

Abstract
Coronary artery disease and associated ischemic heart disease are prevalent disorders worldwide. Further, systemic hypertension is common and markedly increases the risk for heart disease. A common denominator of systemic hypertension of various etiologies is increased myocardial load/mechanical stress. Thus, it is likely that high pressure/mechanical stress attenuates the contribution of cardioprotective but accentuates the contribution of cardiotoxic pathways thereby exacerbating the outcome of an ischemia reperfusion insult to the heart. Critical events which contribute to cardiomyocyte injury in the ischemic-reperfused heart include cellular calcium overload and generation of reactive oxygen/nitrogen species which, in turn, promote the opening of the mitochondrial permeability transition pore, an important event in cell death. Increasing evidence also indicates that the myocardium is capable of mounting a robust inflammatory response which contributes importantly to tissue injury. On the other hand, cardioprotective maneuvers of ischemic preconditioning and postconditioning have led to identification of complex web of signaling pathways (e.g., reperfusion injury salvage kinase) which ultimately converge on the mitochondria to exert cytoprotection. The present review is intended to briefly describe mechanisms of cardiac ischemia reperfusion injury followed by a discussion of our work focused on how pressure/mechanical stress modulates endogenous cardiotoxic and cardioprotective mechanisms to ultimately exacerbate ischemia reperfusion injury.
Keywords: Heart, ischemia-reperfusion, pressure, calcium overload, oxidative/nitrosative stress, signaling mechanisms, inflammation, stem cells
Introduction
Systemic hypertension is a common disorder with global prevalence estimates of 1 billion individuals. It accounts for an estimated 7.6 million deaths each year and for 13.5% of total mortalities, more than any other single risk factor [1-4]. In the United States, the data from 2007-2010 indicate that 33% of adults 20 years or older have hypertension which represents about 78 million American adults; the prevalence is nearly equal between men and women although African-Americans are among those with highest prevalence of hypertension (44%) in the world [5]. Of interest is the fact that the population of the United States continues to increase and the Census Bureau projects it to almost reach 440 million by the year 2050 (an increase of about 130 million from 2010) [6,7]. Importantly, the proportion of patients greater than 65 years of age is increasing at a greater rate than the total population. This is reflected by the data indicating that while the total population increased by 9.7% between 2000 and 2010, those older than 65 years increased by 15.1%. Further, the greatest proportional increases over this 10-year period occurred in the oldest age groups, with a 29.9% increase in those 85-94 years of age and a 25% increase in those greater than 95 years of age [6,8]. Population projection data spanning 2010-2050 also support the general conclusion that greater proportional increase occurs in those 65 years or older and that among them those older than 85 years of age show the greatest increase [7]. Since the prevalence of hypertension increases with age and hypertension represents an accumulation of years of pressure overload on target organs, hypertension-related clinical sequels (e.g., ischemic heart disease and myocardial infarction) will become even more pressing challenges for the health care system.
The propensity of the hypertensive heart to ischemic events is multifactorial including a) epicardial coronary stenosis (e.g., due to atherosclerosis) and b) cardiac microvascular disease and endothelial dysfunction, accompanied with ultrastructural remodeling of cardiac microvessels, that can result in progressive impairment of flow-mediated vasodilation. Other factors can be arterial stiffness with long standing hypertension and accompanying increased left ventricular afterload and central pulse pressure; the concomitant fall in central diastolic pressure reduces coronary perfusion, further exacerbating myocardial ischemia [9-14]. Importantly, alterations in energy metabolism of the hypertensive heart also increase susceptibility to ischemia. This notion is supported by the findings that patients with hypertension have measurably lower phosphocreatine to adenosine triphosphate ratios during stress compared to healthy controls [15].
As discussed above, atherosclerosis is the predominant underlying cause of coronary heart disease which can result in myocardial infarction with ischemic death of cardiomyocytes [16,17]. Reperfusion of the acutely or chronically ischemic myocardium (e.g., via thrombolysis, percutaneous coronary angioplasty and/or coronary bypass) is essential in order to salvage the myocardium [17,18]; yet, injury to the endothelium and cardiomyocytes occurs upon reperfusion [19,20]. Reperfusion-induced injury is also a significant clinical problem in cardiac transplantation or during open heart surgery when the myocardium is subjected to global ischemic cardioplegic arrest [21,22]. The following section provides an overview of some key events in myocardial ischemia reperfusion injury prior to discussion of how pressure overload modulates these mechanisms to exacerbate the outcome of an ischemia reperfusion insult to the heart.
Mechanisms of myocardial ischemia reperfusion injury
Hallmark features of myocardial ischemia reperfusion (IR) injury include marked oxidative/nitrosative stress and intracellular calcium ([Ca2+]i) overload (Figure 1). During ischemia, a reduction in mitochondrial energy production ensues that is accompanied by decreased intracellular pH (pHi) due to increased lactic acid production consequent to anaerobic glycolysis. The reduction in pHi, in turn, causes disruption of ionic homeostasis and subsequent [Ca2+]i overload. This occurs because during ischemia, activation of sarcolemmal Na+/H+ exchanger occurs as the cell attempts to restore its pHi. However, the Na+ that enters the cell on the Na+/H+ exchanger is not pumped out efficiently because a fall in ATP and an increase in phosphate (Pi) inhibit the Na+/K+-ATPase. As a result, the Na+/Ca2+ exchanger, that normally extrudes Ca2+ from the cell, is inhibited or even reversed thereby raising [Ca2+]i. However, it is upon reperfusion that a much greater rise in [Ca2+]i occurs which contributes to the genesis of ventricular arrhythmia and myocardial stunning [20,23-27] (Figure 1). Although the rise in [Ca2+]i is attributed primarily to reversal of the Na+/Ca2+ exchanger and the L-type Ca2+ channel [24-27], T-type Ca2+ channels have also been implicated in this phenomenon [28]. Further, resumption of ATP synthesis upon reperfusion may activate sarcoplasmic reticulum Ca2+ cycling resulting in cytosolic Ca2+ oscillations and propagation of Ca2+ waves [29]. Consequences of [Ca2+]i overload include activation of degradative enzymes including proteases, phospholipases and nucleases that can cause irreversible tissue injury [27].
Figure 1.

Diagram depicts critical events in cardiac ischemia reperfusion injury [27].
Reactive oxygen species (ROS), on the other hand, are generated primarily through mitochondrial respiratory chain, NAD(P)H oxidase and xanthine oxidase during myocardial IR injury [25-27]. Detrimental consequences of ROS which contribute to tissue injury include: a) impairment of respiratory chain activity (e.g., complex I), b) plasma membrane damage with subsequent impairment of ion pumps thereby exacerbating the effects of ATP deprivation on cellular ionic homeostasis and c) peroxidation of unsaturated fatty acid components of the membrane phospholipids; this will render them more susceptible to attack by phospholipase A2 whose activity may already be elevated by [Ca2+]i overload [25-28,30,31].
The large burst of ROS and [Ca2+]i overload upon reperfusion of the ischemic heart are major triggers for the mitochondrial permeability transition (MPT) pore [25,27,30,31]. The MPT pore is a non-specific conduit that is formed at the site of contact between mitochondrial inner and outer membranes which allows for solute flux of less than about 1.5 kDa size. The exact molecular composition of the MPT pore remains controversial although use of genetically modified mice suggest an important regulatory role for cyclophilin D; loss of cyclophilin D reduces the sensitivity of MPT pore to activation by calcium or during ischemia and reperfusion [32,33]. Induction of MPT pore by [Ca2+]i overload and ROS is facilitated by decreased mitochondrial membrane potential and increased Pi levels, conditions that are present during myocardial IR injury. In addition, restoration of pHi at reperfusion also triggers MPT pore induction [20,23,27,34-37]. Opening of the MPT pore allows solutes and water to enter the mitochondria thereby increasing matrix volume. As a result, mitochondrial outer membrane ruptures facilitating release of cytochrome c which, in turn, promotes apoptosis. In addition, MPT pore induction uncouples the mitochondria, leading to inhibition of ATP synthesis and hydrolysis of the ATP that is derived from glycogen breakdown eventually causing cell death by necrosis [25,34,36-39]. Thus, both necrotic and apoptotic cell death occur during IR injury. The pivotal roles of these processes, in mediating myocardial IR injury, are underlined by numerous studies indicating that cardioprotections of both ischemic preconditioning and postconditioning are associated with reductions in generation of ROS, calcium overload and MPT pore opening [26,31,37,39-44]; ischemic preconditioning describes the phenomenon whereby several brief bouts of ischemia and reperfusion prior to a more prolonged ischemic phase (i.e., index ischemia) confers significant protection to the ischemic-reperfused heart while postconditioning refers to cardioprotection conferred by restoration of coronary circulation to the ischemic myocardium in a stuttering fashion [31,40-51] (Figure 2). While considerable attention has focused on cardioprotection of ischemic preconditioning and postconditioning maneuvers [31,40-51], a novel and intriguing paradigm has emerged which advocates that targeted modulation of autophagy could exert beneficial effects in stressful conditions such as IR injury; autophagy is a highly-regulated cellular “housekeeping” process for the degradation and disposal of protein aggregates and dysfunctional/damaged organelles (e.g., mitochondria in a process referred to as mitophagy). Indeed, recognition of the dichotomous “life-or-death” patho (physiological) role of autophagy has led to considerable research focused on harnessing its cardioprotective potential [52]. In support of this notion, upregulation of autophagy has been suggested to play a causal role in infarct-sparring effect of both ischemic-preconditioning and postconditioning [53-55].
