

Abstract
ETS gene fusions, which result in overexpression of an ETS transcription factor, are considered driving mutations in approximately half of all prostate cancers. Dysregulation of ETS transcription factors is also known to exist in Ewing's sarcoma, breast cancer, and acute lymphoblastic leukemia. We previously discovered that ERG, the predominant ETS family member in prostate cancer, interacts with the DNA damage response protein poly (ADP-ribose) polymerase 1 (PARP1) in human prostate cancer specimens. Therefore, we hypothesized that the ERG-PARP1 interaction may confer radiation resistance by increasing DNA repair efficiency and that this radio-resistance could be reversed through PARP1 inhibition. Using lentiviral approaches, we established isogenic models of ERG overexpression in PC3 and DU145 prostate cancer cell lines. In both cell lines, ERG overexpression increased clonogenic survival following radiation by 1.25 (±0.07) fold (mean ± SEM) and also resulted in increased PARP1 activity. PARP1 inhibition with olaparib preferentially radiosensitized ERG-positive cells by a factor of 1.52 (±0.03) relative to ERG-negative cells (P < .05). Neutral and alkaline COMET assays and immunofluorescence microscopy assessing γ-H2AX foci showed increased short- and long-term efficiencies of DNA repair, respectively, following radiation that was preferentially reversed by PARP1 inhibition. These findings were verified in an in vivo xenograft model. Our findings demonstrate that ERG overexpression confers radiation resistance through increased efficiency of DNA repair following radiation that can be reversed through inhibition of PARP1. These results motivate the use of PARP1 inhibitors as radiosensitizers in patients with localized ETS fusion-positive cancers.
Introduction
ETS gene fusions represent the most abundant genetic translocation associated with solid tumors [1,2] and are present in approximately half of all prostate cancers, the majority of Ewing's sarcomas, and subsets of breast cancer and acute lymphoblastic leukemia [1–10]. In prostate cancers, these gene fusions are thought to be driving mutations and result in overexpression of the involved ETS transcription factor [1–7]. Whereas ETS gene fusions and the resultant transcription factor over-expression have been implicated in carcinogenesis and invasion [11–14], the mechanisms by which they mediate their effects are still being elucidated, as are other phenotypes that may be conferred by these fusions.
We recently discovered that the predominant ETS fusion product in prostate cancer, ERG, interacts with poly(ADP-ribose) polymerase 1 (PARP1) [15], a DNA repair protein initially implicated in base excision repair [16,17], but more recently demonstrated to play a role in homologous recombination [18–20], nonhomologous end-joining [21–23], and transcriptional regulation [24]. PARP1 mediates its effects through addition of PAR groups to a subset of nuclear proteins, thereby helping to initiate DNA repair [25,26]. Radiation therapy (RT) is a standard treatment option or component of treatment for many malignancies known to harbor ETS overexpression including prostate cancer. Whereas RT often provides durable long-term responses, a substantial number of patients will experience biochemical recurrence of their disease following treatment, with 5-year rates of biochemical recurrence of approximately 30% [27]. Thus, a need exists to ascertain causes of radioresistance that may lead to recurrences as well as to identify means to improve long-term results following RT. As RT induces DNA damage that leads to tumor cell death, we hypothesized that overexpression of ERG, through its interaction with the DNA repair protein PARP1, would confer radioresistance that would be preferentially reversible through PARP1 inhibition.
To test this hypothesis, we examined, in vitro, whether overexpression of ERG resulted in radioresistance, and whether this radioresistance could be overcome through PARP1 inhibition, and similarly if ERG overexpression resulted in increased PARP1 activity. We further assessed the role of ERG in DNA repair and cell cycle distribution, which are key determinants of response to RT. Finally, we applied our in vitro findings to an in vivo xenograft model.
Materials and Methods
Cell Culture and Cell Lines
PC3 and DU145 prostate cancer cell lines were grown in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Invitrogen) in a 5% CO2 cell culture incubator. All cultures were also maintained with 50 units/ml of penicillin/streptomycin (Invitrogen). Lentiviruses were generated by the University of Michigan Vector Core. PC3 or DU145 cells were infected with the following lentiviral supernatants: pLentilox-CMV-ERG, pLentilox-CMV-ΔETS, pLentilox-CMV-PARG or pLentilox-CMV-green fluorescent protein (GFP) in the presence of 4 µg/ml polybrene (Sigma, St Louis, MO). CMV-GFP, CMV-ΔETS, and CMV-ERG constructs were created as previously described (with the CMV-ΔETS and CMV-ERG constructs containing the most prevalent gene fusion variant) [15], and CMV-PARG was cloned from a cDNA construct purchased from GeneCopoeia (Rockville, MD). Specifically, ΔETS represents an ERG construct in which the ETS domain (which is necessary for the ERG-PARP1 interaction [15]) has been deleted, and it was used as the control. Stable cell lines were selected by sorting at the University of Michigan flow cytometry core. Stable infection was monitored by confirming GFP expression. The genetic identity of each stable cell line was confirmed by genotyping samples as previously described [28]. Experiments were conducted on exponentially growing cells.
PARP Activity Assay
Total protein was isolated from PC3 and DU145 stable cell lines and quantified by the Bradford assay. Twenty-five micrograms of protein was then used to assess PARP activity according to the manufacturer's protocol (Trevigen, Gaithersburg, MD; Cat. No. 4677-096-K). Purified PARP1 enzyme (Trevigen) was used as a positive control for all reactions.
