

Abstract
Atherosclerotic cardiovascular diseases (CVD) are still the leading cause of morbidity and mortality worldwide, although optimal medical therapy has been prescribed for primary and secondary preventions. Residual cardiovascular risk for some population groups is still considerably high although target low density lipoprotein-cholesterol (LDL-C) level has been achieved. During the past few decades, compelling pieces of evidence from clinical trials and meta-analyses consistently illustrate that lipoprotein(a) (Lp(a)) is a significant risk factor for atherosclerosis and CVD due to its proatherogenic and prothrombotic features. However, the lack of effective medication for Lp(a) reduction significantly hampers randomized, prospective, and controlled trials conducting. Based on previous findings, for patients with LDL-C in normal range, Lp(a) may be a useful marker for identifying and evaluating the residual cardiovascular risk, and aggressively lowering LDL-C level than current guidelines' recommendation may be reasonable for patients with particularly high Lp(a) level.
1. Introduction
Atherosclerotic cardiovascular diseases (CVD), in terms of coronary heart disease (CHD), ischemic stroke, and peripheral artery disease, are still the leading cause of morbidity and mortality worldwide, although optimal medical therapy has been prescribed for primary and secondary preventions. According to previous epidemiological studies and meta-analyses [1–7], plasma level of low density lipoprotein-cholesterol (LDL-C) is causally associated with atherosclerosis and CVD, and plasma LDL-C level diminished by hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitor (statins), which leads to incident CVD reduction and further supports the notion that LDL-C plays an important role on atherosclerosis initiation and progression. In line with the outcomes of published clinical trials, American Heart Association/American College of Cardiology (AHA/ACC) and European Society of Cardiology (ESC) guidelines have recommended a target plasma LDL-C level, based on cardiovascular risk stratification, for each individual. However, some researchers observed that the residual cardiovascular risk for some population groups was still considerably high although target LDL-C level has been achieved [8–11] and other risk factors, including lipoprotein(a) (Lp(a)), have been identified to be responsible for these phenomena.
During the past few decades, compelling pieces of evidence from clinical trials and meta-analyses consistently illustrate that Lp(a) is a significant risk factor for atherosclerosis and its manifestations, CHD and ischemic stroke [12–15]. Accordingly, Lp(a) is composed of two key components, named LDL particle and apolipoprotein (Apo(a)), and the assembly of Lp(a) may be both the intracellular and extracellular [16, 17]. Contrary to LDL-C, Lp(a) catabolism is less dependent on LDL-C receptor, and statins have a modest effect on Lp(a) regulation [16]. Substantial studies consistently show that plasma Lp(a) level is predominantly dependent on the isoform size variability of Apo(a) in terms of small isoform size of Apo(a) which results in higher Lp(a) level and vice versa [18–21]. And previous epidemiological studies also firmly demonstrate that, with Lp(a) elevation, the risk for cardiovascular risk is significantly increased [22–24]. For example, Kardys et al. observed that Lp(a) was associated with a higher 1-year risk of cardiovascular events, with an adjusted hazard ratio (HR) of 3.1 (95% confidence interval (CI): 1.1–8.6) for the highest versus the lowest tertile [22]. In another study conducted by Momiyama and colleagues [23], they found out that Lp(a) levels were associated with aortic atherosclerosis as well as coronary atherosclerosis. Therefore, recently, some specialists of lipidology recommend that for patients with established CHD with a history of recurrent events despite appropriate therapy, Lp(a) should be taken into account for future risk stratification [25]. The European specialists of lipidology also recommend screening of elevated Lp(a) in patients with intermediate or high CVD risks [26]. Moreover, some studies also indicate that the Apo(a) isoform may also play an important and independent role on cardiovascular outcomes [21, 27].
Taken together, combined with the state-of-the-art clinical trials published previously and the advancements with regard to the understanding of Lp(a) biological effects on cardiovascular system, our current literature is going to systemically review the following aspects of Lp(a): structural and metabolic characteristics, pathophysiological roles, polymorphisms of Apo(a) gene and related clinical epidemiological studies, and potential approaches in modulating Lp(a), so as to present insights, if any, to future clinical and basic researches.
