

Abstract
The phosphoinositide 3-kinase (PI3K) pathway is activated in most advanced prostate cancers, yet so far treatments with PI3K inhibitors have been at best tumorostatic in preclinical cancer models and do not show significant antitumor efficacy in clinical trials. Results from tissue culture experiments in prostate cancer cells suggest that PI3K inhibitors should be combined with other cytotoxic agents; however, the general toxicity of such combinations prevents translating these experimental data into preclinical and clinical models. We investigated the emerging concept of tumor-targeted synthetic lethality in prostate cancer cells by using the pan-PI3K inhibitor ZSTK474 and the immunotoxin J591PE, a protein chimera between the single-chain variable fragment of the monoclonal antibody J591 against the prostate-specific membrane antigen (PSMA) and the truncated form of the Pseudomonas aeruginosa exotoxin A (PE38QQR). The combination of ZSTK474 and J591PE increased apoptosis within 6 hours and cell death (monitored at 24–48 hours) in the PSMA-expressing cells LNCaP, C4-2, and C4-2Luc but not in control cells that do not express PSMA (PC3 and BT549 cells). Mechanistic analysis suggested that induction of apoptosis requires Bcl-2-associated death promoter (BAD) dephosphorylation and decreased expression of myeloid leukemia cell differentiation protein 1 (MCL-1). A single injection of ZSTK474 and J591PE into engrafted prostate cancer C4-2Luc cells led to consistent and stable reduction of luminescence within 6 days. These results suggest that the combination of a PI3K inhibitor and a PSMA-targeted protein synthesis inhibitor toxin represents a promising novel strategy for advanced prostate cancer therapy that should be further investigated.
Introduction
Prostate cancer is the most common cancer diagnosed in men, representing the 14% of deaths from cancer and 25% of new cases of cancer in the Western world. Although it is usually not fatal, the prognosis for patients with advanced prostate cancer that spreads outside of the prostate gland is poor, because of resistance to available treatments [1–3]. Most advanced prostate cancers have an overactive phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) pathway. This pathway controls cell growth, survival, motility, and angiogenesis, and it is associated with higher Gleason grade, advanced stage, and unfavorable prognosis [4–8].
The PI3K pathway is initiated by a receptor tyrosine kinase that recruits and activates PI3K, resulting in an accumulation of phosphatidylinositol 3,4,5-trisphosphate in the plasma membrane. This lipid second messenger recruits the AKT and the phosphoinositide-dependent protein kinase 1 to the cell membrane, where AKT is phosphorylated by phosphoinositide-dependent protein kinase 1 at threonine 308. The mammalian target of rapamycin forms the mammalian target of rapamycin complex 2, which completes the activation of AKT by phosphorylation at serine 473. Fully activated AKT translocates to the cytoplasm and nucleus, where it phosphorylates downstream substrates [9].
Constitutive activation of the PI3K/AKT pathway in prostate cancer is often led by functional loss of the tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) that dephosphorylates PI3K substrates or by activating mutations in the PI3K itself [4,10,11]. For these reasons, PI3K inhibitors have been considered an adjuvant therapy for advanced prostate cancer, and pharmaceutical companies as well as academic laboratories are actively developing small molecule inhibitors to specifically target the PI3K [7,12–14]. At present, a number of PI3K inhibitors are in phase I clinical trials (i.e., BEZ235, BKM120, and BGT226 from Novartis [New York, NY], XL765 and XL147 from Exelixis [San Francisco, CA], GDC0941 from Piramed/Genentech [San Francisco, CA], GSK1059615 from GlaxoSmithKline [Philadelphia, PA], SF1126 from Semafore [Indianapolis, IN], and ZSTK474 from ZenyakuKogyo [Tokyo, Japan]; source: www.clinicaltrials.gov), because they showed anti-proliferative activity in preclinical models of solid tumors. However, phase II clinical trials have shown only modest anti-tumor efficacy of PI3K inhibitors, suggesting that malignant cells acquire resistance to monotherapy with PI3K inhibitors. Thus, targeting only the PI3K pathway by inhibiting one or more protein kinases on this pathway might not treat established solid tumors [15]. Indeed, increasing number of reports show that combinations of PI3K inhibitors with other therapeutics may be needed for stronger anti-tumor effects [16–19]. Yet such combinations use agents with limited or no specificity in targeting cancer cells, which leads to increased systemic toxicity, emphasizing the need for therapeutics that selectively target prostate tumors.
The prostate-specific membrane antigen (PSMA) is a type II transmembrane protein with folate hydrolase and neurocarboxypeptidase activities, with a short intracellular domain (amino acids 1–18), a transmembrane domain (amino acids 19–43), and a large extracellular domain (amino acids 44–750) [20]. PSMA is expressed in normal prostate epithelial cells at low levels, whereas expression increased by several fold in tumors, especially in high-grade, metastatic, and and rogeninsensitive prostate carcinomas. PSMA is also expressed in the vascular endothelium in a variety of tumors but not in normal tissues, which further broadens its potential utility as a therapeutic target [21]. With its abundant and restricted expression in tumors, its membrane location, and rapid internalization, PSMA represents an attractive target for prostate-selective cancer imaging and therapy. Antibodies to PSMA have been shown to selectively deliver bacterial toxins or radionuclides to prostate cancer cells in xenograft models of prostate cancer and in clinical setting [22–29].
In this report, we treated prostate cancer cells with the PI3K inhibitor ZSTK474 combined with the prostate-targeted immunotoxin J591PE, based on a protein chimera of the single-chain variable fragment of the J591 monoclonal antibody (J591scFv) and the truncated form of the Pseudomonas aeruginosa exotoxin A (PE38QQR) [30]. The combination triggered apoptosis in prostate cancer cells in tissue culture within 6 hours, by inhibiting Bcl-2-associated death promoter (BAD) dephosphorylation and decreasing expression of myeloid leukemia cell differentiation protein 1 (MCL-1). In C4-2Luc cells, a human xenograft model of advanced prostate cancer, the combination of ZSTK474 and J591PE showed a strong and stable reduction of tumor luminescence, compared to transient effects of single-agent administration. These results demonstrate for the first time that the combination of ZSTK474 and J591PE represents an effective therapeutic approach for advanced prostate cancer that should be further investigated.
Materials and Methods
Preparation of J591scFv and J591scFvPE38QQR (J591PE)
The J591scFv sequence was amplified from the SFG-pz1 [31] using the primers 5′-TTATCTAGAATGTCTCATCACCATCACCATCACGAGGTGCAGCTG-3′ (forward, 6x His, XbaI) and 5′-TACCCAAAGCTTTCATCATTTCAGGTCCAGCATGGT-3′ (reverse, HindIII) and cloned into pSF392 expression vector, a pBR322 derivative under the control of the Tac promoter (made in house, unpublished), to generate pSF-J591scFv.
The J591scFv sequence was also amplified from the same template using the primers 5′-TATTATACATATGGAGGTGCAGCTG-3′ (forward, NdeI) and 5′-TGATAAAAGCTTTTTTCAGGTCCAGCA-3′ (reverse, HindIII) and cloned into the pWD-PE38QQR expression vector [32] under the control of the T7 bacteriophage late promoter to generate a pWD-J591scFv-PE38QQR vector. Each clone was sequenced (Wake Forest University Core Facility) and amplified in Escherichia coli DH5′ cells.
