

Abstract
Background
Cardiac hypertrophic remodelling and systolic dysfunction are common in patients with mitochondrial disease and independent predictors of morbidity and early mortality. Endurance exercise training improves symptoms and skeletal muscle function, yet cardiac adaptations are unknown.
Methods and results
Before and after 16-weeks of training, exercise capacity, cardiac magnetic resonance imaging and phosphorus-31 spectroscopy, disease burden, fatigue, quality of life, heart rate variability (HRV) and blood pressure variability (BPV) were assessed in 10 adult patients with m.3243A>G-related mitochondrial disease, and compared to age- and gender-matched sedentary control subjects. At baseline, patients had increased left ventricular mass index (LVMI, p < 0.05) and LV mass to end-diastolic volume ratio, and decreased longitudinal shortening and myocardial phosphocreatine/adenosine triphosphate ratio (all p < 0.01). Peak arterial–venous oxygen difference (p < 0.05), oxygen uptake (VO2) and power were decreased in patients (both p < 0.01) with no significant difference in cardiac power output. All patients remained stable and completed ≥ 80% sessions. With training, there were similar proportional increases in peak VO2, anaerobic threshold and work capacity in patients and controls. LVMI increased in both groups (p < 0.01), with no significant effect on myocardial function or bioenergetics. Pre- and post-exercise training, HRV and BPV demonstrated increased low frequency and decreased high frequency components in patients compared to controls (all p < 0.05).
Conclusion
Patients with mitochondrial disease and controls achieved similar proportional benefits of exercise training, without evidence of disease progression, or deleterious effects on cardiac function. Reduced exercise capacity is largely mediated through skeletal muscle dysfunction at baseline and sympathetic over-activation may be important in pathogenesis.
Keywords: Mitochondrial DNA, Endurance exercise, Autonomic function, Cardio-pulmonary exercise testing, Cardiac magnetic resonance imaging, Cardiac magnetic resonance spectroscopy
1. Introduction
Mitochondrial diseases are a heterogeneous group of genetic disorders resulting in significant morbidity and disability, and affecting up to 8000 adults in the UK [1]. The m.3243A>G mutation in the mt-tRNALeu(UUR) MTTL1 gene is the most common pathogenic mutation, present in ~ 1 in 300 of the general population and causing disease in ~ 1 in 6000 individuals [2]. The clinical phenotype is highly variable: originally described in patients with mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS), the m.3243A>G mutation can also cause maternally inherited diabetes and deafness (MIDD), myopathy, ophthalmoplegia and cardiomyopathy, as isolated clinical features or part of multisystem disease [3]. Despite this phenotypic variability, exercise intolerance and fatigue are common clinical symptoms.
There are currently limited therapeutic options for patients with mitochondrial disease [4]. Endurance exercise training has, however, been demonstrated to improve exercise tolerance, quality of life and skeletal muscle oxidative capacity in this patient group, reversing baseline de-conditioning [5–8]. Resistance exercise training has been linked to similar clinical improvements in patients with mitochondrial disease [9]. While the cellular mechanisms underlying the effects of exercise training are yet to be fully elucidated, no deleterious clinical outcomes related to skeletal muscle function, quality of life or disease progression have been reported in studies from diverse genotypic groups. Yet clinical equipoise exists with regard to cardiac parameters, and understandable concerns of adverse remodelling, in a cohort of patients prone to cardiac abnormalities, may have restricted the widespread uptake of therapeutic endurance exercise training in patients with mitochondrial disease, hampering clinical care. Cardiomyopathy, most commonly with a hypertrophic phenotype, occurs in 20–40% of patients carrying the m.3243A>G mutation [10–14], and is an independent predictor of morbidity and early mortality [13,15]. Impaired cardiac bioenergetics also occur in patients with m.3243A>G-related mitochondrial disease [16,17], and are predictive of prognosis in diverse forms of cardiomyopathy [18]. Cardiovascular autonomic dysfunction has been demonstrated in patients harbouring the m.3243A>G mutation [19,20], and is independently associated with an increased risk of sudden death in the general population, after myocardial infarction [21].
In untrained individuals without mitochondrial disease, endurance exercise training increases left ventricular mass, ventricular cavity dimensions and haemodynamic parameters of cardiac function [22–24]. Similarly, exercise in patients with established heart failure reduces symptoms and improves cardiac function, exercise tolerance, quality of life and daily activity levels, without a significant deleterious effect on impaired cardiac bioenergetics [25–27]. However whether similar effects occur in patients with mitochondrial disease is unknown. Magnetic resonance imaging (MRI) is the gold standard investigation of cardiac morphology and, combined with cardiac tagging and phosphorus-31 (31P) magnetic resonance spectroscopy (MRS) enables detection of subclinical defects in both myocardial deformation and bioenergetics. Using these modalities, we sought to characterize the effects of endurance exercise training on disease burden, resting cardiac function, high energy phosphate metabolism, cardiovascular autonomic function, fatigue and quality of life in a clinically and genetically well-characterized cohort of patients with m.3243A>G-related mitochondrial disease with reference to age- and gender- and habitual physical activity level-matched controls.
2. Methods
2.1. Participants
Ten patients with mitochondrial disease due to the m.3243A>G mutation, but without known cardiac involvement, were recruited from consecutive attendees at a specialist outpatient clinic between August 2010 and July 2011. All eligible patients were verbally invited to participate in the study based on the following clinical inclusion criteria: (i) clinical stability for > 6 months; (ii) ability to use a semi-recumbent stationary bicycle ergometer; and (iii) no current participation in regular physical activity (≥ 1 weekly session). Exclusion criteria were the presence of known cardiac involvement (determined using clinical history, examination, 12-lead ECG and echocardiogram), comorbidities precluding exercise training (e.g. osteoarthritis), and contra-indications to MRI, including the presence of a pacemaker or implantable cardioverter-defibrillator, abnormal renal function (eGFR < 60 ml/min/1.73 m2) or claustrophobia. All 10 patients were matched with respect to age, gender and physical activity level with untrained healthy controls with normal ECG and no history of cardiovascular or metabolic disease, recruited through local advertisement. Institutional ethical approval and written informed consent were obtained.
2.2. Assessments
All baseline and follow-up assessments were completed on a single day. Assessments of exercise parameters, autonomic function, resting venous lactate and creatine kinase (CK) were performed between 0800 and 1000 following an overnight fast (> 10 h), and cardiac MRI took place at the same time of day throughout the study for each subject. All subjects were asked to refrain from smoking, alcohol ingestion, physical exertion and medications that could influence haemodynamic, exercise or autonomic parameters (including β blockers, calcium channel blockers, anti-depressants) for 24 h prior to assessment.
2.3. Exercise testing
Cardiopulmonary exercise testing was performed using analysis of expired air gases (Metalyzer 3B; Cortex, Leipzig, Germany) and non-invasive bioreactance cardiac output (NICOM; Cheetah Medical, Maidenhead, UK) on a calibrated, electronically braked stationary bicycle ergometer (Corival®; Lode, Groningen, Netherlands) at a steady cadence between 60 and 80 bpm. During all visits, a stepped incremental workload test (~ 10–20 W/min) was conducted to elicit a symptom-limited maximum oxygen uptake and heart rate response. Respiratory gas exchange data were collected continuously and the Borg Rating of Perceived Exertion (RPE) score performed every 3 min. Exercise was terminated when subjects developed severe dyspnoea or peripheral muscle fatigue and were physically exhausted (as indicated by respiratory exchange ratio (RER) > 1.1, Borg RPE score > 18 or absence of rise in oxygen consumption with further increases in exercise intensity). Anaerobic threshold was determined using the V-slope method, as previously described [28]. Baseline physical activity was assessed using an armband accelerometer SenseWear® Armband multi-array physical activity monitor (Bodymedia, Pittsburgh, USA) worn for seven days.
Cardiac power output (in Watts) was calculated as the product of mean arterial pressure (in mm Hg), cardiac output (in litres/min) and the conversion factor 2.22 × 10− 3 [29]. Arterial–venous oxygen difference, expressed in ml O2/100 ml of blood, was calculated as the ratio of peak oxygen consumption and cardiac output.
2.4. Disease burden
Subjects underwent physical examination by experienced clinicians (GSG and MGDB). Disease burden in patients was assessed using the Newcastle Mitochondrial Disease Adult Scale (NMDAS), a validated rating system [30], whilst mutation load was determined in urinary epithelial cells, an established marker of clinical status in patients with the m.3243A>G mutation [31].
2.5. Body weight and composition
Subject heights were recorded for body mass index and body surface area calculations. Body weights, body fat percentages and lean body weights were measured using air displacement plethysmography (BODPOD®; COSMED, Rome, Italy).
2.6. Cardiac magnetic resonance imaging
Using a 3-Tesla scanner, cardiac MRI was performed to include: (i) 31P MRS (ii) cine imaging, and (iii) cardiac tagging, as previously described and validated in this patient cohort [17,32].
2.6.1. Cardiac 31P MRS
Subjects were scanned prone with a 31P surface coil using a cardiac-gated, 1-dimensional chemical shift imaging sequence. Sixteen coronal phase-encoding steps (TR = heart rate, 192 averages) yielded spectra from 10 mm slices. The first spectrum arising entirely beyond the chest wall was analysed using AMARES time domain fit [33] to quantify phosphocreatine (PCr), the γ resonance of adenosine triphosphate (ATP), corrected for blood contamination, and their ratio (PCr/ATP), as a marker of cardiac bioenergetics [17].