2. Structural Characteristics of Lp(a)
Lp(a) is a recent evolutionary arrival and was firstly discovered by Berg more than 50 years ago [28]. Lp(a) is only expressed in human, old world nonhuman primates (such as rhesus and baboon), and European hedgehog but not in the commonly used animal models such as rat, mice, and rabbits [16]. After being identified, a series of studies were conducted to further elucidate the characteristics of Lp(a). It has been well recognized that Lp(a) is composed of two main components, named low density lipoprotein (LDL) and Apo(a). These two moieties are covalently tethered by a disulfide bond, which is established between Apo(a) and apolipoprotein B-100 (ApoB-100) motif of lipid core (see Figure 1) [16, 29]. Similar to LDL-C, each Lp(a) is equipped with one molecule of ApoB-100, which is predominantly responsible for the proatherosclerotic feature of Lp(a). Another important motif, Apo(a), is a glycosylated-rich moiety, and the gene sequence of Apo(a) is highly homologous with that of plasminogen (see Figure 1) [30, 31]. Apolipoprotein(a) gene (LPA) locates on chromosome 6q23 and is flanked by plasminogen gene [32]. Accordingly, Apo(a) cDNA contains an inactive protease domain, 1 Kringle V, and 10 types of Kringle IV [33, 34], while the cDNA of plasminogen contains Kringles I to V as well as an active form of protease domain, which is critical for endogenous fibrinolysis [35]. Combined with Kringle V, the inactive protease domain of Apo(a) is structurally similar to the corresponding part of plasminogen (Kringle V plus active protease) while cannot be activated by tissue plasminogen activator and urokinase plasminogen activator [36]. Therefore, when thrombotic event occurs, Lp(a) could interfere with plasminogen activation through a competitive mechanism, which finally leads to thrombosis formation, artery occlusion, and tissue perfusion impairment. Kringle IV is the most mysterious and important domain of Apo(a). Kringle IV comprises 10 domains, named types 1 to 10. KIV-1 and KIV-(3-10) are single copy domains, while KIV-2 is a multiple copy domain (3 to 50) which leads to Apo(a) molecular weight quite variable (approximately ranging from 300 kDa to 800 kDa) [37]. Kringles of Apo(a) are enriched with cysteines and arrayed tandemly like a loop by three disulfide bonds. In brief, each Kringle has its unique binding site for specific substrate wherein special function is related to. For example, the only unpaired cysteine of KIV-9 is special for binding with cysteine of ApoB-100 through a disulfide bond [38]. Furthermore, KIV-9 is found to be related to smooth muscle cells proliferation and migration in an in vitro study, and KIV-(6-7) mediates the process of foam cells formation. KIV-(7-8) is significant for Lp(a) formation, which is believed to be due to the weak lysine binding sites (LBS) among these Kringles [39]. In conjunction with Kringle V, KIV-(9-10) is potential to inhibit angiogenesis. By virtue of unique LBS within Kringles, the capacity of Lp(a) attaching to and retaining in vessel walls is significantly enhanced. Finally, KIV-2, the most critical motif of Apo(a), is not only associated with isoform size heterogeneity of Apo(a) but also determines the synthetic rate, plasma level, and biological function of Lp(a) [34, 40].
Figure 1.
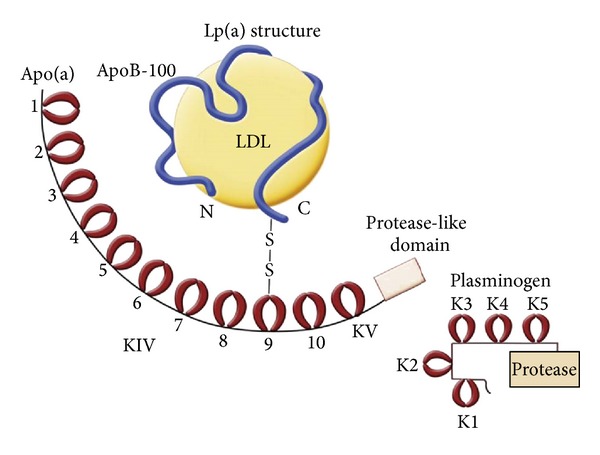
Delineation of structural characteristics of Lp(a) and similarity between Lp(a) and plasminogen. (Adapted from [16].)
