

Abstract
The etiology of Alzheimer’s disease (AD) remains unclear. The emerging view is that cerebrovascular dysfunction is a feature not only of cerebrovascular diseases, such as stroke, but also of neurodegenerative conditions, such as AD. In AD, there is impaired structure and function of cerebral blood vessels and cells in the neurovascular unit. These effects are mediated by vascular oxidative stress. Injury to the neurovascular unit alters cerebral blood flow regulation, depletes vascular reserves, disrupts the blood-brain barrier and reduces the brain’s repair capacity. Such injury can exacerbate the cognitive dysfunction exerted by incident ischemia and coexisting neurodegeneration. This article summarises data regarding cardiovascular risk factors, vascular abnormalities and brain endothelial damage in AD. In view of accumulating evidence of vascular pathology in AD, we also review the literature (MEDLINE, EMBASE) for clinical evidence of impaired endothelial function in AD. A total of 15 articles investigating endothelial dysfunction in AD were identified. 10 of these articles showed impaired endothelial function in AD patients. The current literature suggests endothelial dysfunction may be involved in the pathogenesis of AD. This aspect of AD pathology is particularly interesting in view of its potential for therapeutic intervention. Future research on endothelial function in AD should concentrate on population-based analysis and combine multiple methods to evaluate endothelial function.
Keywords: Alzheimer’s, dementia, endothelium, etiology, endothelial dysfunction, neurodegeneration, pathology, vascular disease
Introduction
Alzheimer’s disease (AD) is a progressive, neurodegenerative disorder characterised by a decline in cognitive function. Over 23.4 million people suffer from dementia today [1]. This figure is predicted to double every 20 years, reaching 81.1 million by 2040. According to numerous epidemiological studies AD is the most common subtype of dementia [2,3]. However, whilst cerebrovascular disease is considered exclusion criterion for AD, lacunar infarcts have been observed in over 70% of AD patients’ suggesting that the differences between vascular dementia (VaD) and AD are not as definitive as initially postulated [4].
Despite thorough research, the pathogenesis of AD remains unclear. The most accepted hypothesis proposes that deposition of insoluble amyloid β protein (Aβ) fragments causes the neurodegeneration in AD [5]. Aβ is thought to contribute to the accumulation of reactive oxygen species (ROS) resulting in oxidative stress, neuronal damage and cognitive decline.
However, there is growing evidence from studies to suggest that vascular dysfunction plays a central role in the development of AD. Amyloid plaques and neurofibrillary tangles (NFTs) may be the result of hypoxia due to inadequate blood supply [6]. In vitro, Aβ has been shown to be toxic to both cerebral and peripheral endothelium [7,8]. It has been postulated that the neurotoxic effects of Aβ may impair normal endothelial function and cause endothelial-dependent vasoconstriction [9,10].
Epidemiological studies, including the Honolulu-Asia Study [11], Rotterdam Study [12,13], EURODEM [14], and Kungsholmen Project [15] report several vascular risk factors (VRFs) associated with AD-type dementia, all of which cause cerebral hypoperfusion. The association of vascular diseases with AD prevalence strongly suggests that vascular mechanisms play a role in the development and/or progression of AD.
The endothelium is the monolayer of endothelial cells lining the lumen of the vascular beds separating the vascular wall from the circulation and the blood components with both metabolic and mechanical roles. Endothelial dysfunction may be reversible so may represent a therapeutic target.
Aims
In this paper, we review:
I) Evidence of vascular involvement in the pathogenesis of AD.
II) In vivo clinical studies investigating the relationship between AD and endothelial dysfunction.
The neurovascular unit
Neurons and glial cells work in concert with endothelial cells (ECs) and smooth muscle cells to maintain homeostasis of the cerebral microenvironment [16,17]. Thus, the neurovascular unit maintains tight control of the chemical milieu of the brain by regulating local cerebral blood flow (CBF) and molecular transport across the blood brain barrier (BBB). Figure 1 shows the structural arrangement of the neurovascular unit.
Figure 1.
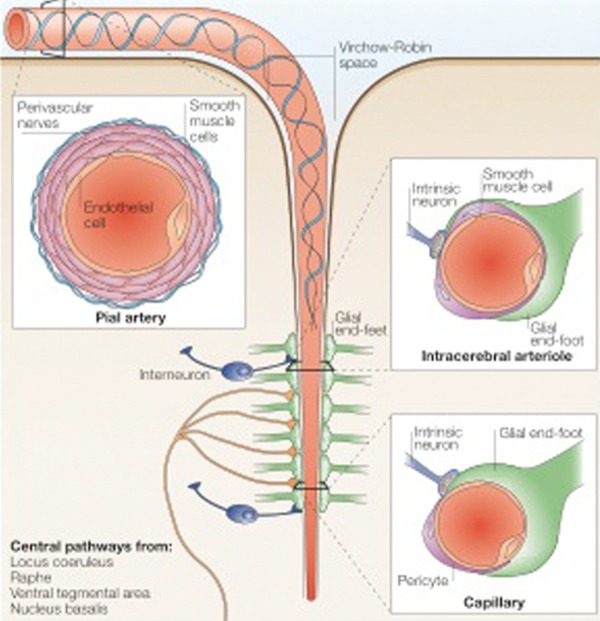
The neurovascular unit. This diagram depicts a cerebral blood vessel and surrounding brain parenchyma that comprise the neurovascular unit. The neurovascular unit encompasses cell-cell and cell-matrix interactions between component endothelial, glial, and neuronal cells. (Image adapted with permission from Iadecola 2004) [268].
Large cerebral arteries branch into smaller pial arteries that run along the surface of the brain across the subarachnoid space [18]. These are composed of an EC layer, a smooth muscle layer and an outer layer of adventitial cells. Penetrating intracerebral arteries are separated from the brain by the Virchow-Robin space [19]. As intracerebral arterioles penetrate deeper into the brain, this space disappears and the vascular basement membrane (VBM) comes into direct contact with the astrocytic end-feet. The VBM separates the endothelium from other cells of the neurovascular unit, including pericytes and astrocytes.
Interestingly, cerebral ECs are not fenestrated but are interconnected by tight junctions forming a continuous cellular structure, the BBB. The BBB prevents passive diffusion of solutes between the blood and brain intracellular fluid and limits uncontrolled entry of neurotoxic substances into the brain [20]. Specialised receptors on the membrane of ECs initiate intracellular signaling cascades in response to agonists that activate specific receptors, or changes in shear stress at the cell surface produced by changes in the rate of blood flow. Gap junctions permit crosstalk between adjacent ECs allowing the transmission of intracellular responses. Once initiated, these cascades trigger the release of potent vasodilator substances such as nitric oxide (NO) and prostacyclin, and vasoconstrictors, such as endothelin and endothelium-derived constrictor factor [21-23]. At low concentrations, reactive oxygen species (ROS) function as vasodilators, however at high concentrations they cause vascular dysfunction [24].
Therefore, neurons, glia, and vascular cells compose a functional unit, which serves to maintain homeostasis of the cerebal microenvironment. Additionally, the neurovascular unit plays an important role in protection against cerebrovascular dysfunction and ischemia [25].
Current hypotheses on Alzheimer’s disease etiology
Much of the literature concerning theories of AD etiology favors one of two hypotheses - the ‘amyloid cascade hypothesis’ (ACH) or the ‘vascular hypothesis’. Whilst the ACH implicates the overproduction and deposition of Aβ fragments as the primary cause of AD [5,26], the vascular hypothesis argues that cerebral hypoperfusion resulting from vascular disease and ageing leads to the neuronal death and cognitive dysfunction in AD (Figure 2).
Figure 2.
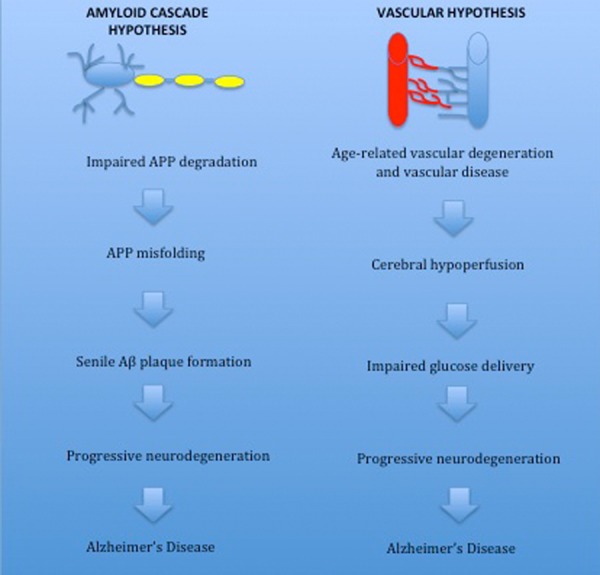
Overview of the vascular and amyloid hypotheses of Alzheimer’s disease. The amyloid hypothesis proposes that overexpression of amyloid precursor protein (APP) is the primary cause of Alzheimer’s disease (AD). Mutations in the presenilin 1, presenilin 2 and APP gene increase amyloid deposition and raise the risk of developing AD. Senile plaques are a key pathological feature of AD and in mouse models plaque formation is associated with neuronal damage. The vascular hypothesis suggests vascular dysfunction associated with ageing is the primary trigger of AD. The prevalence of vascular risk factors is greater in the elderly population and vascular disease is strongly associated with risk of AD. Cerebral hypoperfusion resulting from impaired vasoregulation in vascular disorders causes a neuroglial energy crisis resulting in cognitive. Vascular dysfunction promotes the conversion from mild cognitive impairment to AD.
The amyloid cascade hypothesis
Since the classification of presenile dementia by Alois Alzheimer in 1907, senile plaques and NFTs have been considered key pathological hallmarks of AD [27]. The discovery of Aβ as the main constituent of senile plaques and the identification of gene mutations in amyloid precursor protein (APP), the presenilin 1 (PSEN1) gene and the presenilin 2 (PSEN2) gene causing Aβ accumulation and early onset familial AD led to the ACH [28-30]. The ACH theorises that abnormal Aβ aggregation and deposition initiates the pathogenesis of AD and results in neuronal apoptosis, NFT formation and cognitive decline. Due to the similar phenotype between familial AD and late-onset AD, it was assumed that increases in Aβ deposition were the cause of all types of AD. Despite substantial evidence supporting senile plaque and NFT formation as the primary cause of AD, recent research has greeted this theory with more scepticism [31-35].
A number of observations are used to support the ACH. AD patients have amyloid plaques containing degenerating nerve endings; the plaque count in these patients is greater than in normal aging [36,37]. Furthermore, Down’s syndrome patients invariably develop classical AD pathology by age 50. These individuals also overproduce Aβ from birth and amyloid plaques far precede NFTs and other AD lesions [38,39]. APP over-expression mice initially develop diffuse plaques, which develop into fibrillar plaques associated with damage to neurons and microglia [40]. Similarly, Aβ fibrils reproducibly damage cultured neurons and activate brain microglia [41,42]. Blocking Aβ plaque formation prevents this toxicity [43,44]. A rare coding mutation in the APP gene (A673T) which leads to approximately 40% less APP cleavage actually protects against AD in the elderly [45]. Finally, Apolipoprotein ε4 (APOE-ε4), a significant genetic risk factor for AD, is associated with overproduction of amyloid in the brain before AD symptoms become evident. Each APOE-ε4 allele lowers the age at onset in a dose-dependent fashion [46]. Thus, there is compelling evidence in favor of the ACH.
Due to recent evidence, however, the theory that senile plaque accumulation is the primary cause of neurodegeneration in AD has been criticised by many [31-35]. Senile plaques and NFTs may occur as a reactive response to neurodegeneration rather than being its cause. In head trauma patients, similar pathological hallmarks are seen as in AD patients in neuronal perikarya and in dystrophic neurites surrounding Aβ deposits [47]. Moreover, increased numbers of βAPP-immunoreactive neurons have been observed in the entorhinal cortex after head injury [48]. These findings suggest APP over-expression and amyloid deposition could occur as an acute-phase response to neuronal damage [49]. This concept of amyloid production as a neurotrophic response is supported by the observations that APP shares structural similarities with the precursor for epidermal growth factor [50] and cerebellar Aβ-deposits contain acute phase proteins such as complement factors and α-anti-chymotrypsin [51]. Other evidence contradicting the ACH has also been reported. The degree of amyloid deposition in the brain does not correlate with the severity of cognitive dysfunction. In addition, the brains of many individuals with normal cognitive function have abundant senile plaques or NFTs [52,53]. Transgenic mouse models over-expressing APP show no neuronal loss in the CA1 region [54] and show cognitive dysfunction before Aβ deposition [55]. Furthermore, vaccinations against Aβ over 8 weeks do not improve cognition in mice transgenic for APP and presenilin 1 [56]. Therefore, whilst there is evidence in support of the ACH, there are increasing questions about the validity of this theory.
The vascular hypothesis
The vascular hypothesis of AD suggests that pathology begins with cerebral hypoperfusion. This in turn leads to a crisis among glia and neurons, eventually culminating in neurodegeneration and cognitive impairment. In addition, interactions between vascular dysfunction and increased amyloid production that facilitate the pathogenesis of AD will be discussed.
Vascular disease and Alzheimer’s disease
Several vascular diseases are associated with an increased risk of AD, all of which induce cerebral hypoperfusion [11,13,15]. Recently, Kaffashian et al. found that Framingham general cardiovascular disease (CVD) and stroke risk scores correlate with cognitive decline in late middle age and may be effective for assessing risk of cognitive dysfunction and targeting modifiable risk factors [57]. Furthermore, Li et al. showed that VRFs promote conversion from mild cognitive impairment to AD [58]. In the same study, treatment of VRFs reduced the risk of incident AD. Interaction between AD pathology and VRFs may aggravate perfusion abnormalities in AD (Figure 3).
Figure 3.
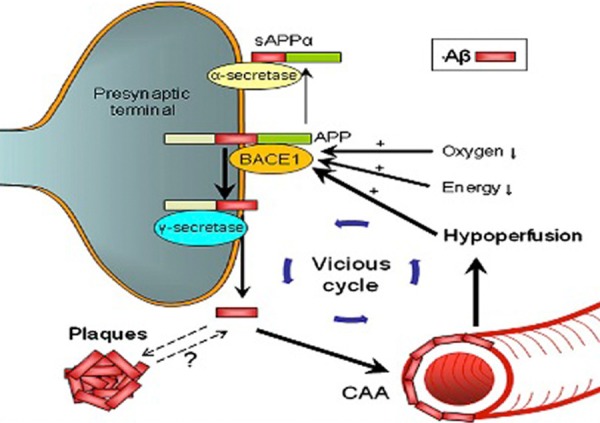
Interaction between vascular disease and AD pathology. Arrows indicate how cerebrovascular factors influence amyloid processing. Hypoxia resulting from vascular disease (e.g atherosclerosis) causes increased expression of hypoxia-inducible factor (HIF)-1α. HIF-1α binds to the hypoxia-responsive element on amyloid cleaving enzyme BACE1. The resulting upregulation of BACE1 mRNA expression increases the production of Aβ fragments. Hypoperfusion resulting from vascular disease and old age further increases BACE1 activity. In addition, hypoperfusion may contribute to oxygen deficiency and cause a neuroglial energy crisis, further ameliorating amyloid plaque production, CAA and ischaemia. CAA = Cerebral amyloid angiopathy, BACE1 = beta secretase 1, sAPP = secreted form of beta-APP, APP = amyloid precursor protein, Aβ = beta amyloid peptide. Image adapted with permission from Kalaria et al. [59].
Hypertension
High blood pressure (BP) has been established as one of the strongest VRFs for AD [11,14,59,60]. The Honolulu-Asia study followed up 3734 men and found a strong correlation between midlife hypertension and AD [14]. This association was greatest in those not undergoing treatment with antihypertensive drugs. Moreover, neuroimaging and post-mortem analysis of AD patients revealed that increased BP in midlife resulted in pathological changes in blood vessels, brain atrophy and an increase in senile Aβ plaques and NFTs in the neocortex and hippocampus [61]. Kivipelto et al. also found an association with midlife hypertension and AD risk [59]. Recently, Rodrigue et al. found that hypertension interacts with the APOE-ε4 genotype to increase amyloid deposition in cognitively healthy middle-aged and older adults [62]. Furthermore, increased pulse pressure was strongly associated with increased mean cortical amyloid level for subjects with at least one ε4 allele. These findings suggest that hypertension greatly exacerbates neuropathological and cognitive brain changes in patients susceptible to AD.