Clonogenic Survival Assays
Exponentially growing cells were treated with siRNAs [nontargeting (NT): Cat. No. D-001810-01-20, PARP1: Cat. No. J-006656-07 or J-006656-08, XRCC1: Cat. No. J009394-06 (Dharmacon, Lafayette, CO)] or olaparib (Axon Medchem, Groningen, The Netherlands) and then replated at cloning densities. Cells were grown for 8 days, fixed, and stained with methanol-acetic acid and crystal violet, respectively, and scored for colonies of 50 cells or more. Drug cytotoxicity was calculated as the ratio of surviving drug-treated cells relative to untreated control cells. Radiation survival data from drug-treated cells were corrected for drug cytotoxicity. Cell survival curves were fit using the linear-quadratic equation, and the mean inactivation dose was calculated according to the method of Fertil and colleagues [29]. The radiation enhancement ratio (RER) was calculated as the ratio of the mean inactivation dose under control conditions divided by the mean inactivation dose under drug-treated conditions.
Immunofluorescence
Cells were cultured on coverslips in 12-well plates and treated with 1.0 µM olaparib (Axon Medchem) for 1 hour and then immediately exposed to radiation. Cells were collected at indicated time points and processed as previously described [15]. Images were collected with a x60 objective lens using an Olympus DP70 camera fitted in an Olympus 1X-71 microscope. The H2AX foci were detected with mouse monoclonal antibodies phosphor γ-H2AX (Millipore, Billerica, MA; #JBW301). For quantitation of γ-H2AX foci, at least 100 cells from each of three independent experiments were visually scored for each condition. Cells with ≥10 γ-H2AX foci were scored as positive and compared for statistical analyses.
Immunoblot Analysis
Cell pellets and mouse xenografts were lysed and immunoblotted as previously described [15]. Proteins were detected with anti-PAR mouse monoclonal antibody (Millipore; Cat. No. MAB3192), anti-ERG rabbit monoclonal antibody (Epitomics, Burlingame, CA; Cat. No. 5115-1), anti-PARP-1 polyclonal antibody (Cell Signaling Technology, Danvers, MA; Cat. No. 9532), PAR glycohydrolase (PARG) polyclonal antibody (Abcam, Cambridge, MA; Cat. No. ab16060), XRCC1 antibody (Abcam; Cat. No. 9147), and anti-β-actin mouse monoclonal antibody (Cell Signaling Technology; Cat. No. 4967).
Single-Cell Gel Electrophoresis
Single-cell gel electrophoresis was performed according to the manufacturer's instructions (Trevigen). Briefly, treated or untreated cells were collected, suspended in ice-cold phosphate-buffered saline (PBS) at 105 cells/ml, mixed with molten LM agarose (1:10 ratio), and spread on CometSlide. After the agarose solidified, the slides were successively placed in lysis and alkaline solutions (Trevigen). The slides were then subjected to electrophoresis. Cells were fixed with 70% ethanol and stained with SYBR Green. Comet tail moments were then assessed using COMETScore v1.5 image processing software as described by the manufacturer (AutoCOMET.com, Sumerduck, VA). More than 100 cells were analyzed in triplicate experiments. Data are reported as tail moments, which assess the fluorescence intensity in the tail relative to the head while accounting for the relative area of both dipoles.
Statistical Analysis
Results are presented as means ± SEM of at least three experiments. Student's t test was used to assess the statistical significance of differences. A significance level threshold of P < .05 was used.
Flow Cytometry
Cells were harvested, washed in cold PBS, fixed in 70% ethanol, and stained with 50 µg/ml propidium iodide and 100 µg/ml RNase A in PBS. The cells were analyzed for their DNA content with a FACS.
Irradiation
Irradiation was carried out using a Philips RT250 (Kimtron Medical, Woodbury, CT) at a dose rate of ∼2 Gy/min in the University of Michigan Comprehensive Cancer Center Experimental Irradiation Core. Dosimetry was carried out using an ionization chamber connected to an electrometer system that is directly traceable to a National Institute of Standards and Technology calibration. For tumor irradiation, animals were anesthetized with isoflurane and positioned such that the apex of each flank tumor was at the center of a 2.4-cm aperture in the secondary collimator, with the rest of the mouse shielded from radiation.
Xenograft Models
PC3-control (0.5 x 106 cells) or PC3-ERG (0.5 x 106 cells) stable cells were suspended in 100 µl of saline with 50% Matrigel (BD Biosciences, Becton Drive, NJ) and were implanted subcutaneously into the left flank region of severe combined immunodeficiency (SCID) mice. Treatment was initiated when the average tumor volume reached 80 mm3. The mice (eight per treatment group) were treated with ABT-888 (100 mg/kg) twice daily (Abbott, Abbott Park, IL) alone, radiation alone (2 Gy for 5 days), or in combination. Growth in tumor volume was recorded three times per week by using digital calipers, and tumor volumes were calculated using the formula (π/6)(L x W2), where L = length of tumor and W = width. Loss of body weight during the course of the study was also monitored weekly. All procedures involving mice were approved by the University Committee on Use and Care of Animals at the University of Michigan and conform to their relevant regulatory standards.
Results
ERG Overexpression Confers Radioresistance that Is Preferentially Reversed by PARP1 Inhibition
Given our previous identification of an interaction between ERG and PARP1 [15], we aimed to assess our hypothesis that ERG over-expression may confer radioresistance and that this radioresistance might be preferentially overcome through PARP1 inhibition. We have previously shown that the conserved ETS domain of ERG is essential for the ERG-PARP1 interaction and that an ERG mutant containing an ETS domain deletion no longer interacts with PARP1 [15]. Having established a lack of interaction between mutated ERG and PARP1, the mutated ERG model previously described was used as the control in this study. Lentiviral approaches were used to establish isogenic models of ERG overexpression (ERG+) and mutated ERG overexpression (control) in PC3 and DU145 prostate cancer cell lines (Figure 1A). To determine if ERG overexpression confers radioresistance, clonogenic survival was compared between ERG+ and control cells in the PC3 (Figure 1B) and DU145 (Figure 1C) cell lines with increasing doses of RT. ERG+ cells had a 1.25 (±0.07)-fold increase (mean ± SEM) in clonogenic survival compared to control cells in the PC3 and DU145 cell lines, suggesting that over-expression of ERG does indeed confers radioresistance (Figure 1D). Next, cells were treated with noncytotoxic concentrations of olaparib (0.5 µM for 1 hour), immediately preceding RT, treated with radiation, and again assessed for clonogenic survival to evaluate if PARP1 inhibition would result in radiosensitization in ERG+ cells. The combination of olaparib with radiation resulted, preferentially, in a substantial decrease in clonogenic survival for ERG+ cells in both cell lines (Figure 1, B–C). The RER of the PARP1 inhibitor was approximately 1.52 (±0.03) (P < .05) in both cell lines with ERG over-expression (Figure 1E).