2.6.2. Cardiac cine imaging
Subjects were scanned supine with steady-state free precession images, using a 6-channel cardiac coil and ECG gating [field of view (FOV) 350 × 350 mm2, repetition time/echo time (TR/TE) = 3.7/1.9 ms, turbo factor 17, flip angle (FA) 40°, slice thickness 8 mm, 25 phases, resolution 1.37 mm]. Left ventricular (LV) mass and systolic and diastolic parameters were calculated as previously described [17,32].
2.6.3. Cardiac tagging
MR signal from myocardium in diastole was cancelled in a rectangular grid pattern and tags were tracked through the cardiac cycle (Fig. 1A) [34]. A multi-shot turbo-field echo sequence was used (TR/TE/FA/number of averages = 4.9/3.1/10°/1, turbo factor 9, SENSE factor 2, FOV 350 × 350 mm2, voxel size 1.37 mm, tag spacing 7 mm, 12 phases). Peak circumferential strain for both the whole myocardial wall and the endocardial third were calculated. Peak torsion between two adjacent short-axis slices was calculated as the circumferential-longitudinal shear angle, defined on the epicardial surface (Fig. 1B).
Fig. 1.
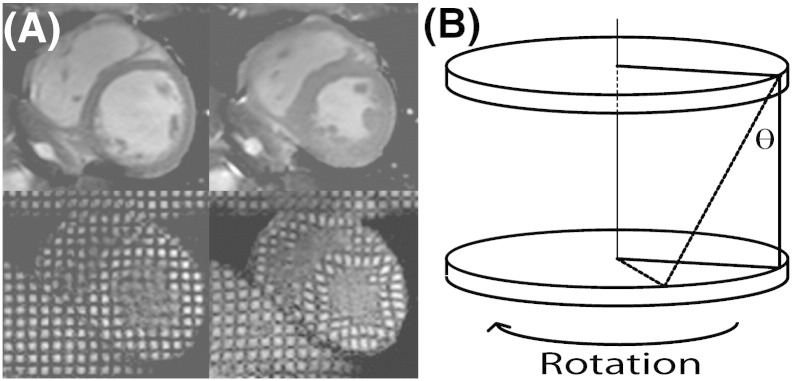
Cardiac tagging analysis. (A) Cine imaging (top panels) and tagging (bottom) at end-diastole (left panels) and end-systole (right). A rectangular grid of nulled myocardium applied in diastole enables tracking of myocardial deformation, including circumferential strain. (B) Tagging of 2 parallel short-axis slices allows calculation of torsion, the longitudinal–circumferential sheer angle (θ) as shown. Torsion describes the twisting motion of the heart due to opposite rotation of base and apex, and maintains homogeneity of strain across the myocardial wall. Increased torsion results from dominant contractile function in the subepicardium, compared to the subendocardium such that the torsion to endocardial circumferential strain ratio (TSR) is a sensitive marker of altered epicardial–endocardial interaction.
2.7. Autonomic function
2.7.1. Apparatus
All participants underwent assessment of autonomic function over 30 min at rest in a cardiorespiratory laboratory using the Task Force Monitor (TFM; CNSystems, Graz, Austria), which provides automated and computed beat-to-beat analysis of heart rate and blood pressure. Biological inputs to the system are provided by: (i) 3-lead ECG; (ii) impedance cardiography; and (iii) continuous, non-invasive blood pressure, recorded using finger plethysmography with intermittently calibration to contralateral oscillometric blood pressure measurements. From these data, the TFM calculates continuous, reliable measurements of haemodynamic and autonomic parameters [35–37].
2.7.2. Heart rate variability (HRV) and blood pressure variability (BPV)
Power spectral analysis for HRV and BPV was conducted using an adaptive auto-regressive (AAR) model as previously described [38]. In addition to total power spectral density (PSD), the low frequency (LF, 0.05–0.17 Hz) and high frequency (HF, 0.17–0.4 Hz) components of the signal were automatically calculated using the TFM. All components are reported in normalized units (nu) for RR interval (RRI), systolic blood pressure (SBP) and diastolic blood pressure (DBP) parameters (LFnu-RRI, HFnu-RRI, LFnu-SBP, HFnu-SBP, LFnu-DBP, and HFnu-DBP). As previously described, all LF components refer to sympathetic modulation of the sinus node activity and vasomotor function, while HF components refer to parasympathetic modulation of such cardiovascular activity. The ratios of LF and HF components provide a marker of sympatho-vagal balance (LF:HF-RRI, LF:HF-SBP, and LF:HF-DBP) [39,40].
2.8. Fatigue and quality of life
Standard, validated self-completion questionnaires were used for the assessment of fatigue (Fatigue Impact Scale (FIS)) and quality of life (the Short-Form 12 (SF-12)).
2.9. Exercise intervention
Participants were instructed to perform 30 min of cycling (excluding 5 min of warming-up and cooling-down) on an upright ergometer, three times per week, for 16 weeks at a self-regulated workload that achieved a heart rate corresponding to 70–80% of the symptom-limited maximum oxygen uptake, and a Borg RPE score 12–14. Target heart rate for all subjects was monitored using an RS100 watch (Polar Electro, Finland) and adjusted at 8 weeks following completion of a maximal graded cardiopulmonary exercise test. Workload was adjusted continuously during all training sessions to achieve target heart rates. Participants were asked to limit training to the prescribed programme, and, although exercise training was unsupervised, all subjects received weekly phone calls to ensure compliance with exercise training, and completed exercise diaries that were regularly reviewed by study investigators.
2.10. Statistical analysis
Data are presented as means ± SD for continuous data and as numbers or percentages for categorical data. Continuous data were tested for normality, linearity and homogeneity of covariance matrices. A mixed model multivariate analysis of variance was performed between patients and controls, before and after completion of the exercise intervention. Group (patient or control) and time (baseline or follow-up) were respected as factors and physiological parameters were taken as the dependent variables. The interaction between group and time conditions permitted detection of any differences in the response to exercise. Where the mixed model main factors or interaction were statistically significant, subsequent between group comparisons were made using unpaired Student's t-tests or Mann–Whitney U tests and within group comparisons using paired Student's t-tests or Wilcoxon signed rank tests. Categorical variables were compared using Fisher's exact test and correlations were executed using Pearson's method. All analysis was performed using SPSS version 17 (SPSS Inc., Chicago, Illinois). All tests were two-sided and statistical significance was assumed at p < 0.05.
3. Results
3.1. Patient group characteristics
The baseline characteristics of 10 patients (9 probands) and 10 control subjects, matched for age, gender and habitual physical activity, are presented in Table 1. Body mass index (BMI) and body surface area (BSA) were significantly lower in patients than controls. Cardiovascular disease features and relevant medications are included in Table 1: five patients had diabetes mellitus and one had treated hypertension. There were no significant differences in current systolic or diastolic blood pressures.
Table 1.
Baseline characteristics.
| Characteristic | Patients (n = 10) |
Controls (n = 10) |
p value |
|---|---|---|---|
| Age (years) | 42.4 ± 10.5 | 39.0 ± 11.8 | 0.391 |
| Male sex, n (%) | 6 (60) | 6 (60) | 1.000 |
| Height (cm) | 171 ± 10 | 172 ± 10 | 0.707 |
| Weight (kg) | 62.1 ± 12.2 | 81.9 ± 12.9 | 0.002 |
| Body mass index (kg/m2) | 21.3 ± 3.4 | 27.7 ± 4.7 | 0.002 |
| Body surface area (m2) | 1.72 ± 0.19 | 1.95 ± 0.16 | 0.010 |
| Habitual physical activity | |||
| Daily energy expenditure (kCal) | 2133 ± 666 | 2529 ± 373 | 0.118 |
| Daily steps (n) | 7949 ± 3968 | 9290 ± 2708 | 0.207 |
| Diabetes mellitus, n (%) | 5 (50) | 0 (0) | N/A |
| Hypertension, n (%) | 1 (10) | 0 (0) | N/A |
| Cardiac clinical parameters | |||
| Heart rate (min− 1) | 75 ± 11 | 64 ± 10 | 0.033 |
| SBP (mm Hg) | 118 ± 11 | 114 ± 8 | 0.430 |
| DBP (mm Hg) | 81 ± 8 | 77 ± 7 | 0.181 |
| Selected medications | |||
| ACE inhibitor/ARB | 4 (40) | 0 (0) | N/A |
| Beta-blocker | 0 (0) | 0 (0) | N/A |
| Calcium channel blocker | 1 (10) | 0 (0) | N/A |
| Insulin | 3 (30) | 0 (0) | N/A |
| Metformin | 2 (20) | 0 (0) | N/A |
| Statin | 4 (40) | 0 (0) | N/A |
| Co-enzyme Q10 | 4 (40) | 0 (0) | N/A |
| Antidepressant | 2 (20) | 0 (0) | N/A |
N/A = not applicable; SBP = systolic blood pressure; DBP = diastolic blood pressure; ACE = angiotensin-converting enzyme; ARB = angiotensin receptor blocker.
Patient details including specific clinical features, mutation loads and baseline NMDAS, FIS and SF-12 scores are presented in Table 2. Disease burden was mild or moderate in all patients with phenotypes consistent with MIDD (five patients), myopathy (three patients), MELAS (one patient) and oligosymptomatic status (one patient). The frequencies of specific clinical features in this group are presented in Supplementary Table 1. Fatigue was a clinical feature of mitochondrial disease in five patients (50%): three patients reported excessive fatigue (FIS score ≥ 40) and one patient reported severe fatigue (FIS score ≥ 80).