Angiotensin II (Ang II) is an important vasoactive peptide hormone associated with hypertension. Angiotensin I (Ang I) is converted to Ang II through removal of two C-terminal residues by angiotensin-converting enzyme (ACE), causing vasoconstriction and increased BF. Ang II also activates NADPH oxidase (NOX), leading to increased ROS production and oxidative stress [63,64].
Several studies have reported increases in ACE activity in patients with AD. Increased binding of radioactively labeled ACE-I to ACE in the temporal cortex and elevated neuronal and perivascular ACE immunoreactivity has been observed in AD patients post-mortem [65,66]. Furthermore, Miners et al. found the strength of the increase in perivascular ACE immunoreactivity correlated not only with the level of cerebral amyloid angiopathy (CAA) but also with AD severity [67]. Not only does ACE catalyse the production of Ang II, it also cleaves Aβ [68]. The cleavage Aβ of by ACE may contribute to pathological Aβ misfolding. Further evidence for the role of Ang II and ACE in AD pathogenesis comes from clinical trials of ACE inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs), which lower the risk of developing AD, alleviate cognitive decline and decrease the load of amyloid plaques in the brain [69-71].
Long-lasting increases in BP may worsen the risk of AD by inducing small vessel disease, structural white matter alterations and cerebral hypoperfusion through impairment of normal vascular regulation or atherosclerosis. An increase in systemic BP can result in BBB dysfunction in cerebral microvasculature, which further damages subcortical vessels [72]. In addition, this causes impairment of solute transport to the brain and a decrease in glucose delivery results in expression leading to an increase in amyloid fibrillogenesis [73]. Therefore, hypertension probably increases the risk of AD by disrupting BBB transport, increasing Ang II production, damaging the endothelium and causing production of amyloid and free radicals.
Obesity
Epidemiological research has shown obesity in midlife is a strong risk factor for AD, however this relationship may not be present as ageing progresses further [74,75]. Singh-Manoux et al. analysed 6401 adults aged 39-63 years and found that increased BP and other metabolic factors resulting from excess body weight causes middle-aged and older adults to experience a rapid decline in cognitive skills [76]. Participants who were both obese and metabolically abnormal showed a 22.5% faster decline in memory and thinking skills than control subjects. The increase in prevalence of AD in obese individuals is, in part, due to insulin resistance. Around 80% of individuals with obesity also suffer from insulin resistance [77]. However, many other risk factors associated with obesity also increase the risk of developing AD.
Plasma free fatty acid (FFA) levels are increased in most obese individuals [78]. Moreover, obesity causes inhibition of the enzyme metalloproteinase, which is associated with amyloid clearance [79,80]. In cultured vascular cells, high FFAs stimulate ROS production through protein kinase C-dependent activation of NOX [81]. These findings suggest that obesity increases oxidative stress and amyloid accumulation in the vasculature which may increase the risk of dementia. Kalmijn et al. found increased FFAs caused increased tau and amyloid expression [82]. Furthermore, elevated FFAs initiate the release of compounds such as Ang II and TNF-α, which may exacerbate the development of AD [83].
The relationship between obesity, atherosclerosis and hypercholesterolemia is well established [84,85]. Hofman et al. found a 3-fold increase in risk of developing AD or VaD in patients with atherosclerosis [13]. Atherosclerosis in carotid arteries produced the strongest risk of developing dementia. In a 6 year follow-up study of elderly individuals with arterial stiffness and atherosclerosis, high pulse pressure (low diastolic and high systolic) was associated with AD and VaD [86]. These findings suggest aortic compliance and stroke volume play important roles in AD pathogenesis. Postmortem grading of Circle of Willis atherosclerotic lesions shows that atherosclerosis is more severe in cases with AD [4,87]. Unsurprisingly, atherosclerosis in the Circle of Willis is positively associated with the formation of neuritic plaques and NFTs [88]. Endothelial and perivascular damage are common features of atherosclerosis, which may result in diminished blood flow and inhibition of EDNO [89]. Accordingly, dysregulation of EDNO resulting from chronic brain hypoperfusion causes spatial memory loss and Aβ40 deposition in the rat hippocampus [90]. Due the slow onset and progression of atherosclerosis, it is probable that the occlusion of coronary arteries prior to cognitive decline precedes AD symptoms. These results may explain the current findings on human cognitive dysfunction and brain hypoperfusion [91]. In patients with carotid artery stenosis who have undergone a carotid endarterectomy and shown no sign of microembolism following surgery cognition is often improved. If however, microembolic events are identified or brain perfusion is not restored, cognition does not tend to improve [92]. If the theory that vessel occlusion precedes cognitive dysfunction is correct, prevention of atherosclerosis may significantly lessen AD prevalence.
Elevated cholesterol levels also seem to contribute to AD pathology. In vitro, high cholesterol leads to overproduction of Aβ [93]. Aβ was found to inhibit cholesterol trafficking and alter cholesterol homeostasis leading to neurodegeneration and the onset of AD pathology, suggesting Aβ has a role in control of cholesterol transport. In addition, cholesterol secosterol aldehydes initiate Schiffs base-formation resulting in enhanced amyloidogenesis [94,95]. Thus, obesity may contribute to AD via a number of mechanisms including increased cholesterol, atherosclerotic occlusion of cerebral blood vessels and insulin resistance.
Diabetes mellitus
A number of population-based studies have found an association between diabetes mellitus and AD with an incidence of AD as high as 2-5 times higher in patients with type-II diabetes [12,96-98]. Both diabetes and AD are associated with localised amyloid accumulation and both increase in prevalence during ageing. Whilst elevated peripheral insulin levels initially result in increased brain insulin, prolonged hyperinsulinemia reduces brain insulin levels via downregulation of BBB insulin receptors and reduced insulin transport into the brain [99,100].
Insulin receptors are expressed in high concentrations in areas of the brain associated with learning and memory such as the hippocampus and limbic system, which may explain the correlation between hyperinsulinemia and cognitive decline [101]. Hyperinsulinemia initiates central inflammation and Aβ production in healthy individuals [102]. In mice with streptozotocin-induced diabetes, impaired insulin-signaling results in tau hyperphosphorylation and elevated Aβ [103]. Furthermore, rat models of spontaneous diabetes elicit pathological changes similar to that observed in AD [104].
Insulin resistance is a key pathological feature of diabetes and there is growing evidence to suggest a link between insulin resistance and AD. Extensive abnormalities in insulin and insulin-like growth factor type I and II signalling mechanisms have been observed in brains with AD, with markedly reduced expression of genes encoding insulin [105]. Hence, insulin resistance in AD has been referred to as ‘type-3 diabetes’. Insulin resistance is also associated with increased oxidative stress, elevated cortisol and mitochondrial dysfunction, which may contribute to cognitive dysfunction [106]. So, diabetes probably increases the risk of AD by increasing senile plaque and NFT production, inducing an inflammatory response, increasing free radical production and causing disruption of nutrient transport in the BBB.
Cerebrovascular disease (stroke)
Numerous studies have found an association between stroke and AD [4,107,108]. Lesions in the white matter surrounding cerebral ventricles closely resembling ischaemic infarcts have been observed in neuropathological studies of AD [109]. Microvascular lesions are a common pathological feature of AD, with similar numbers of lesions being present as there are NFTs and amyloid plaques [110]. Moreover, subcortical infarcts co-exist with AD pathology and increase the risk of developing dementia 4-fold [111].
It has been postulated that cerebrovascular damage (e.g. microinfarcts) precedes the development of AD symptoms. Patients with AD have more pronounced atherosclerotic lesions in the Circle of Willis than age-matched controls and it has been suggested that ischaemic stroke is the main cause of AD in some individuals [87]. Also, AD is more damaging in the presence of cerebral infarction and autopsy studies show around 70% of patients diagnosed with AD have evidence of coexistent cerebrovascular disease [4,107]. A strong correlation between infarcts and AD has been observed, with microinfarcts restricted to watershed cortical zones [112]. Interestingly, CAA was an important risk factor for watershed microinfarcts, suggesting perturbed hemodynamic factors (e.g. decreased BP) play a role in the genesis of cortical watershed microinfarcts, which may further aggravate degeneration and worsen dementia. CAA occurs in over 80% of patients with AD and only around 10% of control subjects. The high incidence of CAA in AD patients suggests it may contribute to the development of AD. CAA is associated with damage to smooth muscle in the cerebral vasculature and reduced amyloid clearance [113]. Pathological amyloid accumulation may have a role in the development of haemorrhagic stroke in AD [114].
In a large population-based study silent stroke doubled the risk of AD in patients aged 60 to 90 years [115]. Lacunar infarcts are small subcortical infarcts that result from occlusion of a single perforating artery [116]. In individuals suffering from silent subcortical lacunar infarcts, cognitive decline correlates with decreased cerebral glucose metabolism and it was found that hypometabolism was a better indicator of cognitive dysfunction than structural changes (e.g. hippocampal atrophy). In conclusion, ischemia, microinfarcts and lesions in AD may provoke stroke triggering the development or progression of AD by reducing blood flow and invoking amyloid overproduction in the brain. It is plausible that a reduction in CBF resulting from stroke facilitates the conversion of preclinical AD to AD. Using effective intervention strategies to prevent VRFs may reduce the risk of AD.
Morphological changes of the cerebral microvasculature in Alzheimer’s disease
There is increasing recognition that structural changes in cerebral blood vessels contribute to the development and progression of AD. Although the presence of cerebrovascular disease is considered exclusion criterion for AD, cerebral capillary degeneration has been found in almost all AD brains examined postmortem and in cortical biopsy material from pathologically confirmed AD [117,118]. Morphological changes in cerebral microvasculature (capillary atrophy, kinking, focal constriction, VBM thickening) have been found to relate to areas of Aβ deposition [119]. The most notable change appears to be thickening of the VBM. The three central components of the VBM are collagen type 1V, laminin, and heparan sulfate proteoglycan (HSPG). Laminin and HSPG are both located at the endothelial (lamina rara interna) and the astrocytic (lamina rara externa) VBM surfaces [120,121]. Both have a role in cell adhesion and attachment and are potent neurite-promoting factors [122,123]. HSPG is negatively charged allowing it to filter anionic and neutral proteins. Charge is crucial in homeostasis of the renal glomerular barrier and may be important for the BBB as well [124,125]. The functional role of the VBM in the maintenance of the BBB is not well established, however it has been postulated that blocked anionic sites could result in disrupted BBB function [126]. Modification of the lamina rara interna could also result in altered attachment and adhesion of the EC. Pathology of the VBM could be associated with impaired solute transport, resulting in leakage of neurotoxic plasma substances or circulating APP into the brain [127,128]. Therefore, VBM pathology In AD may alter the ECs ability to maintain the BBB.
Vascular insufficiency and neurodegeneration
The damaging effect of VRFs and Aβ on cognition suggests a link between ischemia and neurodegeneration. In AD, CBF is reduced and functional hyperaemia is impaired [129,130]. Furthermore, studies indicate that changes in CBF are a powerful predictor of AD [131,132]. Amyloid plaques and NFTs often coexist with vascular pathology in individuals with dementia [133]. Ischaemic conditions may trigger Aβ accumulation by elevating AβPP/β-secretase expression and facilitating amyloidogenic cleavage leading to increased levels of toxic Aβ. In animal models, amyloid deposition is increased in the cortex in response to ischemia [134,135] and a similar process is thought to occur in humans [136]. Indeed, accumulation of both Aβ1-40 and Aβ1-42 has been shown to increase dramatically and consistently after cerebral ischemia [137-139]. It is therefore unsurprising that individuals with cerebrovascular insufficiency or VaD have increased levels of Aβ and senile plaques [140].
Conversely, it has been postulated that the toxic effects of Aβ on cerebral blood vessels may induce cerebral hypoperfusion and increase vulnerability to ischemic damage. In APP over-expression mice focal cerebral ischemia produces more severe infarcts than controls, implicating Aβ as a cause for cerebrovascular dysfunction [141]. Similarly, a higher incidence of cerebrovascular lesions is seen in individuals with AD post-mortem [142]. Paradoxically, VRFs can increase vulnerability to AD without affecting neurodegenerative pathology. Despite a two-fold increase in risk of developing AD in diabetics, no increase in amyloid plaques or tau NFTs is observed. In fact, a higher number of microinfarcts have been identified in these individuals suggesting that microvascular lesions may augment the effects of the neurodegenerative pathology in AD [143]. Since amyloid plaques appear to promote ischemia and vice versa, it is plausible that there is a synergistic relationship between the amyloidogenic and vascular features of AD pathology (Figure 4).
Figure 4.
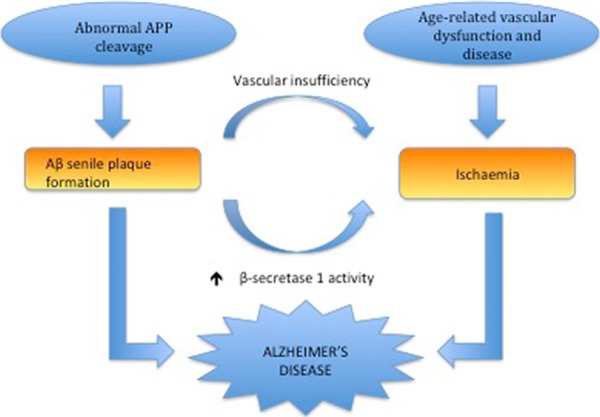
Proposed synergistic relationship between vascular insufficiency and Alzheimer’s disease (Iadecola, 2010 [269]). Vascular insufficiency resulting from vascular risk factors (e.g vascular dementia) can promote β-amyloid (Aβ) production by increasing amyloid cleaving enzyme β secretase-1 and reducing Aβ clearance. Overexpression of amyloid precursor protein in Alzheimer’s disease leads to the formation of senile Aβ plaques, which may induce ROS-mediated vascular damage and compromised vasoregulation. Thus, vascular insufficiency and β-amyloid production may interact via a positive feedback mechanism exacerbating cognitive decline and increasing the risk of dementia.
The research on NFTs and vascular insufficiency is far less conclusive. Elderly patients with hypertension have increased hippocampal NFTs in comparison to control subjects [144]. Furthermore, focal cerebral ischemia enhances tau phosphorylation in rodent stroke models [145]. However, transient hypoperfusion markedly decreases total tau levels in triple transgenic mice expressing amyloid plaques and NFTs. In summary, there is evidence to suggest ischemia contributes to accumulation of Aβ by reducing its degradation and promoting its synthesis whereas the extent to which ischemia regulates tau phosphorylation remains elusive.
Another important question being raised by researchers in this area is whether or not vascular dysfunction and ischemia aggravate cognitive decline in AD. The presence of vascular lesions contributes significantly to the severity of cognitive impairment in AD [146]. Moreover, cerebrovascular disease has a greater capacity to influence cognitive performance at early stages of AD than at more advanced stages of the disease [147] VRFs and lesions also accelerate the progression of cognitive decline in AD [148]. Finally, a study assessing cerebrovascular reactivity to hypercapnia with transcranial Doppler ultrasonography, a measure of cerebrovascular function, found an association between impaired cerebral microvessels functionality and cognitive decline in patients with AD [149]. In conclusion, there is considerable evidence to suggest that vascular dysfunction is linked to cognitive degeneration in AD.
Vascular dysfunction and oxidative stress in Alzheimer’s disease
Vascular oxidative stress induced by elevated Aβ has emerged as a key pathogenic factor in cerebrovascular dysfunction [24,150]. ROS are highly reactive molecules that often have unpaired electrons in their outer orbital. Increased production of free radicals and ROS can disrupt cell membranes and consequently impair cell function (Figure 5).
Figure 5.
