

Abstract
C-reactive protein (CRP) binds with high affinity to fibronectin (Fn), a major component of the extracellular matrix (ECM), but at physiological pH the binding is inhibited by calcium ions (Ca2+). Because CRP circulates in the blood in Ca2+-bound form, the occurrence of CRP-Fn interactions in vivo has been doubtful. To define the basis of inhibition of CRP-Fn interaction by Ca2+ at pH 7.0, we hypothesized that Fn-binding site on CRP consisted of amino acids co-ordinating Ca2+. Site-directed mutagenesis of amino acids co-ordinating Ca2+ drastically decreased the binding of CRP to Fn, indicating that the Ca2+-binding site indeed formed the Fn-binding site. To determine the requirements for possible interaction between Ca2+-bound CRP and Fn, we investigated inhibition of CRP-Fn interaction by Ca2+ as a function of pH. Ca2+ did not inhibit binding of CRP to Fn at pH 6.5 and lower. The contrasting Fn binding properties of CRP at physiological and mildly acidic pH indicated that the interaction of Ca2+-bound CRP with Fn was controlled by pH. We conclude that the inhibition of binding of CRP to Fn by Ca2+ at pH 7.0 is a mechanism to prevent CRP-Fn interactions under normal conditions. CRP, in its Ca2+-bound state, is capable of binding Fn but only at the inflammatory sites and tumors with low pH. CRP, Fn, and the ECM all have been implicated in cancer. Taken together our data raise the possibility that CRP-Fn interactions may change the architecture of ECM to modify the development of tumors.
C-reactive protein (CRP)1 is an evolutionarily conserved plasma protein that is considered to be a multifunctional component of the acute phase response and innate host defense system (1, 2). CRP has been shown to affect the results of the experiments related to diverse pathologic states (3, 4) such as bacterial infection (5, 6), atherosclerosis (7), autoimmune disease (8), and cancer (9). Most proposed functions of CRP depend on its ability to recognize and bind substances of physiologic value (10, 11). By virtue of its wide binding specificities, CRP gets deposited at sites of tissue damage and inflammation (12, 13). Two types of molecular contacts are known for the binding of CRP to its ligands, one that requires calcium ions (Ca2+) and another that does not require Ca2+ and instead is inhibited by Ca2+ (2). The Ca2+-dependent ligands are primarily the phosphocholine (PCh)-containing substances such as C-polysaccharide (PnC) of the cell wall of Streptococcus pneumoniae (10), modified low density lipoprotein (7, 14), and apoptotic cells (14, 15). Ca2+-independent ligands include polycations (16, 17) and the extracellular matrix (ECM) protein fibronectin (Fn) (18, 19). In this paper we report the mechanism of CRP-Fn interactions and discuss their suitability for in vivo circumstances.
CRP is one of the pentraxin proteins composed of 5 identical 206-amino acid-long subunits (20, 21). All five subunits have the same orientation in the pentamer, with a Ca2+-binding site and a PCh-binding site located on the same face of each subunit (2). Each Ca2+ site occupies two Ca2+. Using synthetic peptides derived from CRP, direct binding of Ca2+ to a peptide of residues 134–148 has been shown (22). Crystallography of CRP·Ca2+ · PCh complex (Fig. 1) has demonstrated that two Ca2+ in CRP are co-ordinated by Asp-60, Asn-61, and by residues (Glu-138, Gln-139, Asp-140, Glu-147, and Gln-150) in a loop (23–25). This loop in the absence of bound Ca2+ moves away from the main body of the CRP molecule, exposing an otherwise hidden site of proteolysis (26). Bound Ca2+ ions protect CRP from proteolytic cleavage and are integral structural elements of CRP. It is believed that CRP circulates in the body in its Ca2+-bound form.
Fig. 1. Ca2+-binding site of CRP.

The ViewerPro 4.2 software (Accelrys Inc.) was used to generate ribbon diagram of the crystal structure of CRP·Ca2+ · PCh complex (24) obtained from Brookhaven Protein Data Bank. A portion of one of the five subunits is shown. The side chains of seven amino acids co-ordinating two Ca2+ are highlighted. Ca2+ ions are in gray, and the PCh molecule is in orange.
Fn is a multifunctional glycoprotein present in the blood in addition to being the major component of the ECM (27). Fn is also highly expressed in several tumors (28–30). Fn plays key roles in cell adhesion and migration processes in various physiological events, such as wound healing, thrombosis, tissue remodeling, and cancer (27, 28). Thus, understanding the nature of CRP-Fn interaction and its impact on each other’s functions is of importance.
Some features of CRP-Fn interaction have been reported a decade ago (10, 18, 19). CRP-Fn interaction does not require Ca2+, and the maximal interaction occurs at pH 5.0. At physiological pH it has been shown that Ca2+ inhibits binding of CRP to Fn (18). We hypothesized (31) that the Fn-binding site consisted of amino acids in CRP that co-ordinated Ca2+. In the present study our main goal was to define the mechanism of CRP-Fn interactions so as to validate the occurrence of CRP-Fn interactions in vivo. Our data indicated that the Ca2+ site of CRP participated in binding to Fn and provided an explanation for the inhibitory role of Ca2+ on CRP-Fn interaction. Furthermore, at pH 7.0 Ca2+ inhibited CRP-Fn interaction, whereas at pH 6.5 and lower it did not. These findings support that in vivo Ca2+-bound CRP has the capability of interacting with Fn but exclusively at the ECM of the inflammatory sites including carcinomas where the pH goes down.