Figure 2.
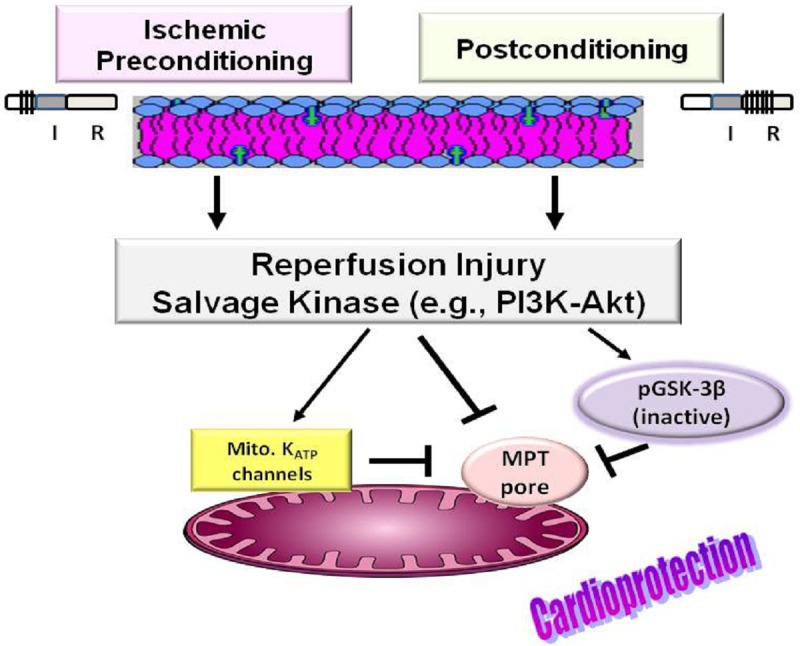
Diagram shows that cardioprotection of ischemic preconditioning (e.g. 3 bouts of ischemia (I) and reperfusion (R), each for 5 min, before a prolonged ischemia phase) and postconditioning (e.g., 6 cycles of I and R, each for 30 sec, at reperfusion) is associated with activation of the reperfusion injury salvage kinase (RISK) pathway. An important outcome relates to upregulation of phosphatidylinositol-3 kinase (PI3K)/Akt pathway which, in turn, leads to phosphorylation and inactivation of glycogen synthase kinase-3β (GSK-3β) culminating in inhibition of the mitochondrial permeability transition (MPT) pore. Also, shown is mitochondrial (mito.) KATP channels whose activation confers cardioprotection, likely through inhibition of MPT pore induction.
The earlier discovery of ischemic preconditioning led to a surge of interest in unraveling cardioprotective mechanisms [37,45-49]; this well-studied phenomenon is also referred to as early or classical preconditioning to distinguish it from the delayed phase or second window of protection [23]. Also the more recently discovered phenomenon of postconditioning has generated much interest in understanding of its underlying mechanisms because it is a more clinically relevant and amenable maneuver than ischemic preconditioning [50-52]. While the exact mechanism(s) of either ischemic preconditioning or postconditioning remains elusive, considerable progress has been made towards a better understanding of the signal transduction pathways that convey the extracellular signal generated by these cardioprotective maneuvers to intracellular targets [30]. As a result, it is now known that cardioprotection involves activation of a diverse array of prosurvival signaling pathways collectively referred to as the reperfusion injury salvage kinase (RISK) pathway [19,30] (Figure 2). Components of RISK include protein kinases C, G, and A, members of the mitogen activated protein kinase family (e.g., extracellular-regulated kinase 1/2, P38, c-jun-N terminal kinase) and the phosphatidylinositol-3 kinase-protein kinase B/Akt (PI3K-PKB/Akt) cascade [30]. While the contribution of individual protein kinases is often the subject of intense debate, it is increasingly recognized that certain survival protein kinases (e.g., PI3K-Akt) are shared by both ischemic preconditioning and postconditioning protocols [19,30], likely accounting for the observation that combination of both protocols does not further reduce infarct size than either maneuver alone [51]. Tyrosine kinase is a well-recognized upstream activator of the PI3K-Akt cascade [30]. In turn, many of the downstream targets phosphorylated by Akt activate various anti-apoptotic pathways (e.g., epsilon isoform of protein kinase C and nitric oxide synthase to generate nitric oxide) [30]. The involvement of these prosurvival pathways is further substantiated by the demonstration that several endogenous cardioprotective agents, such as adenosine, mimic ischemic preconditioning by activating a pathway that is modulated by certain isozymes (e.g., epsilon) of protein kinase C in rabbit or rat heart [47,56-59]. Indeed, generation of adenosine during postconditioning also causes eventual activation of PI3K-Akt and subsequent downstream activation of protein kinase C epsilon [30]. However, debate prevails regarding the nature of the “end effector” of the signaling pathway, with some investigators suggesting the mitochondrial ATP-sensitive K+ (mito. KATP) channels while others further indicate that the opening of mito. KATP channels leads to mild oxidative stress, activating one or more protein kinases that stimulate an “unidentified” end effector [19,30,45-48]. Activation of mito. KATP channels is believed to result in a number of effects including reduction in mitochondrial Ca2+ accumulation and prevention of cytochrome C loss from the intermembrane space [42,43,49]. The importance of this cascade of events is illustrated by the effectiveness of mito. KATP channel inhibitors (e.g., glibenclamide) in blocking the beneficial effects of a wide range of cardioprotective agents and protocols (e.g., adenosine, opioids, ischemic preconditioning and postconditioning) [28,47,48,51]. On the other hand, mito. KATP channel openers (e.g., diazoxide) mimic the effect of preconditioning [47-49,60,61]. Further, studies utilizing isolated mitochondria indicate that pharmacological activation of protein kinase C protects against MPT pore opening under the same conditions in which diazoxide is protective [49]. While the targets of ischemic (or pharmacologic) preconditioning and postconditioning are likely to be multiple (e.g., protein kinase C/mito. KATP channels), the key signaling pathways ultimately must converge to prevent MPT pore induction (e.g., during reperfusion) to reduce infarct size [19,20,27,30,42-46,62-64].