Figure 1.
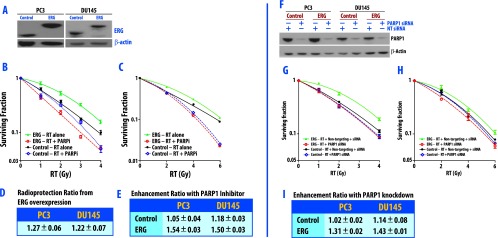
Reduced clonogenic survival of ERG-overexpressing cells (ERG+) after PARP inhibition. As the ETS domain of ERG is essential for the ERG-PARP1 interaction, we established isogenic cell line models of ERG overexpression (ERG+) and mutated ERG overexpression (control) (in which the ETS domain of ERG had been deleted). (A) Western blot analysis of ERG overexpression in PC3 and DU145 prostate cancer cell lines. PC3 cells (B) and DU145 cells (C) were pretreated with 0.5 µM olaparib for 1 hour immediately before IR and then were processed for clonogenic survival 24 hours post-IR. Data are from a single experiment representative of four independent experiments. The effect of ERG overexpression on radioresistance is depicted in D and demonstrates the ability of ERG overexpression to induce radioresistance. After treatment with PARP inhibitor, ERG+ cells are significantly radiosensitized (E). The mean RERs from four independent experiments ± SEM are depicted (E). Additionally, knockdown of PARP1 induces radiation sensitivity in ERG+ cells. Levels of PARP1 protein in total cell extract from cells transfected with NT and PARP-1 siRNA are shown in F. PC3 (G) and DU145 (H) cell lines were treated with PARP1-specific siRNA and NT siRNA, irradiated at 72 hours post-transfection, and then processed for clonogenic survival. Data are from a single experiment representative of three independent experiments. The mean RERs from four independent experiments ± SEM are depicted in I from ERG+ and control cells.
While PARP1 is responsible for the majority of all PARP activities, we wanted to specifically address the contribution of inhibition of PARP1 by olaparib using two independent siRNAs to selectively deplete PARP1 from ERG+ and control cells in both the PC3 and DU145 cell lines and then assess survival following radiation through clonogenic assay. Knockdown of PARP1 with PARP1-specific siRNA resulted in decreased PARP1 levels in ERG+ and control cells in both cell lines, whereas the nonspecific siRNA did not decrease PARP1 levels (Figure 1F). Relative to nonspecific siRNA-treated cells, PARP1 knockdown significantly sensitized ERG+ cells to radiation, as demonstrated by a preferential decrease in clonogenic survival for ERG+ cells in both cell lines (Figure 1, G–H). PARP1 knockdown resulted in an RER of 1.31 (P < .05) and 1.43 (P < .05) in the PC3 and DU145 cell lines, respectively (Figure 1I). Depletion of PARP1 marginally decreased clonogenic survival in the control cells (Figure 1, G–H). These results were PARP1-specific as similar findings were obtained when using a second PARP1 siRNA in DU145 and PC3 cells (see Figure W1).
PARP Activity Is Increased in ERG-Positive Cells
To explore the mechanism of ERG-mediated radioresistance that was preferentially overcome through PARP inhibition, we assessed whether ERG overexpression results in increased PARP activity. In vitro histone ribosylation assays revealed that the rate of PAR synthesis catalyzed by cell extracts from ERG+ cells in the PC3 and DU145 was increased compared to control cells (Figure 2, A and B). Consistent with this result, PAR levels were significantly increased in ERG-overexpressing cells. Treatment with 0.5 µMolaparibfor 24 hours resulted in decreased levels of PAR, with no alteration in ERG and PARP1 expression (Figure 2B). Taken together, these findings demonstrate that ERG overexpression increases PARP activity and that this increased activity can be reversed with the PARP1 inhibitor olaparib.
Figure 2.

PARP activity is increased in ERG+ cells, and overexpression of PARG reverses ERG-mediated radiation resistance. (A) PARP activity assay using Trevigen kits shows that PARP activity is increased in ERG+ PC3 and DU145 cells compared to controls. (B) Western blot analysis of PAR in ERG+ and control cells. Cells were treated with 0.5 µM olaparib for 24 hours and then processed for immunoblot analysis. Treatment with olaparib resulted in decreased levels of PAR, with no alteration in ERG and PARP1 levels. Overexpression of PARG reverses ERG-mediated radiation resistance. (C) PAR levels are reduced in PC3 ERG+ cells with concomitant PARG overexpression. (D) In ERG+ cells, concomitant overexpression of PARG leads to reduced clonogenic survival in PC3 cells.
Indirect Reversal of PARP Activity Preferentially Radiosensitizes ERG+ Cells
Following its activation and autophosphorylation, PARP1 dissociates from DNA strand breaks through charge repulsion [30]. The PAR chains generated by PARP1 are then rapidly degraded by PARG. As ERG+ cells were shown to have increased PARP1 activity, we sought to indirectly reverse the activity of PARP1, through degradation of PAR formed by PARP1 with PARG, and determine if this reversal would also result in radiosensitization of ERG+ cells. The isogenic models of ERG overexpression in the PC3 cell line were modified to create lines with and without overexpression of PARG. As anticipated, overexpression of PARG resulted in decreased PAR levels in ERG+ cells without altering overall levels of ERG (Figure 2C). PARG overexpression additionally reversed ERG-mediated radio-resistance as shown by clonogenic assay (Figure 2D), consistent with our finding that PARP1 inhibition with olaparib preferentially reverses ERG-mediated radioresistance.