EXPERIMENTAL PROCEDURES
Construction and Expression of CRP Mutants
We constructed CRP cDNA encoding E138A, Q139A, D140A, E147A, E147Q, E147K, Q150A, D60A, N61A, and D60A/E147A mutants. The WT human CRP cDNA in the expression vector p91023 (32) was used as the template for construction of cDNA-encoding proteins with single amino acid substitutions. The D60A mutant CRP cDNA was used as a template to construct cDNA for the D60A/E147A CRP. Site-directed mutagenesis was conducted using the QuikChange mutagenesis kit (Stratagene). Mutagenic oligonucleotides (Fig. 2) were designed according to the kit instructions and obtained from Integrated DNA Technologies. Mutations were verified by sequencing performed in our Core Facility. Two clones for each mutant were purified using the maxiprep plasmid isolation kit (Eppendorf). COS cells were used for protein expression and cultured as described previously (31). Cells (2 × 106) were transfected with 5 μg of the plasmids containing mutant CRP cDNA using FuGENE 6 (Roche Applied Science). At 96 h post-transfection, culture media were collected, and the expression of CRP was determined by employing ELISA.
Fig. 2. Pairs of mutagenic oligonucleotides used in constructing CRP mutants.

The sequences were designed based on the published sequence for WT CRP (33). Triplet codons of the mutated amino acids are boxed. Mutated bases are in bold.
Purification of CRP
The WT native CRP (nCRP) was purified from ascitic fluid, and WT recombinant CRP was purified from the culture medium of the stable Chinese hamster ovary cell line (31) by three steps, a Ca2+-dependent affinity chromatography on a PCh-Sepharose column (Pierce) followed by an anion-exchange chromatography on a Mono Q column and a gel filtration on a Superose12 column (GE Healthcare) using the Biologic Duo Flow Protein Purification System (Bio-Rad). Mutants of CRP were purified from culture media by affinity chromatography alone as before (31). Briefly, culture media were passed through the PCh-Sepharose in a Ca2+-containing buffer, and the bound CRP was eluted with an EDTA-containing buffer. CRP-positive fractions, identified by ELISA, were pooled and dialyzed against TBS (10 mM Tris-HCl, pH 7.2, 150 mM NaCl).
CRP ELISA
The concentration of CRP was estimated using ELISA (31). A polyclonal anti-CRP antibody (1/1000 dilution; Sigma) was used as the capture antibody, and the affinity-purified anti-CRP mAb HD2.4 (0.5 μg/ml) was used as the reporter. The HD2.4 mAb was affinity-purified from the cell culture supernatant of a hybridoma cell line (ATCC). Standard curves were constructed with purified nCRP (3.13–100 ng/ml) diluted in TBS, 0.1% gelatin, 0.01% Tween 20. Horseradish peroxidase-conjugated goat anti-mouse IgG H+L (Pierce) was used as the secondary antibody. Color was developed with 300 μg/ml ABTS chromophore, diammonium salt (Calbiochem), 0.03% H2O2 in 0.5 M citrate buffer, pH 4.0, and measured at 405 nm in the microtiter plate reader (Molecular Devices).
PCh Binding Assay
Binding activity of CRP for PCh-containing ligands was evaluated by two assays using PCh-BSA (Biosearch Technologies) and PnC (Statens Seruminstitut) (31). Briefly, microtiter wells were coated with either PCh-BSA (10 μg/ml) or PnC (2 μg/ml). Culture media were diluted to appropriate concentrations in TBS-Ca (TBS, 0.1% gelatin, 0.01% Tween 20, 5 mM CaCl2). Purified nCRP (3.13–100 ng/ml) was used to construct standard curves. Bound CRP was detected as in ELISA. In some experiments binding curves were constructed by using serial dilutions of purified WT and mutant CRP, covering the range from 1.56 to 200 ng/ml.
Fn Binding Assay
The binding of CRP to Fn was assessed under 2 conditions unless otherwise mentioned, pH 7.0 and 5.0, using previously published methods (18, 31) with some modifications. For the assay at pH 5.0, CRP was dialyzed against TBS, pH 5.0, and tested for Fn binding activity (in addition to ELISA and PCh binding) within 72 h. Microtiter wells were coated with Fn (Sigma, F2006) at a concentration of 2 μg/ml in TBS, pH 7.0, overnight at room temperature. Wells were blocked with TBS containing 0.1% BSA. The binding curves were constructed using serial dilutions of purified WT and mutant CRP, covering the concentration range from 0.156 to 10 μg/ml in either buffer A (TBS, pH 7.0, 0.01% Tween 20) or buffer B (TBS, pH 5.0, 0.01% Tween 20) with and without 5 mM CaCl2. After a 3-h incubation at room temperature, the wells were washed with buffer A. Affinity-purified polyclonal rabbit anti-CRP antibody (1 μg/ml) was used as a reporter to detect bound CRP. The reporter antibody was purified from the anti-CRP antibody (Sigma) by affinity chromatography on a CRP-conjugated agarose column prepared by using the AminoLink Immobilization kit (Pierce). Wells were developed with horseradish peroxidase-conjugated goat anti-rabbit IgG H+L (Pierce) as in ELISA.