J591ScFv was expressed in E coli XL1 Blue cells and extracted from the bacterial periplasm as previously reported [33]. Subsequently, we added a purification step consisting of a HisTrap HP column followed by a gel filtration on Superdex 75 100/300 GL size exclusion column (GE Healthcare Life Sciences, Uppsala, Sweden).
The recombinant protein J591scFvPE38QQR (J591PE) was expressed in E coli BL21 as previously described [34]. Briefly, transformed bacteria were grown in Luria-Bertani (LB) medium supplemented with 100 µg/ml ampicillin on an orbital shaker O/N at 37°C and at A600 = 0.7. Recombinant protein expression was induced by addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 90 minutes. J591PE was extracted from inclusion bodies by using 7 M guanidine (MP Biomedicals, Salon, OH) and 1,4-dithiothreitol (Sigma, St Louis, MO). Protein refolding was performed in a 0.1 M Tris buffer containing arginine/l-glutathione oxidase (Sigma-Aldrich, St Louis, MO). The recombinant immunotoxin was further dialyzed and purified by using a Q Sepharose ion exchange liquid chromatography system (GE Healthcare Life Sciences). Purification of J591scFv and J591PE was evaluated using Coomassie staining (PhastGel Blue R; GE Healthcare Bio-Sciences, Uppsala, Sweden) after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10% gels according to the manufacturer's protocol. The most purified preparations were also filtered using a 0.22-µm syringe filter, evaluated for activity, aliquoted, and stored in phosphate-buffered saline (PBS) at -20°C for subsequent experiments.
Cell Cultures
The hormone-responsive prostate cancer LNCaP and breast cancer BT-549 cell lines were purchased from ATCC (Manassas, VA). The androgen-independent prostate cancer LNCaP subline C4-2 and the mouse prostate epithelial WF3 cells were generous gifts from Dr Leland Chung (Cedars-Sinai Medical Center, Los Angeles, CA) and Dr Scott Cramer (University of Colorado Denver, Aurora, CO), respectively. C4-2Luc cells were generated as reported previously [35].
All cells were maintained in a humidified atmosphere (5% CO2) at 37°C, using RPMI 1640 (C4-2, C4-2Luc and BT549) and T-Medium (LNCaP), supplemented with 10% FBS (5% for LNCaP). BT549 medium was also supplemented with 0.023 U/ml insulin. WF3 cells were cultured in PTEN medium [Dulbecco's modified Eagle's medium/F12 supplemented with 1% FBS, 1% BSA, 10 ng/ml cholera toxin, 28 µg/ml BPE, 8 µg/ml insulin, 2.3 µM α-tocopherol, 5 µg/ml transferrin, 1 nM MnCl2, 500 nM Na2SiO3, 1 nM (NH4)6Mo7O24, 5 nM NH4VO3-(Na3VO4), 500 pM NiCl2, 5 pM SnCl2, 20 nM H2SeO3, and 10 ng/ml epidermal growth factor]. All tissue culture reagents were purchased from Invitrogen (Carlsbad, CA).
If not otherwise indicated, cells were seeded as follows: in 96-well plates for the 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)/phenazine methosulfate (PMS) assay (2 x 103/well) and for 3-(4, 5 dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; 5 x 103/well); in 6-well plates for time-lapse video microscopy (1 x 105/well for C4-2, C4-2Luc, PC3, and BT549 and 1.5 x 105/well for LNCaP cells) and 4 x 105 in 6-cm Petri dishes for Western blot analysis and caspase 3 assays. Cells were kept in culture for 24 to 48 hours to reach about 80% confluence before starting any experiments. During experiments, cells were kept in a serum-free medium condition as described previously [36] (except LNCaP cells, which were kept in a medium supplemented with 2.5% FBS).
MTS/PMS and MTT Cell Viability Assays
For MTS/PMS assay, J591PE was administered to C4-2 and PC3 cells at a range of 0.1 to 1000 ng/ml, with or without 1 hour of preincubation with J591scFv (50:1). Cells were incubated for 48 hours, and cell viability was measured using the MTS/PMS dye (Promega, Madison, WI) in triplicate, following the manufacturer's instructions.
For MTT assay, cells were incubated with drugs for 48 hours and cell viability was measured using MTT dye (Sigma-Aldrich) in triplicate, according to the manufacturer's instructions.
Plates were analyzed using a SpectraMax Plus384 plate reader (Molecular Devices, Santa Clara, CA).
Caspase 3 Assay
Apoptosis in whole-cell populations was assessed by monitoring caspase 3 activity with the specific fluorogenic substrate Ac-DEVD-7-amido-4-trifluoromethylcoumarin (Bachem, Torrance, CA), as previously reported [12]. Adherent and floating cells were collected and lysed using a cold lysis buffer (1% NP-40, 150 mM NaCl, 20 mM Hepes, 1 mM EDTA, 1 mM DTT, and 5 µg/ml aprotinin, leupeptin, and pepstatin, respectively). Fluorescence was recorded 15 minutes each for 1 hour using a VersaFluor fluorometer (Bio-Rad Laboratories, Hercules, CA). Caspase 3 activity was expressed in arbitrary units and calculated using Excel 2010 (Microsoft Corporation, Redmond, WA).
Western Blot Analysis
Media were removed, and cells were washed gently twice using cold PBS on ice. Lysates were generated using cold 20 mM Hepes, 150 mM NaCl, 1 mM EDTA, 0.5% Na+ deoxycholate, 1% NP-40, and 1 mM DTT (pH 7.4) buffer containing protease inhibitors (10 µg/ml aprotinin, 10 µg/ml leupeptin, 10 µg/ml pepstatin, 1 mM benzamidine, and 1 mM PMSF) and phosphatase inhibitors (1 mM NaVO4, 50 mMβ-glycerophosphate, 40 mM p-nitrophenylphosphate, 40 mM NaF, and 1 µg/ml microcystin). All reagents were purchased from Sigma-Aldrich. Cell lysates were clarified by centrifugation at 15,000 rpm for 15 minutes at 4°C, and then the supernatant was collected and protein content was measured by a Bradford assay (Bio-Rad Laboratories) according to the manufacturer's directions. Proteins were separated on SDS-PAGE on 12% gels and transferred onto a 0.45-µm nitrocellulose membrane (PerkinElmer, Waltham, MA). Western blot analysis was performed with the Odyssey CLx Infrared Imaging System (LI-COR Biosciences, Lincoln, NE), according to the manufacturer's instructions.