Table 2.
Disease features of patients.
| Patient | Age | Sex | Urinary m.3243A>G mutation load (%) | Clinical features | Baseline clinical scores |
|||
|---|---|---|---|---|---|---|---|---|
| NMDAS | FIS | SF-12 PHC | SF-12 MHC | |||||
| 1a | 39 | M | 80 | SNHL, diabetes mellitus, exercise intolerance, ataxia, migraine, GIT, fatigue, hypothyroidism | 17 | 44 | 38 | 37 |
| 2 | 58 | F | 59 | SNHL, exercise intolerance, mild ataxia, proximal muscle weakness, GIT, low BMI, myalgia | 12 | 2 | 56 | 58 |
| 3a | 42 | M | 82 | SNHL, diabetes mellitus, exercise intolerance, ataxia, migraine, depression | 12 | 9 | 50 | 59 |
| 4 | 47 | M | 63 | SNHL, diabetes mellitus, exercise intolerance, ataxia, proximal and distal muscle weakness, depression, fatigue, myalgia, PEO, ptosis, sensory neuropathy | 28 | 109 | 23 | 29 |
| 5 | 37 | F | 48 | SNHL, diabetes mellitus, exercise intolerance, mild ataxia, mild dysarthria, asthma | 10 | 13 | 52 | 53 |
| 6 | 38 | F | 53 | SNHL, exercise intolerance, proximal muscle weakness, fatigue, migraine, GIT, asthma | 13 | 29 | 32 | 55 |
| 7 | 22 | M | 89 | SNHL, exercise intolerance, ataxia, fatigue, migraine, GIT, low BMI, epilepsy | 18 | 62 | 40 | 46 |
| 8 | 36 | M | 80 | Exercise intolerance, migraine | 4 | 9 | 57 | 54 |
| 9 | 50 | M | 87 | SNHL, exercise intolerance, ataxia, proximal and distal muscle weakness, fatigue, depression, retinopathy, epilepsy, encephalopathy, cognitive decline, stroke-like episodes | 23 | 68 | 57 | 46 |
| 10 | 55 | F | 68 | SNHL, diabetes mellitus, exercise intolerance, ataxia, proximal muscle weakness, GIT, depression, retinopathy, PEO, ptosis, short stature, mild dysphagia, hypertension | 25 | 23 | 36 | 49 |
NMDAS = Newcastle Mitochondrial Disease Adult Scale; M = male; F = female; MIDD = maternally inherited deafness and diabetes; MELAS = mitochondrial encephalopathy, lactic acidosis and stroke-like episodes; FIS = Fatigue Impact Scale; SF-12 = Short Form-12 quality of life questionnaire; PHC = physical health component; MHC = mental health component .
Sibling.
3.2. Exercise training
All patients and control subjects completed ≥ 80% of the 48 scheduled training sessions, and no adverse events were reported. All patients remained clinically stable throughout the study, and there were no changes in medication.
Cardio-pulmonary exercise testing responses for patients and controls before and after completion of the exercise training programme are presented in Table 3. There was no significant effect of subject status (patient or control) on the response to endurance exercise in any haemodynamic, exercise physiology or cardiac parameter.
Table 3.
Exercise parameters before and after training.
| Parameter | Patients |
Controls |
Interaction p valueb | ||||
|---|---|---|---|---|---|---|---|
| Baseline | Follow-up | p valuea | Baseline | Follow-up | p valuea | ||
| Peak exercise | |||||||
| Heart rate | 167 ± 21 | 161 ± 18 | 0.299 | 184 ± 17 | 178 ± 14⁎ | 0.001 | 0.921 |
| SBP (mm Hg) | 183 ± 24 | 193 ± 21 | 0.082 | 188 ± 18 | 194 ± 16 | 0.183 | 0.744 |
| DBP (mm Hg) | 94 ± 17 | 93 ± 15 | 0.880 | 99 ± 12 | 89 ± 17 | 0.069 | 0.222 |
| MAP (mm Hg) | 124 ± 15 | 126 ± 13 | 0.535 | 129 ± 10 | 124 ± 13 | 0.260 | 0.258 |
| Stroke volume (ml) | 91 ± 23 | 95 ± 29 | 0.532 | 116 ± 20⁎⁎ | 118 ± 26 | 0.774 | 0.786 |
| Stroke index (ml/m2) | 53 ± 9 | 54 ± 12 | 0.687 | 59 ± 9 | 60 ± 9 | 0.832 | 0.695 |
| Cardiac output (l/min) | 14.5 ± 3.9 | 15.1 ± 4.8 | 0.398 | 21.3 ± 4.1⁎⁎ | 21.0 ± 4.8⁎ | 0.946 | 0.643 |
| Cardiac index (l/min/m2) | 8.3 ± 1.5 | 8.6 ± 2.1 | 0.681 | 10.6 ± 1.2⁎⁎ | 10.4 ± 1.5 | 0.828 | 0.517 |
| VO2 (ml/min) | 1382 ± 625 | 1596 ± 726 | 0.030 | 2276 ± 670⁎⁎ | 2525 ± 562⁎ | 0.049 | 0.983 |
| VO2 (ml/kg/min) | 21.4 ± 6.3 | 24.8 ± 8.9 | 0.009 | 27.6 ± 6.3⁎ | 30.4 ± 5.9 | 0.047 | 0.965 |
| VO2 (% predicted VO2) | 61 ± 20 | 72 ± 25 | 0.011 | 94 ± 21⁎⁎ | 106 ± 26⁎⁎ | 0.032 | 0.882 |
| CPO | 4.0 ± 1.3 | 4.0 ± 1.2 | 0.708 | 5.7 ± 1.0⁎⁎ | 5.4 ± 1.2⁎ | 0.408 | 0.413 |
| CPOI | 2.3 ± 0.5 | 2.3 ± 0.5 | 0.732 | 2.7 ± 0.4 | 2.8 ± 0.5 | 0.396 | 0.294 |
| Power (W) | 100 ± 45 | 112 ± 55 | 0.044 | 177 ± 40⁎⁎ | 200 ± 39⁎⁎ | 0.001 | 0.621 |
| A–VO2 diff (ml O2/dl) | 9.4 ± 3.0 | 10.5 ± 3.1 | 0.059 | 10.9 ± 2.9⁎ | 11.8 ± 1.9⁎ | 0.242 | 0.736 |
| Anaerobic threshold | |||||||
| ATVO2 (ml/kg/min) | 13.4 ± 5.0 | 16.5 ± 6.4 | 0.014 | 15.4 ± 4.1 | 18.8 ± 4.1 | 0.017 | 0.772 |
| ATVO2 (% predicted peak VO2) | 38 ± 14 | 47 ± 18 | 0.221 | 53 ± 14⁎ | 65 ± 19⁎⁎ | 0.032 | 0.665 |
| ATVO2 (% recorded peak VO2) | 61 ± 8 | 66 ± 5 | 0.128 | 56 ± 7 | 63 ± 5 | 0.049 | 0.758 |
SBP = systolic blood pressure; DBP = diastolic blood pressure; MAP = mean arterial pressure; VO2 = oxygen uptake; AT = anaerobic threshold; CPO = cardiac power output; CPOI = cardiac power output index; A–VO2 diff = arterial–venous oxygen difference; AT = anaerobic threshold.
p value represents the within group comparison of paired before and follow-up time points.
p value represents group by time interaction from mixed model repeated measures analysis.
p < 0.05 for patients vs. controls at equivalent time points.
p < 0.01 for patients vs. controls at equivalent time points.
3.3. Haemodynamic parameters
At peak exercise stroke volume, heart rate, cardiac output, and cardiac index, were all decreased in patients compared to controls (Table 3), achieving statistical significance at either baseline or follow-up or both time-points for each of these parameters. There were no significant differences between the groups in peak systolic, diastolic or mean arterial blood pressures or in cardiac power output, when indexed to BSA (CPO index). Although there was a trend towards reduction in peak heart rate following exercise training in both groups, this only achieved statistical significance in controls (mean decrease 7 bpm, p = 0.001).
3.4. Exercise physiology
At baseline, peak VO2, peak arterial–venous oxygen difference (A–VO2 diff), and peak power were all significantly decreased in patients compared to controls (mean decreases 22%, 30%, 23% and 77% respectively). The anaerobic threshold (AT) at baseline, when expressed as a percentage of predicted peak VO2, was significantly decreased in patients compared to controls, while absolute values showed a supportive trend in the same direction. In both patient and control groups, sixteen weeks of endurance exercise training significantly increased peak work capacity (power, 12% and 13% respectively), peak oxygen uptake (VO2, 16% and 10%), and anaerobic threshold (AT, 23% and 23%) without a significant change in CPO or CPOI. In patients, there was a non-significant trend towards an increase in peak capacity for oxygen extraction (A–VO2 diff, 12%, p = 0.059). Resting venous lactate and creatine kinase were not significantly different at baseline between patients and controls and there was no significant effect of endurance exercise training on these blood parameters, in either group (Supplementary Table 2).
3.5. Body composition
Although significant between-group differences existed at baseline, there were no significant changes in body weight or BMI in patient or control groups in response to exercise training (Supplementary Table 2). In patients, there was a non-significant trend towards an increase in lean body weight (mean increase 1.0 kg, p = 0.056) following the endurance exercise training intervention.