Polycation Binding Assay
Binding activity of CRP for polycationic ligands was evaluated by two assays using poly-L-arginine (Sigma, P-3892, Mr 70,000–150,000) and poly-L-lysine (Sigma, P8920, Mr 150,000–300,000, 0.01% solution). Microtiter wells were coated with either poly-L-arginine (10 μg/ml) or poly-L-lysine (0.002%) in TBS overnight at 4 °C. After blocking the wells with TBS containing 0.5% gelatin, purified WT and mutant CRP that were serially diluted (1.56–100 ng/ml) in TBS containing 0.1% gelatin and 0.01% Tween 20 were added to the wells and incubated overnight at 4 °C. Bound CRP was detected as in ELISA.
Gel Filtration
Gel filtration analyses of purified WT and mutant CRP dialyzed against pH 5.0 buffer was carried out on a Superose12 column (GE Healthcare) using the Biologic Duo Flow Protein Purification System (Bio-Rad). Affinity-purified CRP preparations (250 μl, ~10 μg depending upon the concentrations of the stocks) were injected into the column and eluted with TBS at a flow rate of 200 μl/min. Sixty fractions (300 μl each) were collected, and CRP-containing fractions were located by ELISA. Purified nCRP at pH 7.0 was used to determine the elution volume for the pentameric CRP.
Ca2+-Site Proteolytic Cleavage Assay
The Ca2+-binding site-dependent proteolytic cleavage assay of CRP was conducted as described previously (26) with modifications. WT and mutant CRP (4 μg) in TBS (pH 7.0 or 5.0) were incubated with 1 μg of protease (from Streptococcus griseus, Sigma, P6911) with and without 5 mM CaCl2 for 2 h at 37 °C. Digestion mix was then incubated with 20 μl of packed volume of anti-CRP-Sepharose beads for 1 h at 37 °C to separate CRP from the enzyme. The conjugation of affinity-purified polyclonal anti-CRP antibody to Sepharose was performed using the AminoLink Immobilization kit (Pierce). The beads were then washed with TBS, re-suspended in TBS, and subjected to SDS-PAGE on a 4–20% polyacrylamide gradient gel under reducing conditions. The gels were stained with silver stain (Bio-Rad). The Bio-Rad broad-range marker was used as the molecular weight standard.
RESULTS
Construction, Expression, and Purification of Mutant CRP
Ten CRP cDNAs encoding E138A, Q139A, D140A, E147A, E147Q, E147K, Q150A, D60A, N61A, and the double mutant D60A/E147A were constructed. Expression was determined by ELISA performed on the culture supernatants of transfected COS cells. Despite repeated transfections using duplicate clones, under a wide variety of experimental conditions the CRP mutants Q139A, D140A, and N61A were not expressed at all and could not be pursued further. Expression levels of CRP mutants E138A and Q150A were low (range, 5–100 ng/ml), enough to test their PCh binding activity but not enough to test their Fn binding activity. The other five CRP mutants involving Glu-147 and Asp-60 were successfully expressed, and their expression levels (1000–3000 ng/ml) were similar to that of the WT recombinant CRP.
To determine the effect of mutating the Ca2+-site on the binding of CRP to PCh and to design a purification strategy for the expressed proteins, PCh binding assays using PnC and PCh-BSA were performed on culture media containing WT and mutant CRP. Data from the PnC assays are presented (Fig. 3); PCh-BSA gave similar results. Values shown reflect the avidity of individual CRP species for PCh relative to nCRP, whose PCh binding activity was taken as unity. WT and the five CRP mutants that were equally expressed bound to PCh efficiently. Because binding of CRP to PCh requires binding of Ca2+ to CRP first, these data indicated that the mutation of a single amino acid, Glu-147 or Asp-60, did not affect the binding of CRP to Ca2+. The binding of E138A CRP to PCh was reduced by more than half compared with WT CRP. The Q150A CRP failed to bind to PCh. The mutants E138A and Q150A were not analyzed further. CRP mutants with substituted Glu-147 and Asp-60 could be purified by PCh chromatography for further characterization.
Fig. 3. Binding of CRP to PnC.

Microtiter wells were coated with PnC. Culture media containing various CRP species were diluted to ~50 ng/ml in TBS-Ca buffer and added to the wells. Bound CRP was detected using the anti-CRP mAb HD2.4 as reporter. Values on the x axis represent the ratio of the concentration of CRP measured by the PnC binding assay over that measured by ELISA run in parallel using nCRP to generate standard curves in both assays.
Binding of CRP to Fn
As shown in Fig. 4A, nCRP bound to Fn at pH 7.0 in a dose-dependent manner. In the presence of 5 mM CaCl2, the binding was inhibited throughout the CRP dose-response. At pH 7.0, inhibition of the binding of CRP (10 μg/ml) to Fn was dependent on the concentration of Ca2+ (Fig. 4B), with maximum inhibition at ~2 mM CaCl2. Although 2 mM CaCl2 was sufficient to inhibit binding of CRP to Fn, 5 mM CaCl2 was chosen for subsequent experiments to ensure that Ca2+ was in excess. The binding of various purified CRP mutants to Fn at pH 7.0 was evaluated next (Fig. 4C). Mutations of Asp-60 to Ala and of Glu-147 to Ala/Gln resulted in a 70–95% decrease in the binding of CRP to Fn compared with that of WT CRP. The decrease was most pronounced when Glu-147 was substituted with Lys or when both residues were mutated to Ala.