3. Metabolism of Lp(a)
Accordingly, Apo(a) is synthesized in the liver, and the assembly of Apo(a) and LDL may be either intracellular and extracellular as supported by published basic research papers [41, 42]. Basically, the process of Lp(a) assembly primarily includes two steps [43]. In the first place, establishing two noncovalent couples between KIV-7 of Apo(a) and Lysine680/690 of ApoB-100 and KIV-8 of Apo(a) and Lysine4372 of ApoB-100 and then followed by the formation of a specific disulfide bond between Apo(a) and ApoB-100 [44]. The plasma Lp(a) level is modestly regulated by life style modification and medication therapy, whereas it is significantly dependent on the isoform size of Apo(a), which is determined by the copy number of KIV-2 [16]. An inverse relationship was also identified between the isoform size of Apo(a) and plasma Lp(a) level, and the underlying mechanism is at least partially because (1) the biosynthetic time for small isoform size of Apo(a) is significantly less than that for the large one [45] and (2) large isoform size of Apo(a) is more prone to be degraded than small one [16]. Other factors in regulating LPA gene expression are also considered accountable for the Apo(a) biogenesis [46, 47]. For example, in the transcriptional level, other sequences related to Apo(a) encoding gene may enhance or diminish Apo(a) cDNA transcription as well as mRNA stability [19, 48, 49]. Preceding Apo(a) release from liver, posttranslational modification of Apo(a), in terms of three disulfide bonds formation and N-linked glycan addition in endoplasmic reticulum, is also significantly associated with the rate of Lp(a) assembly [50]. Boffelli reported that estrogen was negatively associated with plasma Lp(a) level in women, which was believed to be due to the negative effect of estrogen on Apo(a) promoter activity [46]. Moreover, many enhancer regions between LPA and plasminogen genes were also identified to be responsible for Apo(a) expression [51, 52]. For example, a regulatory region within LPA and plasminogen genes (accurately 20 Kb upstream of LPA gene) significantly increases the promoter activity of LPA gene thereby enhancing Apo(a) expression [52].
Nevertheless, compared to Lp(a) synthesis, biological catabolism plays a relative modest role on plasma level of Lp(a) [16]. The underlying mechanism through which Lp(a) is removed from circulating system is still not fully understood yet, neither is the site. Lp(a) can be either cleared as wholeness from circulating system or cleaved into LDL and Apo(a) for further degradation [16]. Currently, at least three general pathways have been identified responsible for Lp(a) and its components catabolism [16, 50]. In the first place, Lp(a) can be recognized by LDL receptor and undergo degradation similar to LDL-C. The significance of this process may be less important for Lp(a) than that for LDL-C [53, 54], because it was reported that only 10–25% of Lp(a) clearance from blood takes place via LDL-C receptor [55, 56]. Lp(a) can also be recognized and taken up by fibroblasts, hepatocytes, and macrophages via VLDL receptor [57]. Secondly, Lp(a) can be cleaved into LDL and Apo(a). Knowingly, LDL is abundantly removed from circulating system by its receptor with high affinity. With regard to Apo(a) degradation, the weakness bond of LBS between KIV-4 and KIV-5 is critical for proteolytic enzymes (like neutrophil elastase and metalloproteinase) to cleave Apo(a) into small pieces thereby excreted to urine [50, 58]. Kronenberg and colleagues previously observed an arterial-venous gradient of Lp(a) level after the blood flowed through renal circulating system [59], which suggested that renal secretion might be one of the important approaches for Lp(a) clearance. This finding was further substantiated by other studies in which plasma Lp(a) level in patients with renal dysfunction was significantly increased [60, 61]. Intriguingly, Apo(a) fragment was found in urine, and the volume of Apo(a) in urine was reduced in patients with renal dysfunction, further indicating that kidney was crucial for Apo(a) excretion and catabolism [62, 63]. Last but not the least, Lp(a) and Apo(a) fragments are able to deposit in the vascular wall and exert multiple adverse effects on endothelial cells [57, 64, 65]. Many research papers showed that the volumes of Lp(a) and Apo(a) within the plaque were in proportion to the plasma levels of Lp(a) and Apo(a). Moreover, in comparison with large isoform size of Apo(a), small isoform size seems to have a higher propensity to retain in vessel walls [27, 66]. Typically, Lp(a) and Apo(a) bind to vessel walls predominantly through the coupling of LBS sites of the Kringles with the vessels component such as fibrinogen, fibrin, fibronectin, and laminin [16]. Notably, the affinity of Lp(a) and Apo(a) to bind to extracellular matrix of vessels is higher than that of LDL-C, which may partially explain the previous epidemiological findings that Lp(a) contributed to atherosclerosis and CVD progression was independent of LDL-C [16, 67].
4. Pathophysiological Effects of Lp(a) and Its Moieties on CVD
The pathophysiological effects of Lp(a) on atherosclerotic CVD can be broadly categorized into two aspects, named proatherogenesis and prothrombosis or antifibrinolysis. Similar to LDL-C, cholesterol-enriched lipid core in Lp(a) plays a crucial role in atherogenesis and progression. Importantly, in recent years, many clinical and basic studies observed that, besides LDL, another molecule named oxidized phospholipid (OxPL), binding to ApoB-100 (OxPL/ApoB-100), is also quite critical in mediating adverse effects of Lp(a) on vascular system [68]. OxPL/ApoB is an inflammatory and oxidative compound which leads the atherogenic capacity of Lp(a) to significantly increase [69, 70]. Strong association between OxPL/ApoB and plasma Lp(a) level was also observed (R = 0.86, P < 0.0001) [68]. Furthermore, as reported by Tsimikas et al. [70], the strength of this relationship was dependent on Apo(a) isoform size, in terms of large isoform size that corresponds to weaker association, and vice versa. A series of epidemiological studies also consistently revealed that OxPL/ApoB elevation not only was related to the initiation and progression of CVD but also contributed to the recurrence of CVD and mortality [68, 71–73]. And the underlying mechanisms can be generally ascribed to the following aspects such as amplifying oxidative stress, inducing endothelial cells necrosis, promoting monocytes, macrophages migration, and foam cells formation, and expanding necrotic lipid core [68]. As a consequence, OxPL/ApoB-enriched LDL is considered significantly responsible for the adverse effects of Lp(a) on cardiovascular system.