Rabbit polyclonal anti-p-AKT (T308), anti-AKT, anti-p-BAD (S112), anti-BAD, and anti-cleaved poly(ADP-ribose) polymerase (PARP; Asp214), anti-cleaved caspase 3 (Asp175), and mouse monoclonal anti-caspase 3, anti-caspase 7, anti-p-AKT (S473), and anti-tubulin were purchased from Cell Signaling Technology (Beverly, MA); rabbit anti-MCL-1 and anti-β-actin were purchased from Stressgen Biotechnologies Inc (Philadelphia, PA) and Sigma-Aldrich, respectively. Mouse monoclonal J591 antibody was purchased from Dr N. Bander (Weill Medical College of Cornell University, New York, NY) and used to detect PSMA in prostate cancer cells. Secondary goat anti-mouse IRDye 680 and goat anti-rabbit IRDye 800 were both purchased from LI-COR Biosciences. Protein bands were quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
Metabolic Labeling
Cells were grown in cysteine/methionine-free Dulbecco's modified Eagle's medium with 5% FBS for 5 hours in the presence of either 100 µg/ml cycloheximide, 1 µg/ml J591PE, or 1 µg/ml J591PE and 10 µg/ml J591 antibody followed by addition of 10 µCi/ml S35Cys/Met Easy Tag Express labeling mix (PerkinElmer) for 1 hour in triplicate for each data point. Cells were then washed with PBS and protein lysates were collected as described in the Western Blot Analysis section. Protein lysates (5 µl) were spotted on a nitrocellulose membrane, washed, dried, and placed into scintillation vials with 2 ml of scintillation liquid. Radioactivity was quantified using an LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter, Fullerton, CA). In addition, 25 µg of protein lysates was separated on 10% SDS-PAGE, transferred into a nitrocellulose membrane, and imaged using a Typhoon Trio (GE Healthcare Bio-Sciences, Piscataway, NJ).
MCL-1-Targeted shRNA
An MCL-1-targeted shRNA was generated using the lentiviral vector-based RNAi technology previously described [37]. To find a target sequence, we chose a 23-nucleotide MCL-1 sequence found in MCL-1 isoform 1 that has no homology in the mice and has a high guanine/cytosine (GC) percentage to increase efficiency. Our target sequence (5′-AAT TCA AAA AAT TGT TTA ACT CGC CAG TCC CGT A-3′) targets a region between the transmembrane domain and the carboxyl terminus of human MCL-1. Briefly, polymerase chain reaction (PCR) was used to amplify the U6 promoter region that was digested with BamHI and HindIII restriction sites. Oligonucleotides containing the MCL-1 target sequence, a six-nucleotide hairpin sequence, were annealed and digested by HindIII-EcoRI. The pLL3.7 lentiviral vector was digested with BamHI and EcoRI. The U6 promoter and MCL-1 target oligonucleotide were ligated at HindIII, and the remaining digested ends were ligated to the digested pLL3.7 vector at BamHI and EcoRI sites. A scrambled RNA oligonucleotide sequence 5′-GGTACGGTCAGGCAGCTTCT-3′ was used as a control.
Inducible FLAG-MCL-1
Complementary oligonucleotides encoding FLAG epitope and several restriction sites were synthesized by GenScript (Piscataway, NJ) and cloned into the EcoRI and HindIII sites of pUC57 (termed pUC57-FLAG + ReS). The open reading frame of MCL-1 was PCR amplified from green fluorescent protein (GFP)-MCL-1 with primers containing 5′ BamHI and 3′ XbaI sites and cloned into PCR-BluntII-TOPO vector (Zero Blunt TOPO PCR cloning kit; Invitrogen, Grand Island, NY). Sequence-verified PCR-amplified clones were used to isolate FLAG-MCL-1 fragment (digested with EcoRI and XbaI) and cloned into tetracycline-inducible lentiviral vector, pTET-ON2. Doxycycline induction experiments were performed with TET-FBS-containing medium with 1 mg/ml doxycycline for 24 hours.
Transfections were performed at 60% to 70% confluence using Lipofectamine (Invitrogen) according to the manufacturer's recommendations.
Time-Lapse Video Microscopy
Time-lapse video recording was performed on an Axiovert 100 microscope (Carl Zeiss Microscopy, Thornwood, NY) equipped with a moving stage and climate control chamber (37°C, 5% CO2) and controlled by OpenLab software (Improvision Inc, Lexington, MA). In experiments with cells transiently transfected with FLAG-MCL-1 or shRNA expression vectors that also express GFP, fluorescent images of GFP-positive cells were recorded and analyzed for apoptosis morphology. In an experiment where total population of cells was analyzed, bright fields were recorded. Four randomly chosen fields containing around 100 to 150 cells for each treatment were recorded. Data are expressed as percentage of apoptotic-like morphology cells versus living cells for each hour of treatment.
Prostate Cancer Xenograft Model
Six-week-old BALB/c nude mice (Charles River Laboratories, Wilmington, MA) received three subcutaneous injections of 2 x 106 cells (C4-2Luc) with Matrigel (Sigma-Aldrich). Three injections were performed to increase the probability of generating xenografts, using an insulin syringe with a 29-gauge needle. When tumors reached a diameter of 0.5 cm, intratumoral injections of ZSTK474 (375 µg/kg in 30 µl of DMSO), J591PE (2.5 mg/kg in 30 µl of PBS), or the combination were conducted under anesthesia as reported in the animal protocol. Tumors were also injected with vehicle controls (30 µl of DMSO or 30 µl of PBS) or J591scFv (2.5 mg/kg in 30 µl of PBS). The experimental protocol was approved by the Wake Forest University Institutional Animal Care and Use Committee.
In Vivo Non-Invasive Imaging
Tumor growth was analyzed using a Xenogen IVIS 100 optical imaging system (Caliper Life Sciences, Hopkinton, MA) while animals were anesthetized with 2% isoflurane/O2. To account for background and nonspecific luminescence, mice were imaged before the injection of 100 µl of luciferase substrate (consisting of 3.5 mg/ml luciferin in PBS) and imaged 15 minutes later for 1 minute in the position of interest. Whole-body images were obtained using Living Image software provided with the imaging system. A grayscale photographic image and the bioluminescent color image were superimposed to provide an anatomic registration of the light signal. The region of interest (ROI) was manually selected over the luminescent signal, and the intensity was recorded as photons per second within an ROI.
Statistical Analysis
Probability (P) values were calculated using the one-way analysis of variance test; P values ≤ .05 were considered statistically significant. The results are expressed as means ± SEM.
Results
J951PE Toxin Selectively Inhibits Growth of Cells that Express PSMA
J591 monoclonal antibodies against PSMA have been extensively characterized for selectivity toward prostate cells that express PSMA and used for clinical imaging of prostate tumors and targeted toxin delivery in preclinical models [38–40]. Importantly for therapeutic applications and especially for toxin delivery, these antibodies are rapidly internalized on binding to prostate cancer cells [41]. To selectively inhibit protein synthesis in prostate cancer cells, we generated a chimera of single-chain variable fragment of J591 monoclonal antibody against PSMA and the translocation and catalytic domains of a P aeruginosa exotoxin A (PE38QQR) that ribosylates and inactivates elongation factor 2 (Figures 1A and W1A) [30,42]. PE38QQR is a truncated mutant form of P aeruginosa exotoxin A that lacks cell binding domain 1a and 16 amino acids from cell binding domain 1b, in which lysines 590 and 606 are replaced with glutamines and lysine 613 with arginine. PE38QQR has been used to produce extremely potent immunotoxins that caused complete regression of human carcinoma growth in mice [30,32,33].
Figure 1.