Fig. 4. Binding of CRP to Fn.

Microtiter wells were coated with Fn. A, increasing concentrations of purified nCRP in TBS, pH 7.0, with and without 5 mM CaCl2 were added to the wells. B, nCRP, 10 μg/ml in TBS, pH 7.0, containing increasing concentrations of CaCl2 were added to the wells. Binding of CRP to Fn in the absence of CaCl2 was taken as 100% to calculate percent inhibition. C, increasing concentrations of purified WT and mutant CRP in TBS, pH 7.0, without CaCl2 were added to the wells. D, increasing concentrations of purified WT and mutant CRP in TBS, pH 5.0, without CaCl2 were added to the wells. E, increasing concentrations of purified WT and mutant CRP in TBS, pH 5.0, with and without CaCl2 were added to the wells. F, increasing concentrations of purified nCRP, 10 μg/ml in TBS at increasing pH, with and without Ca2+ were added to the wells. The binding of CRP to Fn in the absence of Ca2+ at various pH was plotted as 100%. In all, bound CRP was detected by using polyclonal anti-CRP antibody as the reporter. All experiments were performed at least three times, and a representative experiment is shown.
Because maximal binding of CRP to Fn occurs at pH 5.0 (18), we tested the binding of CRP mutants to Fn at pH 5.0 (Fig. 4D). Again, the binding of CRP mutants E147K and D60A/E147A to Fn was reduced by 80–90 and 50–60% respectively, and this decrease was observed throughout the dose-response. Unlike at pH 7.0, D60A, E147A, and E147Q CRP mutants efficiently bound to Fn at pH 5.0. We, therefore, determined the effects of Ca2+ on the binding of these CRP mutants to Fn at pH 5.0 (Fig. 4E). For clarity, the results are shown only for the WT CRP and two mutants, E147A and D60A. To our surprise Ca2+ did not inhibit the binding of mutant CRP to Fn at pH 5.0 and did not even inhibit the binding of nCRP to Fn at pH 5.0. Binding of other mutants to Fn at pH 5.0 was also not inhibited by Ca2+ (data not shown). These results clearly indicate that the inhibition of CRP-Fn interaction by Ca2+ occurs only at pH 7.0 and not at pH 5.0. Next, we determined the inhibition of the binding of CRP to Fn by Ca2+ at pH values varying from 4.0 to 7.0 (Fig. 4F). The binding of CRP to Fn was inhibited about 70% at pH 7.0, as before. Unexpectedly, the binding was inhibited by only 10% at pH 6.5, and no inhibition was observed at pH 6.0 and lower.
Assessment of the Overall Structure of CRP
We investigated the possibility that dialysis of CRP against TBS, pH 5.0, might have perturbed the overall structure of CRP. Although the ELISA performed on CRP at pH 5.0 indicated that CRP retained its reactivity toward the mAb, 3 more assays were utilized; PnC binding assay (Fig. 5A), gel filtration (Fig. 5B), and Ca2+-site-dependent proteolytic cleavage assay (Fig. 5C). WT and all mutant CRP bound to PnC in a dose-dependent manner and produced essentially overlapping curves, indicating that their PCh binding activities were retained at pH 5.0. Gel filtration to assess pentameric nature of the dialyzed mutant CRP showed that the elution volume of all CRP mutants at pH 5.0 was similar to that of nCRP at pH 7.0. Data are shown only for 3 mutants at pH 5.0; elution profiles of nCRP and other mutants at pH 5.0 were identical. Last, at both pH 7.0 and 5.0, in the absence of Ca2+, nCRP was cleaved by the protease, generating 3 fragments as reported previously (26), and at both pH values, Ca2+ protected CRP from proteolytic cleavage. Combined results indicated that the overall structure of CRP was not altered at pH 5.0.
Fig. 5. Effect of pH 5.0 on the overall structure of CRP.
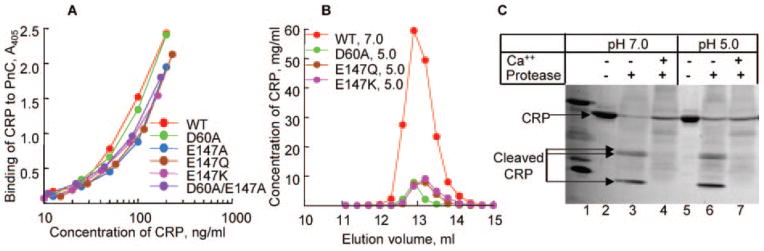
A, binding of purified WT and mutant CRP, dialyzed against TBS, pH 5.0, to PnC. Microtiter wells were coated with PnC. Increasing concentrations of various CRP species in a TBS-Ca were added to the wells. Bound CRP was detected by HD2.4 mAb. A representative of two experiments is shown. B, elution profiles of purified nCRP at pH 7.0 and purified mutant CRP dialyzed against TBS, pH 5.0, from the Superose12 gel filtration column. C, Ca2+ site-dependent proteolytic cleavage of CRP. Purified nCRP at pH 7.0 and 5.0 was subjected to proteolytic cleavage in the absence and presence of Ca2+. A representative of 2–3 experiments is shown.