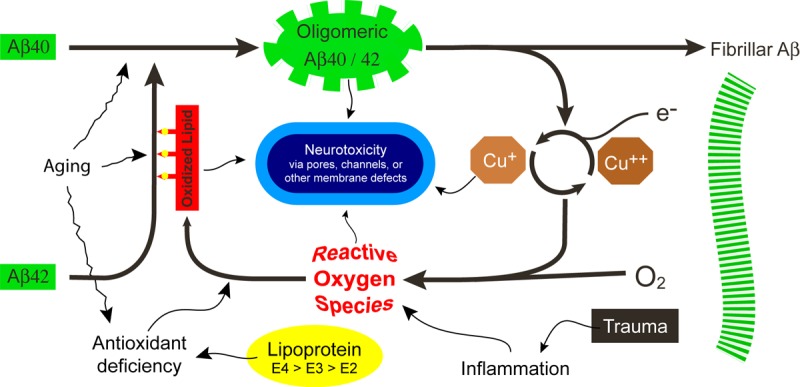
Membrane-related amyloid fibrillogenesis and oxidative stress in the pathogenesis of Alzheimer’s disease. Ageing is associated with reduced tissue antioxidant levels, oxidative lipid damage and enhanced Aβ protein aggregation. These changes are even more pronounced in Alzheimer’s disease. Thiol-mediated antioxidant activity is absent in Apolipoprotein E4 alleles which may allow excess oxidative damage to the lipids in lipoprotein particles. Aβ proteins have a pro-oxidant activity towards the polyunsaturated lipids of cell membranes. Lipid oxidation products may exert direct neurotoxicity or induce structural changes in the His residues of Aβ42, enhancing its membrane affinity. Furthermore, these products may facilitate the oligomerization of Aβ40 and promote fibrillogenesis. Highly reactive oxygen species may be produced as a result of trauma induced-inflammation or fibrillogenesis. Inflammation may also occur in the presence of vascular disease (e.g atherosclerosis). During these processes, Aβ may form neurotoxic pores or channels in endothelial or neuronal membranes. Image adapted from Axelsen et al. [270].
Vascular oxidative stress has been observed in APP over-expression transgenic mice, without any evidence of oxidative stress in other areas of the brain [151]. In the same transgenic models, antioxidant therapy or over-expression of free radical scavenger superoxide dismutase reverses Aβ-mediated cerebrovascular dysfunction, including alterations in both functional hyperemia and endothelial-dependent vasodilatation [152,153]. Furthermore, substitution of methionine 35 with norleucine, a mutation that blocks the ability of Aβ to generate ROS, abolished Aβ1-40 vasoactivity suggesting that Aβ1-40 plays a role in the cerebrovascular alterations observed in AD [154].
Experimental studies suggest that free radicals produced by NOX are responsible for the cerebrovascular alterations induced by Aβ [155,156]. ROS produced by NOX contribute to CVDs associated with vascular oxidative stress including atherosclerosis, hyperhomocysteinaemia and hypertension.
The involvement NOX and oxidative stress in AD is illustrated by an increase in NOX levels or activity in mild cognitively impaired and AD brain tissues [157,158]. Moreover, Aβ induces mitochondrial dysfunction and oxidative stress in astrocytes and microglia through activation of NOX [156,159].
Accumulating evidence suggests ROS impair normal vascular function. In animal models of various disease conditions ROS cause endothelial dysfunction [160]. Reaction of ROS superoxide with NO- generates peroxynitrite, a potentially deleterious oxidant. Several alterations in the function of endothelium are initiated by this reaction. The most established is endothelium-dependent vasorelaxation, which is impaired by a loss of NO- bioactivity in the vessel wall [161,162]. ROS and peroxynitrite can also trigger oxidative damage to proteins and nucleic acids, including strand breaks in DNA and base/nucleotide modifications, particularly in sequences with a higher number of guanosine bases [163]. In addition, peroxynitrite formation inhibits the activity of prostacyclin synthase, reducing the ability of blood vessels to dilate [164]. Tetrahydrobiopterin, a critical cofactor for the NO synthases, is a crucial target for oxidation by peroxynitrite and in its absence these enzymes become uncoupled, producing ROS rather than NO and causing further oxidative stress [165]. Therefore, ROS-mediated vascular dysfunction is caused by decreased availability of NO and changes in enzymes essential for normal vascular function.
Aβ also contributes to neurovascular dysfunction through other mechanisms. In addition to causing endothelial dysfunction, Aβ can damage other components of the neurovascular unit. Aβ causes damage and apoptosis of glia and neurons, which may attenuate CBF response to stimuli [166,167]. Moreover, intracortical microvessels and NO containing neurons in AD are deprived of a cholinergic neurogenic control, which probably leads to a compromised ability to adjust cortical perfusion in response to neuronal activation during functional tasks related to cognition [168]. Thus, Aβ-mediated damage to neurovascular coupling is not solely a result of vascular dysfunction but also involves damage to neurons and glia.
Pharmacotherapy
Current drugs associated with a modest reduction in AD symptoms such as cholinesterase inhibitors also exert beneficial effects on cerebral perfusion [169]. Although these drugs decrease synaptic breakdown of acetylcholine (ACh), it is possible that their effect on CBF also plays a role in their protective action. Considering the vast amount of evidence suggesting cerebral hypoperfusion is important in AD development, drug trials for statins, non-steroidal inflammatory drugs (NSAIDs) and antihypertensives in the prevention or treatment of AD were initially promising.
However, many potential treatments have not been as efficacious as expected. Statins are a class of drugs that inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. HMG-CoA reductase is the rate-limiting enzyme in the cellular cascade of cholesterol biosynthesis. Statins may also reduce Aβ production and inhibit the activation of Rac1, which is critically involved in the activation of NOX [170]. In addition, statins downregulate AT-1 receptor gene expression mediated by destabilization of AT-1 messenger ribonucleic acid.
Due to the possible link between elevated cholesterol and dementia studies have been carried out to test the effect of statins in AD patients. Despite the fact that observational studies suggest statin therapy exerts a benefi cial effect between normal cognitive performance and profound dementia [171], results from randomised control trials have been disappointing in both prevention [172,173] and treatment [174,175] of AD.
The prevalence of AD is decreased in patients with rheumatoid arthritis, suggesting NSAIDs may exert a protective effect against AD. Different NSAIDs inhibit COX isoforms, and the expression levels of COX-1 and COX-2 change in the different stages of AD pathology [176]. Furthermore, COX-1 is heavily expressed in microglia, which are associated with fi brillar Aβ deposits and some NSAIDs inhibit amyloid production via Y-secretase [177]. Indeed, longitudinal observation studies report a reduction in AD symptoms in patients taking NSAIDs [178,179]. Nevertheless, clinical trials found no association between NSAID treatment and cognitive impairment. It is possible that trials were not sustained for a long enough period to yield effective results. More than 2 years of consistent NSAID treatment is likely to be required in order to decrease the risk of AD [179]. Moreover initiation of treatment should occur several years before the onset of AD [180]. However, it is still unclear whether or not infl ammatory mechanisms are invoking damage in AD or whether they are present to remove the debris from other more important pathological processes.
Recent evidence suggests antihypertensive medication may be protective against AD [69-71,181]. In particular, drugs affecting the production of Ang II may have neuroprotective effects. A UK case-control study found that fewer patients prescribed ARBs and ACE-inhibitors went on to develop probable AD [182].
Patients taking ARBs had the lowest risk of developing AD, which may have been due to a lack of ACE-mediated amyloid degradation when taking these drugs [183]. In addition, treatment with ARBs is associated with less AD-related pathology on autopsy evaluations [184]. Large randomised clinical trials are warranted to further investigate the relationship between antihypertensive drugs and dementia. In conclusion, although there is a relationship between VRFs and AD, drug trials have been less successful than anticipated. These findings suggest that the ability of pharmacological intervention in reversing AD pathology is limited.
Endothelial damage and repair in Alzheimer’s disease
Endothelial damage and Amyloid-β
Numerous factors may contribute to endothelial dysfunction in AD. ECs in the cerebral vasculature may contribute to the formation of amyloid deposits surrounding the cerebral blood vessels. Cerebral ECs express APP-cleaving secretases, which may cleave platelet-retained Aβ into fragments leading to increased cerebrovascular amyloid deposition [185-187]. Therefore, brain ECs in AD may produce Aβ causing increased amyloid accumulation and potentially enhancing CAA.
Wu et al. measured gene levels in brain ECs isolated from frontal cortex autopsies in patients with late stage AD, age-matched controls and healthy young adults [188]. Interestingly, the expression of MEOX2, a homeobox gene that is upregulated during vascular differentiation, is significantly reduced in late-stage AD and is required for optimal clearance of Aβ in mice brains. Restoring MEOX2 gene expression in brain ECs from individuals with AD enhanced angiogenesis, transcriptionally suppressed AFX1 forkhead transcription factor-mediated apoptosis and increased the levels of both a major Aβ clearance receptor at the BBB, the low-density lipoprotein receptor- related protein 1 (LRP), and its receptor-associated protein (RAP). They found that in MEOX2 +/- mice, there was a decrease in CBF and capillary length, and that vessels from these mice had an impaired angiogenic response to hypoxia. These findings suggest that MEOX2 may be playing a role in neurovascular dysfunction and other important changes in the AD brain that could contribute to cognitive decline. Thus, MEOX2 could represent a potential new therapeutic target for AD patients.
Aβ has been found to be toxic to cerebral ECs in vitro by causing damage to nuclear and mitochondrial DNA and activation of caspase-8 and caspase-3, which have essential roles in apoptosis [189]. In the same study, Aβ-mediated damage to ECs was inhibited by zVAD-fmk, a broad-spectrum caspase inhibitor or N -acetyl-cysteine, an antioxidant. Numerous other studies have shown the toxic effects of Aβ on ECs, implicating Aβ as the primary cause for impairment of NO production and mitochondrial dysfunction [190,191]. Interestingly, Aβ appears to be dependent on EC state [192]. Moreover, Aβ1-42 induces EC degeneration by raising intracellular calcium via calcium permeable Aβ channels [193]. These findings suggest a number of signaling pathways could be involved in EC dysfunction in AD. Moreover, disruption of normal endothelial function may precede Aβ deposition suggesting that endothelial dysfunction may play a central role in AD pathogenesis.
Both TNF-α and Aβ are over-expressed in AD. It is possible that TNF-α and Aβ induced alterations in the ASK1-JNK/p38 apoptotic signalling pathways are responsible for EC apoptosis and damage [194,195]. There is evidence to suggest protein phosphatase 2A is required for ASK1 activation in ECs [195]. During Aβ-mediated EC apoptosis activation of sphingomyelinase releases ceramide which in turn activates PP2A. Ceramide is involved in the expression of NADPH-oxidase in ECs and may contribute to ROS-induced damage to ECs [196]. Thus, TNF-α and amyloid induced changes in apoptotic signalling may contribute to EC damage and vascular dysregulation in AD.
Regenerative capacity of brain endothelial cells
The process of vasculogenesis was initially thought only to occur during embryonic development, but not in postnatal life [197]. However, there is increasing evidence to suggest that bone derived endothelial progenitor cells (EPCs) contribute to neovascularization and neoendothelialization and differentiate into ECs in response to injury [198]. In patients with AD, decreased levels of circulating EPCs have been recorded [199]. Levels of EPCs have been suggested to correlate with endothelial dysfunction in AD [200]. Moreover, AD-associated peptide Aβ40 blocks endothelial and vascular regeneration and initiates autophagy [201]. A possible explanation for the inhibitory effect of Aβ on endothelial repair was provided by the observation that Aβ blocks vascular endothelial growth receptor (VEGR)-2 [202]. Therefore, the regenerative capacity of ECs may be compromised in AD, perhaps due to the inhibitory actions of Aβ. Since endothelial dysfunction may contribute to Aβ deposition, it is possible that endothelial damage in AD results in a positive feedback loop whereby more Aβ is produced and further aggravates endothelial injury and impairs repair.
The injection of EPCs to induce neovascularization and restore CBF may be a promising treatment opportunity for AD. Indeed, neovascularization can be successfully stimulated by incorporating EPCs into ischaemic sites in animal models [203] and human myocytes [204].
Review of clinical evidence investigating the relationship between endothelial dysfunction and Alzheimer’s disease
Considering the wide range of research favoring a vascular cause for AD discussed in the first section of this review, we believe an analysis of the in vivo evidence of endothelial dysfunction in AD is timely. In this section, we review the literature investigating the relationship between endothelial dysfunction and AD using specific search terms and selection criteria.
In vivo assessment of endothelial function
Despite the evidence supporting the role of endothelial dysfunction in AD, its routine measurement in the clinical setting is challenging. However, due to recent advances in techniques to assess endothelial dysfunction it may now be possible to determine the extent to which vascular impairment correlates with AD. In recent years, a number of non-invasive techniques have been used to assess endothelial function and morphology as an easier and safer alternative to direct assessment of the coronary arteries [205]. Endothelial responses in peripheral vessels correlate well with coronary artery responses [206].
The capacity of blood vessels to respond to mechanical and pharmacological stimuli in the lumen denotes the ability to self-regulate tone and to adjust blood flow and distribution in response to changes in the local environment. NO is probably the most important vasoactive substance produced by the endothelium.
A number of pharmacological (e.g. bradykinin) and mechanical (e.g. sheer stress) stimuli lead to the production of nitric oxide (NO) [207]. Endothelium derived NO (EDNO) is synthesised from the amino acid L-arginine by the endothelial isoform of NO synthase (eNOS), yielding L-citrulline as a byproduct [208]. Specialised calcium-mediated potassium ion channels in the EC membrane open in response to sheer stress [209-211]. Opening K+ channels hyperpolarize the EC, increasing influx of calcium ions (Ca+). Calcium activates eNOS which catalyses the production of endothelial NO [212,213]. NO diffuses across the endothelial cell membrane and enters the vascular smooth muscle cells where it activates guanylate cyclase (GC), leading to increased blood flow. The balance between NO and various endothelium derived vasoconstrictors and the sympathetic nervous system maintains blood vessel tone.
Loss of EDNO would be expected to promote a vascular phenotype more prone to vascular disease. A disturbed synthesis and bioavailability of NO might contribute to the onset of dementia by causing impaired endothelial function leading vascular disease and loss of direct neuroprotective effects [214,215].
Flow-mediated dilatation
Flow-mediated dilatation (FMD) measures the vasodilatation response of blood vessels in response to sheer stress. A principal mediator of FMD is EDNO. Brief ischemia in distal tissue induced by a tourniquet results in reactive hyperemia, local vasodilatation and increased blood flow in the proximal and distal vessels. The increased blood flow in the proximal vessel (brachial artery) results in increased sheer stress and an NO mediated vasodilatation (FMD). Brachial artery imaging coupled with reactive hyperemia is one of the most common techniques for measurement of vascular function [216]. This technique involves the occlusion of conduit vessels for ~ 5 min causing reactive hyperemia of the vascular bed. The subsequent dilatation that is expressed as a percentage increase from the baseline diameter is mainly dependent on NO activity, with a low percentage indicating poor endothelial function [217].
Laser Doppler flowmetry
Laser Doppler flowmetry (LDF) is an inexpensive, non-invasive method for measurement of skin microcirculation. Iontophoresis coupled with LDF makes it possible to assess the real-time changes of the skin blood flow after the administration of different vasoactive substances without systemic effects. ACh introduced into the skin by iontophoresis causes endothelium-dependent vasodilatation, which can be compared with the endothelium-independent vasodilator effect of sodium nitroprusside (SNP), an NO donor [218,219]. Sodium nitroprusside reacts with tissue sulfhydryl groups under physiologic conditions to produce NO directly and thereby stimulate SMC relaxation. ACh mediates vasodilatation via endothelial-dependent production of NO and/or prostanoids with a possible accessory role played by endothelium-derived hyperpolarizing factor. A Laser Doppler Flowmeter works by reading the frequency of the oscillation produced by the Doppler frequency shift of the red blood cells (RBCs) in a peripheral tissue and translates the frequency to an intensity oscillation. The apparatus is composed of a low-powered laser and probe that takes readings and sends results to an analyser. The apparatus can penetrate 1-4 mm of non-pigmented tissue. The light emitted and reflected is fed through optical fibers to the analyser-recorder. The output of the LDF is the flux of RBCs, defined as the number of RBCs times their velocity, which determines circulation.