Given the pivotal role of the MPT pore in myocardial IR injury, intense research has focused on mechanisms regulating the MPT pore; these studies have identified glycogen synthase kinase-3β (GSK-3β) as a critical regulator of the MPT pore (Figure 2). GSK-3β is a serine-threonine kinase which is best known for its regulation of glycogen metabolism. However, GSK-3β is now recognized as a multifunctional kinase responsible for phosphorylation of more than 20 substrates. GSK-3β is primarily localized in the cytosol and is constitutively active. However, multiple kinases (e.g., Akt/protein kinase B) can phosphorylate it at serine 9 residue, rendering it inactive. Indeed, phosphorylation of GSK-3β by multiple signaling pathways (e.g., components of RISK) is believed to increase the activation threshold of MPT pore thereby conferring cardioprotection [63]. For example, Juhaszova and colleagues [64] showed that the threshold for ROS-induced MPT pore opening was elevated by GSK-3β inactivation or by its knockdown using siRNA in isolated cardiomyocytes. Further, Gomez et al. [65] showed that MPT pore opening by [Ca2+]i overload was suppressed in mitochondria isolated from postconditioned wild type mice but not those from mice expressing a mutated form (i.e., GSK-S9A) which is insensitive to phosphorylation at serine 9. Additional evidence in support of phosphoGSK-3β-mediated inhibition of MPT pore opening in response to ROS and calcium overload comes from numerous studies including a) those using pharmacological inhibitors of GSK-3β (e.g., LiCl, SB216763, SB415286), b) those utilizing δ-opioid receptor agonists, adenosine A2b receptor agonists or erythropoietin and c) studies using cardioprotective maneuvers of ischemic preconditioning or postconditioning, among others [63]. Although the role of GSK-3β in regulation of MPT pore is indisputable, it is not yet clear how its inactivation causes MPT pore inhibition. Nonetheless, a number of possibilities have been proposed including mitochondrial translocation of phosphoGSK-3β and complex formation with cyclophilin D ultimately leading to cardioprotection [66]. Importantly, gene targeting studies indicate that cyclophilin D is a component of the MPT pore [67] and cyclosporine A-induced inhibition of cyclophilin D or genetic deletion of cyclophilin D significantly limits infarct size [34,37,63,68].
With that background in mind, the following section describes our studies focused on the impact of pressure overload on myocardial IR injury and the underlying mechanisms.
Impact of myocardial load on the outcome of myocardial IR insult
As mentioned earlier, systemic hypertension is an established risk factor for coronary heart disease and it also adversely affects the outcome of acute myocardial infarction. The latter is corroborated by reports indicating that acute elevation in blood pressure increases while acute reduction in blood pressure reduces susceptibility to IR injury [69-76]. Nonetheless, the interpretation of these studies is confounded by neurohumoral adaptations that accompany changes in blood pressure, some of which are known to impact the outcome of IR injury independent of the blood pressure [77-79]. For our studies focused on pressure-related effects on cardiac IR injury, we have used the isolated heart preparation in order to avoid the confounding influences of neurohumoral changes that accompany chronic or acute elevation of blood pressure. These studies were based on the premise that increase in myocardial load is a common denominator of systemic hypertension of various etiologies. It is noteworthy that for determination of pressure-related effects on the outcome of IR injury we have adjusted the pressure-head of the Langendorff-perfused heart (e.g., 80 or 160 cmH2O). This maneuver significantly increases both the coronary flow and the contractile parameters of the heart. In order to decipher the impact of the increase in coronary flow rate, per se, we carried out additional experiments using the constant flow perfusion protocol as detailed previously [80]. Comparison of data from experiments whereby the pressure-head is adjusted against the heart with those whereby coronary flow rate is adjusted indicates that the primary determinant of the outcome of an IR insult, in our studies, relates to the pressure and associated mechanical stress/load on the myocardium [80].
Our initial studies, using 36-week-old hypertensive and glucose intolerant rats, revealed that isolated hearts subjected to a high pressure (i.e., 160 cmH2O or about 118 mmHg) display a significant increase in infarct size in response to an IR insult compared to those subjected to a low pressure (i.e., 80 cmH2O or about 59 mmHg) [81]. We reasoned that the disease states in the aging rat and pre-existing cardiomyopathy, per se, may have rendered the heart susceptible to the impact of elevated pressure. In order to prevent the confounding influences of disease states and aging, we carried out subsequent IR protocols using healthy adult rats (9-11 weeks of age). Accordingly, we established that pressure overload significantly increases infarct size in association with poorer functional recovery following an IR insult [82]. Subsequent studies explored potential contributing mechanisms to the adverse impact of high pressure on the ischemic-reperfused heart. Thus, we tested the hypothesis that elevated pressure, and associated mechanical stress, accentuates the contribution of cytotoxic pathways (e.g., calcium overload, oxidative stress/nitrosative stress, etc.) and/or attenuates the contribution of cardioprotective pathways (e.g., mito. KATP channels, PI3K-Akt signaling pathway); the net effect of these changes would be increased MPT pore opening and consequent cell death (Figure 3). The details of these studies are presented below.
Figure 3.

Diagram summarizes our working hypothesis that pressure overload accentuates cardiotoxic but attenuates cardioprotective mechanisms thereby causing exacerbation of myocardial ischemia-reperfusion injury.
Effect of myocardial load on calcium overload
As described earlier, a hallmark feature of cardiac IR injury is [Ca2+]i overload which exerts multiple effects including induction of MPT pore thereby contributing to cell death [82]. One consequence of increased myocardial load is activation of the angiotensin II type 1 receptor and nonspecific cation channels with subsequent Ca2+ accumulation via the Na+/H+-Na+/Ca2+ exchanger combination and the T-type or L-type Ca2+ channels. Since [Ca2+]i overload is cytotoxic, in part, by inducing the MPT pore, we also explored the effect of cyclosporine A-induced inhibition of MPT pore in pressure overloaded hearts. Accordingly, the effect of candesartan (angiotensin II type 1 receptor antagonist), cariporide (inhibitor of the Na+/H+ exchanger), mibefradil (T-type Ca2+ channel blocker), diltiazem (L-type Ca2+ channel blocker), and cyclosporine A (inhibitor of MPT pore) were examined. The elevation in perfusion pressure, from 80 to 160 cmH2O, increased baseline myocardial performance but caused larger infarcts and further reduced recovery of mechanical function after ischemia reperfusion. Whereas mibefradil abrogated the effect of high pressure on infarct size, the other agents reduced infarct size at both perfusion pressures. Hearts exposed to mibefradil, diltiazem, or cariporide displayed greater functional recovery than those exposed to candesartan or cyclosporine A, revealing that an uncoupling exists between reduced cell death and recovery of mechanical function of the viable portions of the myocardium. Collectively, the data suggested an important link between pressure-mediated worsening of infarct size and exacerbation of [Ca2+]i overload (e.g., via T type channels). Nonetheless, it is noteworthy that the contribution of sarcoplasmic reticulum to [Ca2+]i overload in the ischemic-reperfused heart is now established [29]. Accordingly, resumption of ATP synthesis upon reperfusion activates sarcoplasmic reticulum Ca2+ cycling. Sarcoplasmic reticulum Ca2+ cycling is promoted by cytosolic Ca2+ overload and consequent Ca2+ uptake through the sarco(endo)plasmic reticulum Ca2+-ATPase followed by Ca2+ release through the ryanodine receptors when the Ca2+ storage capacity of the organelle is exhausted. These changes cause Ca2+ oscillations that propagate as Ca2+ waves and are believed to facilitate partial mitochondrial permeabilization due to close anatomic proximity between the two organelles thereby favoring hypercontracture and cell death [29]. In light of the profound impact of pressure overload on the ischemic-reperfused heart, potential pressure-related regulation of sarcoplasmic reticulum Ca2+ cycling should be established.
Effect of myocardial load on oxidative/nitrosative stress
Excessive ROS generation is a critical event in myocardial IR injury. During ischemia, low levels of ROS are generated which can damage the electron transport chain thereby causing inefficient transfer of electrons with consequent increase in ROS generation. With availability of oxygen during early reperfusion, a large burst of ROS occurs which plays a pivotal role in the genesis of reperfusion-induced injury. Important cardiac sources of ROS (e.g., superoxide) include the mitochondrial respiratory chain distal to complex I (NADH dehydrogenase), xanthine oxidase and NAD(P)H oxidase [26,27,31,83]. While the myocardium possesses endogenous antioxidant defenses, such as the superoxide dismutase (SOD) and catalase, these mechanisms can be overwhelmed following ischemia and reperfusion. In turn, these conditions are conducive to the interaction of superoxide with nitric oxide to produce peroxynitrite, a potent oxidant. Consequently, exacerbated oxidative/nitrosative stress serves as a major trigger for the MPT pore opening and subsequent cell death [25,83].