ERG Overexpression Promotes Increased DNA Repair Following RT that Is Reversed by PARP Inhibition
Having established that ERG+ status confers radioresistance that is preferentially overcome through direct and indirect PARP1 inhibition, we sought to further elucidate the means by which ERG overexpression promotes radioresistance. As radiation causes DNA damage and activation of DNA damage responses, we assessed whether ERG+ cell lines had enhanced ability to repair ionizing radiation (IR)-induced DNA damage, and subsequently, if this enhanced repair response could be inhibited or reversed through PARP1 inhibition. DNA damage and repair in ERG+ and control cells treated with and without PARP1 inhibition was analyzed using neutral (Figure 3A) and alkaline (Figure 3D) COMET assays in the DU145 cell line. Using neutral COMET assays to directly measure DNA double-strand breaks and repair, imaging at 30 minutes after a single 15-Gy dose of RT demonstrated decreased DNA damage in ERG+ cells (Figure 3A). Mean tail moments decreased with increasing time following RT in the ERG+ group compared to the control group. These mean tail moments were statistically different at 10, 30, and 45 minutes following RT, consistent with increased efficiency of short-term DNA repair in ERG+ cells (Figure 3, B and C). Pretreatment with 1.0 µM olaparib for 1 hour before irradiation significantly slowed DNA repair in ERG+ cells, resulting in increased mean tail moments that were dramatically greater than those of the control group (Figure 3C).
Figure 3.

PARP1 inhibition delays DNA double-strand repair as measured by neutral COMET assay and DNA SSBR by alkaline COMET assay. DU145 cells were pretreated with 1.0 µM olaparib for 1 hour before IR (15 Gy) and then processed for neutral COMET assay (A–C) or treated with 100 µM H2O2 in PBS for 20 minutes on ice and then processed for alkaline COMET assay (D–F). (A) Representative image (from neutral COMET assays) of cells under each condition at 30 minutes post-radiation is presented. (B) Quantification of the tail lengths from each condition was calculated from a minimum of 100 cells for each data point. Data are the means ± SEM of three independent experiments. (C) Relative fold change of tail moments by PARP inhibition (comparing mean tail moments following PARP inhibition and radiation versus radiation alone). (D) Representative image (from alkaline COMET assays) of cells under each condition at 15 minutes post-treatment is presented. (E) Quantification of the tail lengths from each condition was calculated from a minimum of 100 cells for each data point. Data are the means ± SEM of three independent experiments. (F) Relative fold change of tail moments by PARP inhibition.
One of the earliest events during single-strand break repair (SSBR) is the rapid synthesis of PAR, followed by its rapid degradation by PARG. Because PARP1 activity is increased in ERG-overexpressing cells, we hypothesized that ERG overexpression may increase SSBR. To specifically measure whether overexpression of ERG increases DNA SSBR, we treated cells with H2O2, a single-strand break generator, and then performed alkaline COMET assays. ERG-overexpressing cells exhibited significantly higher rates of DNA SSBR (Figure 3D). Treatment with olaparib resulted in slower rates of SSBR in ERG-overexpressing cells than the control cells, with significant differences in mean tail length at 8, 15, and 30 minutes following treatment with H2O2 (Figure 3, E and F). Results from the PC3 cell line can be found in Figure W2 and are consistent with the findings in the DU145 cell line. To confirm that SSBR contributes to ERG-mediated radioresistance, we used siRNA approaches to knock down XRCC1, a key protein involved in DNA SSBR including the base excision pathway [31], in both ERG+ and ERG- PC3 prostate cancer cells (Figure W2F) and performed clonogenic survival assays assessing radiation response in these cells (Figure W2E). As shown, XRCC1 knockdown results in partial reversal of the radiation resistance (Figure W2E). These findings suggest that both single- and double-stranded DNA repairs contribute to ERG-mediated radiosensitization by PARP1 inhibition. Together, these results support the conclusion that ERG overexpression increases DNA repair and that PARP1 inhibition preferentially reverses this enhanced DNA repair in ERG-overexpressing cells.
We next assessed the presence of unrepaired DNA damage assessing γ-H2AX foci in ERG+ and control DU145 cells treated with a single 2-Gy dose of RT. RT was administered in the absence and presence of PARP1 inhibition. Treatment with radiation produced γ-H2AX foci as early as 30 minutes post-irradiation, which resolved to baseline levels over time. The quantity of γ-H2AX foci were measured at baseline and then 30 minutes, 2, 6, and 24 hours following RT. In the cells treated with RT in the absence of PARP1 inhibition, ERG+ cells had significantly decreased levels of γ-H2AX compared to the control group at 24 hours post-RT (Figure 4, A–C). The number of cells with >10 γ-H2AX foci were quite similar in the control and ERG+ cells without PARP1 inhibition at 30 minutes and 2 hours following RT. However, as time progressed to 6 and 24 hours post-RT, the ERG+ cells showed dramatically quicker resolution of γ-H2AX foci compared to the control cells (Figure 4B), consistent with improved long-term DNA repair in ERG+ cells.
Figure 4.

The effects of olaparib on γ-H2AX in ERG-positive and ERG-negative cells. DU145 cells were pretreated with 1.0 µM olaparib for 1 hour before IR (2 Gy) and then fixed for immunofluorescence. (A) Representative image of cells under each condition at 24 hours post-radiation is presented. Cells were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; blue) and γ-H2AX (red). (B) Quantification of the foci from each condition was calculated from a minimum of 100 cells for each data point. Data are the mean of three independent experiments ± SEM. (C) Relative fold change in percentage of cells with >10 γ-H2AX foci.