3.6. Cardiac structure and function
Table 4 summarizes the cardiac MRI structural and functional parameters for patient and control groups. The means and ranges of control group parameters are in agreement with a large cohort study using quantitative cardiac MRI [41].
Table 4.
Cardiac parameters before and after exercise training.
| Parameter | Patients |
Controls |
Interaction p valueb | ||||
|---|---|---|---|---|---|---|---|
| Baseline | Follow-up | p valuea | Baseline | Follow-up | p valuea | ||
| Structure and systolic function | |||||||
| EDV (ml) | 96 ± 21 | 101 ± 17 | 0.120 | 136 ± 25⁎⁎ | 139 ± 20⁎⁎ | 0.305 | 0.737 |
| EDI (ml/m2) | 55 ± 8 | 58 ± 6 | 0.138 | 69 ± 10⁎⁎ | 71 ± 8⁎⁎ | 0.224 | 0.992 |
| ESV (ml) | 38 ± 13 | 41 ± 13 | 0.158 | 56 ± 13⁎⁎ | 56 ± 14⁎ | 0.972 | 0.551 |
| ESI (ml/m2) | 22 ± 6 | 23 ± 6 | 0.184 | 29 ± 5⁎ | 29 ± 6 | 0.898 | 0.646 |
| SV (ml) | 58 ± 9 | 60 ± 6 | 0.342 | 79 ± 14⁎⁎ | 83 ± 10⁎⁎ | 0.354 | 0.926 |
| SI (ml/m2) | 33 ± 3 | 35 ± 3 | 0.340 | 41 ± 6⁎⁎ | 43 ± 5⁎⁎ | 0.281 | 0.537 |
| CO (l/min) | 4.3 ± 0.7 | 4.2 ± 0.7 | 0.502 | 5.0 ± 1.0 | 5.1 ± 1.0 | 0.755 | 0.843 |
| CI (l/min/m2) | 2.5 ± 0.2 | 2.4 ± 0.4 | 0.419 | 2.6 ± 0.4 | 2.7 ± 0.5 | 0.689 | 0.528 |
| EF (%) | 61 ± 5 | 60 ± 7 | 0.515 | 59 ± 4 | 60 ± 6 | 0.489 | 0.410 |
| LS (%) | 14.8 ± 0.2 | 13.2 ± 0.3 | 0.213 | 18.1 ± 0.2⁎⁎ | 17.3 ± 0.3⁎ | 0.553 | 0.573 |
| LVM (g) | 124 ± 20 | 140 ± 21 | 0.003 | 116 ± 20 | 134 ± 26 | 0.001 | 0.989 |
| LVMI (g/m2) | 72 ± 13 | 81 ± 10 | 0.004 | 59 ± 8⁎ | 69 ± 11⁎ | 0.002 | 0.666 |
| M/V ratio (g/ml) | 1.35 ± 0.40 | 1.41 ± 0.26 | 0.302 | 0.86 ± 0.07⁎⁎ | 0.97 ± 0.11⁎⁎ | 0.003 | 0.620 |
| Diastolic function | |||||||
| E/A ratio | 1.65 ± 0.62 | 1.45 ± 0.57 | 0.387 | 1.65 ± 0.40 | 1.83 ± 0.43 | 0.320 | 0.451 |
| EFP (%) | 67.7 ± 6.7 | 70.7 ± 14.1 | 0.468 | 72.7 ± 7.6 | 73.9 ± 6.4 | 0.698 | 0.557 |
| Tagging and strains | |||||||
| Torsion (°) | 9.1 ± 3.5 | 7.9 ± 1.0 | 0.337 | 5.9 ± 1.9⁎ | 6.7 ± 1.5⁎ | 0.057 | 0.292 |
| Whole wall circumferential strain (%) | 16.4 ± 1.9 | 16.7 ± 1.4 | 0.624 | 17.8 ± 1.8 | 18.6 ± 3.4 | 0.435 | 0.714 |
| Endocardial circumferential strain (%) | 20.6 ± 2.0 | 20.0 ± 2.0 | 0.353 | 24.7 ± 2.3⁎⁎ | 25.9 ± 5.2⁎ | 0.367 | 0.713 |
| TSR (rad) | 0.80 ± 0.38 | 0.71 ± 0.10 | 0.555 | 0.41 ± 0.12⁎⁎ | 0.46 ± 0.09⁎⁎ | 0.325 | 0.292 |
| Cardiac high energy phosphates | |||||||
| PCr/ATP ratio | 1.45 ± 0.42 | 1.61 ± 0.39 | 0.260 | 1.95 ± 0.34⁎⁎ | 1.97 ± 0.37⁎ | 0.907 | 0.749 |
EDV = end-diastolic volume; ESI = end-diastolic index; ESV = end-systolic volume; ESI = end-systolic index; SV = stroke volume; SI = stroke index; CO = cardiac output; CI = cardiac index; EF = ejection fraction; LS = longitudinal shortening; LVM = left ventricular mass; LVMI = left ventricular mass index; M/V ratio = ratio of left ventricular mass to volume; E/A ratio = ratio of early to late ventricular filling velocity; EFP = early filling percentage; TSR = torsion to (endocardial) strain ratio; PCr = phosphocreatine; ATP = adenosine triphosphate.
p value represents the within group comparison of paired before and follow-up time points.
p value represents group by time interaction from mixed model repeated measures analysis.
p < 0.05 for patients vs. controls at equivalent time points.
p < 0.01 for patients vs. controls at equivalent time points.
At baseline, end-systolic and end-diastolic cardiac volumes were proportionally decreased in patients compared to controls, with no difference in ejection fraction (Table 4). Stroke volume was also decreased in patients: this occurred in association with an increase in heart rate (r = − 0.71, p = 0.021), with no difference in cardiac output. Endurance exercise had no significant effect on cardiac volumes or global systolic or diastolic function in patients or controls.
LV mass index (LVMI) was significantly increased in patients compared to controls (Table 4) at baseline, but remained within the normal range, as LVMI did not fulfil the definition of left ventricular hypertrophy (LVH) in any patient. A significant increase in M/V ratio (57%, p = 0.001) suggested that this subclinical difference represented concentric remodelling. Cardiac mass increased significantly in both patient and control groups following endurance exercise, with similar proportional sizes of effect: LVM (mean increases 13% and 16% respectively, Fig. 2), LVMI (13% and 17%), and M/V ratio (5% and 10%). Subject status (patient or control) had no significant effect on the effect of exercise on LVMI (Fig. 3A).
Fig. 2.

Box-plot of range and quartiles of the increase in LV mass between baseline and 16-week follow-up assessment, after endurance exercise training, expressed as a percentage of the baseline assessment in patient and control groups. LV = left ventricular.
Fig. 3.

Spaghetti plot of the change in (A) LVMI and (B) PCr/ATP ratio in patients (red, open squares) and controls (black, closed circles) following 16 weeks of endurance exercise training. Solid lines represent individual participants; dotted lines represent the corresponding mean group values; and vertical bars show the standard deviation at baseline (left) and following exercise training (right) in the respective groups. LVMI = left ventricular mass index; PCr = phosphocreatine; ATP = adenosine triphosphate; baseline = initial assessment; post-exercise = follow-up assessment after completion of 16 week exercise training.
3.7. Cardiac tagging and myocardial strains
At baseline, there was evidence of altered myocardial strains in patients consistent with subclinical concentric remodelling (Table 4): longitudinal shortening was significantly decreased (18%) and correlated significantly with increased LVMI (r = − 0.61, p = 0.03). Peak torsion was increased (54%) and endocardial circumferential strain was decreased (17%) in patients compared to controls, with a subsequent significant increase in their ratio, the TSR (95%). There were, however, no significant effects of exercise on myocardial strains or torsion in either patients or controls.
3.8. Myocardial bioenergetics
At baseline, PCr/ATP ratio was decreased in patients compared to controls (mean decrease 26%, p = 0.002, Table 4). Seven patients (70%) but no controls had an abnormal PCr/ATP ratio (< 1.6) [18], but there was no significant difference in markers of disease burden, cardiac structure or function between patients with PCr/ATP ratio > 1.6 and those < 1.6.
There was no significant effect of exercise training on the PCr/ATP ratio in patients or controls (Fig. 3B), and no significant difference in the response to exercise between the groups (Table 4). There was a trend towards an increase in the PCr/ATP ratio in patients following exercise training (mean increase 11%, p = 0.260). PCr/ATP ratio increased in six patients and decreased in four patients after exercise training — at follow-up only four patients (40%) had a ratio < 1.6 (Fig. 3B). Representative 31P MR spectra are shown (Fig. 4).
Fig. 4.

Phosphorus-31 magnetic resonance spectroscopy. Representative spectra from a patient carrying the m.3243A>G mutation (left) and a matched control subject (right) at baseline (upper panels) and following completion of 16 week exercise training (lower panels) showing a difference in PCr concentration that is unaffected by exercise. Spectra obtained from patient 3 (A) at baseline with PCr/ATP ratio 1.1, and (B) following exercise training with PCr/ATP 1.2 are displayed alongside spectra from the matched control participant (C) at baseline with PCr/ATP 2.0, and (D) following exercise training with PCr/ATP 1.9. Spectra are presented as acquired before correction for heart rate, flip angle and blood content. PCr = phosphocreatine; ATP = adenosine triphosphate; ppm = parts per million.