In light of the pivotal role of oxidative/nitrosative stress in cardiac IR injury, we sought to determine whether pressure overload exacerbates oxidative/nitrosative stress due to increased generation of reactive substances or reduced ability to scavenge ROS thereby promoting greater MPT pore opening with consequent exacerbation of cell death via necrosis and/or apoptosis. Pressure overload decreased the level of reduced glutathione but increased that of nitrotyrosine (a stable footprint of peroxynitrite) level in ischemic-reperfused hearts. Further, pressure overload increased DNA injury as demonstrated by increased 8-hydroxydeoxyguanosine (an index of oxidative DNA damage) and γH2AX (a sensitive marker of double strand DNA breaks, the most severe form of DNA injury) [80,83]. The activity of catalase, but not SOD, was lower in ischemic-reperfused hearts perfused at higher pressure. Mitochondria isolated from ischemic-reperfused hearts subjected to higher perfusion pressure displayed significantly greater [3H]-2-deoxyglucose-6-Pi entrapment suggestive of greater MPT pore opening and this was consistent with greater necrosis and apoptosis as determined by flow cytometry. Tempol (SOD mimetic) reduced infarct size in hearts subjected to low or high perfusion pressure but it remained greater in the higher pressure group. By contrast, uric acid (peroxynitrite scavenger) markedly reduced infarct size at higher pressure, effectively eliminating the differential between the two groups. Inhibition of xanthine oxidase, with allopurinol, reduced infarct size but did not eliminate the differential between the low and high pressure groups. However, amobarbital (inhibitor of mitochondrial complex I) or apocynin (inhibitor of NAD(P)H oxidase) reduced infarct size at both pressures and also abrogated the differential between the two groups. Consistent with the effect of apocynin, pressure-overloaded hearts displayed significantly higher NAD(P)H oxidase activity. Furthermore, pressure-overloaded hearts displayed increased nitric oxide synthase activity which, along with increased propensity to superoxide generation, may underlie uric acid-induced cardioprotection. Collectively, these observations indicate that increased oxidative/nitrosative stress, coupled with lack of augmented SOD and catalase activities, contributes importantly to the exacerbating impact of pressure overload on MPT pore opening and cell death in ischemic-reperfused hearts [83].
As alluded to above, exacerbated oxidative/nitrosative stress in pressure overloaded ischemic-reperfused heart augments DNA injury. In turn, DNA injury leads to activation of poly (ADP-ribose) polymermase-1 (PARP) in order to facilitate DNA repair in a process which consumes NAD+ [84]. Importantly, however, hyperactivation of PARP has been linked to mitochondria-mediated necrosis although the precise mechanism remains elusive. Nonetheless, a recent study proposes that oxidative stress, MPT pore and PARP activity contribute to a single death pathway in the ischemic-reperfused heart. Accordingly, a provocative mechanism has been proposed whereby PARP-mediated prolongation of mitochondrial depolarization contributes significantly to cell death via an energy crisis (e.g., consequent to depletion of NAD+ thereby limiting ATP generation) rather than by mitochondrial outer membrane rupture. In addition, PARP activity could directly inhibit mitochondrial transport of adenine nucleotides, preventing cytosolic ATP access to the matrix where it could facilitate repolarization. Consequently, ATP depletion would result in sustained depolarization ultimately causing mitochondrial failure and plasma membrane rupture without affecting the integrity of the outer membrane of the mitochondria. In support of the important contribution of PARP hyperactivation to cell death, its inhibition has been shown to exert significant cardioprotection [85-87]. In light of exacerbated DNA injury in pressure overloaded ischemic-reperfused hearts, in a pilot study, we tested the hypothesis that inhibition of PARP would confer greater protection under the high pressure condition. Interestingly, however, our initial observations suggest that while treatment with 4-hydorxyquanozoline (a PARP inhibitor) significantly reduces infarct size of the ischemic-reperfused heart subjected to the low pressure, the treatment seemingly does not confer significant protection under the high pressure condition (unpublished data); the dose-related possibility for this observation is under investigation. Thus, establishing pressure-related regulation of PARP in the ischemic-reperfused heart and its relation to MPT pore status is a fertile ground for exploration.
Effects of myocardial load on cardioprotective mechanisms
The MPT pore may serve as the end-effector of cardioprotective mechanisms, namely the mitochondrial KATP channels and GSK-3β [19,20,30,60,61,63-66,68]. Therefore, in light of our demonstration that augmented MPT pore induction contributes to pressure overload-induced exacerbation of infarct size [82,83], we sought to determine whether elevation in perfusion pressure attenuates cardioprotection associated with activation of mitochondrial KATP channels or inhibition of GSK-3β. Further, we also determined whether perfusion pressure modulates the regulation of the MPT pore by mitochondrial KATP channels and/or GSK-3β. These studies used diazoxide (a mitochondrial KATP channel opener), glibenclamide (inhibitor of KATP channels), lithium chloride (LiCl, a nonselective inhibitor of GSK-3β), SB-216763 (a selective inhibitor of GSK-3β), cyclosporine A (inhibitor of MPT pore induction) and the combination of cyclosporine A and glibenclamide or the combination of glibenclamide and LiCl. As expected, the increase in perfusion pressure in the absence of a drug caused larger infarcts, an effect associated with poorer recovery of function following ischemia reperfusion. Treatment with either diazoxide or cyclosporine A reduced infarct size at both perfusion pressures but cyclosporine A was more protective, than diazoxide, at the higher pressure. On the other hand, LiCl and SB-216763 reduced infarct size at both pressures, with the effect more marked at the higher perfusion pressure. Glibenclamide did not affect infarct size but eliminated the cardioprotective effect of cyclosporine A while having no effect on LiCl-induced cardioprotection [68]. Collectively, the results indicate that perfusion pressure primarily affects GSK-3β-mediated regulation of MPT pore formation in the ischemic reperfused heart.
As mentioned earlier, GSK-3β is downstream of the PI3K/protein kinase B (Akt) pathway. Indeed, the cardioprotection of postconditioning and insulin relates to activation of the PI3K/Akt pathway [88]. Thus, we conjectured that pressure overload attenuates postconditioning- and insulin-induced cardioprotection, an effect caused by reduced PI3K-Akt signaling. The contribution of PI3K/Akt pathway was assessed in the context of determining the levels of relevant proteins and their phosphorylation status including the 3’-phosphoinositide dependent kinase 1 (PDK-1) and phosphatase and tensin homolog on chromosome ten (PTEN); PDK-1 and PTEN are positive and negative regulators of the PI3K/Akt signaling pathway, respectively. To further establish the role of myocardial load, we also determined whether pressure unloading (i.e., switchover from high to low pressure immediately upon reperfusion of the ischemic heart) confers cardioprotection comparable to either postconditioning or insulin treatment [88].
Pressure overload increased infarct size in association with changes in protein levels consistent with reduced PI3K-Akt signaling (i.e., ischemic reperfused vs. normoxic hearts). Postconditioning and insulin treatment reduced infarct size but it was greater in hearts perfused at the higher, than the lower, pressure. Wortmannin (a PI3K inhibitor) partially reversed postconditioning-induced cardioprotection, with infarct size being greater in the high-pressure group. Pressure unloading during reperfusion caused the most marked reduction in infarct size whereas pressure loading abolished postconditioning-induced cardioprotection. Nonetheless, the phospho-Akt/total Akt ratio and phospho-GSK-3beta levels were unaffected by perfusion pressure in insulin-treated or postconditioned hearts. Moreover, protein levels were similar in pressure-unloaded and pressure-loaded hearts. Collectively, these observations indicate that pressure overload reduces PI3K-Akt signaling following IR. However, a differential in PI3K-Akt signaling was not observed in ischemia-reperfused, insulin-treated, and postconditioned hearts, suggesting involvement of pathways other than PI3K-Akt for the effect of pressure on infarct size under these conditions. Potential players include members of mitogen-activated protein kinases which remain to be explored. Importantly, however, these studies also revealed that pressure unloading at reperfusion represents a novel and effective cardioprotective maneuver [88].