Pretreatment of cells with 1.0 µM olaparib for 1 hour immediately before RT resulted in preferential reversal of the increased DNA repair efficiency conferred by ERG overexpression, as indicated by a significant prolongation of γ-H2AX signaling in ERG-overexpressing cells. PARP1 inhibition with olaparib repressed DNA repair more profoundly in ERG-positive cells than the control cells (Figure 4C). Results from the PC3 cell line can be found in Figure W3 and are consistent with the findings in the DU145 cell line. These findings suggest that PARP1 inhibition not only preferentially reverses ERG+ mediated accelerated DNA repair but also decreases DNA repair efficiency compared to control ERG- cells treated only with RT.
Preferential Radiosensitization of ERG+ Cells by PARP1 Inhibition Is Not Due to Cell Cycle Redistribution
Because changes in cell cycle distribution can impact radiosensitivity, we analyzed cell cycle profiles for ERG+ and control cells in the PC3 and DU145 cell lines treated in the absence and presence of olaparib, both before RT and at 24 hours following a single 4-Gy dose of RT (Figure W4). ERG+ cells did not differ from control cells in baseline cell cycle distribution. Additionally, in both ERG+ and control cells, the addition of the PARP1 inhibitor did not affect cell cycle distribution before the administration of RT. Together, these findings demonstrate that the preferential radiosensitization of ERG+ cells by olaparib is not due to cell cycle redistribution. Olaparib-treated cells demonstrated an increase in G2/M arrest following radiation, consistent with the expected response to increased DNA damage.
ERG Overexpression Confers Radioresistance In Vivo that Is Preferentially Reversed by PARP Inhibition
Lastly, we assessed ERG conferred radioresistance and the effect of PARP1 inhibition on this radioresistance using PC3 ERG+ and PC3 control mouse xenograft models. For these experiments, we selected the PARP1 inhibitor ABT-888, as this agent is being combined with cytotoxic agents in ongoing clinical trials specific to prostate cancer (www.clinicaltrials.gov). Two weeks following engraftment, ERG+ and control xenografts were treated with ABT-888 twice daily alone, radiation alone (2 Gy/day for 5 days), or both treatments in combination. Tumor size was measured three times per week for 6 weeks. Eight animals were included in each treatment and control group. After completion of follow-up, time to tumor volume doubling was calculated for each group (Figure 5). The ERG+ and control groups that received no treatment experienced the fastest increase in tumor volume. In both groups, nearly all xenografts doubled in tumor volume by day five. These same two groups treated with ABT-888 alone had the next most rapid increase in tumor volume. In both the ERG+ and control groups receiving ABT-888, nearly all xenografts had doubled in tumor volume by day 20 of treatment. RT significantly prolonged the tumor volume doubling time in both groups; however, the control group treated with RT achieved a better response to RT than the ERG+ group, as one half of the animal models with ERG+ xenografts doubled in tumor volume by day 30, whereas only two xenografts from the control group doubled in tumor volume over the entire course of follow-up. The group with the overall best outcome following treatment were the ERG+ xenografts treated with RT and ABT-888, as not a single xenograft in this group doubled in tumor volume by 6 weeks after treatment initiation. These in vivo findings confirm the findings from the in vitro analyses, suggesting that ERG+ status confers radioresistance, and that this radioresistance can be overcome through inhibition of PARP1. As final confirmation, PAR formation was assessed in tumor xenografts, which again showed increased PAR formation in ERG+ cells that was reversed through PARP inhibition (Figure 5B).
Figure 5.

ERG overexpression confers radiation resistance in vivo, which is reversed with PARP inhibition. Two weeks after engraftment, PC3 ERG+ and control xenografts were treated with ABT888 (100 mg/kg twice daily) alone, radiation alone (2 Gy for 5 days), or in combination. The cumulative incidence plot depicts the percentage of tumors in each treatment group that have doubled in volume as a function of time. (A) ERG overexpression confers radiation resistance as shown by a shorter time needed for ERG+ tumors to achieve volumetric doubling than the control tumors. ERG overexpression causes sensitivity to PARP inhibition that is enhanced in combination with radiation treatment. (B) Western blot analysis of ERG+ PC3 cell xenografts treated with or without 100 mg/kg ABT-888 for 4 hours before harvesting. Total PAR, ERG, PARP1, and actin were assessed.
Discussion
In this study, we have demonstrated that ERG overexpression results in radioresistance that is preferentially overcome through PARP inhibition. ERG overexpression resulted in increased PARP1 activity that subsequently led to an increased efficiency of short- and long-term DNA repairs. Inhibition of PARP1 with the PARP inhibitor olaparib prevented enhanced DNA repair, leading to radiosensitization, without altering cell cycle distribution before treatment with RT. These findings were replicated in an in vivo xenograft model. These results highlight that PARP1 inhibition may be a reasonable means of inducing radiosensitization in cancers overexpressing ETS transcription factors, which we demonstrated confer radioresistance (see Figure 6). This has significant implication, as many cancers known to over-express ETS transcription factors are treated with RT, including prostate cancer, where close to 50% of cancers harbor ETS gene fusions. Thus, a significant number of patients with ETS-driven radioresistant tumors may potentially benefit from the preferential radiosensitization promoted through PARP1 inhibition.
Figure 6.

Proposed mechanism whereby ETS gene fusions lead to an increase in ERG transcription factor activity, upregulating PARP1 activity and increasing DNA repair efficiency after IR, leading to effective radioresistance in prostate cancer. Our data demonstrate that PARP inhibition abrogates the radioresistance conferred by ETS gene fusions and may be an effective strategy to overcome this radioresistance.