Intriguingly, other than OxPL, another important enzyme, named lipoprotein associated-phospholipase A2 (Lp-PLA2), has also been identified noncovalently coupled with ApoB-100. The roles of Lp-PLA2 on cardiovascular system have also drawn many attentions from scientific society in the past decade. Lp-PLA2 is a key enzyme which is mainly responsible for platelet active factor (PAF) catabolism. As it is well known, PAF is a potent proinflammatory factor and is associated with atherosclerotic plaque formation and progression [74]. Many early basic research papers showed that, in comparison with control group, Lp-PLA2 overexpression was beneficial for ameliorating inflammatory reaction and oxidative distress in vessel walls [75–78]. Nevertheless, upon metabolizing oxidative LDL (oxLDL) and other phospholipids such as OxPL, two important and potent proinflammatory factors, named oxidized fatty acid (ox-FAs) and lysophosphatidylcholine (lyso-PC), are abundantly generated [79, 80]. A large number of basic research papers and epidemiological studies consistently indicated that the adverse effects of Lp-PLA2 on cardiovascular system significantly surpassed the so-called cardioprotective benefits of Lp-PLA2 [81, 82]. Basically, apart from metabolizing PAF and oxLDL, Lp-PLA2 is also capable of degrading OxPL/ApoB-100, which is considered at least partially responsible for the cardio-protective effects of Lp(a). For example, in the Bruneck study [66], the authors noted a J-shaped-curve relationship between plasma level of Lp(a) with the incidence of CVD, indicating that at an appropriate plasma level of Lp(a), although the exact value currently is still little known, it may be useful and helpful to degrade OxPL/ApoB-100 and oxLDL within atherosclerotic plaque so as to reduce cardiovascular events. In retrospect, this finding was also indirectly illustrated by the previous 4S study [83], in which the most significant benefits of simvastatin therapy were derived from the second but not the first quartile of plasma Lp(a) level. To our best knowledge, the underlying mechanisms with regard to the cardio-protective effect of Lp(a) may be related to the degradation OxPL/ApoB-100 of by Lp-PLA2. Unfortunately, there is still no randomized, prospective, and controlled trial to document the aforementioned intriguing findings. Because of the complicated and dual effects of Lp-PLA2 on vascular system [84, 85], to our knowledge, prespecifying an appropriate range for plasma Lp(a) level is very useful, helpful, and critical for clinical practices in terms of better stratifying cardiovascular risk and carrying out more appropriate therapy, especially to those whose LDL-C is within target level. Knowingly, Lp-PLA2 activity is also influenced by the isoform size of Apo(a), where an inverse correlation is observed [86, 87], suggesting that the association between Lp-PLA2 and Lp(a) may also partially be determined genetically. This finding may, at least partially, also explain the discrepancies of CVD outcomes among different ethnic populations with comparable plasma Lp(a) levels.
Notably, due to variable and unique characteristics, the pathophysiological roles of Apo(a) have attracted intensive investigation. Apolipoprotein(a) per se is not only a prothrombotic lipoprotein but also a significant mediator in modulating the process of atherogenesis and progression conferred by Lp(a), oxPL/ApoB-100, and Lp-PLA2. As mentioned in the structural section, the negative effect of Apo(a) on fibrinolysis is largely due to competitive mechanisms. Moreover, promoting platelets aggregation and increasing plasminogen activator inhibitor expression are also attributable to the prothrombotic effect of Apo(a). In the study conducted by Hervio and coworkers [88], they revealed that compared with large isoform size, small isoform size of Apo(a) has a higher potential to inhibit endogenous fibrinolysis. In the Bruneck study [66], a strong prothrombotic effect was also found in small isoform size of Apo(a) but not in large isoform size. Overall, the antifibrinolytic and prothrombotic effects are also influenced by its isoform size. In addition, isoform size of Apo(a) is also found significantly associated with the effects of other moieties of Lp(a) and Lp(a) itself. For examples [26, 89], with small isoform size of Apo(a), plasma Lp(a) level, volume of oxPL/ApoB-100, and Lp-PLA2 activity are all significantly increased when compared to large isoform size. Some previous, if not all, epidemiological studies showed that with small isoform size of Apo(a), the risk for premature CVD is profoundly increased, which is independent of LDL-C and other traditional risk factors [27, 90, 91]. In a recent published meta-analysis including 40 studies [89], when compared with KIV-2 repeats > 22 (corresponding to Apo(a) molecular weight ≥ 640 Kda), KIV-2 repeats ≤ 22 results in an increased CVD risk of 2.08 (95% CI: 1.67–2.58). In a study conducted by Kamstrup et al. [92], they revealed that when compared with the highest quartile of KIV-2 repeats (41–99), the lowest quartile of KIV-2 repeats (6–30) was responsible for a higher Lp(a) level as well as cardiovascular events. Taken together, based on previous findings, it is reasonable to make a conclusion that increased plasma Lp(a) level is a significant and independent predictor for incident CVD and mortality, which is profoundly enhanced by coupling with small isoform size of Apo(a) or with small copynumber of KIV-2 repeats.