Binding of CRP to Polycations
To evaluate whether the Ca2+ site binds only Fn or it binds, in general, to all Ca2+-independent ligands of CRP, an assay was set up to determine binding of CRP to polycations. We expected that the Fn site and the polycation site on CRP would be the same since Ca2+ was also found to inhibit CRP-polycation interaction (34). Substitution of Glu-147 with Lys and the double mutation of Asp-60/Glu-147 to Ala abolished the binding of CRP to poly-L-arginine (Fig. 6A). Although D60A CRP bound polycations and the binding of E147A and E147Q to the two polycations was different (Fig. 6B), the results indicated that the Ca2+ site did participate in the binding of CRP to its Ca2+-independent ligands including Fn and polycations.
Fig. 6. Binding of CRP to polycations.
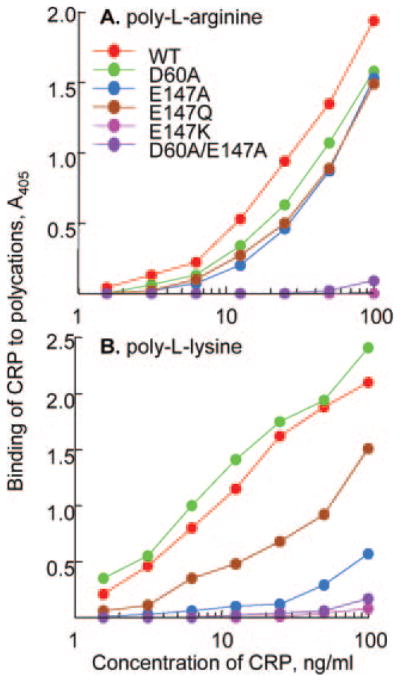
Microtiter wells were coated with either poly-L-arginine (A) or poly-L-lysine (B). Increasing concentrations of purified WT and mutant CRP were added to the wells. Bound CRP was detected by HD2.4 mAb. A representative of two experiments is shown.
Effect of Ca2+ Site Mutations on the Proteolytic Cleavage of CRP
As a result of mutating Asp-60 and Glu-147, we discovered a loss of Fn binding activity and polycation binding activity but not of PCh binding (and, thus, not of Ca2+ binding) activity. This indicated that the structure of the Ca2+ site of Ca2+-bound nCRP was different from that of Ca2+-bound mutant CRP. To support this conclusion, we examined the topology of Ca2+ site by protease cleavage assay (Fig. 7). Protease treatment of nCRP in the absence of Ca2+ generated three fragments (lane 3). The fragments designated A1 and A2 were found to be of 17 and 16 kDa, and fragment B was estimated to be of 6 kDa, consistent with the previous reports (26). nCRP was completely protected from digestion in the presence of Ca2+ (lane 4). The E147A CRP was cleaved in the absence of Ca2+, generating only two fragments (lane 6), and in the presence of Ca2+, cleavage was not protected; instead all three fragments were produced (lane 7). The D60A CRP was not susceptible to protease at all (lanes 9 and 10). The effect of mutating Asp-60 was not apparent in the cleavage of the double mutant (lanes 12 and 13). These data confirmed that the configuration of the Ca2+ site of Ca2+-bound mutants was different from that of Ca2+-bound nCRP.
Fig. 7. Ca2+ site-dependent proteolytic cleavage of CRP.
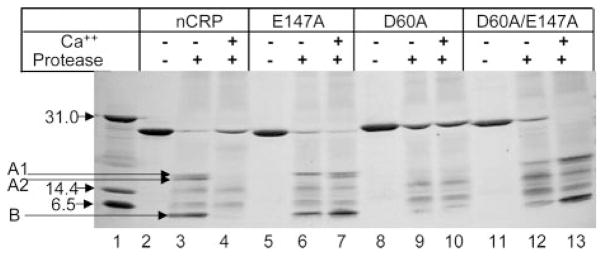
Reducing SDS-PAGE of WT and mutant CRP treated with protease in the absence and presence of Ca2+ was run on a 4–20% polyacrylamide gradient gel. Bands were visualized by silver staining. Before electrophoresis, all CRP samples were incubated with anti-CRP-conjugated agarose beads to remove the protease from CRP. Molecular weight markers are shown in lane 1.
DISCUSSION
CRP circulates in the body in its Ca2+-bound form, and Ca2+ inhibits binding of CRP to Fn. Therefore, the occurrence of CRP-Fn interactions in vivo has been uncertain. We investigated the Fn-binding site on CRP and the mechanism of binding of CRP to Fn to determine the conditions for possible interaction between Ca2+-bound CRP and Fn. Our major findings were as follows. 1) Ca2+ inhibited binding of CRP to Fn at pH 7.0 but not at pH 6.5 and lower. No inhibition was seen at pH 5.0, the pH demonstrated for the maximal binding of CRP to Fn. 2) The Ca2+ site of CRP participated in binding to Fn, and the same site was found to be the polycation-binding site. 3) Mutations of Asp-60 or Glu-147 at the Ca2+ site of CRP abolished Fn binding at pH 7.0 but not at pH 5.0. 4) Mutations of Asp-60 or Glu-147 did not abolish PCh binding and Ca2+ binding. 5) Substitution of Asp-60 with Ala conferred resistance to CRP against proteolytic cleavage. 6) CRP, at a pH as low as 5.0, retained its pentameric form, ability to bind Ca2+ and PCh, reactivity with a mAb, and susceptibility toward the protease.