Characterization of J591PE chimera toxin. (A) Scheme of the J591scFvPE38QQR (J591PE) immunotoxin: a chimera between the single-chain variable fragment of the J591 antibody (J591scFv) and the truncated form of P aeruginosa exotoxin A (PE38QQR) is shown. (B) Analysis of PSMA and PTEN expression in a panel of prostate cancer cells (LNCaP, C4-2, C4-2Luc, PC3, WF3, and DU145) and breast cancer cells (BT549). (C) J591PE selectively inhibits protein synthesis in PSMA-positive C4-2Luc cells. PSMA-negative PC3, DU145, and PSMA-positive C4-2Luc cells were treated with cycloheximide (CX) or J591PE for 5 hours and labeled with S35cystein-methionine for 1 hour. Where indicated, C4-2Luc cells were preincubated with 10 µg/ml J591 antibody for 1 hour before adding J591PE chimera toxin. (D) Cytotoxic effects of J591PE on C4-2 and PC3 cells. J591PE was administered alone and in combination with J591scFv (1 hour of preincubation; 50:1 for each J591PE concentration used) to C4-2 and PC3 cells. After 48 hours, an MTS/PMS assay was performed, measuring cell viability. Data were plotted as percentage of control versus concentration of the toxin used. Results in C and D are expressed as means ± SEM, n = 3.
Analysis of expression of PSMA and PTEN in LNCaP, C4-2, C4-2Luc, PC3, WF3, and DU145 cells (prostate cancer) and in BT549 (breast cancer) cells showed that LNCaP, C4-2, and C4-2Luc cells express PSMA and do not express PTEN; PC3, WF3, and BT549 cells do not express PSMA or PTEN; and DU145 cells express PTEN but do not express PSMA (Figure 1B).
We examined the effects of J591PE on protein synthesis by metabolic labeling of C4-2Luc, PC3, and DU145 cells with S35Cys and S35Met. Incubation with J591PE for 6 hours selectively inhibited S35 incorporation into PSMA-positive C4-2Luc cells but not into PSMA-negative PC3 or DU145 cells (Figures 1C and W1B). In contrast, cycloheximide inhibited protein synthesis in all cell lines.
Consistent with selective inhibition of protein synthesis, cell viability assays showed that J591PE decreased the number of viable C4-2 (PSMA-positive) cells after 48 hours of incubation but had no effect on PSMA-negative PC3 cells (Figure 1D). Specificity of J591PE toxin to PSMA epitope was confirmed by competition assay using the singlechain antibody J591scFv not conjugated with toxin as a competitor in metabolic labeling and cell viability assays. Ten-fold excess of unconjugated J591 antibodies prevented inhibition of protein synthesis and decreased long-term cytotoxic effects in C4-2Luc cells by J591PE (Figures 1, C and D, W1B, and W2). Control treatments with J591scFv did not reduce viability of C4-2 cells, whereas treatments with cycloheximide decreased viability in all cell lines irrespective of PSMA expression (Figure W2).
Combination of the PI3K Inhibitor ZSTK474 with J591PE Induces Apoptosis in Prostate Cancer Cells
Treatment of C4-2 cells with increasing concentrations of PI3K inhibitor ZSTK474 induced minimal apoptosis at 0.2 µg/ml that peaked at 2 µg/ml as judged by measuring caspase 3 activity and cleavage of caspase 3/7 substrate PARP (Figure 2A, left panel); at this concentration, maximal inhibition of PI3K/AKT signaling was achieved (Figure W3). At the same time, treatments with J591PE at the range of 0.001 to 10 µg/ml did not induce detectable apoptosis (Figure 2A, right panel). We used a range of concentrations to find the best combination of ZSTK474 and J591PE for inducing apoptosis (the lowest doses with the strongest pro-apoptotic effects). The combination of 2 µg/ml ZSTK474 and 1 µg/ml J591PE (combination C4; Figure 2B) increased caspase 3 activity by 7-fold compared with a 2.5-fold increase in C4-2 cells treated with ZSTK474 and a 1.2-fold increase in cells treated with J591PE, as single agents. Time course analysis showed that significant increase of caspase activity and PARP cleavage was detected in C4-2 cells 6 hours after treatments with combination C4 (2 µg/ml ZSTK474 and 1 µg/ml J591PE) with further increase at later time points (Figure 2C).
Figure 2.

Optimization of apoptosis induction by the combination of ZSTK474 and J591PE in prostate cancer cells. (A) C4-2 cells treated with either ZSTK474 (left panel) or J591PE (right panel) show modest or no increase of caspase 3 activity/PARP cleavage at 6 hours (green, c-PARP; red, tubulin). (B) Dose-response analysis of caspase 3 activity and PARP cleavage in C4-2 cells treated with the combination of ZSTK474 and J591PE. On the basis of these results, 2 µg/ml ZSTK474 and 1 µg/ml J591PE were used for subsequent experiments (referred to as combination C4). (C) Time-course analysis of the caspase 3 activity (left panel) and PARP cleavage (right panel) in C4-2 cells treated with the combination of ZSTK474 and J591PE. Results are expressed as means ± SEM, n = 2. Representative Western blots are shown.
To further characterize pro-apoptotic effects of the combination of ZSTK474 and J591PE, we followed cell death by time-lapse video microscopy, measured caspase activity, and examined cleavage of effector caspase 3, caspase 7, and PARP in a panel of cell lines. Consistent with the results in C4-2 cells, the combination of ZSTK474 and J591PE increased apoptosis in PSMA-positive cell lines. Induction of apoptosis in LNCaP, C4-2, and C4-2Luc cells treated with the combination of ZSTK474 and J591PE was confirmed by time-lapse video microscopy, detection of the loss of full-length caspase 3, and detection of cleaved forms of caspase 7 and of cleaved PARP (Figures 3A and W4A). In contrast, combined treatments with ZSTK474 and J591PE failed to significantly increase apoptosis in PC3, WF3, and BT549 cell lines that do not express PSMA (Figures 3B, W3, and W4B). However, administration of the protein synthesis inhibitor cycloheximide with ZSTK474 increased caspase 3 activity in PC3, DU145, WF3, and BT549 cells, confirming that the combination of a protein synthesis inhibitor with a PI3K inhibitor can increase apoptosis in PSMA-negative cells (Figure W5). MTT assay supported these data by showing decreased viability of PSMA-positive C4-2 cells after 48 hours of incubation with ZSTK474 and J591PE and further significant decrease of viability in cells treated with the combination of J591PE and ZSTK474 (Figure W2A). In PSMA-negative cells, PC3 and DU145 treatments with J591PE did not reduce viability nor augmented the toxic effects of ZSTK474 in these cells (Figure W2, B and C).
Figure 3.

Combination of ZSTK474 and J591PE promotes apoptosis in C4-2Luc cells. (A) Combination of ZSTK474 and J591PE promotes apoptosis at 6 hours in C4-2Luc cells, as suggested by analysis of caspase 3 activity, cleavage of effector caspases 3 and 7, and cleavage of PARP as well as analysis of cell morphology by time-lapse video recording. (B) Combination of ZSTK474 and J591PE does not promote apoptosis in PC3 cells, when compared to single-agent treatments. Results are expressed as means ± SEM, n = 2 (caspase 3 assay) and n = 4 (time-lapse video microscopy). Representative Western blots and time-lapse video microscopy frames (original magnification, x20) are shown (vehicle, DMSO; combination, ZSTK474 + J591PE).