5. Effects of Apo(a) Gene Polymorphism on CVD and Related Epidemiological Studies
Interestingly, compared to LDL-C, which is highly modifiable by lipid lowering drugs and lifestyle change, plasma Lp(a) level is predominantly determined by its encoding gene (LPA), which locates on chromosome 6q23, and the underlying mechanism is largely due to the copy-number variation of KIV-2 [48, 65]. Isoform size variability of Apo(a), determined by KIV-2 copy-number, was reported to be responsible for 40% to 70% of interindividual variation of plasma level of Lp(a) [20]. Other than KIV-2 copy-number variation, copy variation of pentanucleotide repeats (TTTTA) in the promoter area and C/T polymorphism in the coding area were also associated with plasma Lp(a) level [16]. In the past decades, some single nucleotide polymorphisms (SNPs) of Apo(a) gene associated with copy-number variability of KIV-2 have been identified [27, 93–96]. Nonetheless, after adjusting for other variables, only a minority of these SNPs have been documented to contribute to the elevated plasma Lp(a) level and increased risk for incident CVD. Accordingly, rs3798220 and rs10455872 may be the most intensively investigated variants [17, 97]. For example, in the study conducted by Luke and colleagues [98], they observed that compared with noncarriers, carriers of the variant rs3798220 have both increased 5-fold higher median plasma Lp(a) level and an adjusted odds ratio for severe CHD of 3.14. In the Cardiovascular Health study [99], rs3798220 was also observed associated with incidence of myocardial infarction in Caucasian. Interestingly, in the Women's Health Study [27], after a 9.9-year followup, aspirin seemed to render benefits to the carrier of rs3798220 in the healthy Caucasian women, in which cardiovascular risks were reduced more than 2-fold when compared with control group. Another common variant rs10455872 was concomitantly identified to be strongly correlated with both increased plasma Lp(a) level and an odds ratio for CHD of 1.70. Lower copynumber of KIV-2 and small isoform size of Apo(a) were considered attributing to these findings [95]. In a prospective study [100], rs10455872 SNP was found associated with incident CHD, and in case-control studies [95, 101], a relationship between rs10455872 SNP and incident CHD and myocardial infarction was also observed. In our recent study (paper has been recently accepted by Journal of Lipids in Health and Disease) in investigating the relationship between 3 variants (rs3798220, rs10455872, and rs6415984), plasma Lp(a) level, and future cardiovascular risk in Chinese Han populations with previously documented CHD, we observed that only the variant of rs6415084 was associated with plasma Lp(a) level and no correlation was found between these 3 SNPs with future cardiovascular risk. To our knowledge, these discrepancies among different studies may be at least partially due to the differences of ethnic background, the prevalence of minor allele of SNPs, and the different clinical protocols.