The presence of ETS gene fusions in prostate cancer has not consistently been demonstrated to be associated with patient outcomes. Two studies assessing outcomes in non-prostate-specific antigen (PSA)-screened watchful waiting cohorts found that patients harboring ETS gene fusions were at higher risk for prostate cancer-specific mortality [32,33]. Many retrospective analyses have also assessed the clinical impact of ETS gene fusion status following radical prostatectomy in PSA-screened populations, with variable results. Several have identified an association between the presence of ETS gene fusions and increased risk for PSA recurrence following radical prostatectomy [34–37], while others have noted no association [4,38–41], or even improved outcomes in the presence of ETS gene fusions [42,43]. These conflicting results make it difficult to draw conclusions concerning the clinical implications associated with positive ETS gene fusion status following radical prostatectomy. More recently, ETS factors have been proposed to cause subtle transcriptional changes to the androgen receptor (AR) cistrome [44,45], which may be dependent on the presence of additional genetic lesions [44], suggesting that the interpretation of the associative value of ERG with clinical outcome may also be confounded by the presence of additional genetic lesions.
The uncertainty surrounding the prognostic ability of ETS gene fusions following radical prostatectomy or watchful waiting does not lessen their potential as predictive biomarkers in the setting of radiation. In fact, it is likely that predictive biomarkers for RT would indeed differ from prognostic biomarkers following radical prostatectomy or watchful waiting, as prognosticators following surgery or observation are potentially prognostic independent of therapy. Despite their potential, limited data exist regarding the ability of ETS gene fusions to predict response to RT. Given that approximately 50% of patients with prostate cancer, as well as patients with other cancers, overexpress ETS transcription factors and are treated with RT, the ability to predict response to RT from these biomarkers is clinically relevant and may assist in guiding therapy and future decision making, particularly in the context of more aggressive prostate cancers. Similar to the inconsistent findings in prostatectomy series, Swanson et al. found that the presence of the TMPRSS2:ERG fusion resulted in radiosensitization in PC3 prostate cancer cells and radioresistance in DU145 prostate cancer cells [46]. The methods used to select cells for clonogenic survival in their analysis may provide a possible explanation for their conflicting findings. Clonogenic survival was performed on single monoclonal cell populations from cell lines [46]. By using a highly selected monoclonal population as opposed to a polyclonal cell population, it is difficult to discern whether the findings regarding radiosensitivity are truly a reflection of ETS status or due to other unknown genetic confounders of the clone. Our findings in polyclonal cell populations from both the PC3 and DU145 prostate cancer cell lines suggest that the presence of ETS gene fusions does confer radioresistance and that this radioresistance can be preferentially overcome with PARP inhibition.
As the prototypical ETS gene fusion in prostate cancer, TMPRSS2: ERG, combines the androgen-responsive transmembrane protease TMPRSS2 with the ETS family member ERG, we intentionally performed our analyses in the AR-negative prostate cancer model cell lines PC3 and DU145 to differentiate the effects of PARP inhibition on ETS+ cell lines from AR-related effects. Androgens and the AR play an integral role in normal prostate tissue differentiation and function, as well as in prostate carcinogenesis and progression [47]. Androgen deprivation therapy (ADT) is commonly employed as treatment for both localized and metastatic prostate cancer. Additionally, ADT is a standard component of treatment for high-risk prostate cancer patients receiving RT. Multiple phase III clinical trials have shown improved survival in this population with the addition of ADT to RT [48,49]. However, the underlying mechanism by which ADT synergistically interacts with RT to improve outcomes is not fully understood. More recently, androgen stimulation has been implicated in the formation of ETS gene fusions in prostate cancer by inducing genomic spatial rearrangements and DNA breaks that promote these fusions [50–52]. ETS gene fusions have also been demonstrated to modulate androgen signaling in prostate cancer [44,45] but in a context-specific manner based on the genetic background. Given the known interactions between the AR and ETS gene fusions in prostate cancer, it is not unreasonable to hypothesize that the mechanism through which ADT administration concurrently with RT improves outcomes for high-risk prostate cancer patients is through an ETS gene fusion-AR interaction. Future research is needed to clearly define the mechanism by which ADT improves RT outcomes and if this is dependent on an ETS gene fusion-AR interaction.
In addition to the interaction between ETS gene fusions and the AR, we have previously identified an interaction between these gene fusions and PARP1 [15], which led to our hypothesis that ETS over-expression results in radioresistance that might be overcome through PARP inhibition. Herein, we confirm our hypothesis, further establishing an ETS-PARP relationship. While others, including us, have previously demonstrated that PARP1 inhibition adds to the effectiveness of cytotoxic therapies in ETS fusion-positive cancers [15,53–55], our study is the first to employ both genetic knockdown and pharmacologic inhibition to prove the specificity of this finding, to demonstrate a mechanism through increased PARP1 activity resulting in increased DNA repair efficiency, and to demonstrate this finding in prostate cancer in vivo. Along with the interaction with ETS gene fusions, PARP has also been shown to interact with the AR, assisting in AR transcriptional activity and overall function [24]. Therefore, PARP inhibition may serve as a potential strategy to simultaneously target ETS overexpression and aberrant AR function in prostate cancer and thereby improve outcomes in patients with prostate cancer treated with RT. Further work is required to elucidate the exact mechanisms through which PARP and ETS gene fusions interact, but clearly, PARP inhibition has the potential to significantly improve radiation results in prostate cancer, as well as other ETS-overexpressing tumors treated with RT.
In summary, our findings suggest that ETS overexpression results in radioresistance and that PARP inhibition serves as a potentially novel strategy to improve patient outcomes following RT for tumors with ETS overexpression. These findings gain further clinical relevance given that ETS gene fusions serve as strong biomarkers. First, they are highly specific, as ETS gene fusions and resultant overexpression does not occur in normal tissue. Additionally, ETS gene fusion status can be determined through tissue biopsy and, in the case of prostate cancer, can be detected through noninvasive urine assays, allowing for easy identification of patients with inherently radioresistant tumors [56]. Finally, while transcription factor rearrangements, such as ETS gene fusions, are pharmacologically difficult to target directly, we show that PARP inhibition is a viable method to reverse the radioresistance generated through ETS overexpression. Clinical trials targeting ERG overexpression with the addition of PARP inhibition to RT are needed, as is further revelation of the mechanisms through which ETS gene fusions and PARP interact. Reversal of radioresistance through PARP inhibition in patients identified to harbor tumors with ETS gene fusions would be a significant step in the direction toward personalized cancer therapy.