Based on the expression levels of CRP mutants, 7 amino acids forming the Ca2+ site can be divided into two groups; group 1 with Asp-60 and Glu-147 and group 2 with Asn-61, Glu-138, Gln-139, Asp-140, and Gln-150. CRP mutants of group 1 residues were expressed and bound PCh, reflecting their binding to Ca2+ also. CRP mutants of group 2 residues were either not expressed or poorly expressed with the loss of Ca2+ binding and PCh binding ability. These results indicated that the group 2 residues were critical for binding to Ca2+ and for maintaining the native structure of CRP. Expression pattern of various CRP mutants favor the notion that binding of Ca2+ to CRP occurs during its biosynthesis and is a prerequisite for the correct folding and assembly of the protein, and perhaps this may be a general phenomenon for other Ca2+-binding proteins.
The inability of group 2 CRP mutants to be expressed was not surprising in view of the crystallographic data on Ca2+-bound CRP (23–25). Each CRP subunit binds 2 Ca2+, Ca2+-1 and Ca2+-2. Ca2+-1 is co-ordinated by Asp-60, Asn-61, Glu-138, Gln-139, and Asp-140. Asp-60 provides only one oxygen ligand to Ca2+-1. Ca2+-2 is co-ordinated by Glu-138, Asp-140, Glu-147, and Gln-150. In the crystal packing, CRP was still bound to one Ca2+ when Glu-147 was not co-ordinated to Ca2+-2 but was co-ordinated to Phe-66 from the neighboring pentamer. Thus, Asn-61, Glu-138, Gln-139, Asp-140, and Gln-150 are involved more than Asp-60 and Glu-147 in co-ordinating Ca2+.
Analyses of group 1 CRP mutants, D60A and E147A, were sufficient to provide data to support the hypothesis that the Ca2+ site of CRP participated in binding not only to Fn but also to polycations (31, 35). The loss of Fn binding activity of these CRP mutants at pH 7.0 suggested that the amino acids in CRP that coordinated Ca2+ were also required for binding to Fn. On the other hand, it was unlikely that these 2 amino acids were the contact residues because their mutations did not abolish binding of CRP to Fn at pH 5.0. Loss of binding of CRP to Fn at pH 7.0 due to these mutations could be due to a change in the disposition of the Ca2+ binding loop in the mutant CRP. This conclusion is supported by the data showing altered susceptibility of these mutants to proteolytic cleavage.
The definition of the Fn-binding site of CRP can be derived from the observed effects of acidic pH on CRP-Fn interaction. Although we did not find a difference in the Ca2+ site of CRP at pH 7.0 versus 5.0 using PCh binding and proteolytic cleavage assays, a pH 6.3-dependent conformational change in CRP has been shown previously by circular dichroism and fluorescence spectra analyses (36). Assuming that the pH lower than 6.0 results in the loss of binding of one Ca2+ per CRP subunit, creating half of the Ca2+ site vacant, and that Fn utilizes that portion of the Ca2+ site to bind, then CRP can bind to PCh and to Fn despite mutations of Asp-60 and Glu-147. It is not known whether two Ca2+ per CRP subunit is an absolute necessity for CRP-PCh interaction. The finding that the CRP-Fn interaction is inhibited in a dose-dependent manner by relatively higher concentrations of Ca2+ at pH 7.0 also supports that the vacancy of entire Ca2+ site is not required for binding to Fn. The Fn-binding site may also include the hydrophobic pocket located adjacent to the PCh-binding site of CRP (23–25). We hypothesize that the Fn-binding site of CRP is composed of the Ca2+ site occupied with only one Ca2+, and formation of such a site is the consequence of low pH-induced conformational change in and around the Ca2+ site. A similar proposal involving a conformational change has been proposed for the binding of Fn to vascular endothelial growth factor whose binding to Fn also increases at acidic pH (37).
We propose that the inhibition of binding of CRP to Fn by Ca2+ at physiological pH serves as a mechanism to prevent CRP-Fn interaction in the ECM of normal tissues. The Ca2+-bound CRP gains the capacity to interact with Fn, exclusively at the ECM of the inflammatory sites. One characteristic feature of the inflammatory sites, also of the tumors, is local acidosis, resulting in a pH that can be as low as 5.8 (36, 38). The architecture of the ECM in tumors is different from normal tissues. Possible CRP-Fn interaction at the ECM of tumor sites is relevant because both Fn (28) and CRP (39–41) are present at tumor sites. Recently, a polymeric form of Fn has been shown to have antitumor properties (42). The ability of CRP to convert Fn into polymeric Fn is not known, but CRP may modify the ECM around the tumors to look it more like ECM of normal tissues. CRP, monomeric CRP and the CRP-derived peptides, all have been shown to inhibit tumor metastases in experimental models of cancer (9, 43, 44). Involvement of Fn in these in vivo antitumor actions of CRP is yet to be discovered, but while investigating the regulation of CRP-Fn interaction by pH, we made an unexpected observation that the denatured CRP bound Fn more avidly than pentameric CRP did.2 These findings strongly raise the possibility that CRP-Fn interaction might play a role in modulating the progression of cancer.