6. Potential Approaches to Modulate Plasma Lp(a) Level
As it has been intensively discussed, plasma level of Lp(a) is less affected by statins, life style, and dietary than LDL-C [16, 17]; however, some studies showed that nicotinic acid as well as estrogen and/or progestin hormone replacement might be one of the most potential useful approaches to reduce plasma Lp(a) level [102–106]. In the late 1980s, Carlson LA treated hyperlipidemic patients with 4 g nicotinic acid daily [102]. Six weeks later, they observed that Lp(a) was decreased concomitantly with the same percentage reduction of LDL-C when compared with baseline levels, which they speculated to be due to inhibition of ApoB-100 production, a common protein to Lp(a) and LDL-C. In 2010, Bruckert et al. published a meta-analysis regarding the effects of nicotinic acid alone or in combination with lipid lower drugs on the incident CVD [106]. This meta-analysis included 11 studies with more than 5000 participants, and the primary outcomes indicated that nicotinic acid therapy not only reduced the incidence of cardiovascular events but also reversed coronary and carotid atherosclerosis. In line with previous compelling pieces of evidence, the European Society of Atherosclerosis recommended that a patient with increased plasma level of Lp(a) is reasonably considered for nicotinic acid (1–3 g/day) therapy [26]. Accordingly, hormone replacement with estrogen and/or progestin is also potential to decrease plasma Lp(a) level [24, 103, 105]. Furthermore, reduction of incident CVD and mortality rate deriving from Lp(a) reduction with hormone replacement has also confirmed the potential efficacy with this strategy. For example, the study from Shlipak and coworkers revealed that estrogen and progestin therapy contributed to a more favorable outcome on CVD than control group, particularly in participants with initially higher plasma Lp(a) level [107]. In another study, the authors showed that hormone replacement not only reduced plasma Lp(a) level but also markedly attenuated future CVD risk among women with hormone replacement in comparison with the control group [24]. Other less intensively investigative approaches for Lp(a) reduction involve nonsteroid anti-inflammatory drug aspirin, statins, IL-6 inhibitor, and PCSK9 inhibitor [16, 108]. For example, some researchers reported that interleukin-6 (IL-6) pathway was positively correlated with plasma Lp(a) level and Apo(a) expression in humans and monkey hepatocytes and inhibition of IL-6 pathway may be possible to reduce Lp(a) level [109, 110]. In the Women's Health Study [27], carriers of an Apo(a) variant had elevated Lp(a) and doubled cardiovascular risk in healthy Caucasian women, while aspirin therapy conferred a benefit for cardiovascular outcomes. Statins may reduce Lp(a) level by the means of increasing hepatic clearance of circulating LDL and decreasing Lp(a) assembly. PCSK9 inhibitor may also be useful for lower Lp(a) level via the clearance of LDL particle [111]. Until now, the most robust approach may be blood apheresis, which has been validated to be highly efficient in removing Lp(a) from the circulating system. However, this technique is neither noninvasive nor cheap. Therefore, this strategy seems less practical in clinic [112]. Finally, some previous epidemiological studies suggested that populations with both Lp(a) and LDL-C elevations have a higher cardiovascular risk [66, 90], and these population groups should be given a more intensive lipid lowering drug therapy so as to achieve a lower LDL-C level than those with only Lp(a) elevation. In the future, to our knowledge, exploring a unique medication specific for Lp(a) modulation must be a top priority.
7. Conclusion
With respect to its proatherosclerotic and prothrombotic effects, Lp(a) is believed to be a promising and critical biomarker for cardiovascular risk estimation. Causal relationship between Lp(a) and CVD has been recognized and demonstrated in the past decades. In patients with established CVD and with target LDL-C level achievement, Lp(a) measurement may add addictive value for cardiovascular risk stratification.
Conflict of Interests
All authors declare that there is no conflict of interests.
Acknowledgments
The authors appreciate very much the supports from the following grants: the Technology Project Foundation of Guangdong province, China (2009A030301004, 2011B031800021, and 2011B031800263), the Research Grant of Cardiovascular Medication of Guangdong province (2011X25), and Medical Scientific Research Grant of the Health Ministry of Guangdong province, China (B2011310, A2012663). Anping Cai and Liwen Li are cofirst authors.