Supplementary Material
Acknowledgments
We thank Steven Kronenberg for his assistance in graphic design for the figures.
Footnotes
F.Y.F. is supported by the Prostate Cancer Foundation, the Department of Defense (W81XWH-10-1-0582), the National Institutes of Health Specialized Program of Research Excellence (NIH SPORE) (P50 CA69568), and the Fund for Cancer Research. J.C.B., K.E.K., and A.M.C. are also supported by the Prostate Cancer Foundation. A.M.C. is a co-inventor on a patent issued to the University of Michigan on ETS fusions in prostate cancer. The diagnostic field of use has been licensed to Gen-Probe, Inc, who has sublicensed certain rights to Ventana Medical Systems. J.C.B. and A.M.C. are listed as co-inventors on a patent filed by the University of Michigan covering the use of poly(ADP-ribose) polymerase inhibitors in ETS-positive cancers.
This article refers to supplementary materials, which are designated by Figures W1 to W4 and are available online at www.neoplasia.com.
References
- 1.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 3.Brenner JC, Chinnaiyan AM. Translocations in epithelial cancers. Biochim Biophys Acta. 2009;1796:201–215. doi: 10.1016/j.bbcan.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapointe J, Kim YH, Miller MA, Li C, Kaygusuz G, van de Rijn M, Huntsman DG, Brooks JD, Pollack JR. A variant TMPRSS2 isoform and ERG fusion product in prostate cancer with implications for molecular diagnosis. Mod Pathol. 2007;20:467–473. doi: 10.1038/modpathol.3800759. [DOI] [PubMed] [Google Scholar]
- 5.Mehra R, Tomlins SA, Shen R, Nadeem O, Wang L, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM, et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–544. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- 6.Mosquera JM, Perner S, Demichelis F, Kim R, Hofer MD, Mertz KD, Paris PL, Simko J, Collins C, Bismar TA, et al. Morphological features of TMPRSS2-ERG gene fusion prostate cancer. J Pathol. 2007;212:91–101. doi: 10.1002/path.2154. [DOI] [PubMed] [Google Scholar]
- 7.Soller MJ, Isaksson M, Elfving P, Soller W, Lundgren R, Panagopoulos I. Confirmation of the high frequency of the TMPRSS2/ERG fusion gene in prostate cancer. Genes Chromosomes Cancer. 2006;45:717–719. doi: 10.1002/gcc.20329. [DOI] [PubMed] [Google Scholar]
- 8.Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, Becker L, Carneiro F, MacPherson N, Horsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 9.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. 2012;1826:1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuka G, Kauer M, Kofler R, Haas OA, Panzer-Grumayer R. The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PLoS One. 2011;6:e26348. doi: 10.1371/journal.pone.0026348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino PT, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, Nelson PS, Vasioukhin V. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci USA. 2008;105:2105–2110. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–678. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dantzer F, de La Rubia G, Ménissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 17.Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Ménissier-de Murcia J, de Murcia G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999;81:69–75. doi: 10.1016/s0300-9084(99)80040-6. [DOI] [PubMed] [Google Scholar]
- 18.Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34:1685–1691. doi: 10.1093/nar/gkl108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci USA. 2010;107:2201–2206. doi: 10.1073/pnas.0904783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saberi A, Hochegger H, Szuts D, Lan L, Yasui A, Sale JE, Taniguchi Y, Murakawa Y, Zeng W, Yokomori K, et al. RAD18 and poly(ADP-ribose) polymerase independently suppress the access of nonhomologous end joining to double-strand breaks and facilitate homologous recombination-mediated repair. Mol Cell Biol. 2007;27:2562–2571. doi: 10.1128/MCB.01243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchell J, Smith GC, Curtin NJ. Poly(ADP-ribose) polymerase-1 and DNA-dependent protein kinase have equivalent roles in double strand break repair following ionizing radiation. Int J Radiat Oncol Biol Phys. 2009;75:1520–1527. doi: 10.1016/j.ijrobp.2009.07.1722. [DOI] [PubMed] [Google Scholar]
- 22.Nowsheen S, Bonner JA, Yang ES. The poly(ADP-ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99:331–338. doi: 10.1016/j.radonc.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–1149. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 26.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 27.Feng FY, Blas K, Olson K, Stenmark M, Sandler H, Hamstra DA. Retrospective evaluation reveals that long-term androgen deprivation therapy improves cause-specific and overall survival in the setting of dose-escalated radiation for high-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2013;86:64–71. doi: 10.1016/j.ijrobp.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 28.Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, Bradford CR, Carey TE. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2009;32:417–426. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fertil B, Dertinger H, Courdi A, Malaise EP. Mean inactivation dose: a useful concept for intercomparison of human cell survival curves. Radiat Res. 1984;99:73–84. [PubMed] [Google Scholar]
- 30.Ferro AM, Olivera BM. Poly(ADP-ribosylation) in vitro. Reaction parameters and enzyme mechanism. J Biol Chem. 1982;257:7808–7813. [PubMed] [Google Scholar]