All studies on CRP-Fn interaction so far have utilized plasma Fn representing ECM Fn. However, under inflammatory conditions there are many alternatively spliced forms of Fn expressed at various other sites in vivo (45). For example, in atherosclerotic lesions, alternatively spliced Fn have been found (46) in addition to the deposition of CRP at such lesions (3, 4). Alternatively spliced forms of Fn are also expressed on activated T lymphocytes (47), and interestingly, CRP binds to activated T lymphocytes (48). We do not know if the mechanisms we report here for the interaction between CRP and plasma Fn would be the same for the interaction between CRP and alternatively spliced forms of Fn. It is a possibility that Ca2+-bound CRP might interact with alternatively spliced forms of Fn even at physiological pH. Existence of low pH at tumor ECM and expression of Fn at sites other than ECM such as atherosclerotic lesions and the T lymphocytes provide opportunity to investigate the functions of CRP-Fn complexes.
In summary, the interaction of circulating Ca2+-bound CRP with Fn is controlled by pH. In normal physiological conditions, binding of Ca2+ to CRP prevents interaction with Fn at the ECM. At the ECM of the inflammatory sites with low pH, such as in tumors, Ca2+-bound CRP gains the capability to interact with Fn. Because CRP, Fn, and the ECM all have been implicated in cancer, we propose that CRP-Fn interactions may change the architecture of ECM to modify the course of the disease progression.
Footnotes
This work was supported by National Institutes of Health Grant HL071233.
The abbreviations used are: CRP, C-reactive protein; nCRP, native CRP; Fn, fibronectin; PCh, phosphocholine; PnC, pneumococcal C-polysaccharide; ECM, extracellular matrix; WT, wild type; ELISA, enzyme-linked immunosorbent assay; TBS, Tris-buffered saline; mAb, monoclonal antibody; BSA, bovine serum albumin.
M. V. Suresh and A. Agrawal, unpublished data.
References
- 1.Kushner I. Ann N Y Acad Sci. 1982;382:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x. [DOI] [PubMed] [Google Scholar]
- 2.Volanakis JE. Mol Immunol. 2001;38:189–197. doi: 10.1016/s0161-5890(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 3.Hirschfield GM, Pepys MB. QJM. 2003;96:793–807. doi: 10.1093/qjmed/hcg134. [DOI] [PubMed] [Google Scholar]
- 4.Jialal I, Devaraj S, Venugopal SK. Hypertension. 2004;44:6–11. doi: 10.1161/01.HYP.0000130484.20501.df. [DOI] [PubMed] [Google Scholar]
- 5.Mold C, Rodic-Polic B, Du Clos TW. J Immunol. 2002;168:6375–6381. doi: 10.4049/jimmunol.168.12.6375. [DOI] [PubMed] [Google Scholar]
- 6.Yother J, Volanakis JE, Briles DE. J Immunol. 1982;128:2374–2376. [PubMed] [Google Scholar]
- 7.Bhakdi S, Torzewski M, Paprotka K, Schmitt S, Barsoom H, Suriyaphol P, Han SR, Lackner KJ, Husmann M. Circulation. 2004;109:1870–1876. doi: 10.1161/01.CIR.0000124228.08972.26. [DOI] [PubMed] [Google Scholar]
- 8.Szalai AJ, Weaver CT, McCrory MA, van Ginkel FW, Reiman RM, Kearney JF, Marion TN, Volanakis JE. Arthritis Rheum. 2003;48:1602–1611. doi: 10.1002/art.11026. [DOI] [PubMed] [Google Scholar]
- 9.Deodhar SD, James K, Chiang T, Edinger M, Barna BP. Cancer Res. 1982;42:5084–5088. [PubMed] [Google Scholar]
- 10.Agrawal A, Xu Y, Ansardi D, Macon KJ, Volanakis JE. J Biol Chem. 1992;267:25352–25358. [PMC free article] [PubMed] [Google Scholar]
- 11.Hack CE, Wolbink GJ, Schalkwijk C, Speijer H, Hermens WT, van den Bosch H. Immunol Today. 1997;18:111–115. doi: 10.1016/s0167-5699(97)01002-5. [DOI] [PubMed] [Google Scholar]
- 12.Du Clos TW, Mold C, Paterson PY, Alroy J, Gewurz H. Clin Exp Immunol. 1981;43:565–573. [PMC free article] [PubMed] [Google Scholar]
- 13.Kushner I, Kaplan MH. J Exp Med. 1961;114:961–973. doi: 10.1084/jem.114.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang MK, Binder CJ, Torzewski M, Witztum JL. Proc Natl Acad Sci U S A. 2002;99:13043–13048. doi: 10.1073/pnas.192399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gershov D, Kim S, Brot N, Elkon KB. J Exp Med. 2000;192:1353–1363. doi: 10.1084/jem.192.9.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Camelli R, Potempa LA, Siegel J, Suyehira L, Petras K, Gewurz H. J Immunol. 1980;125:1933–1938. [PubMed] [Google Scholar]
- 17.Lee RT, Takgahara I, Lee YC. J Biol Chem. 2002;277:225–232. doi: 10.1074/jbc.M106039200. [DOI] [PubMed] [Google Scholar]
