of Consensus sequence and identification of their rate of conservation:
Consensus sequence of all the miRNA was created by taking their respective matching regions with the help of CLC MAIN WORKBENCH 6.8.2. The rate of conservation of a particular base at a particular position was also identified using CLC MAIN WORKBENCH 6.8.2.
Prediction of secondary structure of the miRNAs:
The stem loop sequences of the miRNA are downloaded from miRBase for prediction of the secondary structures. The program developed by M.ZUKER (based on thermodynamic score) was used for the construction of secondary structure.
Results & Discussion
Prediction of Submergence responsive miRNA:
Our prediction method predicts the targets by combining the statistical score and sequence information (Figure 1). Before applying our perl script against the genomic data of Oryza sativa we first investigated whether our script gives true result with their exact position by using a small nucleotide sequence. It has been reported that miRNAs affect the expression of a number of target genes involved in different developmental processes and stresses. We expect that both the complementarity and the statistical scores are informative enough to discriminate targets from non-targets. To achieve a good prediction it is important to find out which gene expressed at high rate during the submergence. With the help of GRAMENE database it has been found that the gene ABA 8'-hydroxylase1 (ABA8ox1), Submergence tolerance1 (SUB 1), Oryza sativa cation transport protein (osCTP) are highly expressed during submergence. The nucleotide sequence of these genes were downloaded from NCBI database and used as a user submitted transcript in psRNA target: a plant small RNA target analysis online tool to find out the submergence responsive miRNAs. The results of the search that had been carried out to find the submergence responsive miRNA are given in the Table 3 (see supplementary material). Out of eleven (11) miRNAs it was found that the sequence of osa-miR396a-5p / osa-miR396b-5p and osa-miR821a/osa-miR821b/osa-miR821c were similar, but they have different stem loop sequences. Since this project is sequence based only one of the similar miRNA sequence are taken in to consideration. MicroRNA shows dynamic expression pattern i.e. a given miRNA may have multiple different mRNA targets, and a given target might similarly be targeted by multiple miRNAs. By coordinating the expression of multiple genes, miRNAs are responsible for guiding the embryonic development, immune and related inflammatory responses, as well as the cellular growth and proliferation. So, with the help of bioinformatics applications our aim is to identify all the possible submergence responsive miRNA targets within the whole genome of Oryza sativa and thereby their cellular response.
Figure 1.
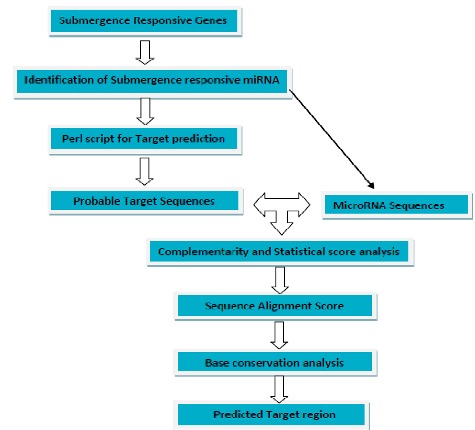
The procedure for computational prediction of miRNA targets within the genome of Oryza sativa
Genome-wide identification of submergence responsive miRNA targets:
Depending on the sequence information, perl script was written for each of these miRNAs to find out their binding position within the genome of Oryza sativa. The position based feature of the script increases the overall specificity of the program. Our perl script performed well on the genomic data of Oryza sativa and produced significant results (allowing mismatch not more than 5 bases) with their positions. The script was written in such a way that it extracts the nucleotide sequence for the entire possible binding region within the genome. The binding region and the target sequence for osa-mir1867 are given in the Table 4 (see supplementary material). So, further analysis of the target sequence can be done along with its respective miRNA. We then perform the analysis to compare the frequency of nucleotides, weight and rate of base conservation for all possible targets with their miRNAs. These predicted binding sequences are aligned with their respective miRNA using the software CLC MAIN WORKBENCH 6.8.2 to find out the consensus sequence. We scored the alignment using a position dependent, mismatch penalty system to find out the most probable binding region. We also identified the rate of conservation of bases at a particular position for all the predicted binding regions and it was found that the all the predicted binding regions maintain above 70% rate of conservation of bases. The frequency (Figure 2), base conservation rate (Figure 3) and weight analysis (Figure 4) was done for all the predicted targets given by the perl program. At the end of all the analysis we have predicted the following targets within the genome of Oryza sativa for submergence responsive miRNAs. We also predicted the secondary structure of the submergence responsive miRNAs (given in the supplementary file) using the algorithm developed by M. Zuker (based on thermodynamics). The algorithm showed 97.99% specificity and 2.01% false positive rate during the prediction of secondary structure of miRNAs.