Laser Doppler perfusion imaging
Laser Doppler perfusion imaging (LDPI) is based on the recording of Doppler shift caused by movements of RBCs in the backscattered light of a laser beam that successively scans a certain tissue area [220]. A laser Doppler imager generates a color-coded image of the spatial distribution of tissue perfusion. This allows measurement of tissue blood flow over a set area. Iontophoresis of small charged molecules is used to assess the skin microcirculation [221].
This technique is increasingly being adopted as a clinical measure of endothelial function, solely in the microcirculation rather than in the large conduit arteries. This is useful as the microcirculation may well be the initial site for endothelial damage [222,223].
Strain gauge plethysmography
Forearm blood flow can be assessed using strain gauge plethysmography [224]. ACh interacts with endothelial muscarinic receptors and stimulates the release of NO. Intra-arterial (brachial) infusion of ACh and SNP can be used to assess local endothelial-dependent and independent responses [225]. The rate of forearm swelling during the infusion, measured by strain gauge plethysmography, gives an estimate of the endothelial function.
Pulse wave velocity
Pulse wave velocity (PWV) is a measure of regional arterial stiffness of the arterial area between two measurement sites. PWV is related not only to the elastic modulus of the arterial wall (which represents the intrinsic stiffness of the wall), but also to the arterial geometry and blood density [226]. Although structural changes in the vessel wall are a major component of arterial stiffness (increased collagen and decreased elastin), the endothelium also appears to play an important dynamic role in arterial stiffness [227]. Evidence from both animal [228,229] and human studies [230,231] suggests that the endothelium is an important regulator of arterial stiffness, both functionally and structurally. Various interventions that reduce arterial stiffness may also improve endothelial function, particularly ACE inhibitors and statins.
Serum biomarkers
Many serum biomarkers have been shown to correlate with endothelial function including interleukin-6 (IL-6), tumor-necrosis factor-α (TNF-α), soluble P-selectin and soluble intercellular adhesion molecule-1 [232]. Plasma nitrite can be used an indicator of EDNO generation [233]. In addition, impaired tissue plasminogen activator (t-PA) secretion by the endothelium has been suggested as a useful measure of endothelial function [234]. The sensitivity of t-PA in this setting is apparently greater than that for other markers of endothelial dysfunction, including agonist- or flow-mediated vasodilatation, because it is measurable in some clinical conditions (e.g., hypertension) in which agonist-induced vasodilatation is unaltered. CRP is associated with several processes involved in endothelial dysfunction, these include increasing ET-1 synthesis, down-regulating eNOS, increasing the release of PAI-1 from ECs, and influencing endothelial progenitor cells [235,236]. Cellular adhesion molecules (CAMs) are expressed on the surface of activated ECs, and increased plasma concentrations of soluble CAMs are seen in patients with atherosclerosis [237].
Plasma asymmetric dimethylarginine (ADMA) is increased in patients with vascular disease, as well as in patients with risk factors for vascular disease [238-240]. In individuals with hypercholesterolemia, plasma ADMA levels provide a more accurate indication of endothelial dysfunction than LDL cholesterol levels [240]. ADMA inhibits the production of NO in cultured ECs and isolated blood vessels [241,242]. Hydrolysis of ADMA by the enzyme dimethylaminohydrolase (DDAH) produces l-citruline and methylamine [243]. It has been postulated that DDAH regulates NO synthase activity by controlling the concentration of ADMA [244]. Homocysteine inhibits the activity of endothelial DDAH, causing the accumulation of ADMA and the inhibition of NO synthesis [245]. Thus, plasma ADMA appears to be a promising indicator of endothelial dysfunction.
Endothelial function in Alzheimer’s disease: available evidence
Search strategy
A search of the published literature from January 1966 (MEDLINE) and from January 1989 (EMBASE) was conducted. By way of the advanced search method, the following terms were combined: ‘Alzheimer’s disease’, ‘cognitive impairment’, and ‘cognitive decline’, using the Boolean operator ‘OR’. The Boolean operator ‘AND’ was used to link these to the terms ‘endothelial dysfunction’, ‘vascular reactivity’, ‘flow-mediated dilation’, ‘plethysmography’, ‘doppler flowmetry’, and ‘pulse wave velocity’. Papers published until February 2013 were included. Books, abstracts and conference proceedings were excluded.
Eligibility of studies
This review sought publications with original data on humans suffering from dementia of the Alzheimer’s type. Articles without proper AD classification were excluded. Only articles in English were included, as the authors were familiar with these languages. Only papers investigating human subjects without a history of cardiovascular events or major VRFs were included. Since the endothelium possesses different physiologic functions - i.e. regulation of vessel tone and expression of adhesion molecules - the studies were organised into subheadings. The references in the selected articles were scanned for other relevant articles, using the same criteria.
Results
By searching MEDLINE and EMBASE 568 papers were found. Applying the above-mentioned criteria yielded a total of 15 articles. The main reasons for excluding papers were animal models, reviews and vascular or mixed dementia participants. Main findings of the studies for each subheading are discussed below. A summary of results is provided in Tables 1 and 2.
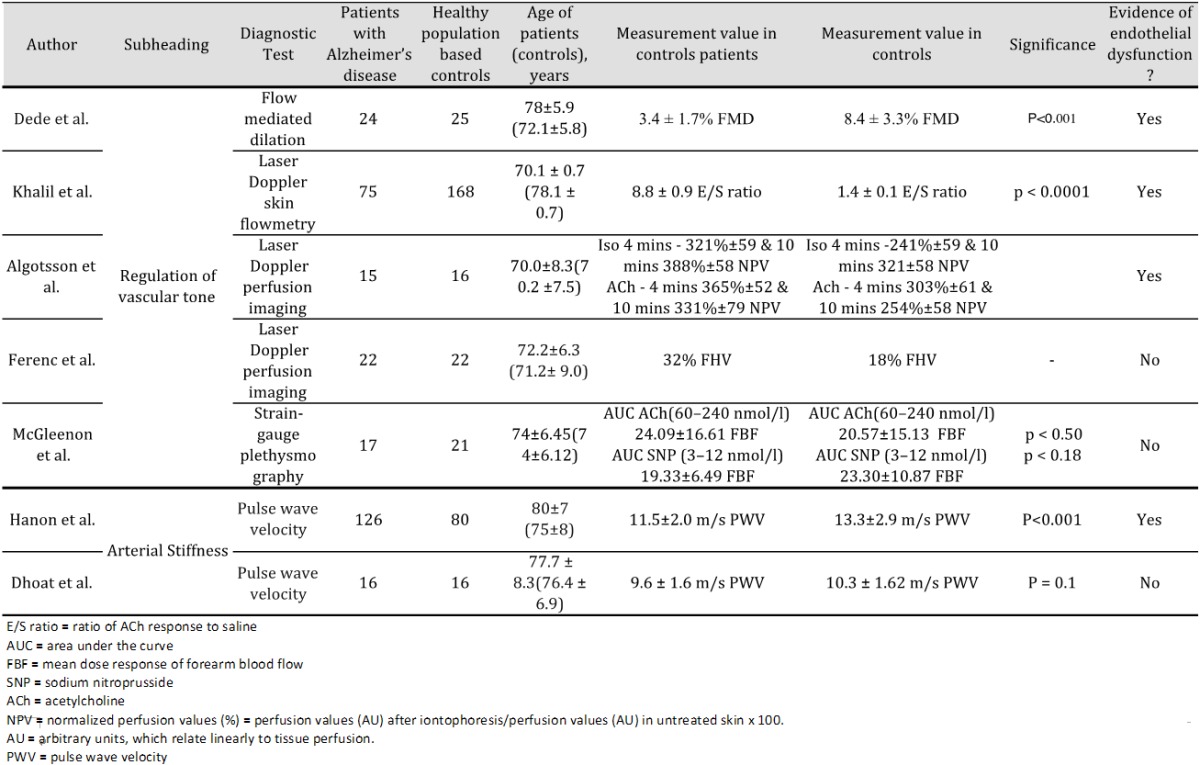
Table 1.
Selected papers using functional assessment to measure endothelial dysfunction in Alzheimer’s disease
|
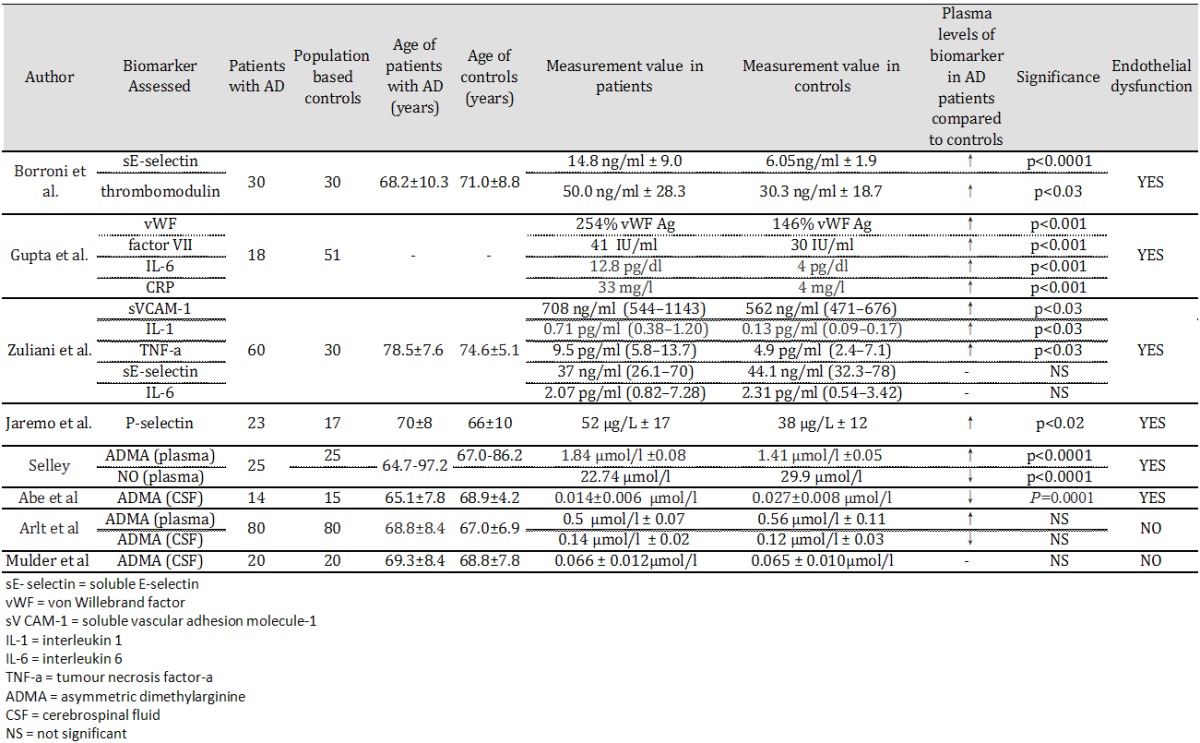
Table 2.
Selected papers using serum biomarkers to measure endothelial function in Alzheimer’s disease
|
Regulation of vascular tone
Dede et al. evaluated endothelial function in 24 patients with AD-type dementia and 25 age-matched controls using flow-mediated dilatation [246]. Significantly lower FMD was found in the AD group, which indicated endothelial dysfunction (mean FMD: AD group, 3.4±1.7%; control group, 8.4±3.3%; P<0.001). When the correlation between Clinical Dementia Rating (CDR) and Mini-Mental State Examination (MMSE) scores and FMD values of the total sample (n = 49) was examined, FMD had a negative correlation with CDR score (r = -0.605, P<0.001) and a positive correlation with MMSE score (r = 0.560, P<0.001). Algotsson et al. measured skin vessel reactivity of 15 patients with AD and 16 age-matched controls using LDPI [247]. Compared to the controls, the skin vessel vasodilatation of the AD group was significantly impaired with respect to the reactions to ACh and isoprenaline but not SNP in all three tests. Khalil et al. measured microvascular responsiveness in 75 patients with AD and 168 age-matched controls using LDF [248]. The ratio of ACh response to saline (ratio E/S) was determined. Mean±SEM of ratios E/S were 8.8±0.9 for controls and 1.4±0.1 for AD patients, thus endothelial dysfunction was present in AD patients. Ferenc et al. evaluated flow motion pattern in forehead skin using LDF. No significant difference in was seen in occurrence of forehead vasomotion pattern between patients with AD (18%) and age-matched controls (32%) [249]. Occlusion (biological zero) values for control subjects and AD patients were 5.5±2.2 and 4.7±0.9 respectively. McGleenon et al. measured endothelial function in 9 male and 8 female patients with AD and 9 male and 12 female control subjects [250]. The basal FBF values were not significantly different for AD and controls (mean±(SE), 2.89±1.0 and 2.81±1.42 d, respectively).
Arterial stiffness
Hanon et al. measured the relationship between arterial stiffness and cognitive function in 126 AD patients and 80 age-matched controls and found significantly higher PWV in AD patients (13.3±2.9 m/s) than individuals without cognitive impairment (11.5±2.0 m/s; P<0.001) [251]. Dhoat et al. also assessed PWV in 16 patients with late onset-AD and 16 age-matched controls [252]. PWV in the muscular and elastic arteries were not statistically different between the two groups.
Expression of serum biomarkers
Borroni et al. found a significant difference in thrombomodulin levels (mean SD, control = 6.05±1.9; AD = 14.9±9.0; p<0.0001) and soluble E-selectin (sE-Selectin) (control = 30.3±18.2; AD = 50.0±28.5; p<0.001) between 30 patients with mild AD and 30 age-matched controls [253]. There was an inverse correlation between plasmatic TM levels and MMSE scores. Gupta et al. studied serum markers of coagulation and inflammation in patients suffering from dementia [254]. Serum concentrations of vWF, factor VII, IL-6, CRP and FDP were significantly higher in all three dementia groups (including the AD-type group) as compared to controls (p<0.001). Zuliani et al. tested endothelial dysfunction in 60 patients with late onset AD by measuring plasma levels of E-selectin and vascular cell adhesion molecule 1 (VCAM-1) [245]. VCAM-1 levels were significantly increased in patients with late onset AD than controls (control = 562 ng/ml (544-1143); AD = 708 ng/ml (471-676); p<0.03) suggesting endothelial function is impaired in AD [255]. Järemo et al. investigated P-selectin behavior in 23 persons diagnosed with moderate AD and 17 healthy elders without obvious memory problems [256]. Circulating P-selectin was analysed using an ELISA technique and flow cytometry was used to measure surface-bound P-selectin. Soluble P-selectin was augmented in AD (p = 0.019) but platelet membrane-attached P-selectin was not significantly different from controls. AD diagnosis was associated with less surface-bound P-selectin after provocation. Significant results were obtained when 74 μmol/L TRAP-6 (a thrombin-receptor activating peptide) was used as a platelet agonist (p = 0.0008). Selley measured plasma homocysteine, ADMA, and NO (nitrite and nitrate) concentrations in patients with AD and healthy controls [257]. AD patients showed higher plasma ADMA (AD = 1.84±0.08; control = 1.41±0.05 μmol/L, p<0.0001) and homocysteine concentrations, and lower NO concentrations, than controls. Abe et al. measured ADMA concentrations in the cerebrospinal fluid in AD patients and healthy controls [258]. AD patients had significantly lower ADMA concentrations in the cerebrospinal fluid (AD = 0.014±0.006; controls = 0.027±0.008 μmol/L, p = 0.0001). There was a significant correlation between lower cerebrospinal concentrations of ADMA and lower MMSE in patients with AD (r = 0.58, p<0.05). Mulder et al. also measured ADMA concentrations in the cerebrospinal fluid in AD patients and healthy controls [259]. Unlike the study by Abe et al. no significant differences were observed between the 2 groups (AD = 0.066±0.012; control = 0.065±0.010 μmol/L; p = 0.91) [258]. Finally, Arlt et al. measured ADMA concentrations both in plasma and in the cerebrospinal fluid in AD patients and healthy controls [260]. AD patients had higher plasma ADMA concentrations (AD = 0.65±0.11; control = 0.54±0.11 μmol/L; p<0.001), but lower cerebrospinal fluid ADMA concentrations (AD = 0.15±0.03; control = 0.22±0.12 μmol/L; P = 0.022), in comparison to healthy controls. However, these differences were not significant.