Effect of myocardial load on inflammation and the role of GSK-3β
The contribution of systemic immune and inflammatory mechanisms to the outcome of myocardial IR injury is well-established [89-94]. Importantly, however, it is increasingly recognized that the myocardium can mount a robust inflammatory response to an IR insult [95,96]. The growth arrest- and DNA-damage inducible protein 153 (GADD153) regulates both apoptosis and inflammatory response [97,98]. Importantly, GSK-3β may provide a mechanistic link for cellular expression of GADD153, inflammatory response and cell death [99,100]. In light of our demonstration that pressure overload exacerbates myocardial IR injury associated with significant reduction in phosphorylated (inactive) GSK-3β level, we conjectured that pressure overload, through a GSK-3β-dependent mechanism, increases GADD153 expression, thereby upregulating inflammatory cytokine production and contributing to worsening of myocardial IR injury [80]. In the ischemic-reperfused hearts, pressure overload reduced the anti-inflammatory cytokine, interleukin (IL)-10, but increased pro-inflammatory cytokine, IL-17 without affecting IL-23 (a pro-inflammatory cytokine). Subsequent immunofluorescent labeling studies showed colocalization of IL-17 immunostaining with the brain natriuretic peptide indicating that the cardiomyocyte is a major source of IL-17. Subsequently, using flow cytometry, we have shown co-expression of IL-17 and IL-23 suggestive of cardiomyocyte generation of IL-23 too (Figure 4). These observations substantiate the robust ability of endogenous cardiac mechanisms to mount an inflammatory response following an IR insult. Other effects of the pressure overload in the ischemic-reperfused heart included increased expression of GADD153, decreased JC-1 aggregates but increased JC-1 monomers (suggestive of reduced mitochondrial membrane potential (ψm)) in association with increased annexin V immunostaining as well as apoptotic and necrotic cell death. Importantly, treatment with LiCl (an inhibitor of GSK-3β) caused a robust increase in IL-10, preserved ψm and markedly decreased other parameters (e.g., IL-17 and GADD153) with the effect being most prominent for hearts perfused at the high pressure. Collectively, these observations indicate that pressure overload, via a GSK-3β-dependent mechanism, exacerbates cell death in the isolated ischemic-reperfused heart involving regulation of GADD153 expression and inflammatory response [80]. It is noteworthy that while both an IR insult and pressure overload regulate cardiac cytokine production, the link and mechanisms between cytokine production and cell death remain to be established. However, of interest is a recent report suggesting synergistic interaction between IL-17 ad tumor necrosis factor-α (TNF-α) in augmenting oxidative stress and apoptosis of oligodendrocytes [101]. TNF-α generation also increases in the isolated ischemic-reperfused heart [102]. Therefore, further studies should explore pressure-related regulation of mitochondrial death pathway by pro-inflammatory cytokines in the ischemic-reperfused heart.
Figure 4.

Panel shows a dot matrix from flow cytometry-based assessment of cardiac cells from ischemic-reperfused hearts that co-express IL-17 and IL-23 (upper right quadrant; indicated by asterisk).
Effect of bone marrow-derived stem cells on the outcome of IR injury
The heart is now known to have resident stem cells; yet, the endogenous reparative capacity of the myocardium seems unable to replenish marked loss of cardiomyocytes which occur following acute myocardial infarction [103,104]. Consequently, in view of the prevailing ethical considerations about use of embryonic stem cells, attention has focused on the potential usefulness of adult stem cells particularly given the demonstration that bone marrow-derived stem cells (BMDSCs) can be recruited into the heart and transdifferentiate into cardiomyocytes and cells of vascular lineage [105-110]. Indeed, the therapeutic usefulness of BMDSCs in the setting of acute myocardial infarction is now well-established. Nonetheless, it is increasingly recognized that the principal mechanism underlying the beneficial effects of BMDSCs does not relate to their ability to transdifferentiate to cardiomyocytes, smooth muscle and endothelial cells. Rather, BMDSCs release a whole host of cytokines, chemokines and growth factors which then exert their effects via paracrine fashion [106,109,110]. As a result, they promote a local microenvironment and cytokine milieu conducive to reducing initial damage to the injurious stimulus (i.e., ischemia and/or reperfusion insult) and also promote repair and recovery of the damaged tissue.
In light of the above, we have carried out pilot studies to determine whether administration of BMDSCs would confer protection to the pressure-overloaded ischemic-reperfused heart. We utilized a protocol which has been extensively used by Meldrum and colleagues whereby Langendorff-perfused rat heart is transplanted via intracoronary administration of stem cells prior to induction of global ischemia [111-114]. As expected, pressure overload increased cell death in vehicle-treated ischemic-reperfused hearts. Treatment with Sca1+ cells reduced cell death with the effect more prominent for hearts subjected to high, than low, perfusion pressure (Figure 5). Thus, our ongoing studies are focused on establishing the impact of BMDSCs on pressure-related cardiac cytokine production along with assessment of mitochondrial status and cell death. In this context, it is of interest to establish whether empowering BMDSCs (through up- or down-regulation of relevant genes) would abrogate the adverse impact of pressure overload on the ischemic-reperfused heart.
Figure 5.

Scatter plots depict early apoptotic (green), late apoptotic (blue) and necrotic (red) cell death in cardiac cell preparations of ischemic-reperfused hearts that were subjected to either 80 or 160 cmH2O. Immediately before the ischemic phase, hearts were transplanted (through the coronary arteries) with Sca1+ cells.
Conclusion
Systemic hypertension is a very common disorder worldwide. Further, since the prevalence of systemic hypertension increases with age, it represents an accumulation of years of pressure overload on target organs. Thus, hypertension-related clinical sequels, such as ischemic heart disease and myocardial infarction, will continue to present pressing challenges. While the myocardium can develop adaptive mechanisms to cope with stress, such mechanisms usually fail in the long-term thereby increasing its venerability to insults including ischemic events. Surprisingly, however, the vast majority of studies focusing on mechanisms of ischemia reperfusion injury have not taken into consideration the impact of myocardial load/mechanical stress. Our studies over the last decade indicate critical dependency of key components of endogenous cardiac mechanisms on myocardial load. Accordingly, reduced contribution of cardioprotective pathways coupled with augmented activity of cardiotoxic pathways predispose the pressure overloaded heart to exacerbated ischemia reperfusion injury (Figure 6). Further elucidation of mechanisms that are differentially regulated by myocardial load should lead to identification of novel therapeutic target(s).
Figure 6.

Schematic diagram showing major relevant pathways involved in the effect of pressure overload on the ischemic reperfused heart. An ischemia reperfusion insult exerts multiple and diverse effects including a) increased oxidative/nitrosative stress, b) intracellular calcium overload, c) downregulation of cardioprotection of PI3K-Akt/GSK-3β pathway and d) enhanced inflammatory responses, in part, through a GSK-3β-dependent mechanism involving increased GADD153 expression. Consequently, dysregulation of mitochondrial membrane potential leads to induction of the MPT pore. These changes are augmented by pressure overload, culminating in exacerbation of cell death/infarct size.
Acknowledgements
The studies presented in this manuscript were supported by the National Institutes of Health, American Heart Association, Southeast Affiliate and Institutional Seed Funds.
Disclosure of conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Tulman DB, Stawicki SP, Papadimos TJ, Murphy CV, Bergese SD. Advances in management of acute hypertension: a concise review. Discov Med. 2012;13:375–383. [PMC free article] [PubMed] [Google Scholar]
- 2.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–223. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 3.Deaton C, Froelicher ES, Wu LH, Ho C, Shishani K, Jaarsma T. The global burden of cardiovascular disease. J Cardiovasc Nurs. 2011;26(Suppl 4):S5–14. doi: 10.1097/JCN.0b013e318213efcf. [DOI] [PubMed] [Google Scholar]
- 4.Arima H, Barzi F, Chalmers J. Mortality patterns in hypertension. J Hypertens. 2011;29(Suppl 1):S3–S7. doi: 10.1097/01.hjh.0000410246.59221.b1. [DOI] [PubMed] [Google Scholar]
- 5.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Michol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive Summary: Heart Disease and Stroke Statistics—2013 Update: A Report From the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 6.Vigen R, Maddox TM, Allen LA. Aging of the United States Population: Impact on Heart Failure. Curr Heart Fail Rep. 2012;9:369–374. doi: 10.1007/s11897-012-0114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.United States Census Bureau. Projections of the Population by Selected Age Groups and Sex for the United States: 2010 to 2050. 2008. Available at www.census.gov.