Altogether these results confirm specificity of apoptosis induction by the combination of ZSTK474 and J591PE in PSMA-expressing cells.
Inhibition of the PI3K/AKT/BAD Pathway and Loss of MCL-1 Is Needed to Induce Apoptosis
BAD has been recognized as a sentinel of apoptosis and a target responsible for pro-apoptotic effects of the PI3K/AKT inhibitors in numerous cell lines, including prostate cancer cells [43,44]. Analysis of AKT and BAD phosphorylation by Western blot analysis confirmed that in all PTEN-deficient cell lines, the PI3K/AKT pathway was constitutively active and BAD was phosphorylated, whereas no phosphorylation of AKT or BAD was detected in PTEN-positive DU145 cells (Figures 4 and W5C). Administration of ZSTK474 inhibited both AKT and BAD phosphorylation. However, robust apoptosis was induced only in C4-2, C4-2Luc, and LNCaP cells that express PSMA (a target of J591PE), when cells were treated with the combination of ZSTK474 and J591PE, whereas treatment with ZSTK474 alone did not induce substantial apoptosis (Figures 3A and W4). These results suggested that inhibition of the PI3K/AKT/BAD axis alone is not sufficient to induce apoptosis that may promote cell death in PTEN-deficient prostate cancer cells.
Figure 4.
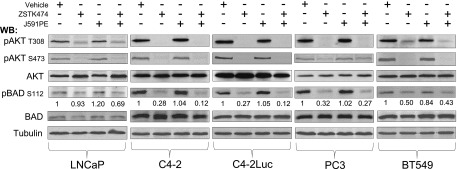
ZSTK474 alone or in combination with J591PE inhibits the PI3K/AKT pathway and BAD phosphorylation in PTEN-deficient cell lines. Inhibition of the PI3K/AKT pathway by ZSTK474 was followed in LNCaP, C4-2, C4-2Luc, PC3, and BT549 cells by monitoring the phosphorylation levels of AKT (T308 and S473) and BAD (S112). Representative Western blots are shown (vehicle, DMSO).
Recently, it was proposed that loss of induced MCL-1, an antiapoptotic Bcl-2 family protein characterized by a short half-life, provides a possible mechanism for the pro-apoptotic effect of protein synthesis inhibitors [45–47]. To test whether loss of MCL-1 in human prostate cancer cells may be responsible for apoptosis induction by the combination of ZSTK474 and J591PE, we assessed MCL-1 protein levels in these cell lines. Loss of MCL-1 expression was detected in C4-2Luc cells treated with the C4 combination (1 µg/ml J591PE and 2 µg/ml ZSTK474) and in cells treated with higher doses of both agents (Figure 5A). In the comparison of MCL-1 levels in a panel of cell lines treated with pro-apoptotic agents, C4-2Luc cells showed strongest decrease ofMCL-1 expression, followed by C4-2 and LNCaP cells, whereas no apparent change in MCL-1 expression was detected in PC3 and BT549 cells (Figure 5B). Thus, levels of MCL-1 expression are inversely proportional to levels of apoptosis in these cell lines. To test the role of MCL-1 in apoptosis induced by the combination of ZSTK474 and J591PE, we compared apoptosis in cells in which MCL-1 was knocked down with an shRNA and in cells that ectopically expressed doxycycline-inducible FLAG-MCL-1. Analysis of apoptosis in cells that express an MCL-1-specific shRNA or a scrambled shRNA showed that loss of MCL-1 expression sensitized cells to ZSTK474 administration, resulting in increased apoptosis as determined by timelapse video microscopy (Figure 5C). Conversely, ectopic expression of FLAG-MCL-1 antagonized the effects of ZSTK474 and J591PE, decreasing the levels of apoptosis when cells were treated with both agents (Figure 5D).
Figure 5.

Loss of MCL-1 contributes to apoptosis induction by the combination of ZSTK474 and J591PE. (A) Analysis of MCL-1 expression in C4-2 cells treated with a range of combinations (C1-C6) of ZSTK474 and J591PE. Concentrations of each agent are shown in Figure 1B. (B) Analysis of MCL-1 expression in a panel of PTEN-positive (LNCaP, C4-2, and C4-2Luc) and PTEN-negative (PC3 and BT549) cells treated with ZSTK474, J591PE, or the combination of both for 6 hours. Representative Western blots are presented; numbers show fold change of MCL-1 band intensity compared to cells treated with vehicle (DMSO). (C) Knockdown of MCL-1 with shRNA sensitizes C4-2Luc cells treated with ZSTK474 to apoptosis. C4-2Luc cells were infected with either MCL-1-specific shRNA or scrambled shRNA expressed in a lentiviral vector with GFP marker, and apoptosis morphology in GFP-positive cells was followed by time-lapse video microscopy. Data for the 6-hour time point are shown. Insert shows Western blot analysis of MCL-1 expression (green) in GFP-positive (left) and GFP-negative (right) cells. β-Actin (red) was used as a loading control. (D) Expression of FLAG-MCL-1 protects C4-2Luc cells from apoptosis. C4-2Luc cells were transfected with vectors encoding FLAG-MCL-1 under doxycycline-inducible promoter and GFP and were treated and analyzed as in C. Insert shows doxycycline-inducible expression of FLAG-MCL-1. Results are expressed as means ± SEM, n = 3.
Combination of the PI3K Inhibitor ZSTK474 and the Immunotoxin J591PE Induces Regression of Human Prostate Cancer Xenografts
To determine whether the combination of ZSTK474 and J591PE inhibits tumor growth in vivo, experiments were conducted in C4-2Luc xenografts that express firefly luciferase. Recently, we have shown that injection of PI3K inhibitor ZSTK474 into C4-2Luc xenografts induced apoptosis and in parallel decreased tumors luminescence; thus, growth of C4-2Luc xenografts could be monitored by noninvasive optical imaging [48].
ZSTK474 injected in C4-2Luc tumors in combination with J591PE reduced the luminescence levels for at least 5 days, whereas single injections with either ZSTK474 or J591PE led to a transient reduction of luminescence (Figure 6, A and C). C4-2Luc tumors injected with J591scFv (antibody without toxin) or PBS (solvent for J591PE) continue to grow (Figure W6, A and B), whereas injections of DMSO (solvent for ZSTK474) stabilized luminescence for 5 days. Analysis of C4-2Luc xenograft tissues for MCL-1 expression, BAD phosphorylation, p-AKT, and apoptosis markers (cleaved PARP and cleaved caspase 3; Figure 6B) showed decreased MCL-1 expression in C4-2Luc tumors treated with either agent, with further decrease in C4-2Luc xenografts injected with ZSTK474 and J591PE. Consistent with a long-term reduction of luminescence, more cleaved PARP and cleaved caspase 3 was observed in C4-2Luc xenografts treated with the combination of J591PE and ZSTK474, supporting the hypothesis that simultaneous loss of MCL-1 expression and BAD dephosphorylation is requested to increase apoptosis in PSMA-expressing prostate tumors in vivo, as previously observed in vitro.