Discussion
The evidence reviewed suggests vascular involvement plays a role in the pathogenesis of AD. Morphological changes in the endothelium and the VBM, as well as diminished endothelial repair capacity have been recorded in AD [117]. Vascular abnormalities may be attributed to vascular disorders that frequently develop and progress with increasing age [57]. These vascular alterations probably facilitate several factors that are potentially neurotoxic. In particular, amyloid has received a great amount of interest as it is overproduced in AD and has been found to be neurotoxic [40,41]. In ageing, it is possible that vascular dysfunction may occur early on or even precede the pathological features of AD. As AD is a neurodegenerative disorder in which amyloid accumulation is believed a key event, it may be that an association exists between vascular changes and amyloid production.
Aβ may be produced by endothelial, neuronal and glial cells [187]. It has been postulated that Aβ deposition stimulates the production of ROS, which in turn cause vascular oxidative stress and cerebrovascular dysfunction [261]. In vitro findings are supported by studies indicating that cerebral ischemia causes stress-induced amyloid deposition in highly vascularized cortical brain regions [137,139]. These theories suggesting a possible synergistic relationship between Aβ deposition and vascular dysfunction may provide an explanation for endothelial dysfunction in AD.
In our systematic search, we found that studies investigating a possible relationship between endothelial dysfunction and AD in vivo were limited. The results of published studies show evidence of endothelial dysfunction in AD except for the studies by McGleenon et al., Malt et al., Dhoat et al., Arlt et al. and Mulder et al.. Although the measurement of endothelial dysfunction was quite diverse, elevated levels of serum biomarkers suggestive of endothelial dysfunction (TM, vWF, ADMA etc.), abnormal values of FMD and blood flow abnormalities assessed using Doppler shift were all associated with AD. These findings suggest that, independent of any other VRFs, endothelial function is present in AD, which could indicate a systemic element in AD pathology.
Another hypothesis is that only cerebral vascular function is compromised in AD [149]. The techniques reviewed in this study are all limited in their assessment of the peripheral circulation. However, there is currently no effective method of measuring endothelial function in the cerebral circulation. Moreover, McGleenon et al. found that cerebral vessels and peripheral vessels show similar responses to ACh infusion [250]. Nevertheless, the argument remains that whilst the peripheral circulation remains functional the cerebral circulation may be compromised in AD.
In vivo studies of endothelial function are fraught with difficulty and currently no gold standard exists. Many measurements that are purported to measure endothelial function actually measure “vascular reactivity.” Although many researchers consider FMD the gold standard for clinical research [207], it is not without its limitations [262]. FMD provides little mechanistic data and structural alterations of blood vessels in vascular disease which affect their ability to dilate may affect results. Moreover, lack of standardisation and variations in positioning of the arm cuffs and measurement of vessel diameter make comparing study results challenging [263]. Despite the similarities in vascular responses between the skin microcirculation and larger arteries, whether skin blood-flow studies (Doppler imaging and Doppler flowmetry) are indicative of other vascular beds is unclear and requires further research [264]. Strain gauge plethysmography is an invasive technique involving cannulation of the brachial artery, which is not without some risk. Thus, this is a specialist technique that is not suitable for widespread use. Further studies are required to fully comprehend the link between endothelial function and arterial stiffness. Although most studies suggest that the endothelium regulate stiffness, impaired elastic function itself might have adverse effects on endothelial function, and future work needs to address this issue [265]. Finally, the use of serum biomarkers to measure vascular disease or prognosis is still at an early stage in development.
The studies published so far cannot establish whether endothelial dysfunction in AD is causative or coincidental. It would be ideal to collect measurements before symptoms of AD occur in a population-based design. However, this would require a very large study population. Furthermore, none of the markers described are brain-specific endothelial markers and endothelial function may vary across vascular beds [264,266].
A major limitation in the literature on AD and endothelial dysfunction is the relative paucity of studies and small number of subjects in each study. A much larger population is necessary to prove any clinical significance. It may be difficult to recruit volunteers with AD for invasive studies due to volunteer frailty and lack of independent decision-making capacity in these individuals. Patients with AD often have vascular risk factors (VRFs) and it is challenging to find subjects without accompanying vascular conditions. However, only patients without existing VRFs were included in these investigations. Whilst this eliminates the possibility of the endothelial dysfunction of subjects being caused by conditions other than AD, it is well known that a high proportion of the AD population has vascular conditions, which may be associated with the pathogenesis of AD [57,267]. The evidence, therefore, may not be generalisable to the AD population as a whole. Future research investigating the relationship between AD and endothelial dysfunction should include subjects with and without VRFs to give a more accurate representation of the general AD population.
A final limitation of the reviewed studies was that they focused on serum markers of endothelial dysfunction (e.g. vWF) or on function assessment (e.g. FMD). For the future, it would be ideal to combine both methods to provide as robust an evidence base as possible.
Although this review was not a full systematic review, it aimed to provide a thorough analysis of evidence of endothelial dysfunction in AD. Strict selection criteria were employed with the aim to provide enough information to allow repeatability and enhance the validity of findings. However, it is possible that some of the published literature was not included. Whilst the studies reviewed suggest endothelial dysfunction may be present in AD, more research is required to prove this link.
Conclusion
There is conflicting evidence regarding the pathogenesis of AD. The vascular hypothesis challenges the long established neuron-centric view of AD. It is likely that AD has both neurodegenerative and vascular elements. Based on our findings, we suggest endothelial dysfunction may play a role in AD pathogenesis. More basic and clinical research on the vascular features of AD is required. Further study may have implications for therapeutic intervention with the potential to reverse or prevent endothelial dysfunction and relieve or prevent the symptoms of AD.
Acknowledgements
Dr Soiza is funded by an NRS Career Research Fellowship. The authors are grateful to Alzheimer’s Research UK for providing funding.
Disclosure of conflict of interest
None.
References
- 1.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thies W, Bleiler L Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9:208–45. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Herbert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the U. S. population: Prevalence estimates using the 2000 Census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 4.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–7. [PubMed] [Google Scholar]
- 5.Hardy JA, Higgins GA. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 6.Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci. 2006;103:18727–18732. doi: 10.1073/pnas.0606298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas T, Thomas G, Mclendon C, Sutton T, Mullan M. Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 8.Sutton ET, Hellermann GR, Thomas T. Beta- amyloid-induced endothelial necrosis and inhibition of nitric oxide production. Exp Cell Res. 1997;230:368–376. doi: 10.1006/excr.1996.3440. [DOI] [PubMed] [Google Scholar]
- 9.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 10.Crawford F, Suo Z, Fang C, Mullan M. Characteristics of the in vitro vasoactivity of beta-amyloid peptides. Exp Neurol. 1998;150:159–168. doi: 10.1006/exnr.1997.6743. [DOI] [PubMed] [Google Scholar]
- 11.Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, Havlik RJ. Midlife blood pressure and dementia: the Honolulu-Asia Aging Study. Neurobiol Aging. 2000;21:49–55. doi: 10.1016/s0197-4580(00)00096-8. [DOI] [PubMed] [Google Scholar]
- 12.Ott A, Stolk RP, Hofman A, Van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- 13.Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, van Duijn CN, Van Broeckhoven C, Grobbee DE. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam study. Lancet. 1997;349:151–4. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- 14.Launer LJ, Andersen K, Dewey ME, Letenneur L, Ott A, Amaducci LA, Brayne C, Copeland JR, Dartigues JF, Kragh-Sorensen P, Lobo A, Martinez-Lage JM, Stijnen T, Hofman A. Rates and risk factors for dementia and Alzheimer’s disease: results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups. European Studies of Dementia. Neurology. 1999;52:78–84. doi: 10.1212/wnl.52.1.78. [DOI] [PubMed] [Google Scholar]
- 15.Fratiglioni L, Winblad B, Von Strauss E. Prevention of Alzheimer’s disease and dementia. Major findings from the Kungsholmen Project. Physiol Behav. 2007;92:98–104. doi: 10.1016/j.physbeh.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 16.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nature Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 17.Woolsey TA, Rovainen CM, Cox SB, Henegar MH, Liang GE, Liu D, Moskalenko YE, Sui J, Wei L. Neuronal units linked to microvascular modules in cerebral cortex: response elements for imaging the brain. Cereb Cortex. 1996;6:647–660. doi: 10.1093/cercor/6.5.647. [DOI] [PubMed] [Google Scholar]
- 18.Jones EG. On the mode of entry of blood vessels into the cerebral cortex. J Anat. 1970;106:507–520. [PMC free article] [PubMed] [Google Scholar]
- 19.Peters A, Palay S, Webster HD. The Fine Structure of the Nervous System. Oxford: Oxford University Press; 1991. [Google Scholar]
- 20.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 21.Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 22.Golding EM, Marrelli SP, You J, Bryan RM. Endothelium-derived hyperpolarizing factor in the brain: a new regulator of cerebral blood flow? Stroke. 2002;33:661–663. [PubMed] [Google Scholar]
- 23.Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- 24.Faraci FM. Reactive oxygen species: influence on cerebral vascular tone. J Appl Physiol. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- 25.Del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- 26.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 27.Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET. Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1147–1155. doi: 10.1097/00005072-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;34:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 29.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–7. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 30.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K Tsuda T, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–8. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 31.Davis JN, Chisolm JC. The ‘amyloid’ cascade hypothesis of AD: decoy or real McCoy? Trends Neurosci. 1997;20:558–559. doi: 10.1016/s0166-2236(97)85989-9. [DOI] [PubMed] [Google Scholar]
- 32.Neve R, Robakis NK. Alzheimer’s disease: a re-examination of the amyloid hypothesis. Trends Neurosci. 1998;21:15–19. doi: 10.1016/s0166-2236(97)01168-5. [DOI] [PubMed] [Google Scholar]
- 33.Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging. 2002;23:1101–1105. doi: 10.1016/s0197-4580(02)00050-7. [DOI] [PubMed] [Google Scholar]
- 34.de la Torre JC. Alzheimer’s disease as a vascular disorder: nosological evidence. Stroke. 2002;33:1152–1162. doi: 10.1161/01.str.0000014421.15948.67. [DOI] [PubMed] [Google Scholar]
- 35.de la Torre JC. Alzheimer’s disease: how does it start? J Alzheimers Dis. 2002;4:497–512. doi: 10.3233/jad-2002-4606. [DOI] [PubMed] [Google Scholar]
- 36.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Younkin LH, Suzuki N, Younkin SG. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43) J Biol Chem. 1995;270:7013–6. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 37.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 38.Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 39.Querfurth HW, Wijsman EM, St George-Hyslop PH, Selkoe DJ. Beta APP mRNA transcription is increased in cultured fibroblasts from the familial Alzheimer’s disease-1 family. Brain Res Mol Brain Res. 1995;28:319–337. doi: 10.1016/0169-328x(94)00224-3. [DOI] [PubMed] [Google Scholar]
- 40.Miguel-Hidalgo JJ, Cacabelos R. Beta-amyloid (1-40)-induced neurodegeneration in the rat hippocampal neurons of the CA1 subfield. Acta Neuropathol. 1998;95:455–65. doi: 10.1007/s004010050825. [DOI] [PubMed] [Google Scholar]
- 41.Meda L, Cassatella MA, Szendrei GI, Otvos L, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 42.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 45.Jonsson T, Atwal JK, Steinberg S, Snædal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 46.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 47.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–44. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 48.Mckenzie JE, Gentleman SM, Roberts GW, Graham DI, Royston MC. Increased numbers of beta APP-immunoreactive neurones in the entorhinal cortex after head injury. Neuroreport. 1994;6:161–4. doi: 10.1097/00001756-199412300-00041. [DOI] [PubMed] [Google Scholar]
- 49.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. β Amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Regland B, Gottfries CG. The role of amyloid β-protein in Alzheimer’s disease. Lancet. 1992;340:467–469. doi: 10.1016/0140-6736(92)91780-c. [DOI] [PubMed] [Google Scholar]
- 51.Kalaria RN, Perry G. Amyloid P component and other acute-phase proteins associated with cerebellar Aβ-deposits in Alzheimer’s disease. Brain Res. 1993;631:151–5. doi: 10.1016/0006-8993(93)91202-4. [DOI] [PubMed] [Google Scholar]
- 52.Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol. 1999;58:376–388. doi: 10.1097/00005072-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 53.Terry RD, Masliah E, Salmon DP, Butters N, Deteresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;4:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 54.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–73. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 55.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, A-beta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 56.Austin L, Arendash GW, Gordon MN, Diamond DM, DiCarlo G, Dickey C, Ugen K, Morgan D. Short-term beta-amyloid vaccinations do not improve cognitive performance in cognitively impaired APP + PS1 mice. Behav Neurosci. 2003;117:478–84. doi: 10.1037/0735-7044.117.3.478. [DOI] [PubMed] [Google Scholar]
- 57.Kaffashian S, Dugravot A, Elbaz A, Shipley MJ, Sabia S, Kivimäki M, Singh-Manoux A. Predicting cognitive decline: A dementia risk score vs the Framingham vascular risk scores. Neurology. 2013;80:1300–1306. doi: 10.1212/WNL.0b013e31828ab370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Wang YJ, Zhang M, Xu ZQ, Gao CY, Fang CQ, Yan JC, Zhou HD. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology. 2011;76:1485–1491. doi: 10.1212/WNL.0b013e318217e7a4. [DOI] [PubMed] [Google Scholar]