Figure 2.
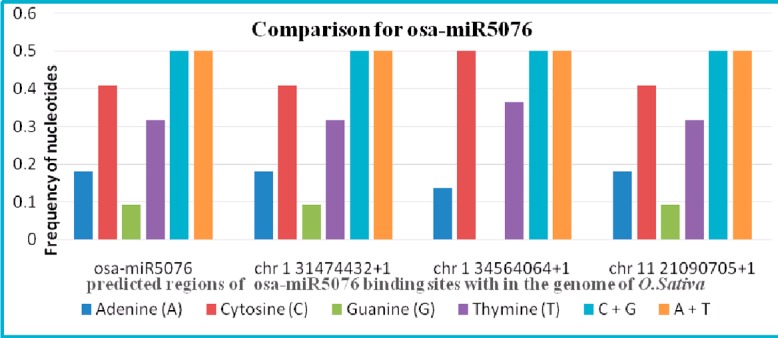
Differences in the frequency of nucleotides among all the predicted binding regions of miR5076 within the genome of O.sativa (Results of nucleotide frequency analysis for all the miRNAs with their target sites are given in the supplementary file)
Figure 3.
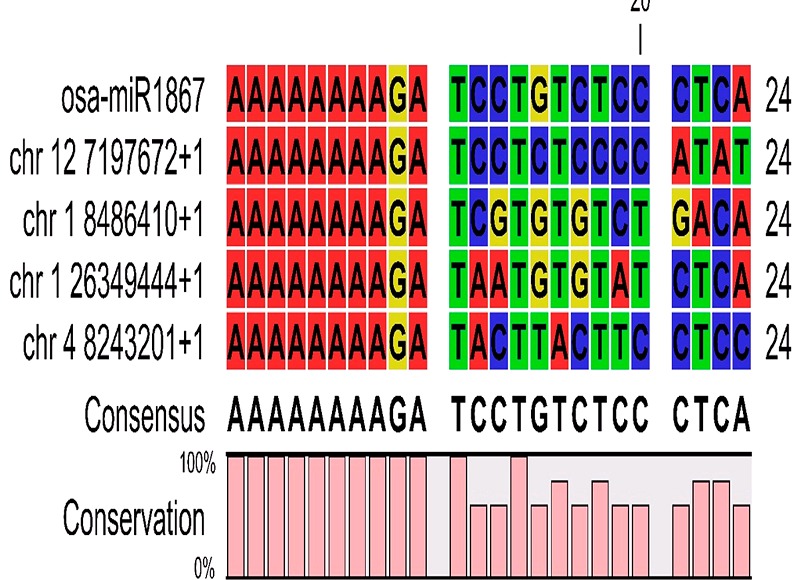
Consensus sequence for Osa-miR1867 and rate of conservation of bases at a particular position of that consensus sequence among all probable binding region present within the genome of O.sativa (Consensus sequence and base conservation analysis results for all the miRNAs are given in the supplementary file)
Figure 4.