- 18.Salonen EM, Vartio T, Hedman K, Vaheri A. J Biol Chem. 1984;259:1496–1501. [PubMed] [Google Scholar]
- 19.Tseng J, Mortensen RF. Mol Immunol. 1988;25:679–686. doi: 10.1016/0161-5890(88)90103-4. [DOI] [PubMed] [Google Scholar]
- 20.Osmand AP, Friedenson B, Gewurz H, Painter HP, Hofmann T, Shelton E. Proc Natl Acad Sci U S A. 1977;74:739–743. doi: 10.1073/pnas.74.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gotschlich EC, Edelman GM. Proc Natl Acad Sci U S A. 1965;54:558–566. doi: 10.1073/pnas.54.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullenix MC, Mortensen RF. Mol Immunol. 1994;8:615–622. doi: 10.1016/0161-5890(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 23.Shrive AK, Cheetham GMT, Holden D, Myles DAA, Turnell WG, Volanakis JE, Pepys MB, Bloomer AC, Greenhough TJ. Nat Struct Biol. 1996;3:346–354. doi: 10.1038/nsb0496-346. [DOI] [PubMed] [Google Scholar]
- 24.Thompson D, Pepys MB, Wood SP. Structure Fold Des. 1999;7:169–177. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 25.Ramadan MAM, Shrive AK, Holden D, Myles DAA, Volanakis JE, DeLucas LJ, Greenhough TJ. Acta Crystallogr Sect D. 2002;58:992–1001. doi: 10.1107/s0907444902005693. [DOI] [PubMed] [Google Scholar]
- 26.Kinoshita CM, Ying SC, Hugli TE, Siegel JN, Potempa LA, Jiang H, Houghten RA, Gewurz H. Biochemistry. 1989;28:9840–9848. doi: 10.1021/bi00451a044. [DOI] [PubMed] [Google Scholar]
- 27.Magnusson MK, Mosher DF. Arterioscler Thromb Vasc Biol. 1998;18:1363–1370. doi: 10.1161/01.atv.18.9.1363. [DOI] [PubMed] [Google Scholar]
- 28.Ruoslahti E. Adv Cancer Res. 1999;76:1–20. doi: 10.1016/s0065-230x(08)60772-1. [DOI] [PubMed] [Google Scholar]
- 29.David L, Nesland JM, Holm R, Sobrinho-Simoes M. Cancer. 1994;73:518–527. doi: 10.1002/1097-0142(19940201)73:3<518::aid-cncr2820730305>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 30.Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis DL, Agnantis NJ, Pavlidis N. Eur J Cancer. 2002;38:2362–2370. doi: 10.1016/s0959-8049(02)00210-1. [DOI] [PubMed] [Google Scholar]
- 31.Agrawal A, Simpson MJ, Black S, Carey MP, Samols D. J Immunol. 2002;169:3217–3222. doi: 10.4049/jimmunol.169.6.3217. [DOI] [PubMed] [Google Scholar]
- 32.Agrawal A, Lee S, Carson M, Narayana SVL, Greenhough TJ, Volanakis JE. J Immunol. 1997;158:345–350. [PubMed] [Google Scholar]
- 33.Woo P, Korenberg JR, Whitehead AS. J Biol Chem. 1985;260:13384–13388. [PubMed] [Google Scholar]
- 34.Potempa LA, Siegel JN, Gewurz H. J Immunol. 1981;127:1509–1514. [PubMed] [Google Scholar]
- 35.Black S, Agrawal A, Samols D. Mol Immunol. 2003;39:1045–1054. doi: 10.1016/s0161-5890(03)00031-2. [DOI] [PubMed] [Google Scholar]
- 36.Miyazawa K, Inoue K. J Immunol. 1990;145:650–654. [PubMed] [Google Scholar]
- 37.Goerges AL, Nugent MA. J Biol Chem. 2004;279:2307–2315. doi: 10.1074/jbc.M308482200. [DOI] [PubMed] [Google Scholar]
- 38.Wike-Hooley JL, Haveman J, Reinhold HS. Radiother Oncol. 1984;2:343–366. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 39.Berman MA, Burnham JA, Sheahan DG. Hum Pathol. 1988;19:784–794. doi: 10.1016/s0046-8177(88)80261-2. [DOI] [PubMed] [Google Scholar]
- 40.Hurlimann J, Gardiol D. Am J Surg Pathol. 1991;15:280–288. doi: 10.1097/00000478-199103000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Nozoe T, Korenaga D, Futatsugi M, Saeki H, Maehara Y, Sugimachi K. Cancer Lett. 2003;192:89–95. doi: 10.1016/s0304-3835(02)00630-4. [DOI] [PubMed] [Google Scholar]
- 42.Yi M, Ruoslahti E. Proc Natl Acad Sci U S A. 2001;98:620–624. doi: 10.1073/pnas.98.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kresl JJ, Potempa LA, Anderson B, Radosevich JA. Tumour Biol. 1999;20:72–87. doi: 10.1159/000030050. [DOI] [PubMed] [Google Scholar]
- 44.Barna BP, Eppstein DA, Thomassen MJ, Nestor JJ, Jr, Ho T, Medendorp SV, Deodhar SD. Cancer Immunol Immunother. 1993;36:171–176. doi: 10.1007/BF01741088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pankov R, Yamada KM. J Cell Sci. 2002;115:3881–3883. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 46.Tan MH, Sun Z, Opitz SL, Schmidt TE, Peters JH, George EL. Blood. 2004;104:11–18. doi: 10.1182/blood-2003-09-3363. [DOI] [PubMed] [Google Scholar]
- 47.Wagner C, Pioch M, Meyer C, Iking-Konert C, Andrassy K, Hänsch GM. J Mol Med. 2000;78:337–345. doi: 10.1007/s001090000107. [DOI] [PubMed] [Google Scholar]
- 48.Mortensen RF, Osmand AP, Gewurz H. J Exp Med. 1975;141:821–839. [PMC free article] [PubMed] [Google Scholar]