Figure 6.


Combined administration of ZSTK474 and J591PE induces regression of C4-2Luc prostate cancer tumors in nude mice. (A) Dynamics of luminescence of C4-2Luc tumors after a single local injection of DMSO, ZSTK474 (375 µg/kg), J591PE (2.5 mg/kg), or the combination of ZSTK474 and J591PE. Luminescence was monitored for 6 days by optical imaging and expressed as fold change compared to luminescence at day 0. Results are expressed as means ± SEM, n = 4 (DMSO) and n = 10 (ZSTK474, J591PE, combination). (B) Western blot analysis of C4-2Luc tumors excised 6 hours after injections of drugs. Combination of ZSTK474 and J591PE decreases MCL-1 protein levels and promotes apoptosis (increased c-caspase 3 and c-PARP) when compared to vehicle (DMSO). ZSTK474 alone or in combination with J591PE inhibits the phosphorylation of p-AKT (S473) and p-BAD (S112). Representative Western blots are shown. (C) Representative images of nude mice recorded on IVIS 100 luminescent imaging station showing ROI, 15 minutes after intraperitoneal (i.p.) luciferin injection. Representative images of background luminescence outside the tumors are shown in Figure W6C.
Discussion
Castration-resistant prostate cancer is a late-stage and lethal form of prostate cancer for which no curative treatments are available at present. Despite several ongoing trials, there is an urgent need for new agents or rational drug combinations to eliminate growth of androgen-insensitive malignant cells [49,50]. Accumulating evidence supports the role of anti-apoptotic signaling cascades through protein kinases in the therapeutic resistance of castration-resistant prostate cancer. In this disease, the PI3K/AKT pathway is prominent as a result of gain-of-function mutations in PI3Kα or loss of PTEN, which is associated with cancer progression, high Gleason grade, and poor prognosis [11,51]. Thus, PI3K inhibitors were considered a suitable form of therapy, but their potential as anti-tumor agents has yet to be realized. Early clinical trials suggest that these drugs are well tolerated in patients and at least partially inhibit the PI3K pathway in tumors. However, clinical response is poor, even in patients with PTEN loss or activating mutations of the PI3K [52,53].
Consistent with the results of these clinical trials, our earlier experiments in prostate cancer cells (and experimental results depicted in Figure 2) show that PI3K inhibitors induce modest apoptosis in PTEN-negative prostate cancer cells, but the combination of PI3K inhibitors and protein synthesis inhibitors increases the levels of apoptosis able to promote prostate cancer cell death (Yancey et al., in press) [43].
Although general protein synthesis inhibitors have been used in the clinic, they have substantial side effects [54,55]. To reduce these agents' toxicity, we produced a novel prostate cancer-targeted immunotoxin (J591PE), by generating a recombinant protein chimera between the single-chain variable fragment of the monoclonal antibody J591 (J591scFv) and the truncated form of the P aeruginosa exotoxin A (PE38QQR). The result is a prostate-specific protein synthesis inhibitor that targets PSMA-expressing cells. A combination of J591PE with the pan-PI3K inhibitor ZSTK474 promotes apoptosis both in vitro and in vivo in xenograft tumors, when compared with single-agent treatments. A single injection of the combination of ZSTK474 and J591PE is sufficient to induce apoptosis and prolonged inhibition of C4-2Luc xenograft luminescence.
Anti-apoptotic signaling by a hyperactive PI3K pathway has been suggested as the likely cause of therapeutic resistance of prostate cancer cells [7]. Analysis of downstream effectors identified BAD as a major target of the constitutively active PI3K pathway in prostate cancer cells and a convergence node for several other anti-apoptotic signaling pathways [43,56]. Subsequent experiments, including the results presented above, argue that BAD dephosphorylation is necessary but not sufficient to induce apoptosis. However, when BAD dephosphorylation is combined with decreased MCL-1 expression, prostate cancer cells undergo apoptosis. BAD cannot bind MCL-1; therefore, MCL-1 expression prevents apoptosis in cells with dephosphorylated BAD [57]. Analysis of MCL-1 expression in cells that undergo apoptosis on treatment with J591PE has shown that J591PE triggers loss of MCL-1 expression, whereas experiments with shRNA-mediated knockdown and ectopic expression of FLAG-MCL-1 confirmed the determinant role of MCL-1 in apoptosis induction in prostate cancer cells (Figure 4). Taken together, these experiments point at MCL-1 loss as the primary mechanism behind the pro-apoptotic effects of the immunotoxin J591PE.
Increased MCL-1 expression has been associated with prostate cancer progression and may represent a mechanism of tumor resistance to apoptosis-inducing therapies [58].
In summary, our results demonstrate synthetic lethality of a PI3K inhibitor and a prostate-directed protein synthesis inhibitor immunotoxin, suggesting that BAD phosphorylation and MCL-1 expression could be used as biomarkers to predict anti-tumor efficacy in prostate cancer cells with a constitutive activated PI3K/AKT pathway. Future experiments will determine optimal choices and delivery regiments of inhibitors in preclinical models of prostate cancer, by evaluating their general toxicity, adverse effects, and specificity in inducing apoptosis and regression of prostate tumors. Possible directions to increase specificity by reducing systemic nonspecific effects may include the use of prostate-selective PI3K inhibitor prodrugs and a new generation of prostate-specific toxins in which antibody targeting is represented by prostate-selective aptamers or other small molecule ligands [12,59].
Supplementary Material
Acknowledgments
We are grateful to Dr John Wilkinson and Dr Kazushi Inoue for sharing their expertise and reagents and to Karen Klein for manuscript editing.
Abbreviations
- J591PE
a protein chimera between the single-chain variable fragment of the monoclonal antibody J591 against the prostate-specific membrane antigen and the truncated form of the Pseudomonas aeruginosa exotoxin A (PE38QQR)
- PSMA
prostate-specific membrane antigen
Footnotes
Project described was supported by Award No. R01CA118329 from the National Cancer Institute to G.K. D.B. is currently supported by a T32 CA079448 training grant from the National Cancer Institute. D.Y. was supported in part by Research Supplement to Promote Diversity in Health-Related Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
This article refers to supplementary materials, which are designated by Figures W1 to W6 and are available online at www.neoplasia.com.