- 59.Kivipelto M, Helkala EL, Laasko MP, Hanninen T, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A. Midlife vascular factors and Alzheimer’s disease in later life: longitudinal population based study. BMJ. 2001;822:1447–1451. doi: 10.1136/bmj.322.7300.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kennelly SP, Lawlor BA, Kenny RA. Blood pressure and the risk for dementia: a double edged sword. Ageing Res Rev. 2009;8:61–70. doi: 10.1016/j.arr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 61.Petrovich H, White LR, Izmirillian G, Ross GW, Havlik RJ, Markesbery W, Nelson J, Davis DG, Hardman J, Foley DJ, Launer LJ. Midlife blood pressure and neuritic plaques, neurofibrillary tangles and brain weight at death: the Honolulu Asia Aging study. Neurobiol Aging. 2000;21:57–62. doi: 10.1016/s0197-4580(00)00106-8. [DOI] [PubMed] [Google Scholar]
- 62.Rodrigue KM, Rieck JR, Kennedy KM, Devous MD, Diaz-Arrastia R, Park DC. Risk Factors for β-Amyloid Deposition in Healthy Aging: Vascular and Genetic Effects. JAMA Neurol. 2013;70:600–606. doi: 10.1001/jamaneurol.2013.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 64.Touyz RM. Oxidative stress and vascular damage in hypertension. Curr Hypertens Rep. 2000;2:98–105. doi: 10.1007/s11906-000-0066-3. [DOI] [PubMed] [Google Scholar]
- 65.Barnes NM, Cheng CH, Costall B, Naylor RJ, Williams TJ, Wischik CM. Angiotensin converting enzyme density is increased in temporal cortex from patients with Alzheimer’s disease. Eur J Pharmacol. 1991;200:289–292. doi: 10.1016/0014-2999(91)90584-d. [DOI] [PubMed] [Google Scholar]
- 66.Savaskan E, Hock C, Olivieri G, Bruttel S, Rosenberg C, Hulette C, Müller-Spahn F. Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiol Aging. 2001;22:541–546. doi: 10.1016/s0197-4580(00)00259-1. [DOI] [PubMed] [Google Scholar]
- 67.Miners JS, Ashby E, Van Helmond Z, Chalmers KA, Palmer LE, Love S, Kehoe PG. Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer’s disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2008;34:181–193. doi: 10.1111/j.1365-2990.2007.00885.x. [DOI] [PubMed] [Google Scholar]
- 68.Toropygin IY, Kugaevskaya EV, Mirgorodskaya OA, Elisseeva YE, Kozmin YP, Popov IA, Nikolaev EN, Makarov AA, Kozin SA. The N-domain of angiotensin-converting enzyme specifi cally hydrolyzes the Arg-5-His-6 bond of Alzheimer’s Abeta-(1-16) peptide and its isoAsp-7 analogue with different effi ciency as evidenced by quantitative matrix-assisted laser desorption/ionization time-of-fl ight mass spectrometry. Rapid Commun Mass Spectrom. 2008;2:231–9. doi: 10.1002/rcm.3357. [DOI] [PubMed] [Google Scholar]
- 69.Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM. Valsartan lowers brain Beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117:3393–3402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsukuda K, Mogi M, Iwanami J, Min LJ, Sakata A, Jing F, Iwai M, Horiuchi M. Cognitive deficit in amyloid-b-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-g activation. Hypertension. 2009;54:782–787. doi: 10.1161/HYPERTENSIONAHA.109.136879. [DOI] [PubMed] [Google Scholar]
- 71.Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, Wolozin B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. doi: 10.1136/bmj.b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, Persson G, Odén A, Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 73.Webster NJ, Green KN, Peers C, Vaughan PF. Altered processing of human amyloid precursor protein in human neuroblastoma SH-SY5Y. J Neurochem. 2002;83:1262–71. doi: 10.1046/j.1471-4159.2002.01236.x. [DOI] [PubMed] [Google Scholar]
- 74.Anstey KJ, Cherbuin N, Budge M, Young J. Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev. 2011;12:e426–37. doi: 10.1111/j.1467-789X.2010.00825.x. [DOI] [PubMed] [Google Scholar]
- 75.Xu WL, Atti AR, Gatz M, Pedersen NL, Johansson B, Fratiglioni L. Midlife overweight and obesity increase late-life dementia risk: a population-based twin study. Neurology. 2011;76:1568–1574. doi: 10.1212/WNL.0b013e3182190d09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh-Manoux A, Czernichow S, Elbaz A, Dugravot A, Sabia S, Hagger-Johnson G, Kaffashian S, Zins M, Brunner EJ, Nabi H, Kivimäki M. Obesity phenotypes in midlife and cognition in early old age: the Whitehall II cohort study. Neurology. 2012;79:755–62. doi: 10.1212/WNL.0b013e3182661f63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lobo A, Launer LJ, Fratiglioni L, Andersen K, Di Carlo A, Breteler MM, Copeland JR, Dartigues JF, Jagger C, Martinez-Lage J, Soininen H, Hofman A. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54:S4–9. [PubMed] [Google Scholar]
- 78.Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37:1020–1024. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 79.Belo VA, Souza-Costa DC, Luizon MR, Lanna CM, Carneiro PC, Izidoro-Toledo TC, Ferraz KC, Tanus-Santos JE. Matrix metalloproteinase-9 genetic variations affect MMP-9 levels in obese children. Int J Obes (Lond) 2012;36:69–75. doi: 10.1038/ijo.2011.169. [DOI] [PubMed] [Google Scholar]
- 80.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–45. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 82.Kalmijn S, Foley D, White L, Burchfiel CM, Curb JD, Petrovich H, Ross GW, Havlik RJ, Launer LJ. Metabolic cardiovascular syndrome and risk of dementia in Japanese American elderly men. The Honolulu-Asia ageing study. Arterioscler Thromb Vasc Biol. 2000;20:2255–2260. doi: 10.1161/01.atv.20.10.2255. [DOI] [PubMed] [Google Scholar]
- 83.Grundy SM. Metabolic syndrome: a multiplex cardiovascular risk factor. J Clin Endocrinol Metab. 2007;92:399–404. doi: 10.1210/jc.2006-0513. [DOI] [PubMed] [Google Scholar]
- 84.Kawada T. Body mass index is a good predictor of hypertension and hyperlipidemia in a rural Japanese population. Int J Obes Relat Metab Disord. 2002;26:725–729. doi: 10.1038/sj.ijo.0801984. [DOI] [PubMed] [Google Scholar]
- 85.Kivimäki M, Davey-Smith M, Timpson NJ, Lawlor DA, Batty GD, Kähönen M, Juonala M, Rönnemaa T, Viikari JS, Lehtimäki T, Raitakari OT. Lifetime BMI and later atherosclerosis risk in young adults: Examining causal links using Mendelian randomisation in the Cardiovascular Risk in Young Finns Study. Eur Heart J. 2008;29:2552–2560. doi: 10.1093/eurheartj/ehn252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Qiu C, Winblad B, Viitanen M And Fratiglioni L. Pulse pressure and risk of Alzheimer disease in persons aged 75 years and older: a community-based, longitudinal study. Stroke. 2003;34:594–599. doi: 10.1161/01.STR.0000060127.96986.F4. [DOI] [PubMed] [Google Scholar]
- 87.Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI, Beach TG. Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003;23:2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- 88.Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AE. Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;113:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- 89.Aliev G, Seyidova D, Neal ML, Shi J, Lamb BT, Siedlak SL, Vinters HV, Head E, Perry G, Lamanna JC, Friedland RP, Cotman CW. Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels as a central target for the development of human AD and AD-like pathology in aged transgenic mice. Ann N Y Acad Sci. 2002;977:45–64. doi: 10.1111/j.1749-6632.2002.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 90.de la Torre JC, Pappas BA, Prevot V, Emmerling MR, Mantione K, Fortin T, Watson MD, Stefano GB. Hippocampal nitric oxide upregulation precedes memory loss and Aβ1-40 accumulation after chronic brain hypoperfusion in rats. Neurol Res. 2003;25:635–641. doi: 10.1179/016164103101201931. [DOI] [PubMed] [Google Scholar]
- 91.Sinforiani E, Curci R, Fancellu R, Facchinetti P, Mille T, Bono G. Neuropsychological changes after carotid endarterectomy. Funct Neurol. 2001;16:329–336. [PubMed] [Google Scholar]
- 92.Heyer EJ, Adams D, Solomon RA, Todd GJ, Quest DO, Mcmahon DJ, Steneck SD, Choudhri TF, Connolly ES. Neuropsychometric changes in patients after carotid endarterectomy. Stroke. 1998;29:1110–1115. doi: 10.1161/01.str.29.6.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yao ZX, Papadopoulos V. Function of beta-amyloid in cholesterol transport: a lead to neurotoxicity. Faseb J. 2002;16:1677–1679. doi: 10.1096/fj.02-0285fje. [DOI] [PubMed] [Google Scholar]
- 94.Nieva J, Shafton A, Altobell LJ 3rd, Tripuraneni S, Rogel JK, Wentworth AD, Lerner RA, Wentworth P Jr. Lipid-derived aldehydes accelerate light chain amyloid and amorphous aggregation. Biochemistry. 2008;47:7695–705. doi: 10.1021/bi800333s. [DOI] [PubMed] [Google Scholar]
- 95.Duthie A, Chew D, Soiza RL. Non-psychiatric comorbidity associated with Alzheimer’s disease. QJM. 2011;104:913–920. doi: 10.1093/qjmed/hcr118. [DOI] [PubMed] [Google Scholar]
- 96.Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, Ohmori S, Nomiyama K, Kawano H, Ueda K. Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: the Hisayama Study. Neurology. 1995;45:1161–1168. doi: 10.1212/wnl.45.6.1161. [DOI] [PubMed] [Google Scholar]
- 97.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’brien PC, Palumbo PJ. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Ann N Y Acad Sci. 1997;826:422–427. doi: 10.1111/j.1749-6632.1997.tb48496.x. [DOI] [PubMed] [Google Scholar]
- 98.Kuusisto J, Koivisto K, Mykkanen L, Helkala EL, Vanhanen M, Hanninen T, Kervinen K, Kesäniemi YA, Riekkinen PJ, Laakso M. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ. 1997;315:1045–1049. doi: 10.1136/bmj.315.7115.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cavalieri M, Ropele S, Petrovic K, Pluta-Fuerst A, Homayoon N, Enzinger C, Grazer A, Katschnig P, Schwingenschuh P, Berghold A, Schmidt R. Metabolic syndrome, brain magnetic resonance imaging, and cognition. Diabetes Care. 2010;33:2489–2495. doi: 10.2337/dc10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Craft S. The role of metabolic disorders in Alzheimer’s disease and vascular dementia: two roads converged? Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Young SE, Mainous AG, Carnemolla M. Hyperinsulinemia and cognitive decline in middle aged cohort. Diabetes Care. 2006;29:2688–2693. doi: 10.2337/dc06-0915. [DOI] [PubMed] [Google Scholar]
- 102.Fishel MA, Watson GS, Montine SJ, Wang Q, Green PS, Kulstad JJ, Cook DG, Peskind ER, Baker LD, Goldgaber D, Nie W, Asthana S, Plymate SR, Schwartz MW, Craft S. Hyperinsulinemia provokes synchronous increase in central inflammation and beta-amyloid in normal adults. Arch Neurol. 2005;62:1539–1544. doi: 10.1001/archneur.62.10.noc50112. [DOI] [PubMed] [Google Scholar]
- 103.Jolivalk CG, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochem Biophys Acta. 2009;1792:482–496. doi: 10.1016/j.bbadis.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 104.Li ZG, Zhang W, Sima AA. Alzheimer-like change in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–24. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 105.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 106.Meigs JB, Larson MG, Fox CS, Keaney JF Jr, Vasan RS, Benjamin EJ. Association of oxidative stress, insulin resistance, and diabetes risk phenotypes: the Framingham Offspring Study. Diabetes Care. 2007;30:2529–2535. doi: 10.2337/dc07-0817. [DOI] [PubMed] [Google Scholar]
- 107.Kokmem EM, Shisnant JP, O’fallon WM, Chu CP, Beard CM. Dementia after ischaemic stroke: a population based study in Rochester Minnesota (1960-1984) Neurology. 1996;46:154–159. doi: 10.1212/wnl.46.1.154. [DOI] [PubMed] [Google Scholar]
- 108.Pohjasvaara T, Erkinjuntti T, Ylikoski R, Hietanen M, Vataja R, Kaste M. Clinical determinants of poststroke dementia. Stroke. 1998;29:75–81. doi: 10.1161/01.str.29.1.75. [DOI] [PubMed] [Google Scholar]
- 109.Brun A, Englund E. A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol. 1986;19:253–262. doi: 10.1002/ana.410190306. [DOI] [PubMed] [Google Scholar]
- 110.White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW, Masaki K, Launer L, Markesbery WR. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 111.Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA. Subcortical infarcts, Alzheimer’s disease pathology, and memory function in older persons. Ann Neurol. 2007;62:59–66. doi: 10.1002/ana.21142. [DOI] [PubMed] [Google Scholar]
- 112.Suter OC, Sunthorn T, Kraftsik R, Straubel J, Darekar P, Khalili K, Miklossy J. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke. 2002;33:1986–1992. doi: 10.1161/01.str.0000024523.82311.77. [DOI] [PubMed] [Google Scholar]
- 113.Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW. Pathogenic effects of D23N Iowa mutant amyloid b-protein. J Biol Chem. 2001;276:32860–32866. doi: 10.1074/jbc.M104135200. [DOI] [PubMed] [Google Scholar]
- 114.Vinters HV, Farag ES. Amyloidosis of cerebral arteries. Adv Neurol. 2003;92:105–112. [PubMed] [Google Scholar]
- 115.Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348:1215–22. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- 116.Ay H, Oliveira-Filho J, Buonanno FS, Ezzeddine M, Schaefer PW, Rordorf G, Schwamm LH, Gonzalez RG, Koroshetz WJ. Diffusion-weighted imaging identifies a subset of lacunar infarction associated with embolic source. Stroke. 1999;30:2644–50. doi: 10.1161/01.str.30.12.2644. [DOI] [PubMed] [Google Scholar]
- 117.Kalaria RN. Cerebrovascular degeneration is related to amyloid-beta protein deposition in Alzheimer’s disease. Ann N Y Acad Sci. 1997;826:263–71. doi: 10.1111/j.1749-6632.1997.tb48478.x. [DOI] [PubMed] [Google Scholar]
- 118.Claudio L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol. 1996;91:6–14. doi: 10.1007/s004010050386. [DOI] [PubMed] [Google Scholar]
- 119.Kalaria RN, Hedera PJ. Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. Neuroreport. 1995;6:477–480. doi: 10.1097/00001756-199502000-00018. [DOI] [PubMed] [Google Scholar]
- 120.Garbisa S, Negro A. Macromolecule organization and functional architecture of basement membranes. Appl Pathol. 1984;2:217–22. [PubMed] [Google Scholar]
- 121.Martinez-Hemandez A, Amenta P. The basement membrane in pathology. Lab Invest. 1983;48:656–77. [PubMed] [Google Scholar]
- 122.Dow KE, Mirski SE, Roder JC, Riopelle RJ. Neuronal proteoglycans: biosynthesis and functional interaction with neurons in vitro. J Neurosci. 1988;8:3278–3289. doi: 10.1523/JNEUROSCI.08-09-03278.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lander AD, Fujii DK, Reichardt LF. Purification of a factor that promotes neurite outgrowth: isolation of laminin and associated molecules. J Cell Biol. 1985;101:898–913. doi: 10.1083/jcb.101.3.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hart MN, Vandyk L, Moore SA, Shasby DM, Canciila PA. Differential opening of the brain endothelial barrier following neutralization of the endothelial luminal anionic charge in vitro. J Neuropathol Exp Neurol. 1987;46:141–153. doi: 10.1097/00005072-198703000-00003. [DOI] [PubMed] [Google Scholar]
- 125.Rennke HG, Cotran RS, Venkatachalam MA. Role of molecular charge in glomerular permeability. Tracer studies with cationized ferritins. J Cell Biol. 1975;67:638–46. doi: 10.1083/jcb.67.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fillit HM, Kemeny E, Luine V, Weksler ME, Zabrislcie JB. Antivascular antibodies in the sera of patients with senile dementia of the Alzheimer’s type. J Gerontol. 1987;42:180–184. doi: 10.1093/geronj/42.2.180. [DOI] [PubMed] [Google Scholar]
- 127.Hardy JA, Mann DM, Webster P, Winblad B. An integrative hypothesis concerning the patbogenesis and progression of Alzheimer’s disease. Neurobiol Aging. 1986;7:489–502. doi: 10.1016/0197-4580(86)90086-2. [DOI] [PubMed] [Google Scholar]
- 128.Miyakawa T, Shimoji A, Kuramoto R, Higuchi Y. The relationship between senile plaques and cerebral blood vessels in Alzheimer’s disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Arch B Cell Pathol Incl Mol Pathol. 1982;40:121–129. doi: 10.1007/BF02932857. [DOI] [PubMed] [Google Scholar]