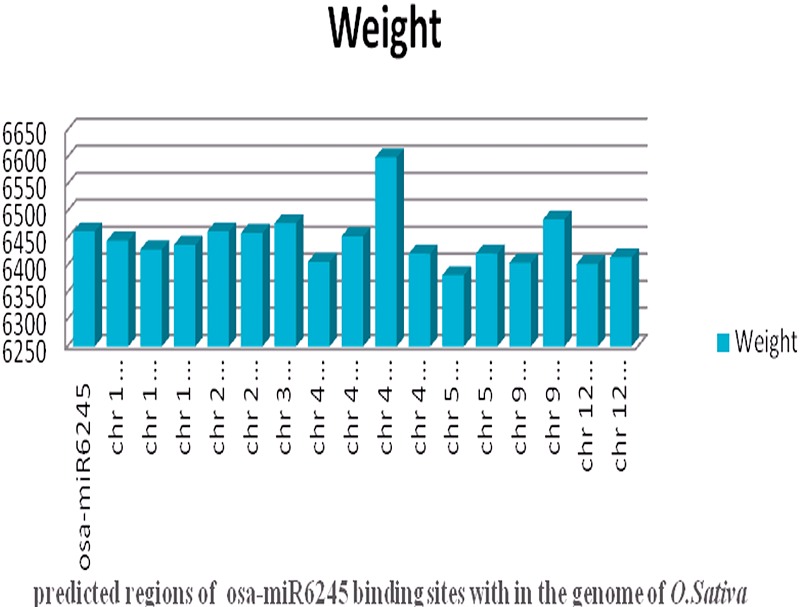
Comparison of weight of all predicted binding regions for osa-miR6245 (Results of weight comparison for all the miRNAs with their target regions are given in the supplementary file)
Discussion
Most rice varieties are intolerant to flood but some varieties have the ability to survive after the occurrence of flood. Submergence tolerance is a genetically determined trait with relatively high stability. However, the duration of survival is also influenced by environmental factors such as water turbidity, temperature, light, and other factors such as seedling age. Plants become more tolerant as they get older. It was observed that microRNA plays crucial role in response to adverse biotic and abiotic stresses at the post transcriptional level (Sunkar et al., 2008). MicroRNAs (miRNAs) are small noncoding RNA gene products about 22-nt long that are found in diverse organisms, including plant and animals. Submergence tolerant rice varieties contain several genes like submergence 1 (SUB1), ABA8ox1, osCTP which allow the plant to survive under 10-14 days of complete submergence and to renew growth when the water recedes. The present study was carried out with the objectives: (a) To identify the genes involved in submergence tolerance.(b)To identify the miRNA that is involved in delivering resistance to plants during submergence.(c)To identify the binding position of these miRNA within the genome of Oryza sativa.(d)To predict the pre-miRNA secondary structure of these miRNAs.
As per our mentioned objectives in this present study, we had predicted a total of eleven (11) submergence responsive miRNAs (osa-miR396a-5p, osa-miR396b-5p, osa-miR396c-5p, osamiR821a, osa-mi8521b, osa-miR821c, osa-miR2919, osa-miR1867, osa-miR5076, osa-miR6245, osa-miR6248) from the genes expressed during submergence. The miRNAs were identified using the online miRNA tool psRNA Target. The parameters used for the prediction were almost same for all the genes except that of the maximum expectation value (range 0.0-5.0). The maximum expectation value 4, 2, 1, 3.5 was used for the genes osCTP (hspsize 18), SUB1A (hspsize 18), SUB1C (hspsize 20) and ABA8ox1 (hspsize 18) respectively. The mature and stem loop sequence of these miRNAs were downloaded from miRBase database. Out of eleven (11) miRNAs it was found that the mature sequence of osa-miR396a-5p / osa-miR396b-5p and osamiR821a/ osa-miR821b/osa-miR821c were similar, but they have different stem loop sequences. Since this project is sequencebased only one of the similar miRNA sequences is taken into consideration. MicroRNA shows dynamic expression i.e. a given miRNA may have multiple different mRNA targets, and a given target might similarly be targeted by multiple miRNAs. The mature miRNA sequences were used as query sequence in this study for the identification of their most probable binding region within the genome of O. sativa. The genomic data of Oryza sativa was downloaded from GRAMENE database. In this study we used a perl script to predict the submergence responsive miRNA, binding sites within the genome of Oryza sativa. The perl script was written in such a way that it keeps the first eight to ten bases of the miRNA constant (depending on the length of the miRNA) to find out the miRNA binding site within the genome. The position based feature of the script increases the overall specificity of the program. Our perl script performed well on the genomic data of Oryza sativa and produced significant results (allowing mismatch not more than 5 bases) with their positions. The script gives 16 probable binding sites for osa-miR396a /b, 22 binding sites for osa-miR396c, 13 binding sites for osamiR821a/ b/c, 4 binding sites for osa-miR1867, 100 binding sites for osa-miR2919, 3 binding sites for osa-miR5076, 16 binding sites for