References
- 1.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 2.Toren PJ, Gleave ME. Evolving landscape and novel treatments in metastatic castrate-resistant prostate cancer. Asian J Androl. 2013;15:342–349. doi: 10.1038/aja.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang TY, Agarwal N, Sonpavde G, DiLorenzo G, Bellmunt J, Vogelzang NJ. Management of castrate resistant prostate cancer—recent advances and optimal sequence of treatments. Curr Urol Rep. 2013;14(3):174–183. doi: 10.1007/s11934-013-0322-0. [DOI] [PubMed] [Google Scholar]
- 4.Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 2009;9:237–249. doi: 10.2174/156800909787580999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999;59:4291–4296. [PubMed] [Google Scholar]
- 6.Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, Hicks JL, Park BH, Humphreys E, Partin AW, et al. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res. 2011;17:6563–6573. doi: 10.1158/1078-0432.CCR-11-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer. 2013;20(3):R83–R99. doi: 10.1530/ERC-12-0394. [DOI] [PubMed] [Google Scholar]
- 8.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 10.Bouali S, Chrétien AS, Ramacci C, Rouyer M, Becuwe P, Merlin JL. PTEN expression controls cellular response to cetuximab by mediating PI3K/AKT and RAS/RAF/MAPK downstream signaling in KRAS wild-type, hormone refractory prostate cancer cells. Oncol Rep. 2009;21:731–735. [PubMed] [Google Scholar]
- 11.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–4805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 12.Baiz D, Pinder TA, Hassan S, Karpova Y, Salsbury F, Welker ME, Kulik G. Synthesis and characterization of a novel prostate cancer-targeted phosphatidylinositol-3-kinase inhibitor prodrug. J Med Chem. 2012;55:8038–8046. doi: 10.1021/jm300881a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yaguchi S, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H, Hirono S, Yamazaki K, Yamori T. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer Inst. 2006;98:545–556. doi: 10.1093/jnci/djj133. [DOI] [PubMed] [Google Scholar]
- 14.Mueller A, Bachmann E, Linnig M, Khillimberger K, Schimanski CC, Galle PR, Moehler M. Selective PI3K inhibition by BKM120 and BEZ235 alone or in combination with chemotherapy in wild-type and mutated human gastrointestinal cancer cell lines. Cancer Chemother Pharmacol. 2012;69:1601–1615. doi: 10.1007/s00280-012-1869-z. [DOI] [PubMed] [Google Scholar]
- 15.Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA. Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget. 2012;3:371–394. doi: 10.18632/oncotarget.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR, Watson J, Sauveur-Michel M, Garcia-Echeverria C, Cho CY, et al. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin Cancer Res. 2010;16:5692–5702. doi: 10.1158/1078-0432.CCR-10-1601. [DOI] [PubMed] [Google Scholar]
- 17.Wempe SL, Gamarra-Luques CD, Telleria CM. Synergistic lethality of mifepristone and LY294002 in ovarian cancer cells. Cancer Growth Metastasis. 2013;6:1–13. doi: 10.4137/CGM.S11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gedaly R, Angulo P, Chen C, Creasy KT, Spear BT, Hundley J, Daily MF, Shah M, Evers BM. The role of PI3K/mTOR inhibition in combination with sorafenib in hepatocellular carcinoma treatment. Anticancer Res. 2012;32:2531–2536. [PubMed] [Google Scholar]
- 19.Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci USA. 2013;110:4015–4020. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Guo Z, Du T, Chen J, Wang W, Xu K, Lin T, Huang H. Prostate specific membrane antigen (PSMA): a novel modulator of p38 for proliferation, migration, and survival in prostate cancer cells. Prostate. 2013;73:835–841. doi: 10.1002/pros.22627. [DOI] [PubMed] [Google Scholar]
- 21.Milowsky MI, Nanus DM, Kostakoglu L, Sheehan CE, Vallabhajosula S, Goldsmith SJ, Ross JS, Bander NH. Vascular targeted therapy with anti-prostate-specific membrane antigen monoclonal antibody J591 in advanced solid tumors. J Clin Oncol. 2007;25:540–547. doi: 10.1200/JCO.2006.07.8097. [DOI] [PubMed] [Google Scholar]
- 22.Elsässer-Beile U, Bühler P, Wolf P. Targeted therapies for prostate cancer against the prostate specific membrane antigen. Curr Drug Targets. 2009;10:118–125. doi: 10.2174/138945009787354601. [DOI] [PubMed] [Google Scholar]
- 23.Morris MJ, Divgi CR, Pandit-Taskar N, Batraki M, Warren N, Nacca A, Smith-Jones P, Schwartz L, Kelly WK, Slovin S, et al. Pilot trial of unlabeled and indium-111-labeled anti-prostate-specific membrane antigen antibody J591 for castrate metastatic prostate cancer. Clin Cancer Res. 2005;11:7454–7461. doi: 10.1158/1078-0432.CCR-05-0826. [DOI] [PubMed] [Google Scholar]
- 24.Kuroda K, Liu H, Kim S, Guo M, Navarro V, Bander NH. Saporin toxin-conjugated monoclonal antibody targeting prostate-specific membrane antigen has potent anticancer activity. Prostate. 2010;70:1286–1294. doi: 10.1002/pros.21164. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, Nedrow-Byers JR, Hopkins MR, Wu LY, Lee J, Reilly PT, Berkman CE. Targeting prostate cancer cells with a multivalent PSMA inhibitor-guided streptavidin conjugate. Bioorg Med Chem Lett. 2012;22:3931–3934. doi: 10.1016/j.bmcl.2012.04.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osborne JR, Akhtar NH, Vallabhajosula S, Anand A, Deh K, Tagawa ST. Prostate-specific membrane antigen-based imaging. Urol Oncol. 2013).;31:144–154. doi: 10.1016/j.urolonc.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tagawa ST, Beltran H, Vallabhajosula S, Goldsmith SJ, Osborne J, Matulich D, Petrillo K, Parmar S, Nanus DM, Bander NH. Anti-prostate-specific membrane antigen-based radioimmunotherapy for prostate cancer. Cancer. 2010;116:1075–1083. doi: 10.1002/cncr.24795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang F, Shan L, Liu Y, Neville D, Woo JH, Chen Y, Korotcov A, Lin S, Huang S, Sridhar R, et al. An anti-PSMA bivalent immunotoxin exhibits specificity and efficacy for prostate cancer imaging and therapy. Adv Healthc Mater. 2013;2:736–744. doi: 10.1002/adhm.201200254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frigerio B, Fracasso G, Luison E, Cingarlini S, Mortarino M, Coliva A, Seregni E, Bombardieri E, Zuccolotto G, Rosato A, et al. A single-chain fragment against prostate specific membrane antigen as a tool to build theranostic reagents for prostate cancer. Eur J Cancer. 2013 doi: 10.1016/j.ejca.2013.01.024. pii: S0959-8049(13)00087-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 30.Debinski W, Pastan I. An immunotoxin with increased activity and homogeneity produced by reducing the number of lysine residues in recombinant Pseudomonas exotoxin. Bioconjug Chem. 1994;5:40–46. doi: 10.1021/bc00025a006. [DOI] [PubMed] [Google Scholar]
- 31.Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 32.Debinski W, Pastan I. Monovalent immunotoxin containing truncated form of Pseudomonas exotoxin as potent antitumor agent. Cancer Res. 1992;52:5379–5385. [PubMed] [Google Scholar]