- 129.Jagust WJ, Haan MN, Reed BR, Eberling Jl. Brain perfusion imaging predicts survival in Alzheimer’s disease. Neurology. 1998;51:1009–1013. doi: 10.1212/wnl.51.4.1009. [DOI] [PubMed] [Google Scholar]
- 130.Johnson KA, Albert MS. Perfusion abnormalities in prodromal AD. Neurobiol Aging. 2000;21:289–292. doi: 10.1016/s0197-4580(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 131.Lee BC, Mintun M, Buckner RL, Morris JC. Imaging of Alzheimer’s disease. J Neuroimaging. 2003;13:199–214. [PubMed] [Google Scholar]
- 132.Rapoport SI. Functional brain imaging to identify affected subjects genetically at risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:5696–5698. doi: 10.1073/pnas.120178897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jellinger KA. Morphologic diagnosis of “vascular dementia” - a critical update. J Neurol Sci. 2008;270:1–12. doi: 10.1016/j.jns.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 134.Abe K, Tanzi RE, Kogure K. Selective induction of Kunitz-type protease inhibitor domain containing amyloid precursor protein mRNA after persistent focal ischemia in the rat cerebral cortex. Neurosci Lett. 1991;125:172–174. doi: 10.1016/0304-3940(91)90020-t. [DOI] [PubMed] [Google Scholar]
- 135.Tomimoto H, Wakita H, Akiguchi I, Nakamura S, Kimura J. Temporal profiles of accumulation of amyloid beta/A4 protein precursor in the gerbil after graded ischemic stress. J Cereb Blood Flow Metab. 1994;14:565–573. doi: 10.1038/jcbfm.1994.70. [DOI] [PubMed] [Google Scholar]
- 136.Jendroska K, Poewe W, Daniel SE, Pluess J, Iwerssen-Schmidt H, Paulsen J, Barthel S, Schelosky L, Cervós-Navarro J, DeArmond SJ. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995;90:461–6. doi: 10.1007/BF00294806. [DOI] [PubMed] [Google Scholar]
- 137.Qi JP, Wu H, Yang Y, Wang DD, Chen YX, Gu YH, Liu T. Cerebral ischemia and Alzheimer’s disease: the expression of amyloid-beta and apolipoprotein E in human hippocampus. J Alzheimers Dis. 2007;12:335–341. doi: 10.3233/jad-2007-12406. [DOI] [PubMed] [Google Scholar]
- 138.Nihashi T, Inao S, Kajita Y, Kawai T, Sugimoto T, Niwa M, Kabeya R, Hata N, Hayashi S, Yoshida J. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir. 2001;143:287–295. doi: 10.1007/s007010170109. [DOI] [PubMed] [Google Scholar]
- 139.Li L, Zhang X, Yang D, Luo G, Chen S, Le W. Hypoxia increases Abeta generation by altering beta- and gamma-cleavage of APP. Neurobiol Aging. 2009;30:1091–8. doi: 10.1016/j.neurobiolaging.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 140.Lewis H, Beher D, Cookson N, Oakley A, Piggott M, Morris CM, Jaros E, Perry R, Ince P, Kenny RA, Ballard CG, Shearman MS, Kalaria RN. Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol Appl Neurobiol. 2006;32:103–118. doi: 10.1111/j.1365-2990.2006.00696.x. [DOI] [PubMed] [Google Scholar]
- 141.Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jellinger KA, Mitter-Ferstl E. The impact of cerebrovascular lesions in Alzheimer disease—a comparative autopsy study. J Neurol. 2003;250:1050–1055. doi: 10.1007/s00415-003-0142-0. [DOI] [PubMed] [Google Scholar]
- 143.Sonnen JA, Larson EB, Brickell K, Crane PK, Woltjer R, Montine TJ, Craft S. Different patterns of cerebral injury in dementia with or without diabetes. Arch Neurol. 2009;66:315–22. doi: 10.1001/archneurol.2008.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sparks DL, Scheff SW, Liu H, Landers TM, Coyne CM, Hunsaker JC. Increased incidence of neurofibrillary tangles (NFT) in non-demented individuals with hypertension. J Neurol Sci. 1995;131:162–169. doi: 10.1016/0022-510x(95)00105-b. [DOI] [PubMed] [Google Scholar]
- 145.Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007;1772:473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 146.Riekse RG, Leverenz JB, Mccormick W, Bowen JD, Teri L, Nochlin D, Simpson K, Eugenio C, Larson EB, Tsuang D. Effect of vascular lesions on cognition in Alzheimer’s disease: a community-based study. J Am Geriatr Soc. 2004;52:1442–1448. doi: 10.1111/j.1532-5415.2004.52405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;35:919–920. doi: 10.1016/S0140-6736(99)02355-7. [DOI] [PubMed] [Google Scholar]
- 148.Helzner EP, Luchsinger JA, Scarmeas N, Cosentino S, Brickman AM, Glymour MM, Stern Y. Contribution of vascular risk factors to the progression in Alzheimer disease. Arch Neurol. 2009;66:343–348. doi: 10.1001/archneur.66.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Silvestrini M, Pasqualetti P, Baruffaldi R, Bartolini M, Handouk Y, Matteis M, Moffa F, Provinciali L, Vernieri F. Cerebrovascular reactivity and cognitive decline in patients with Alzheimer disease. Stroke. 2006;37:1010–1015. doi: 10.1161/01.STR.0000206439.62025.97. [DOI] [PubMed] [Google Scholar]
- 150.Simpson JE, Ince PG, Haynes LJ, Theaker R, Gelsthorpe C, Baxter L, Forster G, Lace GL, Shaw PJ, Matthews FE, Savva GM, Brayne C, Wharton SB. Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol Appl Neurobiol. 2010;36:25–40. doi: 10.1111/j.1365-2990.2009.01030.x. [DOI] [PubMed] [Google Scholar]
- 151.Park L, Anrather J, Forster C, Kazama K, Carlson GC, Constantino I. Aβ-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab. 2004;24:334–342. doi: 10.1097/01.WCB.0000105800.49957.1E. [DOI] [PubMed] [Google Scholar]
- 152.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nature Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 153.Rosa-Neto P, Tong XK, Hamel E. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor g agonist. J Neurosci. 2008;28:9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Niwa K, Carlson GA, Iadecola C. Exogenous Aβ1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab. 2000;20:1659–1668. doi: 10.1097/00004647-200012000-00005. [DOI] [PubMed] [Google Scholar]
- 155.Abramov AY, Duchen MR. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos Trans R Soc Lond B Biol S. 2005;360:2309–2314. doi: 10.1098/rstb.2005.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. Beta-Amyloid activates the O2 forming NADPH oxidase in microglia, monocytes, and neutrophils. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 157.Bruce-Keller AJ, Gupta S, Parrino TE, Knight AG, Ebenezer PJ, Weidner AM, Levine H, Keller JN, Markesbery WR. NOX activity is increased in mild cognitive impairment. Antioxid Redox Signal. 2010;12:1371–1382. doi: 10.1089/ars.2009.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimot S. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun. 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 159.Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24:565–75. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 161.Mugge A, Elwell JH, Peterson TE, Hofmeyer TG, Heistad DD, Harrison DG. Chronic treatment with polyethylene-glycolated superoxide dismutase partially restores endothelium-dependent vascular relaxations in cholesterol-fed rabbits. Circ Res. 1991;69:1293–1300. doi: 10.1161/01.res.69.5.1293. [DOI] [PubMed] [Google Scholar]
- 162.Villa LM, Salas E, Darley-Usmar VM, Radomski MW, Moncada S. Peroxynitrite induces both vasodilatation and impaired vascular relaxation in the isolated perfused rat heart. Proc Natl Acad Sci U S A. 1994;91:12383–7. doi: 10.1073/pnas.91.26.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Burney S, Niles JC, Dedon PC, Tannenbaum SR. DNA damage in deoxynucleosides and oligonucleotides treated with peroxynitrite. Chem Res Toxicol. 1999;12:513–520. doi: 10.1021/tx980254m. [DOI] [PubMed] [Google Scholar]
- 164.Hink U, Oelze M, Kolb P, Bachschmid M, Zou MH, Daiber A, Mollnau H, August M, Baldus S, Tsilimingas N, Walter U, Ullrich V, Münzel T. Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J Am Coll Cardiol. 2003;42:1826–1834. doi: 10.1016/j.jacc.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 165.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 166.Orellana JA, Shoji KF, Abudara V, Ezan P, Amigou E, Sáez PJ, Jiang JX, Naus CC, Sáez JC, Giaume C. Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J Neurosci. 2011;31:4962–4977. doi: 10.1523/JNEUROSCI.6417-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Smale G, Nichols NR, Brady DR, Finch CE, Horton WE Jr. Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 168.Tong XK, Hamel E. Regional cholinergic denervation of cortical microvessels and nitric oxide synthase-containing neurons in Alzheimer’s disease. Neuroscience. 1999;92:163–175. doi: 10.1016/s0306-4522(98)00750-7. [DOI] [PubMed] [Google Scholar]
- 169.Nobili F, Koulibaly M, Vitali P, Migneco O, Mariani G, Ebmeier K, Pupi A, Robert PH, Rodriguez G, Darcourt J. Brain perfusion follow-up in Alzheimer’s patients during treatment with acetylcholinesterase inhibitors. J Nucl Med. 2002;43:983–90. [PubMed] [Google Scholar]
- 170.Ostrowski SM, Wilkinson BL, Golde TE, Landreth G. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J Biol Chem. 2007;282:26832–26844. doi: 10.1074/jbc.M702640200. [DOI] [PubMed] [Google Scholar]
- 171.Sparks L. Statins and cognitive function. J Neurol Neurosurg Psych. 2009;80:1–2. doi: 10.1136/jnnp.2008.160291. [DOI] [PubMed] [Google Scholar]
- 172.Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(02)09327-3. [DOI] [PubMed] [Google Scholar]
- 173.Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, Ford I, Gaw A, Hyland M, Jukema JW, Kamper AM, Macfarlane PW, Meinders AE, Norrie J, Packard CJ, Perry IJ, Stott DJ, Sweeney BJ, Twomey C, Westendorp RG. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 174.Simons M, Schwarzler F, Lutjohann D, Von Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T, Schulz JB. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol. 2002;52:346–350. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- 175.Sano M. Multi-center, randomized, double-blind, placebo-controlled trial of simvatatin to slow the progression of Alzheimer’s disease. Alz Dement. 2008;4:T200. doi: 10.1016/j.trci.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hoozemans JJ, Rozemuller JM, Van Haastert ES, Veerhuis R, Eikelenboom P. Cyclooxygenase-1 and -2 in the different stages of Alzheimer’s disease pathology. Curr Pharm Des. 2008;14:1419–1427. doi: 10.2174/138161208784480171. [DOI] [PubMed] [Google Scholar]
- 177.Sastre M, Gentleman SM. NSAIDs: how they work and their prospects as therapeutics in Alzheimer’s disease. Front Aging Neurosci. 2010;2:20. doi: 10.3389/fnagi.2010.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28:639–647. doi: 10.1016/j.neurobiolaging.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 179.Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hayden KM, Zandi PP, Khachaturian AS, Szekely CA, Fotuhi M, Norton MC, Tschanz JT, Pieper CF, Corcoran C, Lyketsos CG, Breitner JC, Welsh-Bohmer KA Cache County Investigators. Does NSAID use modify cognitive trajectories in the elderly? Neurology. 2007;69:275–82. doi: 10.1212/01.wnl.0000265223.25679.2a. [DOI] [PubMed] [Google Scholar]
- 181.Forette F, Seux ML, Staessen JA, Thijs L, Birkenhager WH, Babarskiene MR, Babeanu S, Bossini A, Gil-Extremera B, Girerd X, Laks T, Lilov E, Moisseyev V, Tuomilehto J, Vanhanen H, Webster J, Yodfat Y, Fagard R. Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet. 1998;352:1347–1351. doi: 10.1016/s0140-6736(98)03086-4. [DOI] [PubMed] [Google Scholar]
- 182.Davies NM, Kehoe PG, Ben-shlomo Y, Martin RM. Associations of anti-hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis. 2011;26:699–708. doi: 10.3233/JAD-2011-110347. [DOI] [PubMed] [Google Scholar]
- 183.Kehoe P, Miners S, Love S. Angiotensins in Alzheimer’s disease-friend or foe? Trends Neurosci. 2009;32:619–628. doi: 10.1016/j.tins.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 184.Hajjar I, Brown L, Mack WJ, Chui H. Impact of Angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol. 2012;69:1632–1638. doi: 10.1001/archneurol.2012.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Miao J, Vitek MP, Xu F, Previti ML, Davis J, Van Nostrand WE. Reducing cerebral microvascular amyloid-beta protein deposition diminishes regional neuroinflammation in vasculotropic mutant amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6271–7. doi: 10.1523/JNEUROSCI.1306-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Simons ER, Marshall DCL, Long HJ, Otto K, Billingslea A, Tibbles H, Wells J, Eisenhauer P, Fine RE, Cribbs DH, Davies TA, Abraham CR. Blood brain barrier endothelial cells express candidate amyloid precursor protein-cleaving secretases. Amyloid. 1998;5:153–162. doi: 10.3109/13506129809003841. [DOI] [PubMed] [Google Scholar]
- 187.Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain Res Mol Brain Res. 1992;16:128–134. doi: 10.1016/0169-328x(92)90202-m. [DOI] [PubMed] [Google Scholar]
- 188.Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, Brooks AI, Kanagala S, Rubio A, Sagare A, Liu D, Li F, Armstrong D, Gasiewicz T, Zidovetzki R, Song X, Hofman F, Zlokovic BV. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- 189.Xu J, Chen S, Ku G, Ahmed SH, Chen H, Hsu CY. Amyloid β peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J Cereb Blood Flow Metab. 2001;21:702–710. doi: 10.1097/00004647-200106000-00008. [DOI] [PubMed] [Google Scholar]
- 190.Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B, Mcphie DL, Walsh K, Querfurth H. Aβ42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK-3β signaling-dependent mechanism. Neurobiol Aging. 2003;24:437–451. doi: 10.1016/s0197-4580(02)00135-5. [DOI] [PubMed] [Google Scholar]
- 191.Grammas P, Botchlet T, Fugate R, Ball MJ, Roher AE. Alzheimer disease amyloid proteins inhibit brain endothelial cell proliferation in vitro. Dementia. 1995;6:126–130. doi: 10.1159/000106934. [DOI] [PubMed] [Google Scholar]
- 192.Balcells M, Wallins JS, Edelman ER. Amyloid beta toxicity dependent upon endothelial cell state. Neurosci Lett. 2008;441:319–322. doi: 10.1016/j.neulet.2008.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bhatia R, Lin H, Lal R. Fresh and globular amyloid β protein (1-42) induces rapid cellular degeneration: Evidence for AβP channel-mediated cellular toxicity. Faseb J. 2000;14:1233–1243. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- 194.Tsou HK, Chen HT, Chang CH, Yang WY, Tang CH. Apoptosis signal-regulating kinase 1 is mediated in TNF-α-induced CCL2 expression in human synovial fibroblasts. J Cell Biochem. 2012;113:3509–19. doi: 10.1002/jcb.24227. [DOI] [PubMed] [Google Scholar]
- 195.Hsu MJ, Hsu CY, Chen BC, Chen MC, Ou G, Lin CH. Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. J Neurosci. 2007;27:5719–5729. doi: 10.1523/JNEUROSCI.1874-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Matsunaga T, Kotamraju S, Kalivendi SV, Dhanasekaran A, Joseph J, Kalyanaraman B. Ceramide-induced intracellular oxidant formation, iron signaling, and apoptosis in endothelial cells: protective role of endogenous nitric oxide. J Biol Chem. 2004;279:28614–28624. doi: 10.1074/jbc.M400977200. [DOI] [PubMed] [Google Scholar]
- 197.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 198.Timmermans F, Plum J, Yöder MC, Ingram DA, Vandekerckhove B, Case J. Endothelial progenitor cells: identity defined? J Cell Mol Med. 2009;13:87–102. doi: 10.1111/j.1582-4934.2008.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lee ST, Chu K, Jung KH, Park HK, Kim DH, Bahn JJ, Kim JH, Oh MJ, Lee SK, Kim M, Roh JK. Reduced circulating angiogenic cells in Alzheimer disease. Neurology. 2009;72:1858–63. doi: 10.1212/WNL.0b013e3181a711f4. [DOI] [PubMed] [Google Scholar]
- 200.Kong XD, Zhang Y, Liu L, Sun N, Zhang MY, Zhang JN. Endothelial progenitor cells with Alzheimer’s disease. Chin Med J. 2011;124:901–906. [PubMed] [Google Scholar]
- 201.Hayashi S, Sato N, Yamamoto A, Ikegame Y, Nakashima S, Ogihara T, Morishita R. Alzheimer disease-associated peptide, amyloid beta40, inhibits vascular regeneration with induction of endothelial autophagy. Arterioscler Thromb Vasc Biol. 2009;29:1909–15. doi: 10.1161/ATVBAHA.109.188516. [DOI] [PubMed] [Google Scholar]