3.9. Quality of life and disease burden
At baseline, patients had NMDAS scores averaging 16 ± 7, which correlated with FIS score (r = 0.784, p = 0.007), but not urinary m.3243A>G mutation load (r = 0.465, p = 0.176) or SF-12 scores (r = 0.234, p = 0.477). After exercise training there was no significant change in NMDAS or SF-12 quality-of-life scores in patients (Supplementary Table 3).
3.10. Fatigue and autonomic function
FIS scores, both at baseline and after exercise training, were significantly increased in patients compared to controls (Table 5), but there was no significant effect of exercise on FIS score in either group; the proportion of patients reporting excessive or severe fatigue at baseline (40%) was unchanged following exercise training.
Table 5.
Autonomic parameters and fatigue before and after exercise training.
| Parameter | Patients |
Controls |
Interaction p valueb | ||||
|---|---|---|---|---|---|---|---|
| Baseline | Follow-up | p valuea | Baseline | Follow-up | p valuea | ||
| Heart rate variability | |||||||
| Mean RRI (ms) | 808 ± 101 | 854 ± 112 | 0.220 | 891 ± 102⁎ | 948 ± 119⁎ | 0.128 | 0.568 |
| LFnu-RRI | 68 ± 18 | 64 ± 19 | 0.711 | 54 ± 12⁎⁎ | 48 ± 17⁎ | 0.086 | 0.591 |
| HFnu-RRI | 46 ± 18 | 48 ± 19 | 0.711 | 49 ± 22 | 58 ± 25 | 0.086 | 0.591 |
| PSD-RRI | 1472 ± 923 | 1976 ± 979 | 0.083 | 1248 ± 824 | 2084 ± 1094 | 0.051 | 0.769 |
| LF:HF-RRI | 3.4 ± 0.9 | 2.8 ± 1.5 | 0.249 | 1.9 ± 1.7⁎ | 1.9 ± 1.8 | 0.994 | 0.380 |
| Blood pressure variability | |||||||
| LFnu-SBP | 47 ± 9 | 44 ± 14 | 0.613 | 42 ± 13 | 38 ± 19 | 0.274 | 0.843 |
| HFnu-SBP | 18 ± 10 | 20 ± 15 | 0.729 | 26 ± 8 | 28 ± 11 | 0.950 | 0.852 |
| PSD-SBP | 9.0 ± 2.7 | 11.8 ± 7.1 | 0.169 | 10.9 ± 6.3 | 12.5 ± 9.8 | 0.716 | 0.543 |
| LF:HF-SBP | 3.9 ± 1.8 | 3.7 ± 2.7 | 0.330 | 3.0 ± 2.6 | 2.8 ± 3.5 | 0.830 | 0.503 |
| LFnu-DBP | 51 ± 14 | 47 ± 15 | 0.317 | 48 ± 15 | 43 ± 17 | 0.382 | 0.969 |
| HFnu-DBP | 13 ± 9 | 18 ± 11 | 0.081 | 20 ± 11⁎ | 29 ± 13⁎⁎ | 0.006 | 0.767 |
| PSD-DBP | 4.9 ± 2.0 | 6.0 ± 3.7 | 0.088 | 7.1 ± 2.4⁎ | 10.6 ± 5.6⁎⁎ | 0.341 | 0.430 |
| LF:HF-DBP | 6.1 ± 2.5 | 5.6 ± 2.1 | 0.278 | 4.2 ± 2.1⁎ | 3.9 ± 2.5 | 0.278 | 0.127 |
| Fatigue | |||||||
| FIS score | 37 ± 34 | 34 ± 37 | 0.350 | 9 ± 11⁎ | 5 ± 8⁎⁎ | 0.087 | 0.928 |
FIS = Fatigue Impact Scale; LF = low frequency; HF = high frequency; SBP = systolic blood pressure; DBP = diastolic blood pressure; nu = normalized units; PSD = power spectral density; RRI = RR interval. a p value represents the within group comparison of paired before and follow-up time points. b p value represents group by time interaction from mixed model repeated measures analysis. * p < 0.05 for patients vs. controls at equivalent time points. ** p < 0.01 for patients vs. controls at equivalent time points.
Consistent with the significant elevation in resting heart rate, mean RR interval was significantly decreased in patients compared to controls (Table 5). LF:HF-RRI was significantly increased (mean increase 79%, p = 0.03) in patients compared to controls, providing evidence of a shift in sympatho-vagal balance, with effects predominantly driven through an increase in sympathetic function (LFnu-RRI). Total diastolic BPV (PSD-DBP) was significant decreased in patients compared to controls both before and after exercise training (Table 5). A significant shift in sympatho-vagal balance was again evident in patients compared to controls (LF:HF-DPB), but driven here by a decrease in high frequency components, representing parasympathetic function (HFnu-DBP). There was no significant effect of exercise training on any HRV or BPV parameter in either group.
4. Discussion
The principal findings of this study of the effects of a 16-week endurance exercise training programme in patients harbouring the m.3243A>G mutation compared to controls are: 1) endurance exercise is safe with no deleterious effects on cardiac morphology and function; 2) patients achieved similar proportional benefits of training on cardio-pulmonary and haemodynamic parameters including peak work capacity, peak oxygen uptake and anaerobic threshold, with no evidence of disease progression or increased fatigue compared to sedentary controls; 3) reduced exercise capacity detected at baseline, is predominately mediated through peripheral skeletal muscle dysfunction, rather than central cardiac factors; and 4) increased sympathetic and decreased parasympathetic cardiovascular autonomic activity pre- and post-exercise training suggests a role for sympathetic over-activation in disease pathogenesis.
4.1. Reduced exercise capacity and skeletal muscle function
Consistent with a previous case–control study of patients with mitochondrial disease [8], peak oxygen consumption, peak arterial–venous oxygen difference and peak work capacity at baseline were significantly decreased in patients compared to untrained, sedentary controls, with proportional reductions in peak exercise haemodynamic parameters. Despite these facts, all patients completed the 16-week endurance exercise training intervention (≥ 80% of the 48 scheduled sessions) and no adverse events were reported. Participants remained medically stable throughout the study with no deleterious changes in markers of disease burden, skeletal muscle dysfunction or quality of life.
Limitations in exercise parameters at baseline in patients were not reflected in indexed peak cardiac power output, which was not statistically different to controls. This suggests that the reduction in maximal exercise capacity and the associated symptom of exercise intolerance in patients with the m.3243A>G mutation is related to reduced peripheral ability of the skeletal muscle to extract oxygen during exercise, rather than to a central cardiac limitation. This potential mechanism is consistent with previous studies that have reported preservation, or even elevation, of skeletal muscle oxygen delivery [42–52], endothelial function, and oxygen unloading mechanisms [42] in similar cohorts of patients with mitochondrial disease. To our knowledge, this is the first study to definitively exclude central cardiac factors as a mediator of reduced exercise capacity in this cohort of patients. Moreover, a similar blunted response to exercise resulting from limited peripheral muscle oxygen extraction has been reported in other disease states, where skeletal muscle dysfunction, and indeed peripheral mitochondrial abnormalities, have been implicated in the aetiology of reduced exercise capacity, including stroke [53], heart failure [54–56], chronic obstructive pulmonary disease (COPD) and chronic renal failure [57]. Interestingly, while the anaerobic threshold of patients in our study was reduced compared to controls when expressed relative to predicted peak oxygen consumption, there was no significant difference in the absolute values or those relative to the actual peak oxygen consumption between the groups. This implies that skeletal muscle oxidative consumption may be similar between the groups, suggesting that peripheral factors other than mitochondrial oxidative metabolism may contribute to reduced, exercise tolerance seen in patients with mitochondrial disease. Biopsy studies have previously demonstrated an increase in skeletal muscle oxidative capacity and respiratory chain enzymatic complex activities with exercise training in patients with mitochondrial disease [8,9], but further interventional studies would be necessary to examine other skeletal muscle factors in this patient group.
Endurance exercise training improved peak work capacity, peak oxygen consumption and anaerobic threshold in both patient and control groups. This expected result is similar to previous aerobic exercise training studies in patients with mitochondrial disease [6,9], and occurred without an improvement in CPO or indexed CPO. This further suggests that training improvements in exercise capacity, as well as the baseline restrictions in these parameters, are manifestations of skeletal muscle rather than cardiac involvement in mitochondrial disease. The trend towards an improvement in peak arterio–venous oxygen difference at peak exercise in patients following a 16-week aerobic exercise training intervention suggests an increased ability of the skeletal muscle to extract and utilise oxygen during aerobic exercise as a result of the training programme. The ability of skeletal muscle to extract oxygen during exercise represents an important and physiologically-relevant surrogate marker of skeletal muscle mitochondrial function, and our data suggest that although this parameter is blunted in patients with mitochondrial disease due to the m.3243A>G mutation, it can be improved by exercise training and that the magnitude of this improvement is similar to that in untrained, sedentary controls. Similarly, despite a much lower weight at baseline, patients showed a trend towards an increase in lean body mass following exercise training, which may represent morphological changes in skeletal muscle. Importantly, although all patients in this study reported the clinical feature of exercise intolerance, and fatigue was noted in half, there was no deleterious effect of exercise training on fatigue or reported quality of life. This finding occurred despite an increase in exercise capacity in patients, suggesting a clinical benefit in this group.