- 8.Werner CA. The Older Population: 2010. The 2010 Census Briefs. 2011 Nov [Google Scholar]
- 9.Raman SV. The hypertensive heart. An integrated understanding informed by imaging. J Am Coll Cardiol. 2010;55:91–96. doi: 10.1016/j.jacc.2009.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lapu-Bula R, Ofili E. From hypertension to heart failure: role of nitric oxide-mediated endothelial dysfunction and emerging insights from myocardial contrast echocardiography. Am J Cardiol. 2007;99:7D–14D. doi: 10.1016/j.amjcard.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Pilz G, Klos M, Ali E, Hoefling B, Scheck R, Bernhardt P. Angiographic correlations of patients with small vessel disease diagnosed by adenosine-stress cardiac magnetic resonance imaging. J Cardiovasc Magn Reson. 2008;10:8. doi: 10.1186/1532-429X-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bigi R, Bestetti A, Strinchini A, Conte A, Gregori D, Brusoni B, Fiorentini C. Combined assessment of left ventricular perfusion and function by gated single-photon emission computed tomography for the risk stratification of high-risk hypertensive patients. J Hypertens. 2006;24:767–773. doi: 10.1097/01.hjh.0000217861.12617.33. [DOI] [PubMed] [Google Scholar]
- 13.Schuijf JD, Bax JJ, Jukema JW, Lamb HJ, Vliegen HW, van der Wall EE, de Roos A. Noninvasive evaluation of the coronary arteries with multislice computed tomography in hypertensive patients. Hypertension. 2005;45:227–232. doi: 10.1161/01.HYP.0000152201.79955.e4. [DOI] [PubMed] [Google Scholar]
- 14.London GM, Guerin AP. Influence of arterial pulse and reflected waves on blood pressure and cardiac function. Am Heart J. 1999;138:220–224. doi: 10.1016/s0002-8703(99)70313-3. [DOI] [PubMed] [Google Scholar]
- 15.Lamb HJ, Beyerbacht HP, van der Laarse A, Stoel BC, Doornbos J, van der Wall EE, de Roos A. Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation. 1999;99:2261–2267. doi: 10.1161/01.cir.99.17.2261. [DOI] [PubMed] [Google Scholar]
- 16.Leuschner F, Nahrendorf M. Molecular imaging of coronary atherosclerosis and myocardial infarction: considerations for the bench and perspectives for the clinic. Circ Res. 2011;108:593–606. doi: 10.1161/CIRCRESAHA.110.232678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 18.Magro M, Garg S, Serruys PW. Revascularization treatment of stable coronary artery disease. Expert Opin Pharmacother. 2011;12:195–212. doi: 10.1517/14656566.2010.517522. [DOI] [PubMed] [Google Scholar]
- 19.Gross GJ, Auchampach JA. Reperfusion injury: does it exist? J Mol Cell Cardiol. 2007;42:12–8. doi: 10.1016/j.yjmcc.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honda HM, Korge P, Weiss JN. Mitochondria and Ischemia/Reperfusion Injury. Ann NY Acad Sci. 2005;1047:248–258. doi: 10.1196/annals.1341.022. [DOI] [PubMed] [Google Scholar]
- 21.Luciani GB, Forni A, Rigatelli G, Chiominto B, Cardaioli P, Mazzucco A, Faggian G. Myocardial protection in heart transplantation using blood cardioplegia: 12-year outcome of a prospective randomized trial. J Heart Lung Transplant. 2011;30:29–36. doi: 10.1016/j.healun.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 22.Laubach VE, French BA, Okusa MD. Targeting of adenosine receptors in ischemia-reperfusion injury. Expert Opin Ther Targets. 2011;15:103–118. doi: 10.1517/14728222.2011.541441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2005;14:170–175. doi: 10.1016/j.carpath.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Khandoudi N, Bernard M, Cozzone P, Feuvray D. Intracellular pH and role of Na+/Ca2+ exchange during ischaemia and reperfusion of normal and diabetic rat hearts. Cardiovasc Res. 1990;24:873–878. doi: 10.1093/cvr/24.11.873. [DOI] [PubMed] [Google Scholar]
- 25.Wong R, Steenbergen C, Murphy E. Mitochondrial permeability transition pore and calcium handling. Methods Mol Biol. 2012;810:235–242. doi: 10.1007/978-1-61779-382-0_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vanden Hoek TL, Shao Z, Li C, Schumacker PT, Becker LB. Myocardial electron transport can be a significant source of oxidative injury in cardiomyocytes. J Mol Cell Cardiol. 1997;29:2441–2450. doi: 10.1006/jmcc.1997.0481. [DOI] [PubMed] [Google Scholar]
- 27.Suleiman MS, Halestrap AP, Griffiths EJ. Mitochondria: a target for myocardial protection. Pharmacol Ther. 2001;89:29–46. doi: 10.1016/s0163-7258(00)00102-9. [DOI] [PubMed] [Google Scholar]
- 28.Mocanu MM, Gadgil S, Yellon DM, Baxter GF. Mibefradil, a T-type and L-type calcium channel blocker, limits infarct size through a glibenclamide-sensitive mechanism. Cardiovasc Drugs Ther. 1999;13:115–122. doi: 10.1023/a:1007732025184. [DOI] [PubMed] [Google Scholar]
- 29.Ruiz-Meana M, Abellan A, Miro-Casas E, Agullo E, Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol. 2009;297:H1281–H1289. doi: 10.1152/ajpheart.00435.2009. [DOI] [PubMed] [Google Scholar]
- 30.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 31.Zweier JL, Talukder MAH. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 32.Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–1191. doi: 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- 33.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 34.Borutaite V, Brown GC. Mitochondria in apoptosis of ischemic heart. FEBS Lett. 2003;541:1–5. doi: 10.1016/s0014-5793(03)00278-3. [DOI] [PubMed] [Google Scholar]
- 35.Schild L, Keilhoff G, Augustin W, Reiser G, Striggow F. Distinct Ca2+ thresholds determine cytochrome c release or permeability transition pore opening in brain mitochondria. FASEB J. 2001;15:565–567. doi: 10.1096/fj.00-0551fje. [DOI] [PubMed] [Google Scholar]
- 36.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia—reperfusion injury. Cardiovasc Res. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 37.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–543. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 38.Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84:143–152. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- 39.Walters AM, Porter GA Jr, Brookes PS. Mitochondria as a Drug Target in Ischemic Heart Disease and Cardiomyopathy. Circ Res. 2012;111:1222–1236. doi: 10.1161/CIRCRESAHA.112.265660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Lisa F, Bernardi P. Mitochondria and ischemia—reperfusion injury of the heart: Fixing a hole. Cardiovasc Res. 2006;70:191–199. doi: 10.1016/j.cardiores.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res. 2004;94:7–16. doi: 10.1161/01.RES.0000108082.76667.F4. [DOI] [PubMed] [Google Scholar]
- 42.Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–524. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004;287:H841–H849. doi: 10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- 44.Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111:194–197. doi: 10.1161/01.CIR.0000151290.04952.3B. [DOI] [PubMed] [Google Scholar]
- 45.Schulz R, Cohen MV, Behrends M, Downey JM, Heusch G. Signal transduction of ischemic preconditioning. Cardiovasc Res. 2001;52:181–198. doi: 10.1016/s0008-6363(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 46.Sommerschild HT, Kirkeboen KA. Preconditioning-endogenous defense mechanisms of the heart. Acta Anaesthesiol Scand. 2002;46:123–137. doi: 10.1034/j.1399-6576.2002.460202.x. [DOI] [PubMed] [Google Scholar]
- 47.O’Rourke B. Myocardial KATP channels in preconditioning. Circ Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- 48.Fryer RM, Hsu AK, Gross GJ. Mitochondrial KATP channel opening is important during index ischemia and following myocardial reperfusion in ischemic preconditioned rat hearts. J Mol Cell Cardiol. 2001;33:831–834. doi: 10.1006/jmcc.2001.1350. [DOI] [PubMed] [Google Scholar]