- 33.Debinski W, Karlsson B, Lindholm L, Siegall CB, Willingham MC, FitzGerald D, Pastan I. Monoclonal antibody C242-Pseudomonas exotoxin A. A specific and potent immunotoxin with antitumor activity on a human colon cancer xenograft in nude mice. J Clin Invest. 1992;90:405–411. doi: 10.1172/JCI115875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madhankumar AB, Mintz A, Debinski W. Alanine-scanning mutagenesis of α-helix D segment of interleukin-13 reveals new functionally important residues of the cytokine. J Biol Chem. 2002;277:43194–43205. doi: 10.1074/jbc.M205047200. [DOI] [PubMed] [Google Scholar]
- 35.Smith AJ, Karpova Y, D'Agostino R, Jr, Willingham M, Kulik G. Expression of the Bcl-2 protein BAD promotes prostate cancer growth. PLoS One. 2009;4:e6224. doi: 10.1371/journal.pone.0006224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sastry KS, Karpova Y, Prokopovich S, Smith AJ, Essau B, Gersappe A, Carson JP, Weber MJ, Register TC, Chen YQ, et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J Biol Chem. 2007;282:14094–14100. doi: 10.1074/jbc.M611370200. [DOI] [PubMed] [Google Scholar]
- 37.Sui G, Shi Y. Gene silencing by a DNA vector-based RNAi technology. Methods Mol Biol. 2005;309:205–218. doi: 10.1385/1-59259-935-4:205. [DOI] [PubMed] [Google Scholar]
- 38.Ross JS, Gray KE, Webb IJ, Gray GS, Rolfe M, Schenkein DP, Nanus DM, Millowsky MI, Bander NH. Antibody-based therapeutics: focus on prostate cancer. Cancer Metastasis Rev. 2005;24:521–537. doi: 10.1007/s10555-005-6194-0. [DOI] [PubMed] [Google Scholar]
- 39.Gong MC, Chang SS, Sadelain M, Bander NH, Heston WD. Prostate-specific membrane antigen (PSMA)-specific monoclonal antibodies in the treatment of prostate and other cancers. Cancer Metastasis Rev. 1999;18:483–490. doi: 10.1023/a:1006308826967. [DOI] [PubMed] [Google Scholar]
- 40.Akhtar NH, Pail O, Saran A, Tyrell L, Tagawa ST. Prostate-specific membrane antigen-based therapeutics. Adv Urol. 2012;2012:973820. doi: 10.1155/2012/973820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu H, Rajasekaran AK, Moy P, Xia Y, Kim S, Navarro V, Rahmati R, Bander NH. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998;58:4055–4060. [PubMed] [Google Scholar]
- 42.Hwang J, Fitzgerald DJ, Adhya S, Pastan I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell. 1987;48:129–136. doi: 10.1016/0092-8674(87)90363-1. [DOI] [PubMed] [Google Scholar]
- 43.Sastry KS, Smith AJ, Karpova Y, Datta SR, Kulik G. Diverse anti-apoptotic signaling pathways activated by vasoactive intestinal polypeptide, epidermal growth factor, and phosphatidylinositol 3-kinase in prostate cancer cells converge on BAD. J Biol Chem. 2006;281:20891–20901. doi: 10.1074/jbc.M602928200. [DOI] [PubMed] [Google Scholar]
- 44.Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, Sun J, Danial NN, Liu J, Lin A. Inactivation of BAD by IKK inhibits TNFα-induced apoptosis independently of NF-κB activation. Cell. 2013;152:304–315. doi: 10.1016/j.cell.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindqvist LM, Vikström I, Chambers JM, McArthur K, Ann AM, Henley KJ, Happo L, Cluse L, Johnstone RW, Roberts AW, et al. Translation inhibitors induce cell death by multiple mechanisms and Mcl-1 reduction is only a minor contributor. Cell Death Dis. 2012;3:e409. doi: 10.1038/cddis.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zang C, Eucker J, Liu H, Müller A, Possinger K, Scholz CW. Concurrent inhibition of PI3-kinase and mTOR induces cell death in diffuse large B cell lymphomas, a mechanism involving down regulation of Mcl-1. Cancer Lett. 2012 doi: 10.1016/j.canlet.2012.11.013. pii: S0304-3835(12)00660-X. E-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 47.Mattoo AR, Fitzgerald DJ. Combination treatments with ABT-263 and an immunotoxin produce synergistic killing of ABT-263-resistant small cell lung cancer cell lines. Int J Cancer. 2013;132:978–987. doi: 10.1002/ijc.27732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hassan S, Karpova Y, Baiz D, Yancey D, Pullikuth A, Flores A, Register T, Cline JM, D'Agostino R, Jr, Danial N, et al. Behavioral stress accelerates prostate cancer development in mice. J Clin Invest. 2013;123:874–886. doi: 10.1172/JCI63324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loriot Y, Massard C, Fizazi K. Recent developments in treatments targeting castration-resistant prostate cancer bone metastases. Ann Oncol. 2012;23(5):1085–1094. doi: 10.1093/annonc/mdr573. [DOI] [PubMed] [Google Scholar]
- 50.Fizazi KS, Higano CS, Nelson JB, Gleave M, Miller K, Morris T, Nathan FE, McIntosh S, Pemberton K, Moul JW. Phase III, randomized, placebo-controlled study of docetaxel in combination with zibotentan in patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2013;31(14):1740–1747. doi: 10.1200/JCO.2012.46.4149. [DOI] [PubMed] [Google Scholar]
- 51.Cohen MB, Rokhlin OW. Mechanisms of prostate cancer cell survival after inhibition of AR expression. J Cell Biochem. 2009;106:363–371. doi: 10.1002/jcb.22022. [DOI] [PubMed] [Google Scholar]
- 52.Shuttleworth SJ, Silva FA, Cecil AR, Tomassi CD, Hill TJ, Raynaud FI, Clarke PA, Workman P. Progress in the preclinical discovery and clinical development of class I and dual class I/IV phosphoinositide 3-kinase (PI3K) inhibitors. Curr Med Chem. 2011;18:2686–2714. doi: 10.2174/092986711796011229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmes D. pathway inhibitors approach junction. Nat Rev Drug Discov. 2011;10:563–564. doi: 10.1038/nrd3527. [DOI] [PubMed] [Google Scholar]
- 54.Robert F, Carrier M, Rawe S, Chen S, Lowe S, Pelletier J. Altering chemosensitivity by modulating translation elongation. PLoS One. 2009;4:e5428. doi: 10.1371/journal.pone.0005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wetzler M, Segal D. Omacetaxine as an anticancer therapeutic: what is old is new again. Curr Pharm Des. 2011;17:59–64. doi: 10.2174/138161211795049778. [DOI] [PubMed] [Google Scholar]
- 56.Sastry KS, Karpova Y, Kulik G. Epidermal growth factor protects prostate cancer cells from apoptosis by inducing BAD phosphorylation via redundant signaling pathways. J Biol Chem. 2006;281:27367–27377. doi: 10.1074/jbc.M511485200. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 58.Jackson RS, Placzek W, Fernandez A, Ziaee S, Chu CY, Wei J, Stebbins J, Kitada S, Fritz G, Reed JC, et al. Sabutoclax, a Mcl-1 antagonist, inhibits tumorigenesis in transgenic mouse and human xenograft models of prostate cancer. Neoplasia. 2012;14:656–665. doi: 10.1593/neo.12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rockey WM, Hernandez FJ, Huang SY, Cao S, Howell CA, Thomas GS, Liu XY, Lapteva N, Spencer DM, McNamara JO, et al. Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther. 2011;21:299–314. doi: 10.1089/nat.2011.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.