4.2. Endurance exercise and cardiac remodelling
Our group has previously demonstrated, in similar cohorts, that patients harbouring the m.3243A>G mutation, without known cardiac involvement on standard screening, display evidence of sub-clinical hypertrophic remodelling compared to age- and gender-matched controls [17,58]. The baseline results of this study support these findings with decreased blood pool volumes, increased LVMI and M/V ratio, and subtle abnormalities of systolic myocardial strains in patients compared to controls. The direction and magnitude of exercise-induced changes in LVM, LVMI and M/V ratio in both patient and control groups are consistent with normal physiological responses to aerobic exercise in untrained healthy controls [22–24]. Importantly, there was no significant effect of group status on the proportional degree of hypertrophic remodelling observed: patients and controls had similar responses to exercise. We found no significant effect of a 16-week endurance exercise training programme on left ventricular volumes or cardiac systolic or diastolic function, but acknowledge that these additional physiological changes may not have been appreciated in this time frame.
Consistent with previous studies, we found abnormal cardiac bioenergetics at baseline in patients harbouring the m.3243A>G mutation [17]. In mitochondrial disease and other forms of inherited hypertrophic cardiomyopathy, several groups have suggested the primacy of bioenergetic defects by detection of abnormalities in mutation carriers without evidence of LVH [16,59,60]. Given that we also detected significant differences in cardiac remodelling, known to cause a reduction in PCr/ATP ratio, we cannot comment on the temporal relationship of these findings, but a reduced ratio has already been demonstrated to have prognostic importance in a variety of forms of cardiomyopathy [18]. Importantly therefore, we were able to demonstrate, using 31P cardiac MRS, that endurance exercise training had no further deleterious effect on the abnormal myocardial bioenergetics in our patients, despite the expected and modest physiological increase in LVMI. Indeed, following aerobic exercise training, fewer patients had an abnormal PCr/ATP ratio and the decreased mean ratio of the patient group was less evident compared to controls. Taken together, these morphological, functional and bioenergetic results suggest that a 16-week endurance exercise training programme has no deleterious effects on the heart in patients with mitochondrial disease due to the m.3243A>G mutation. Given previous demonstrations of the beneficial effects of endurance exercise on exercise tolerance, quality of life and skeletal muscle function in this cohort of patients, this important result should allay fears of any detrimental cardiac response to endurance exercise and potentially enable more widespread adoption of a proven treatment in a population with currently limited therapeutic options.
4.3. Skeletal muscle oxidative capacity and sympathetic activation
Significant differences between patients and controls in resting heart rate and mean RR interval together with directionally opposing changes in low and high frequency components of both HRV and diastolic BPV provide evidence of a shift in the sympatho-vagal balance in patients harbouring the m.3243A>G mutation. This finding is supported by an increased LF:HF ratio for both RR interval and diastolic blood pressure, suggesting increased sympathetic activity and a parallel reduction in parasympathetic function in patients. This is consistent with previous observations of resting sympathetic over-activity both in small studies of patients with mitochondrial disease and in larger cohorts of patients with other chronic diseases characterized by similar skeletal muscle dysfunction and exercise intolerance [61]. Our findings further corroborate reports of a hyperadrenergic state in patients with mitochondrial disease indicated by increased plasma concentration of noradrenaline in patients [42], and the high prevalence of sympathetic symptoms in this population [62]. Chronic sympathetic activation is recognised as detrimental contributing to vasoconstriction, tissue hypoxia, inflammation, oxidative stress, and impairment of muscle proton homeostasis; all factors that may lead to increased skeletal myopathy, hypertension and elevated cardiovascular risk [32,61,63]. The mechanisms underlying the effects of chronic sympathetic activation are largely unknown, but sympathetic outflow to skeletal muscle, which is dependent on both baroreceptor and chemoreceptor regulation, is heightened selectively in patients with other chronic diseases in which skeletal myopathy is recognised such as heart failure, COPD, essential hypertension and end stage renal disease [64–67]. It has been postulated that overactivity of skeletal muscle somatic afferents maintains an increased sympathetic drive both during exercise and at rest, contributing to skeletal myopathy [68]. These nerve fibres, which mediate the exercise pressor reflex, include metaboreceptors that are sensitive to ischaemic metabolites during exercise, including lactic acid, and mechanoreceptors that are primarily sensitive to stretch. Yet it has been shown that exercise intolerance and sympathetic activation are independent of lactic acidosis in patients with mitochondrial disease [69,70]. We propose that the increased sympathetic tone evident at baseline in patients, and the exaggerated neurovascular responses to exercise, with increased heart rate at equivalent cardiopulmonary work capacities and time intervals, may be at least partly due to increased skeletal muscle afferent mechanoreceptor sensitivity as seen in other chronic diseases with sympathetic overactivity and exercise intolerance [71]. Indeed our data also suggest a physiological upregulation of parasympathetic activity in patients in response to exercise training, similar in magnitude to the effect in control subjects. Further evaluation of the role of autonomic dysregulation and impact on disease burden, phenotypic expression and exercise intolerance in patients with mitochondrial disease is warranted. Such studies may help to discern the major limitations of exercise capacity in other chronic diseases associated with skeletal myopathy, in which mitochondrial dysfunction has been implicated, directing future therapeutic interventions.
4.4. Limitations
Although this study is the largest investigation of cardiac adaptations to exercise performed to date in this patient group, it remains limited in sample size, and duration, and was not designed to investigate pathogenetic mechanisms or disease progression. In an already extensive protocol, we did not make direct assessments of skeletal muscle function or bioenergetics. We studied a relatively homogenous cohort of patients, harbouring the single commonest mtDNA point mutation, without known cardiac involvement: such patients account for ~ 25% of our specialist clinic attendees yet we recognize that our findings may not be generalizable to all patients with mtDNA point mutations.
5. Conclusions
Our data represent the first comprehensive evaluation of the cardiac safety and efficacy profile of aerobic, endurance exercise training in patients with m.3243A>G-related mitochondrial disease. We have determined that endurance exercise training is safe in patients harbouring the m.3243A>G mutation, and produces improvements in clinical parameters of a similar magnitude and direction to those observed in untrained, sedentary controls, and holds potential therapeutic benefits for patients with mitochondrial disease. We also provide evidence of altered sympatho-vagal balance with sympathetic over-activation in patients harbouring the m.3243A>G mutation. This represents a further potential therapeutic target for pharmacological intervention or improved vagal tone with exercise. Identification of an effective intervention that could slow or reverse progression of skeletal muscle involvement in mitochondrial disease has the potential to achieve significant health gains with a substantial impact on patient health and well-being. Our findings underpin the future translation of effective exercise therapy into the clinical care of patients with mitochondrial disease.
Acknowledgements
We thank all patients and volunteers involved in this study. We are also grateful to Jessie Pairman, for assistance with the rating scales, and to Louise Morris, Tamsin Gaudie and Tim Hodgson, Research Radiographers.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution–NonCommercial–No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was supported by the Wellcome Trust [BH092142 to MGDB, 096919Z/11/Z and 074454/Z/04/Z to DMT and RWT]; the Medical Research Council [G1100160 to KGH, G0601943 to DMT and G0800674 to DMT and RWT]; the UK National Institute for Health Research Biomedical Research Centre for Ageing and Age-related Diseases award to Newcastle upon Tyne Hospitals NHS Foundation Trust [for DMT, GSG and cardiac tagging software]; the British Heart Foundation [CH/07/001 to BDK]; and the UK NHS Specialized Services and Newcastle upon Tyne Hospitals NHS Foundation Trust that support the ‘Rare Mitochondrial Disorders of Adults and Children’ Diagnostic Service [http://www.mitochondrialncg.nhs.uk].
All authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ijcard.2013.05.062.
Contributor Information
Matthew G.D. Bates, Email: m.g.d.bates@newcastle.ac.uk.
Gráinne S. Gorman, Email: grainne.gorman@newcastle.ac.uk.
Appendix 1. Abbreviations
- 1-D
1-dimensional
- 31P
phosphorus-31
- 2,3-DPG
2,3-disphosphoglycerate
- AMARES
accumulation method for accurate, robust and efficient spectra
- ATP
adenosine triphosphate
- CSI
chemical shift imaging
- DNA
deoxyribonucleic acid
- DPG
diphosphoglycerate
- E/A
early/late filling velocity
- ECG
electrocardiograph
- FA
flip angle
- FOV
field of view
- HCM
hypertrophic cardiomyopathy
- LV
left ventricular
- LVH
left ventricular hypertrophy
- LVMI
left ventricular mass index
- MELAS
mitochondrial encephalopathy with lactic acidosis and stroke-like episode
- MIDD
maternally inherited diabetes and deafness
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- mt-tRNA
mitochondrial transfer RNA
- mtDNA
mitochondrial DNA
- M/V
left ventricular mass/end-diastolic volume
- NMDAS
Newcastle Mitochondrial Disease Adult Scale
- PCr
phosphocreatine
- PDE
phosphodiesters
- ppm
parts per million
- RNA
ribonucleic acid
- SD
standard deviation
- SENSE
sensitivity encoding
- TE
echo time
- TR
repetition time
- TSR
torsion to endocardial circumferential strain ratio
Appendix 2. Supplementary data
Supplementary tables.