osa-miR6245, and 26 binding sites for osa-miR6248 within the genome of O.sativa. These results were analyzed on the basis of complementarity and the statistical scores are used to find out the most probable binding regions. The predicted target location for all the submergence responsive miRNAs are given in the Table 5 (see supplementary material). These predicted binding regions are aligned with their respective miRNAs using the software BioEdit to find out the consensus sequence. We scored the alignment using a position dependent, mismatch penalty system. We also identified the rate of conservation of bases at a particular position for all the predicted binding regions and it was found that the all the predicted binding regions maintain above 70% rate of conservation of bases. However, the predicted regions need to be verified in wet lab. It is important here to mention that the function of miRNA closely resembles its structure. Proper understanding of the structure function relationship requires knowledge about the three dimensional structure of the miRNA molecule, which is very difficult to obtain and time consuming. Moreover, it has been reported that MIRgenes are more conserved in the secondary structure than in primary structure [11]. We predicted the secondary structure of the submergence responsive miRNAs using the algorithm developed by M. Zuker (based on thermodynamics). The algorithm showed 97.99% specificity and 2.01% false positive rate during the prediction of secondary structure of miRNAs.
Conclusion
Our results suggested that the perl script we used for analysis has the potential to discriminate miRNA targets from nontargets. The combination of complementarity and statistical scoring based method ensures retrieval of true targets. We have shown in O. sativa that the target region related to the miRNA was successfully extracted with their positions by the program. The above study could be significantly helpful in understanding the dynamic expression pattern of the miRNAs associated with submergence condition. Further research work might be taken up for identifying the exact location of the target site of the specific miRNA within the genome. If the miRNA binds to the exonic region of a gene within the genome, it might be concluded that the miRNA could play a significant role in the expression of the gene. However, if the miRNA binds to the intronic or intergenic site (junk) within the genome, it might be involved in either up-regulation or down-regulation of gene expression in the downstream region.
Supplementary material
Footnotes
Citation:Paul & Chakraborty, Bioinformation 9(17): 858-863 (2013)
References
- 1.Klok, et al. Plant Cell. 2002;14:2481. doi: 10.1105/tpc.004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu F, et al. Plant Physiol. 2005;137:1115. doi: 10.1104/pp.104.055475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang, et al. Russian J Plant Physl. 2005;52:43. [Google Scholar]
- 4.Zhang, et al. Environ Exp Bot. 2006;58:53. [Google Scholar]
- 5.Fire A, et al. Nature. 1998;391:806. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 6.Hammond SM, et al. Nature. 2000;404:293. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 7.Sunkar R, et al. BMC Plant Biol. 2008;8:25. doi: 10.1186/1471-2229-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang HW, Mendell JT. Br J Cancer. 2007;96:R40. [PubMed] [Google Scholar]
- 9.Ovanovic M, Hengartner MO. Oncogene. 2006;25:6176. doi: 10.1038/sj.onc.1209912. [DOI] [PubMed] [Google Scholar]
- 10.Sunkar R, Zhu JK. Plant Cell. 2004;16:2001. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Wang X. Nucleic Acids Res. 2006;34:1646. doi: 10.1093/nar/gkl068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuker M, et al. Nucleic Acids Res. 2003;31:3406. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.