References
- 1.Gibson CM, Pride YB, Hochberg CP, Sloan S, Sabatine MS, Cannon CP. Effect of intensive statin therapy on clinical outcomes among patients undergoing percutaneous coronary intervention for acute coronary syndrome. PCI-PROVE IT: a PROVE IT-TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22) substudy. Journal of the American College of Cardiology. 2009;54(24):2290–2295. doi: 10.1016/j.jacc.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18, 686 people with diabetes in 14 randomised trials of statins: a meta-analysis. The Lancet. 2008;371(9607):117–125. doi: 10.1016/S0140-6736(08)60104-X. [DOI] [PubMed] [Google Scholar]
- 3.Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. Journal of the American College of Cardiology. 2006;48(3):438–445. doi: 10.1016/j.jacc.2006.04.070. [DOI] [PubMed] [Google Scholar]
- 4.Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12, 064 survivors of myocardial infarction: a double-blind randomised trial. The Lancet. 2010;376(9753):1658–1669. doi: 10.1016/S0140-6736(10)60310-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170, 000 participants in 26 randomised trials. The Lancet. 2010;376(9753):1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. The New England Journal of Medicine. 2004;350(15):1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 7.LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. The New England Journal of Medicine. 2005;352(14):1425–1435. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 8.Chapman MJ. Beyond the statins: new therapeutic perspectives in cardiovascular disease prevention. Cardiovascular Drugs and Therapy. 2005;19(2):135–139. doi: 10.1007/s10557-005-1049-z. [DOI] [PubMed] [Google Scholar]
- 9.Schachter M. Strategies for modifying high-density lipoprotein cholesterol: a role for nicotinic acid. Cardiovascular Drugs and Therapy. 2005;19(6):415–422. doi: 10.1007/s10557-005-5685-0. [DOI] [PubMed] [Google Scholar]
- 10.Shah PK. Inhibition of CETP as a novel therapeutic strategy for reducing the risk of atherosclerotic disease. European Heart Journal. 2007;28(1):5–12. doi: 10.1093/eurheartj/ehl392. [DOI] [PubMed] [Google Scholar]
- 11.Campbell CY, Rivera JJ, Blumenthal RS. Residual risk in statin-treated patients: future therapeutic options. Current Cardiology Reports. 2007;9(6):499–505. doi: 10.1007/BF02938395. [DOI] [PubMed] [Google Scholar]
- 12.Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the National Heart, Lung, and Blood Institute Workshop on Lipoprotein(a) and Cardiovascular Disease: recent advances and future directions. Clinical Chemistry. 2003;49(11):1785–1796. doi: 10.1373/clinchem.2003.023689. [DOI] [PubMed] [Google Scholar]
- 13.Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease: meta-analysis of prospective studies. Circulation. 2000;102(10):1082–1085. doi: 10.1161/01.cir.102.10.1082. [DOI] [PubMed] [Google Scholar]
- 14.Ariyo AA, Thach C, Tracy R. Lp(a) lipoprotein, vascular disease, and mortality in the elderly. The New England Journal of Medicine. 2003;349(22):2108–2115. doi: 10.1056/NEJMoa001066. [DOI] [PubMed] [Google Scholar]
- 15.Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. Journal of the American Medical Association. 2009;302(4):412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoover-Plow J, Huang M. Lipoprotein(a) metabolism: potential sites for therapeutic targets. Metabolism. 2013;62(4):479–491. doi: 10.1016/j.metabol.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamstrup PR. Lipoprotein(a) and ischemic heart disease—a causal association? A review. Atherosclerosis. 2010;211(1):15–23. doi: 10.1016/j.atherosclerosis.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 18.Marcovina SM, Albers JJ, Jacobs DR, Jr., et al. Lipoprotein(a) concentrations and apolipoprotein(a) phenotypes in Caucasians and African Americans: the CARDIA study. Arteriosclerosis and Thrombosis. 1993;13(7):1037–1045. doi: 10.1161/01.atv.13.7.1037. [DOI] [PubMed] [Google Scholar]
- 19.Rubin J, Kim HJ, Pearson TA, Holleran S, Berglund L, Ramakrishnan R. The apolipoprotein(a) gene: linkage disequilibria at three loci differs in African Americans and Caucasians. Atherosclerosis. 2008;201(1):138–147. doi: 10.1016/j.atherosclerosis.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boerwinkle E, Menzel HJ, Kraft HG, Utermann G. Genetics of the quantitative Lp(a) lipoprotein trait. III. Contribution of Lp(a) glycoprotein phenotypes to normal lipid variation. Human Genetics. 1989;82(1):73–78. doi: 10.1007/BF00288277. [DOI] [PubMed] [Google Scholar]