References
- 1.Schaefer A.M., McFarland R., Blakely E.L. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 2.Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates M.G.D., Bourke J.P., Giordano C., d'Amati G., Turnbull D.M., Taylor R.W. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33:3023–3033. doi: 10.1093/eurheartj/ehs275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfeffer G., Majamaa K., Turnbull D.M., Thorburn D., Chinnery P.F. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4 doi: 10.1002/14651858.CD004426.pub3. [CD004426] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taivassalo T., De Stefano N., Argov Z. Effects of aerobic training in patients with mitochondrial myopathies. Neurology. 1998;50:1055–1060. doi: 10.1212/wnl.50.4.1055. [DOI] [PubMed] [Google Scholar]
- 6.Taivassalo T., Gardner J.L., Taylor R.W. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. 2006;129:3391–3401. doi: 10.1093/brain/awl282. [DOI] [PubMed] [Google Scholar]
- 7.Cejudo P., Bautista J., Montemayor T. Exercise training in mitochondrial myopathy: a randomized controlled trial. Muscle Nerve. 2005;32:342–350. doi: 10.1002/mus.20368. [DOI] [PubMed] [Google Scholar]
- 8.Jeppesen T.D., Schwartz M., Olsen D.B. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain. 2006;129:3402–3412. doi: 10.1093/brain/awl149. [DOI] [PubMed] [Google Scholar]
- 9.Murphy J.L., Blakely E.L., Schaefer A.M. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain. 2008;131:2832–2840. doi: 10.1093/brain/awn252. [DOI] [PubMed] [Google Scholar]
- 10.Hirano M., Pavlakis S.G. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9:4–13. doi: 10.1177/088307389400900102. [DOI] [PubMed] [Google Scholar]
- 11.Anan R., Nakagawa M., Miyata M. Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation. 1995;91:955–961. doi: 10.1161/01.cir.91.4.955. [DOI] [PubMed] [Google Scholar]
- 12.Majamaa-Voltti K., Peuhkurinen K., Kortelainen M.L., Hassinen I.E., Majamaa K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc Disord. 2002;2:12. doi: 10.1186/1471-2261-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmgren D., Wahlander H., Eriksson B., Oldfors A., Holme E., Tulinius M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J. 2003;24:280–288. doi: 10.1016/s0195-668x(02)00387-1. [DOI] [PubMed] [Google Scholar]
- 14.Vydt T.C.G., de Coo R.F.M., Soliman O.I.I. Cardiac involvement in adults with m.3243A>G MELAS gene mutation. Am J Cardiol. 2007;99:264–269. doi: 10.1016/j.amjcard.2006.07.089. [DOI] [PubMed] [Google Scholar]
- 15.Majamaa-Voltti K., Turkka J., Kortelainen M.L., Huikuri H., Majamaa K. Causes of death in pedigrees with the 3243A>G mutation in mitochondrial DNA. J Neurol Neurosurg Psychiatry. 2008;79:209–211. doi: 10.1136/jnnp.2007.122648. [DOI] [PubMed] [Google Scholar]
- 16.Lodi R., Rajagopalan B., Blamire A.M., Crilley J.G., Styles P., Chinnery P.F. Abnormal cardiac energetics in patients carrying the A3243G mtDNA mutation measured in vivo using phosphorus MR spectroscopy. Biochim Biophys Acta. 2004;1657:146–150. doi: 10.1016/j.bbabio.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Bates M.G., Hollingsworth K.G., Newman J.H. Concentric hypertrophic remodelling and subendocardial dysfunction in mitochondrial DNA point mutation carriers. Eur Heart J Cardiovasc Imaging. 2012 doi: 10.1093/ehjci/jes226. (accessed 28th January 2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neubauer S., Krahe T., Schindler R. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Altered cardiac high-energy phosphate metabolism in heart failure. Circulation. 1992;86:1810–1818. doi: 10.1161/01.cir.86.6.1810. [DOI] [PubMed] [Google Scholar]
- 19.Momiyama Y., Suzuki Y., Ohtomo M. Cardiac autonomic nervous dysfunction in diabetic patients with a mitochondrial DNA mutation: assessment by heart rate variability. Diabetes Care. 2002;25:2308–2313. doi: 10.2337/diacare.25.12.2308. [DOI] [PubMed] [Google Scholar]
- 20.Majamaa-Voltti K., Majamaa K., Peuhkurinen K., Makikallio T.H., Huikuri H.V. Cardiovascular autonomic regulation in patients with 3243A>G mitochondrial DNA mutation. Ann Med. 2004;36:225–231. doi: 10.1080/07853890410028456. [DOI] [PubMed] [Google Scholar]
- 21.Bigger J.T., Jr., Fleiss J.L., Steinman R.C., Rolnitzky L.M., Kleiger R.E., Rottman J.N. Frequency domain measures of heart period variability and mortality after myocardial infarction. Circulation. 1992;85:164–171. doi: 10.1161/01.cir.85.1.164. [DOI] [PubMed] [Google Scholar]
- 22.Douglas P.S., O'Toole M.L., Katz S.E., Ginsburg G.S., Hiller W.D., Laird R.H. Left ventricular hypertrophy in athletes. Am J Cardiol. 1997;80:1384–1388. doi: 10.1016/s0002-9149(97)00693-0. [DOI] [PubMed] [Google Scholar]
- 23.Pelliccia A., Culasso F., Di Paolo F.M., Maron B.J. Physiologic left ventricular cavity dilatation in elite athletes. Ann Intern Med. 1999;130:23–31. doi: 10.7326/0003-4819-130-1-199901050-00005. [DOI] [PubMed] [Google Scholar]
- 24.Scharhag J., Schneider G., Urhausen A., Rochette V., Kramann B., Kindermann W. Athlete's heart: right and left ventricular mass and function in male endurance athletes and untrained individuals determined by magnetic resonance imaging. J Am Coll Cardiol. 2002;40:1856–1863. doi: 10.1016/s0735-1097(02)02478-6. [DOI] [PubMed] [Google Scholar]
- 25.Beer M., Wagner D., Myers J. Effects of exercise training on myocardial energy metabolism and ventricular function assessed by quantitative phosphorus-31 magnetic resonance spectroscopy and magnetic resonance imaging in dilated cardiomyopathy. J Am Coll Cardiol. 2008;51:1883–1891. doi: 10.1016/j.jacc.2007.09.075. [DOI] [PubMed] [Google Scholar]
- 26.Stolen K.Q., Kemppainen J., Ukkonen H. Exercise training improves biventricular oxidative metabolism and left ventricular efficiency in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2003;41:460–467. doi: 10.1016/s0735-1097(02)02772-9. [DOI] [PubMed] [Google Scholar]
- 27.Belardinelli R., Georgiou D., Cianci G., Purcaro A. Randomized, controlled trial of long-term moderate exercise training in chronic heart failure: effects on functional capacity, quality of life, and clinical outcome. Circulation. 1999;99:1173–1182. doi: 10.1161/01.cir.99.9.1173. [DOI] [PubMed] [Google Scholar]
- 28.Beaver W.L., Wasserman K., Whipp B.J. A new method for detecting anaerobic threshold by gas exchange. J Appl Physiol. 1986;60:2020–2027. doi: 10.1152/jappl.1986.60.6.2020. [DOI] [PubMed] [Google Scholar]
- 29.Williams S.G., Cooke G.A., Wright D.J. Peak exercise cardiac power output; a direct indicator of cardiac function strongly predictive of prognosis in chronic heart failure. Eur Heart J. 2001;22:1496–1503. doi: 10.1053/euhj.2000.2547. [DOI] [PubMed] [Google Scholar]
- 30.Schaefer A.M., Phoenix C., Elson J.L., McFarland R., Chinnery P.F., Turnbull D.M. Mitochondrial disease in adults: a scale to monitor progression and treatment. Neurology. 2006;66:1932–1934. doi: 10.1212/01.wnl.0000219759.72195.41. [DOI] [PubMed] [Google Scholar]
- 31.Whittaker R.G., Blackwood J.K., Alston C.L. Urine heteroplasmy is the best predictor of clinical outcome in the m.3243A>G mtDNA mutation. Neurology. 2009;72:568–569. doi: 10.1212/01.wnl.0000342121.91336.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones D.E., Hollingsworth K., Fattakhova G. Impaired cardiovascular function in primary biliary cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2010;298:G764–G773. doi: 10.1152/ajpgi.00501.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanhamme L., Van Huffel S., Van Hecke P., van Ormondt D. Time-domain quantification of series of biomedical magnetic resonance spectroscopy signals. J Magn Reson. 1999;140:120–130. doi: 10.1006/jmre.1999.1835. [DOI] [PubMed] [Google Scholar]
- 34.Lumens J., Delhaas T., Arts T., Cowan B.R., Young A.A. Impaired subendocardial contractile myofiber function in asymptomatic aged humans, as detected using MRI. Am J Physiol Heart Circ Physiol. 2006;291:H1573–H1579. doi: 10.1152/ajpheart.00074.2006. [DOI] [PubMed] [Google Scholar]
- 35.Fortin J., Marte W., Grullenberger R. Continuous non-invasive blood pressure monitoring using concentrically interlocking control loops. Comput Biol Med. 2006;36:941–957. doi: 10.1016/j.compbiomed.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 36.Gratze G., Fortin J., Holler A. A software package for non-invasive, real-time beat-to-beat monitoring of stroke volume, blood pressure, total peripheral resistance and for assessment of autonomic function. Comput Biol Med. 1998;28:121–142. doi: 10.1016/s0010-4825(98)00005-5. [DOI] [PubMed] [Google Scholar]