- 31.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair (Amst) 2003;2:955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 32.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, Berney D, Foster CS, Fletcher A, Gerald WL, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–263. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demichelis F, Fall K, Perner S, Andrén O, Schmidt F, Setlur SR, Hoshida Y, Mosquera JM, Pawitan Y, Lee C, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–4599. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 34.Nam RK, Sugar L, Yang W, Srivastava S, Klotz LH, Yang LY, Stanimirovic A, Encioiu E, Neill M, Loblaw DA, et al. Expression of the TMPRSS2: ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br J Cancer. 2007;97:1690–1695. doi: 10.1038/sj.bjc.6604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perner S, Rupp NJ, Braun M, Rubin MA, Moch H, Dietel M, Wernert N, Jung K, Stephan C, Kristiansen G. Loss of SLC45A3 protein (prostein) expression in prostate cancer is associated with SLC45A3-ERG gene rearrangement and an unfavorable clinical course. Int J Cancer. 2013;132:807–812. doi: 10.1002/ijc.27733. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Cai Y, Ren C, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 37.Perner S, Svensson MA, Hossain RR, Day JR, Groskopf J, Slaughter RC, Jarleborn AR, Hofer MD, Kuefer R, Demichelis F, et al. ERG rearrangement metastasis patterns in locally advanced prostate cancer. Urology. 2010;75:762–767. doi: 10.1016/j.urology.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, Fine SW, Eastham JA, Scardino PT, Scher HI, Tickoo SK, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res. 2009;69:1400–1406. doi: 10.1158/0008-5472.CAN-08-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoogland AM, Jenster G, van Weerden WM, Trapman J, van der Kwast T, Roobol MJ, Schroder FH, Wildhagen MF, van Leenders GJ. ERG immunohistochemistry is not predictive for PSA recurrence, local recurrence or overall survival after radical prostatectomy for prostate cancer. Mod Pathol. 2012;25:471–479. doi: 10.1038/modpathol.2011.176. [DOI] [PubMed] [Google Scholar]
- 40.Minner S, Enodien M, Sirma H, Luebke AM, Krohn A, Mayer PS, Simon R, Tennstedt P, Muller J, Scholz L, et al. ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin Cancer Res. 2011;17:5878–5888. doi: 10.1158/1078-0432.CCR-11-1251. [DOI] [PubMed] [Google Scholar]
- 41.Toubaji A, Albadine R, Meeker AK, Isaacs WB, Lotan T, Haffner MC, Chaux A, Epstein JI, Han M, Walsh PC, et al. Increased gene copy number of ERG on chromosome 21 but not TMPRSS2-ERG fusion predicts outcome in prostatic adenocarcinomas. Mod Pathol. 2011;24:1511–1520. doi: 10.1038/modpathol.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Säramaki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, Visakorpi T. TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res. 2008;14:3395–3400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- 43.Petrovics G, Liu A, Shaheduzzaman S, Furusato B, Sun C, Chen Y, Nau M, Ravindranath L, Dobi A, Srikantan V, et al. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene. 2005;24:3847–3852. doi: 10.1038/sj.onc.1208518. [DOI] [PubMed] [Google Scholar]
- 44.Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;19:1023–1029. doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, Gong Y, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swanson TA, Krueger SA, Galoforo S, Thibodeau BJ, Martinez AA, Wilson GD, Marples B. TMPRSS2/ERG fusion gene expression alters chemo- and radio-responsiveness in cell culture models of androgen independent prostate cancer. Prostate. 2011;71:1548–1558. doi: 10.1002/pros.21371. [DOI] [PubMed] [Google Scholar]
- 47.Debes JD, Tindall DJ. The role of androgens and the androgen receptor in prostate cancer. Cancer Lett. 2002;187:1–7. doi: 10.1016/s0304-3835(02)00413-5. [DOI] [PubMed] [Google Scholar]
- 48.Horwitz EM, Bae K, Hanks GE, Porter A, Grignon DJ, Brereton HD, Venkatesan V, Lawton CA, Rosenthal SA, Sandler HM, et al. Ten-year follow-up of radiation therapy oncology group protocol 92-02: a phase III trial of the duration of elective androgen deprivation in locally advanced prostate cancer. J Clin Oncol. 2008;26:2497–2504. doi: 10.1200/JCO.2007.14.9021. [DOI] [PubMed] [Google Scholar]
- 49.Lawton CA, Winter K, Murray K, Machtay M, Mesic JB, Hanks GE, Coughlin CT, Pilepich MV. Updated results of the phase III Radiation Therapy Oncology Group (RTOG) trial 85–31 evaluating the potential benefit of androgen suppression following standard radiation therapy for unfavorable prognosis carcinoma of the prostate. Int J Radiat Oncol Biol Phys. 2001;49:937–946. doi: 10.1016/s0360-3016(00)01516-9. [DOI] [PubMed] [Google Scholar]
- 50.Wu D, Zhang C, Shen Y, Nephew KP, Wang Q. Androgen receptor-driven chromatin looping in prostate cancer. Trends Endocrinol Metab. 2011;22:474–480. doi: 10.1016/j.tem.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bastus NC, Boyd LK, Mao X, Stankiewicz E, Kudahetti SC, Oliver RT, Berney DM, Lu YJ. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70:9544–9548. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, Palanisamy N, Chinnaiyan AM. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chatterjee P, Choudhary GS, Sharma A, Singh K, Heston WD, Ciezki J, Klein EA, Almasan A. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS One. 2013;8:e60408. doi: 10.1371/journal.pone.0060408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee HJ, Yoon C, Schmidt B, Park DJ, Zhang AY, Erkizan HV, Toretsky JA, Kirsch DG, Yoon SS. Combining poly(ADP-ribose) polymerase 1 (PARP-1) inhibition and radiation in Ewings sarcoma results in lethal DNA damage. Mol Cancer Ther. 2013 doi: 10.1158/1535-7163.MCT-13-0338. E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, Liu M, Lonigro R, Prensner JR, Tomlins SA, et al. PARP-1 inhibition as a targeted strategy to treat Ewing's sarcoma. Cancer Res. 2012;72:1608–1613. doi: 10.1158/0008-5472.CAN-11-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomlins SA, Aubin SM, Siddiqui J, Lonigro RJ, Sefton-Miller L, Miick S, Williamsen S, Hodge P, Meinke J, Blase A, et al. Urine TMPRSS2: ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA. Sci Transl Med. 2011;3:94ra72. doi: 10.1126/scitranslmed.3001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.