- 49.Korge P, Honda HM, Weiss JN. Protection of cardiac mitochondria by diazoxide and protein kinase C: Implications for ischemic preconditioning. Proc Natl Acad Sci U S A. 2002;99:3312–3317. doi: 10.1073/pnas.052713199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 51.Zhao ZQ, Vinten-Johansen J. Postconditioning: reduction of reperfusion-induced injury. Cardiovasc Res. 2006;70:200–211. doi: 10.1016/j.cardiores.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 52.Przyklenk K, Dong Y, Undyala VV, Whittaker P. Autophagy as a therapeutic target for ischaemia/reperfusion injury? Cardiovasc Res. 2012;94:197–205. doi: 10.1093/cvr/cvr358. [DOI] [PubMed] [Google Scholar]
- 53.Gurusamy N, Leki I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13:373–387. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM Jr, Gottlieb RA. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2012;3:365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inagaki K, Hahn HS, Dorn GW, Mochly-Rosen D. Additive protection of the ischemic heart Ex vivo by combined treatment with delta–protein kinase C inhibitor and Є-protein kinase C activator. Circulation. 2003;108:869–875. doi: 10.1161/01.CIR.0000081943.93653.73. [DOI] [PubMed] [Google Scholar]
- 57.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular distribution of total protein kinase C activity. Circ Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 58.Zhao TC, Kukreja RC. Protein kinase C-delta mediates adenosine A3 receptor-induced delayed cardioprotection in mouse. Am J Physiol Heart Circ Physiol. 2003;285:H434–441. doi: 10.1152/ajpheart.00095.2003. [DOI] [PubMed] [Google Scholar]
- 59.Hassouna A, Matata BM, Galinanes M. PKC-Є is upstream and PKC-α is downstream of mitoKATP channels in the signal transduction pathway of ischemic preconditioning of human myocardium. Am J Physiol Cell Physiol. 2004;287:C1418–C1425. doi: 10.1152/ajpcell.00144.2004. [DOI] [PubMed] [Google Scholar]
- 60.Baines CP, Liu GS, Birincioglu M, Critz SD, Cohen MV, Downey JM. Ischemic preconditioning depends on interaction between mitochondrial KATP channels and actin cytoskeleton. Am J Physiol. 1999;276:H1361–H1368. doi: 10.1152/ajpheart.1999.276.4.H1361. [DOI] [PubMed] [Google Scholar]
- 61.Garlid KD, Santos PD, Xie ZJ, Costa ADT, Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochem Biophys Acta. 2003;1606:1–21. doi: 10.1016/s0005-2728(03)00109-9. [DOI] [PubMed] [Google Scholar]
- 62.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore during myocardial reperfusion- a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 63.Miura T, Tanno T. Mitochondria and GSK-3β in cardioprotection against ischemia/reperfusion injury. Cardiovasc Drugs Ther. 2010;24:255–263. doi: 10.1007/s10557-010-6234-z. [DOI] [PubMed] [Google Scholar]
- 64.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3 beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–2168. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- 66.Xi J, Wang H, Mueller RA, Norfleet EA, Xu Z. Mechanism for resveratrol-induced cardioprotection against reperfusion injury involves glycogen synthase kinase 3β and mitochondrial permeability transition pore. Eur J Pharmacol. 2009;604:111–116. doi: 10.1016/j.ejphar.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baines CP. The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2009;46:850–857. doi: 10.1016/j.yjmcc.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mozaffari MS, Schaffer SW. Effect of pressure overload on cardioprotection of mitochondrial KATP channels and GSK-3beta: interaction with the MPT pore. Am J Hypertens. 2008;21:570–575. doi: 10.1038/ajh.2008.25. [DOI] [PubMed] [Google Scholar]
- 69.Lawes CM, Bennett DA, Lewington S, Rodgers A. Blood pressure and coronary heart disease: a review of the evidence. Semin Vasc Med. 2002;2:355–368. doi: 10.1055/s-2002-36765. [DOI] [PubMed] [Google Scholar]
- 70.Gustafsson F, Kober L, Torp-Pedersen C, Hildebrandt P, Ottesen MM, Sonne B, Carlsen J. Long-term prognosis after acute myocardial infarction in patients with a history of arterial hypertension. Eur Heart J. 1998;19:588–594. doi: 10.1053/euhj.1997.0822. [DOI] [PubMed] [Google Scholar]
- 71.Njolstad I, Arnesen E. Preinfarction blood pressure and smoking are determinants for a fatal outcome of myocardial infarction: a prospective analysis from the Finnmark Study. Arch Intern Med. 1996;158:1326–1332. doi: 10.1001/archinte.158.12.1326. [DOI] [PubMed] [Google Scholar]
- 72.Herlitz J, Bang A, Karlson BW. Five-year prognosis after acute myocardial infarction in relation to a history of hypertension. Am J Hypertens. 1996;9:70–76. doi: 10.1016/0895-7061(95)00302-9. [DOI] [PubMed] [Google Scholar]
- 73.Rutherford JD, Pfeffer MA, Moye LA, Davis BR, Flaker GC, Kowey PR, Lamas GA, Miller HS, Packer M, Roulens JL. Effects of captopril on ischemic events after myocardial infarction. Results of the Survival and Ventricular Enlargement trial. Circulation. 1994;90:1731–1738. doi: 10.1161/01.cir.90.4.1731. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Dorado D, Theroux P, Elizaga J, Fernandez Aviles F, Alonso J, Solares J. Influence of tachycardia and arterial hypertension on infarct size in the pig. Cardiovasc Res. 1988;22:620–626. doi: 10.1093/cvr/22.9.620. [DOI] [PubMed] [Google Scholar]
- 75.Dellsperger KC, Clothier JL, Hartnett JA, Haun LM, Marcus ML. Acceleration of the wavefront of myocardial necrosis by chronic hypertension and left ventricular hypertrophy in dogs. Circ Res. 1988;63:87–96. doi: 10.1161/01.res.63.1.87. [DOI] [PubMed] [Google Scholar]
- 76.Yoshiyama M, Kamimori K, Shimada Y, Omura T, Kino N, Yoshikawa J. Left ventricular remodeling after myocardial infarction in antecedent hypertensive patients. Hypertens Res. 2005;28:293–299. doi: 10.1291/hypres.28.293. [DOI] [PubMed] [Google Scholar]
- 77.Broadley KJ, Penson PE. The roles of alpha- and beta-adrenoceptor stimulation in myocardial ischaemia. Auton Autacoid Pharmacol. 2004;24:87–93. doi: 10.1111/j.1474-8673.2004.00324.x. [DOI] [PubMed] [Google Scholar]
- 78.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–1453. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 79.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 80.Baban B, Liu JY, Mozaffari MS. Pressure Overload Regulates Expression of Cytokines, γH2AX, an Growth Arrest- and DNA-Damage Inducible Protein 153 via Glycogen Synthase Kinase-3β in Ischemic-Reperfused Hearts. Hypertension. 2013;61:95–104. doi: 10.1161/HYPERTENSIONAHA.111.00028. [DOI] [PubMed] [Google Scholar]
- 81.Mozaffari MS, Schaffer SW. Effect of hypertension and hypertension-glucose intolerance on myocardial ischemia-reperfusion injury. Hypertension. 2003;42:1042–1049. doi: 10.1161/01.HYP.0000095614.91961.40. [DOI] [PubMed] [Google Scholar]
- 82.Mozaffari MS, Patel C, Schaffer SW. Mechanisms underlying afterload-induced exacerbation of myocardial infarct size. Role of T-type Ca2+ channel. Hypertension. 2006;47:1–8. doi: 10.1161/01.HYP.0000209940.65941.46. [DOI] [PubMed] [Google Scholar]
- 83.Mozaffari MS, Baban B, Liu JY, Abebe W, Sullivan JC, El-Marakby A. Mitochondrial Complex I and NADP(H) Oxidase Are Major Sources of Exacerbated Oxidative Stress in Pressure-Overloaded Ischemic-Reperfused Hearts. Basic Res Cardiol. 2011;106:287–297. doi: 10.1007/s00395-011-0150-7. [DOI] [PubMed] [Google Scholar]