- 37.Parati G., Ongaro G., Bilo G. Non-invasive beat-to-beat blood pressure monitoring: new developments. Blood Press Monit. 2003;8:31–36. doi: 10.1097/00126097-200302000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Bianchi A.M., Mainardi L.T., Meloni C., Chierchia S., Cerutti S. Continuous monitoring of the sympatho-vagal balance through spectral analysis. IEEE Eng Med Biol Mag. 1997;16:64–73. doi: 10.1109/51.620497. [DOI] [PubMed] [Google Scholar]
- 39.Stauss H.M. Heart rate variability. Am J Physiol Regul Integr Comp Physiol. 2003;285:R927–R931. doi: 10.1152/ajpregu.00452.2003. [DOI] [PubMed] [Google Scholar]
- 40.Akselrod S., Gordon D., Madwed J.B., Snidman N.C., Shannon D.C., Cohen R.J. Hemodynamic regulation: investigation by spectral analysis. Am J Physiol. 1985;249:H867–H875. doi: 10.1152/ajpheart.1985.249.4.H867. [DOI] [PubMed] [Google Scholar]
- 41.Alfakih K., Plein S., Thiele H., Jones T., Ridgway J.P., Sivananthan M.U. Normal human left and right ventricular dimensions for MRI as assessed by turbo gradient echo and steady-state free precession imaging sequences. J Magn Reson Imaging. 2003;17:323–329. doi: 10.1002/jmri.10262. [DOI] [PubMed] [Google Scholar]
- 42.Jeppesen J., Kiens B. Regulation and limitations to fatty acid oxidation during exercise. J Physiol. 2012;590:1059–1068. doi: 10.1113/jphysiol.2011.225011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taivassalo T., Jensen T.D., Kennaway N., DiMauro S., Vissing J., Haller R.G. The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain. 2003;126:413–423. doi: 10.1093/brain/awg028. [DOI] [PubMed] [Google Scholar]
- 44.Taivassalo T., Abbott A., Wyrick P., Haller R.G. Venous oxygen levels during aerobic forearm exercise: an index of impaired oxidative metabolism in mitochondrial myopathy. Ann Neurol. 2002;51:38–44. doi: 10.1002/ana.10027. [DOI] [PubMed] [Google Scholar]
- 45.Jensen T.D., Kazemi-Esfarjani P., Skomorowska E., Vissing J. A forearm exercise screening test for mitochondrial myopathy. Neurology. 2002;58:1533–1538. doi: 10.1212/wnl.58.10.1533. [DOI] [PubMed] [Google Scholar]
- 46.Taivassalo T., Shoubridge E.A., Chen J. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol. 2001;50:133–141. doi: 10.1002/ana.1050. [DOI] [PubMed] [Google Scholar]
- 47.Abe K., Matsuo Y., Kadekawa J., Inoue S., Yanagihara T. Measurement of tissue oxygen consumption in patients with mitochondrial myopathy by noninvasive tissue oximetry. Neurology. 1997;49:837–841. doi: 10.1212/wnl.49.3.837. [DOI] [PubMed] [Google Scholar]
- 48.Bank W., Chance B. Diagnosis of defects in oxidative muscle metabolism by non-invasive tissue oximetry. Mol Cell Biochem. 1997;174:7–10. [PubMed] [Google Scholar]
- 49.Bank W., Chance B. An oxidative defect in metabolic myopathies: diagnosis by noninvasive tissue oximetry. Ann Neurol. 1994;36:830–837. doi: 10.1002/ana.410360606. [DOI] [PubMed] [Google Scholar]
- 50.Vissing J., Galbo H., Haller R.G. Exercise fuel mobilization in mitochondrial myopathy: a metabolic dilemma. Ann Neurol. 1996;40:655–662. doi: 10.1002/ana.410400416. [DOI] [PubMed] [Google Scholar]
- 51.Ozawa M., Goto Y., Sakuta R., Tanno Y., Tsuji S., Nonaka I. The 8,344 mutation in mitochondrial DNA: a comparison between the proportion of mutant DNA and clinico-pathologic findings. Neuromuscul Disord. 1995;5:483–488. doi: 10.1016/0960-8966(95)00009-c. [DOI] [PubMed] [Google Scholar]
- 52.Haller R.G., Lewis S.F., Estabrook R.W., DiMauro S., Servidei S., Foster D.W. Exercise intolerance, lactic acidosis, and abnormal cardiopulmonary regulation in exercise associated with adult skeletal muscle cytochrome c oxidase deficiency. J Clin Invest. 1989;84:155–161. doi: 10.1172/JCI114135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jakovljevic D.G., Moore S.A., Tan L.B., Rochester L., Ford G.A., Trenell M.I. Discrepancy between cardiac and physical functional reserves in stroke. Stroke. 2012;43:1422–1425. doi: 10.1161/STROKEAHA.111.649434. [DOI] [PubMed] [Google Scholar]
- 54.Mancini D.M., Coyle E., Coggan A. Contribution of intrinsic skeletal muscle changes to 31P NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure. Circulation. 1989;80:1338–1346. doi: 10.1161/01.cir.80.5.1338. [DOI] [PubMed] [Google Scholar]
- 55.Mancini D.M., Walter G., Reichek N. Contribution of skeletal muscle atrophy to exercise intolerance and altered muscle metabolism in heart failure. Circulation. 1992;85:1364–1373. doi: 10.1161/01.cir.85.4.1364. [DOI] [PubMed] [Google Scholar]
- 56.Harrington D., Anker S.D., Chua T.P., Webb-Peploe K.M., Ponikowski P.P., Poole-Wilson P.A., Coats A.J. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J Am Coll Cardiol. 1997;30:1758–1764. doi: 10.1016/s0735-1097(97)00381-1. [DOI] [PubMed] [Google Scholar]
- 57.Troosters T., Gosselink R., Decramer M. Chronic obstructive pulmonary disease and chronic heart failure: two muscle diseases? J Cardiopulm Rehabil. 2004;24:137–145. doi: 10.1097/00008483-200405000-00001. [DOI] [PubMed] [Google Scholar]
- 58.Hollingsworth K.G., Gorman G.S., Trenell M.I. Cardiomyopathy is common in patients with the mitochondrial DNA m.3243A>G mutation and correlates with mutation load. Neuromuscul Disord. 2012;22:592–596. doi: 10.1016/j.nmd.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashrafian H., Redwood C., Blair E., Watkins H. Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet. 2003;19:263–268. doi: 10.1016/S0168-9525(03)00081-7. [DOI] [PubMed] [Google Scholar]
- 60.Crilley J.G., Boehm E.A., Blair E. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776–1782. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 61.Middlekauff H.R. Making the case for skeletal myopathy as the major limitation of exercise capacity in heart failure. Circ Heart Fail. 2010;3:537–546. doi: 10.1161/CIRCHEARTFAILURE.109.903773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parsons T., Weimer L., Engelstad K. Autonomic symptoms in carriers of the m.3243A>G mitochondrial DNA mutation. Arch Neurol. 2010;67:976–979. doi: 10.1001/archneurol.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Syme P.D., Brunotte F., Green Y., Aronson J.K., Radda G.K. The effect of beta 2-adrenoceptor stimulation and blockade of l-type calcium channels on in vivo Na +/H + antiporter activity in rat skeletal muscle. Biochim Biophys Acta. 1991;1093:234–240. doi: 10.1016/0167-4889(91)90128-k. [DOI] [PubMed] [Google Scholar]
- 64.Middlekauff H.R., Hamilton M.A., Stevenson L.W., Mark A.L. Independent control of skin and muscle sympathetic nerve activity in patients with heart failure. Circulation. 1994;90:1794–1798. doi: 10.1161/01.cir.90.4.1794. [DOI] [PubMed] [Google Scholar]
- 65.Grassi G., Colombo M., Seravalle G., Spaziani D., Mancia G. Dissociation between muscle and skin sympathetic nerve activity in essential hypertension, obesity, and congestive heart failure. Hypertension. 1998;31:64–67. doi: 10.1161/01.hyp.31.1.64. [DOI] [PubMed] [Google Scholar]
- 66.Hering D., Zdrojewski Z., Krol E. Tonic chemoreflex activation contributes to the elevated muscle sympathetic nerve activity in patients with chronic renal failure. J Hypertens. 2007;25:157–161. doi: 10.1097/HJH.0b013e3280102d92. [DOI] [PubMed] [Google Scholar]
- 67.Park J., Campese V.M., Nobakht N., Middlekauff H.R. Differential distribution of muscle and skin sympathetic nerve activity in patients with end-stage renal disease. J Appl Physiol. 2008;105:1873–1876. doi: 10.1152/japplphysiol.90849.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clark A.L., Poole-Wilson P.A., Coats A.J. Exercise limitation in chronic heart failure: central role of the periphery. J Am Coll Cardiol. 1996;28:1092–1102. doi: 10.1016/S0735-1097(96)00323-3. [DOI] [PubMed] [Google Scholar]
- 69.Vissing J., Gansted U., Quistorff B. Exercise intolerance in mitochondrial myopathy is not related to lactic acidosis. Ann Neurol. 2001;49:672–676. [PubMed] [Google Scholar]
- 70.Vissing J., Vissing S.F., MacLean D.A., Saltin B., Quistorff B., Haller R.G. Sympathetic activation in exercise is not dependent on muscle acidosis. Direct evidence from studies in metabolic myopathies. J Clin Invest. 1998;101:1654–1660. doi: 10.1172/JCI555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith S.A., Mitchell J.H., Garry M.G. The mammalian exercise pressor reflex in health and disease. Exp Physiol. 2006;91:89–102. doi: 10.1113/expphysiol.2005.032367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary tables.