- 202.Patel NS, Mathura VS, Bachmeier C, Beaulieu-Abdelahad D, Laporte V, Weeks O, Mullan M, Paris D. Alzheimer’s amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR-2. J Neurochem. 2010;112:66–76. doi: 10.1111/j.1471-4159.2009.06426.x. [DOI] [PubMed] [Google Scholar]
- 203.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 204.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 205.Tio RA, Monnink SH, Amoroso G, Jessurun GA, Veeger N, Volkers C, Hautvast R, Tan ES, van Gilst WH, van Boven AJ. Safety evaluation of routine intracoronary acetylcholine infusion in patients undergoing a first diagnostic coronary angiogram. J Investig Med. 2002;50:133–139. doi: 10.2310/6650.2002.31305. [DOI] [PubMed] [Google Scholar]
- 206.Anderson TJ, Uehata A, Gerhard MD, Meredith IT, Knab S, Delagrange D, Lieberman EH, Ganz P, Creager MA, Yeung AC. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26:1235–1241. doi: 10.1016/0735-1097(95)00327-4. [DOI] [PubMed] [Google Scholar]
- 207.Deanfield JE, Halcox JP, Rabelink TJ. Endothelial Function and Dysfunction: Testing and Clinical Relevance. Circulation. 2007;115:1285–1295. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- 208.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 209.Cooke JP, Rossitch EJR, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J Clin Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Miura H, Wachtel RE, Liu Y, Loberiza FR Jr, Saito T, Miura M, Gutterman DD. Flow-induced dilation of human coronary arterioles: important role of Ca2+-activated K+ channels. Circulation. 2001;103:1992–1998. doi: 10.1161/01.cir.103.15.1992. [DOI] [PubMed] [Google Scholar]
- 211.Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in endothelial cells. Nature. 1988;331:168–170. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- 212.Pohl U, Holtz J, Busse R, Bassenge E. Crucial role of the endothelium in the vasodilator response to flow in vivo. Hypertension. 1985;8:37–44. doi: 10.1161/01.hyp.8.1.37. [DOI] [PubMed] [Google Scholar]
- 213.Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, Lüscher TF. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91:1314–1319. doi: 10.1161/01.cir.91.5.1314. [DOI] [PubMed] [Google Scholar]
- 214.Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T. Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:906–916. doi: 10.1093/jnen/60.9.906. [DOI] [PubMed] [Google Scholar]
- 215.Steinert JR, Chernova T, Forsythe ID. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist. 2010;16:435–452. doi: 10.1177/1073858410366481. [DOI] [PubMed] [Google Scholar]
- 216.Patel S, Celermajer DS. Assessment of vascular disease using arterial flow mediated dilatation. Pharmacol Rep. 2006;58:3–7. [PubMed] [Google Scholar]
- 217.Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R International Brachial Artery Reactivity Task Force. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–65. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 218.Morris SJ, Shore AC, Tooke JE. Responses of the skin microcirculation to acetylcholine and sodium nitroprusside in patients with NIDDM. Diabetologia. 1995;38:1337–1344. doi: 10.1007/BF00401767. [DOI] [PubMed] [Google Scholar]
- 219.Newton DJ, Khan F, Belch JF. Assessment of microvascular endothelial function in human skin. Clin Sci. 2001;101:567–572. [PubMed] [Google Scholar]
- 220.Wardell K, Jacobsson AJ, Nilsson GE. Laser Doppler perfusion imaging by dynamic light scattering. IEEE Trans Biomed Eng. 1993;40:309–316. doi: 10.1109/10.222322. [DOI] [PubMed] [Google Scholar]
- 221.Cracowski JL, Minson CT, Salvat-Melis M, Halliwill JR. Methodological issues in the assessment of skin microvascular endothelial function in humans. Trends Pharmacol Sci. 2006;27:503–508. doi: 10.1016/j.tips.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 222.Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, Gross SS, Nasjletti A, Goligorsky MS. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ Res. 2004;94:377–384. doi: 10.1161/01.RES.0000111802.09964.EF. [DOI] [PubMed] [Google Scholar]
- 223.Liao D, Wong TY, Klein R, Jones D, Hubbard L, Sharrett AR. Relationship between carotid artery stiffness and retinal arteriolar narrowing in healthy middle-aged persons. Stroke. 2004;35:837–842. doi: 10.1161/01.STR.0000120310.43457.AD. [DOI] [PubMed] [Google Scholar]
- 224.Leslie SJ, Attina T, Hultsch E, Bolscher L, Grossman M, Denvir MA, Webb DJ. Comparison of two plethysmography systems in assessment of forearm blood flow. J Appl Physiol. 2004;96:1794–1799. doi: 10.1152/japplphysiol.00567.2002. [DOI] [PubMed] [Google Scholar]
- 225.Benjamin N, Calver A, Collier J, Robinson B, Vallance P, Webb D. Measuring forearm blood fl ow and interpreting the responses to drugs and mediators. Hypertension. 1995;25:918–923. doi: 10.1161/01.hyp.25.5.918. [DOI] [PubMed] [Google Scholar]
- 226.Boutouyrie P, Briet M, Collina C, Vermeersch S, Pannier B. Assessment of pulse wave velocity. Art Res. 2009;3:3–8. [Google Scholar]
- 227.Kinlay S, Behrendt D, Wainstein M, Beltrame J, Fang JC, Creager MA, Selwyn AP, Ganz P. Role of endothelin-1 in the active constriction of human atherosclerotic coronary arteries. Circulation. 2001;104:1114–1118. doi: 10.1161/hc3501.095707. [DOI] [PubMed] [Google Scholar]
- 228.Boutouyrie P, Bezie Y, Lacolley P, Challande P, Chamiot-Clerc P, Benetos A, De La Faverie JF, Safar M, Laurent S. In vivo/in vitro comparison of rat abdominal aorta wall viscosity: influence of endothelial function. Arterioscler Thromb Vasc Biol. 1997;17:1346–1355. doi: 10.1161/01.atv.17.7.1346. [DOI] [PubMed] [Google Scholar]
- 229.Safar M, Chamiot-Clerc P, Dagher G, Renaud JF. Pulse pressure, endothelium function, and arterial stiffness in spontaneously hypertensive rats. Hypertension. 2001;38:1416–1421. doi: 10.1161/hy1201.096538. [DOI] [PubMed] [Google Scholar]
- 230.Wilkinson IB, Maccallum H, Cockcroft JR, Webb DJ. Inhibition of basal nitric oxide synthesis increases aortic augmentation index and pulse wave velocity in vivo. Br J Clin Pharmacol. 2002;53:189–192. doi: 10.1046/j.1365-2125.2002.1528adoc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Joannides R, Richard V, Haefeli WE, Benoist A, Linder L, Luscher TF, Thuillez C. Role of nitric oxide in the regulation of the mechanical properties of peripheral conduit arteries in humans. Hypertension. 1997;30:1465–1470. doi: 10.1161/01.hyp.30.6.1465. [DOI] [PubMed] [Google Scholar]
- 232.Armstrong EJ, Morrow DA, Sabatine MS. Inflammatory biomarkers in acute coronary syndromes, part II: acute-phase reactants and biomarkers of endothelial cell activation. Circulation. 2006;113:e152–5. doi: 10.1161/CIRCULATIONAHA.105.595538. [DOI] [PubMed] [Google Scholar]
- 233.Heiss C, Lauer T, Dejam A, Kleinbongard P, Hamada S, Rassaf T, Matern S, Feelisch M, Kelm M. Plasma nitroso compounds are decreased in patients with endothelial dysfunction. J Am Coll Cardiol. 2006;47:573–579. doi: 10.1016/j.jacc.2005.06.089. [DOI] [PubMed] [Google Scholar]
- 234.Oliver JJ, Webb DJ, Newby DE. Stimulated tissue plasminogen activator release as a marker of endothelial function in humans. Arterioscler Thromb Vasc Biol. 2005;25:2470–2479. doi: 10.1161/01.ATV.0000189309.05924.88. [DOI] [PubMed] [Google Scholar]
- 235.Verma S, Wang CH, Li SH, Dumont AS, Fedak PW, Badiwala MV, Dhillon B, Weisel RD, Li RK, Mickle DA, Stewart DJ. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106:913–9. doi: 10.1161/01.cir.0000029802.88087.5e. [DOI] [PubMed] [Google Scholar]
- 236.Devaraj S, Xu DY, Jialal I. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation. 2003;107:398–404. doi: 10.1161/01.cir.0000052617.91920.fd. [DOI] [PubMed] [Google Scholar]
- 237.Parker C, Vita JA, Freedman JE. Soluble adhesion molecules and unstable coronary artery disease. Atherosclerosis. 2001;156:417–424. doi: 10.1016/s0021-9150(00)00672-9. [DOI] [PubMed] [Google Scholar]
- 238.Holden D, Fickling S, Whitley G, Nussey S. Plasma concentrations of asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and preeclampsia. Am J Obstet Gynecol. 1998;178:551–556. doi: 10.1016/s0002-9378(98)70437-5. [DOI] [PubMed] [Google Scholar]
- 239.Surdacki A, Nowicki M, Sandmann J, Tsikas D, Boeger RH, Bode-Boeger SM, Kruszelnicka-Kwiatkowska O, Kokot F, Dubiel JS, Froelich JC. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol. 1999;33:652–8. doi: 10.1097/00005344-199904000-00020. [DOI] [PubMed] [Google Scholar]
- 240.Böger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98:1842–1847. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- 241.Böger RH, Bode-Böger SM, Tsao PS, Lin PS, Chan JR, Cooke JP. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. J Am Coll Cardiol. 2000;36:2287–2295. doi: 10.1016/s0735-1097(00)01013-5. [DOI] [PubMed] [Google Scholar]
- 242.Faraci FM, Brian JE, Heistad DD. Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am J Physiol. 1995;269:H1522–H1527. doi: 10.1152/ajpheart.1995.269.5.H1522. [DOI] [PubMed] [Google Scholar]
- 243.Böger RH. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res. 2003;59:824–833. doi: 10.1016/s0008-6363(03)00500-5. [DOI] [PubMed] [Google Scholar]
- 244.Macallister RJ, Parry H, Kimoto M, Ogawa T, Russell RJ, Hodson H, Whitley GS, Vallance P. Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br J Pharmacol. 1996;119:1533–1540. doi: 10.1111/j.1476-5381.1996.tb16069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Stühlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation. 2001;104:2569–75. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- 246.Dede DS, Yavuz B, Yavuz BB, Cankurtaran M, Halil M, Ulger Z, Cankurtaran ES, Aytemir K, Kabakci G, Ariogul S. Assessment of endothelial function in Alzheimer’s disease: is Alzheimer’s disease a vascular disease? J Am Geriatr Soc. 2007;55:1613–1617. doi: 10.1111/j.1532-5415.2007.01378.x. [DOI] [PubMed] [Google Scholar]
- 247.Algotsson A, Nordberg A, Almkvist O, Winblad B. Skin vessel reactivity is impaired in Alzheimer’s disease. Neurobiol Aging. 1995;16:577–582. doi: 10.1016/0197-4580(95)00077-r. [DOI] [PubMed] [Google Scholar]
- 248.Khalil Z, Logiudice D, Khodr B, Maruff P, Masters C. Impaired peripheral endothelial microvascular responsiveness in Alzheimer’s disease. J Alzheimers Dis. 2007;11:25–32. doi: 10.3233/jad-2007-11106. [DOI] [PubMed] [Google Scholar]
- 249.Bari F, Tóth-Szuki V, Domoki F, Kálmán J. Flow motion pattern differences in the forehead and forearm skin: Age-dependent alterations are not specific for Alzheimer’s disease. Microvasc Res. 2005;70:121–8. doi: 10.1016/j.mvr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 250.McGleenon BM, Passmore AP, Mcauley DF, Johnston GD. No evidence of endothelial dysfunction in patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2000;11:197–201. doi: 10.1159/000017237. [DOI] [PubMed] [Google Scholar]
- 251.Hanon O, Haulon S, Lenoir H, Seux ML, Rigaud AS, Safar M, Girerd X, Forette F. Relationship between arterial stiffness and cognitive function in elderly subjects with complaints of memory loss. Stroke. 2005;36:2193–2197. doi: 10.1161/01.STR.0000181771.82518.1c. [DOI] [PubMed] [Google Scholar]
- 252.Dhoat S, Ali K, Bulpitt CJ, Rajkumar C. Vascular compliance is reduced in vascular dementia and not in Alzheimer’s disease. Age Ageing. 2008;37:653–659. doi: 10.1093/ageing/afn158. [DOI] [PubMed] [Google Scholar]
- 253.Borroni B, Volpi R, Martini G, Del Bono R, Archetti S, Colciaghi F, Akkawi NM, Di Luca M, Romanelli G, Caimi L, Padovani A. Peripheral blood abnormalities in Alzheimer disease: evidence for early endothelial dysfunction. Alzheimer Dis Assoc Disord. 2002;16:150–155. doi: 10.1097/00002093-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 254.Gupta A, Watkins A, Thomas P, Majer R, Habubi N, Morris G, Pansari K. Coagulation and inflammatory markers in Alzheimer’s and vascular dementia. Int J Clin Pract. 2005;59:52–57. doi: 10.1111/j.1742-1241.2004.00143.x. [DOI] [PubMed] [Google Scholar]
- 255.Zuliani G, Cavalieri M, Galvani M, Passaro A, Munari MR, Bosi C, Zurlo A, Fellin R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J Neurol Sci. 2008;272:164–170. doi: 10.1016/j.jns.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 256.Järemo P, Milovanovic M, Buller C, Nilsson S, Winblad B. P-selectin paradox and dementia of the Alzheimer type: circulating P-selectin is increased but platelet-bound P-selectin after agonist provocation is compromised. Scand J Clin Lab Invest. 2013;73:170–4. doi: 10.3109/00365513.2013.764572. [DOI] [PubMed] [Google Scholar]
- 257.Selley ML. Increased concentrations of homocysteine and asymmetric dimethylarginine and decreased concentrations of nitric oxide in the plasma of patients with Alzheimer’s disease. Neurobiol Aging. 2003;24:903–907. doi: 10.1016/s0197-4580(03)00007-1. [DOI] [PubMed] [Google Scholar]
- 258.Abe T, Tohgi H, Murata T, Isobe C, Sato C. Reduction in asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in the cerebrospinal fluid during aging and in patients with Alzheimer’s disease. Neurosci Lett. 2001;312:177–179. doi: 10.1016/s0304-3940(01)02214-5. [DOI] [PubMed] [Google Scholar]
- 259.Mulder C, Wahlund LO, Blomberg M, De Jong S, Van Kamp GJ, Scheltens P, Teerlink T. Alzheimer’s disease is not associated with altered concentrations of the nitric oxide synthase inhibitor asymmetric dimethylarginine in cerebrospinal fluid. J Neural Transm. 2002;109:1203–1208. doi: 10.1007/s00702-002-0760-1. [DOI] [PubMed] [Google Scholar]
- 260.Arlt S, Schwedhelm E, Kölsch H, Jahn H, Linnebank M, Smulders Y, Jessen F, Böger RH, Popp J. Dimethylarginines, homocysteine metabolism, and cerebrospinal fluid markers for Alzheimer’s disease. J Alzheimers Dis. 2012;31:751–758. doi: 10.3233/JAD-2012-112138. [DOI] [PubMed] [Google Scholar]
- 261.Roy S, Rauk A. Alzheimer’s disease and the ‘ABSENT’ hypothesis: Mechanism for amyloid beta endothelial and neuronal toxicity. Med Hypotheses. 2005;65:123–137. doi: 10.1016/j.mehy.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 262.Sejda T, Pit’ha J, Svandová E, Poledne R. Limitations of non-invasive endothelial function assessment by brachial artery flow-mediated dilatation. Clin Physiol Funct Imaging. 2005;25:58–61. doi: 10.1111/j.1475-097X.2004.00590.x. [DOI] [PubMed] [Google Scholar]
- 263.Le Brocq M, Leslie SJ, Milliken P, Megson IL. Endothelial dysfunction: from molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid Redox Signal. 2008;10:1631–1674. doi: 10.1089/ars.2007.2013. [DOI] [PubMed] [Google Scholar]
- 264.Maltz RL, De Sotomayor MA, Schott C, Stoclet JC, Andriantsitohaina R. Vascular bed heterogeneity in age-related endothelial dysfunction with respect to NO and eicosanoids. Br J Pharmacol. 2000;131:303–311. doi: 10.1038/sj.bjp.0703568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Oliver JJ, Webb DJ. Noninvasive assessment of arterial stiffness and risk of atherosclerotic events. Arterioscler Thromb Vasc Biol. 2003;23:554–566. doi: 10.1161/01.ATV.0000060460.52916.D6. [DOI] [PubMed] [Google Scholar]
- 266.Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Arterioscler Thromb Vasc Biol. 1997;17:1193–1202. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- 267.Asif M, Soiza RL, McEvoy M, Mangoni AA. Asymmetric dimethylarginine: a possible link between vascular disease and dementia. Curr Alzheimer Res. 2013;10:347–56. doi: 10.2174/1567205011310040001. [DOI] [PubMed] [Google Scholar]
- 268.Iadecola C. Neurovascular regukation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 269.Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120:287–296. doi: 10.1007/s00401-010-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Axelsen PH, Komatsu H, Murray IV. Oxidative stress and cell membranes in the pathogenesis of Alzheimer’s disease. Physiology (Bethesda) 2011;26:54–69. doi: 10.1152/physiol.00024.2010. [DOI] [PubMed] [Google Scholar]