- 84.Schriewer JM, Peek CB, Bass J, Schumacker PT. ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia reperfusion. J Am Heart Assoc. 2013;2:e000159. doi: 10.1161/JAHA.113.000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oh KS, Lee S, Yi KY, Seo HW, Koo HN, Lee BH. A novel and orally active poly(ADP-ribose) polymerase inhibitor, KR-33889 [2-[methoxycarbonyl(4-methoxyphenyl) methylsulfanyl] -1H-benzimidazole-4-carboxylic acid amide] , attenuates injury in in vitro model of cell death and in vivo model of cardiac ischemia. J Pharmacol Exp Ther. 2009;328:10–8. doi: 10.1124/jpet.108.143719. [DOI] [PubMed] [Google Scholar]
- 86.Song ZF, Ji XP, Li XX, Wang SJ, Wang SH, Zhang Y. Inhibition of the activity of poly (ADP-ribose) polymerase reduces heart ischaemia/reperfusion injury via suppressing JNK-mediated AIF translocation. J Cell Mol Med. 2008;12:1220–1228. doi: 10.1111/j.1582-4934.2008.00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang LQ, Qi GX, Jiang DM, Tian W, Zou JL. Increased poly ADP-ribosylation in peripheral leukocytes and the reperfused myocardium tissue of rats with ischemia/reperfusion injury: prevention by 3-aminobenzamide treatment. Shock. 2012;37:492–500. doi: 10.1097/SHK.0b013e31824989d7. [DOI] [PubMed] [Google Scholar]
- 88.Mozaffari MS, Liu JY, Schaffer SW. Effect of pressure overload on cardioprotection via PI3K-Akt: Comparison of postconditioning, insulin, and pressure unloading. Am J Hypertens. 2010;23:668–674. doi: 10.1038/ajh.2010.43. [DOI] [PubMed] [Google Scholar]
- 89.Arslan F, de Kleijn D, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 90.Hohensinner PJ, Niessner A, Huber K, Weyand CM, Wojta J. Inflammation and cardiac outcome. Curr Opin Infect Dis. 2011;24:259–264. doi: 10.1097/QCO.0b013e328344f50f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Valeur HS, Valen G. Innate immunity and myocardial adaptation to ischemia. Basic Res Cardiol. 2009;104:22–32. doi: 10.1007/s00395-008-0756-6. [DOI] [PubMed] [Google Scholar]
- 92.Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011;108:1133–1145. doi: 10.1161/CIRCRESAHA.110.226936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ha T, Liu L, Kelley J, Kao R, Williams D, Li C. Toll-like receptors: new players in myocardial ischemia/reperfusion injury. Antioxid Redox Signal. 2011;15:1875–1893. doi: 10.1089/ars.2010.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taqueti VR, Mitchell RN, Lichtman AH. Protecting the pump: controlling myocardial inflammatory responses. Annu Rev Physiol. 2006;68:67–95. doi: 10.1146/annurev.physiol.68.040104.124611. [DOI] [PubMed] [Google Scholar]
- 95.Wang M, Baker L, Tsai BM, Meldrum KK, Meldrum DR. Sex differences in the myocardial inflammatory response to ischemia-reperfusion injury. Am J Physiol Endocrinol Metab. 2005;288:E321–E326. doi: 10.1152/ajpendo.00278.2004. [DOI] [PubMed] [Google Scholar]
- 96.Hermann JL, Markel TA, Abarbanell AM, Weil BR, Wang M, Wang Y, Tan J, Meldrum DR. Pro-inflammatory stem cell signaling in cardiac ischemia. Antioxid Redox Signal. 2009;11:1883–1896. doi: 10.1089/ars.2009.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Meares GP, Mines MA, Beurel E, Eom TY, Song L, Zmijewska AA, Jope RS. Glycogen synthase kinase-3 regulates endoplasmic reticulum (ER) stress-induced CHOP expression in neuronal cells. Exp Cell Res. 2011;317:1621–1628. doi: 10.1016/j.yexcr.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, Ogawa H, Oike Y. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- 99.Klamer G, Song E, Ko KH, O’Brien TA, Dolnikov A. Using small molecule GSK3β inhibitors to treat inflammation. Curr Med Chem. 2010;17:2873–2881. doi: 10.2174/092986710792065090. [DOI] [PubMed] [Google Scholar]
- 100.Gao HK, Yin Z, Zhou N, Feng XY, Gao F, Wang HC. Glycogen synthase kinase 3 inhibition protects the heart from acute ischemia-reperfusion injury via inhibition of inflammation and apoptosis. J Cardiovasc Pharmacol. 2008;52:286–292. doi: 10.1097/FJC.0b013e318186a84d. [DOI] [PubMed] [Google Scholar]
- 101.Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic activity of interleukin-17 and tumor necrosis factor-α enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem. 2011;116:508–521. doi: 10.1111/j.1471-4159.2010.07136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Poynter JA, Herrmann JL, Manukyan MC, Wang Y, Abarbanell AM, Weil BR, Brewster BD, Meldrum DR. Intracoronary mesenchymal stem cells promote postischemic myocardial functional recovery, decrease inflammation, and reduce apoptosis via a signal transducer and activator of transcription 3 mechanism. J Am Coll Surg. 2011;213:253–260. doi: 10.1016/j.jamcollsurg.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 103.Dawn B, Bolli R. Bone marrow cells for cardiac regeneration: the quest for the protagonist continues. Cardiovasc Res. 2005;65:293–295. doi: 10.1016/j.cardiores.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 104.Leri A, Kajstura J, Anversa P. Role of cardiac stem cells in cardiac pathophysiology: A paradigm shift in human myocardial biology. Circ Res. 2011;109:941–961. doi: 10.1161/CIRCRESAHA.111.243154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105.Herrmann JL, Abarbanell AM, Weil BR, Manukyan MC, Poynter JA, Brewster BJ, Wang Y, Meldrum DR. Optimizing stem cell function for the treatment of ischemic heart disease. J Surg Res. 2011;166:138–145. doi: 10.1016/j.jss.2010.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms ina dult stem cell signaling and therapy. Circ Res. 2008;103:1204–1219. doi: 10.1161/CIRCRESAHA.108.176826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shi Y, Hu G, Su J, Li W, Chen Q, Shou P, Xu C, Chen X, Huang Y, Zhu Z, Huang X, Han X, Xie N, Ren G. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell Res. 2010;20:510–518. doi: 10.1038/cr.2010.44. [DOI] [PubMed] [Google Scholar]
- 108.Yagi H, Soto-Gutierrez A, Parekkadan B, Kitagawa Y, Tompkins RG, Kobayashi N, Yarmush ML. Mesenchymal stem cells: mechanisms of immunomodulation and homing. Cell Transplant. 2010;19:667–679. doi: 10.3727/096368910X508762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wojakowski W, Ratajczak MZ, Tendera M. Mobilization of very small embryonic-like stem cells in acute coronary syndromes and stroke. Herz. 2010;35:467–473. doi: 10.1007/s00059-010-3389-0. [DOI] [PubMed] [Google Scholar]
- 110.Ii M, Nishimura H, Iwakura A, Wecker A, Eaton E, Asahara T, Losordo DW. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via “imported“ nitric oxide synthase activity. Circulation. 2005;111:1114–1120. doi: 10.1161/01.CIR.0000157144.24888.7E. [DOI] [PubMed] [Google Scholar]
- 111.Herrmann JL, Wang Y, Abarbanell AM, Weil BR, Tan J, Meldrum DR. Preconditioning mesenchymal stem cells with transforming growth factor-alpha improves mesenchymal stem cell-mediated cardioprotection. Shock. 2010;33:24–30. doi: 10.1097/SHK.0b013e3181b7d137. [DOI] [PubMed] [Google Scholar]
- 112.Kelly ML, Wang M, Crisostomo PR, Abarbanell AM, Herrmann JL, Weil BR, Meldrum DR. TNF receptor 2, not TNF receptor 1, enhances mesenchymal stem cell-mediated cardiac protection following acute ischemia. Shock. 2010;33:602–607. doi: 10.1097/SHK.0b013e3181cc0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Markel TA, Wang Y, Herrmann JL, Crisostomo PR, Wang M, Novotny NM, Herring CM, Tan J, Lahm T, Meldrum DR. VEGF is critical for stem cell-mediated cardioprotection and a crucial paracrine factor for defining the age threshold in adult and neonatal stem cell function. Am J Physiol Heart Circ Physiol. 2008;295:H2308–H2314. doi: 10.1152/ajpheart.00565.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crisostomo PR, Abarbanell AM, Wang M, Lahm T, Wang Y, Meldrum DR. Embryonic stem cells attenuate myocardial dysfunction and inflammation after surgical global ischemia via paracrine actions. Am J Physiol Heart Circ Physiol. 2008;295:H1726–H1735. doi: 10.1152/ajpheart.00236.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]