- 21.Erqou S, Thompson A, Di Angelantonio E, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58, 000 participants. Journal of the American College of Cardiology. 2010;55(19):2160–2167. doi: 10.1016/j.jacc.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 22.Kardys I, Oemrawsingh RM, Kay IP, et al. Lipoprotein(a), interleukin-10, C-reactive protein, and 8-year outcome after percutaneous coronary intervention. Clinical Cardiology. 2012;35(8):482–489. doi: 10.1002/clc.21988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Momiyama Y, Ohmori R, Fayad ZA, et al. Associations between serum lipoprotein(a) levels and the severity of coronary and aortic atherosclerosis. Atherosclerosis. 2012;222(1):241–244. doi: 10.1016/j.atherosclerosis.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Suk DJ, Rifai N, Buring JE. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. Journal of the American College of Cardiology. 2008;52(2):124–131. doi: 10.1016/j.jacc.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidson MH, Ballantyne CM, Jacobson TA, et al. Clinical utility of inflammatory markers and advanced lipoprotein testing: advice from an expert panel of lipid specialists. Journal of Clinical Lipidology. 2011;5(5):338–367. doi: 10.1016/j.jacl.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. European Heart Journal. 2010;31(23):2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chasman DI, Shiffman D, Zee RYL, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203(2):371–376. doi: 10.1016/j.atherosclerosis.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berg K. A new serum type system in man—the Lp system. Acta Pathologica et Microbiologica Scandinavica. 1963;59:369–382. doi: 10.1111/j.1699-0463.1963.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 29.Koschinsky ML, Cote GP, Gabel B, van der Hoek YY. Identification of the cysteine residue in apolipoprotein(a) that mediates extracellular coupling with apolipoprotein B-100. The Journal of Biological Chemistry. 1993;268(26):19819–19825. [PubMed] [Google Scholar]
- 30.McLean JW, Tomlinson JE, Kuang W-J, et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330(6144):132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 31.Miles LA, Plow EF. Lp(a): an interloper into the fibrinolytic system? Thrombosis and Haemostasis. 1990;63(3):331–335. [PubMed] [Google Scholar]
- 32.Malgaretti N, Acquati F, Magnaghi P, et al. Characterization by yeast artificial chromosome cloning of the linked apolipoprotein(a) and plasminogen genes and identification of the apolipoprotein(a) 5′ flanking region. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(23):11584–11588. doi: 10.1073/pnas.89.23.11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Hoek YY, Wittekoek ME, Beisiegel U, Kastelein JJP, Koschinsky ML. The apolipoprotein(a) kringle IV repeats which differ from the major repeat kringle are present in variably-sized isoforms. Human Molecular Genetics. 1993;2(4):361–366. doi: 10.1093/hmg/2.4.361. [DOI] [PubMed] [Google Scholar]
- 34.Lackner C, Cohen JC, Hobbs HH. Molecular definition of the extreme size polymorphism in apolipoprotein(a) Human Molecular Genetics. 1993;2(7):933–940. doi: 10.1093/hmg/2.7.933. [DOI] [PubMed] [Google Scholar]
- 35.Berglund L, Ramakrishnan R. Lipoprotein(a): an elusive cardiovascular risk factor. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(12):2219–2226. doi: 10.1161/01.ATV.0000144010.55563.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabel BR, Koschinsky ML. Analysis of the proteolytic activity of a recombinant form of apolipoprotein(a) Biochemistry. 1995;34(48):15777–15784. doi: 10.1021/bi00048a023. [DOI] [PubMed] [Google Scholar]
- 37.Utermann G, Menzel HJ, Kraft HG, Duba HC, Kemmler HG, Seitz C. Lp(a) glycoprotein phenotypes. Inheritance and relation to Lp(a)-lipoprotein concentrations in plasma. The Journal of Clinical Investigation. 1987;80(2):458–465. doi: 10.1172/JCI113093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koschinsky ML, Cote GP, Gabel B, van der Hoek YY. Identification of the cysteine residue in apolipoprotein(a) that mediates extracellular coupling with apolipoprotein B-100. The Journal of Biological Chemistry. 1993;268(26):19819–19825. [PubMed] [Google Scholar]
- 39.Becker L, Cook PM, Wright TG, Koschinsky ML. Quantitative evaluation of the contribution of weak lysine-binding sites present within apolipoprotein(a) kringle IV types 6-8 to lipoprotein(a) assembly. The Journal of Biological Chemistry. 2004;279(4):2679–2688. doi: 10.1074/jbc.M309414200. [DOI] [PubMed] [Google Scholar]
- 40.Marcovina SM, Hobbs HH, Albers JJ. Relation between number of apolipoprotein(a) kringle 4 repeats and mobility of isoforms in agarose gel: basis for a standardized isoform nomenclature. Clinical Chemistry. 1996;42(3):436–439. [PubMed] [Google Scholar]
- 41.Bonen DK, Hausman AML, Hadjiagapiou C, Skarosi SF, Davidson NO. Expression of a recombinant apolipoprotein(a) in HepG2 cells. Evidence for intracellular assembly of lipoprotein(a) The Journal of Biological Chemistry. 1997;272(9):5659–5667. doi: 10.1074/jbc.272.9.5659. [DOI] [PubMed] [Google Scholar]
- 42.Lobentanz E, Krasznai K, Gruber A, et al. Intracellular metabolism of human apolipoprotein(a) in stably transfected Hep G2 cells. Biochemistry. 1998;37(16):5417–5425. doi: 10.1021/bi972761t. [DOI] [PubMed] [Google Scholar]
- 43.Trieu VN, McConathy WJ. A two-step model for lipoprotein(a) formation. The Journal of Biological Chemistry. 1995;270(26):15471–15474. doi: 10.1074/jbc.270.26.15471. [DOI] [PubMed] [Google Scholar]
- 44.McCormick SP. Lipoprotein(a): biology and clinical importance. The Clinical Biochemist Reviews. 2004;25(1):69–80. [PMC free article] [PubMed] [Google Scholar]
- 45.Brunner C, Lobentanz E, Pethö-Schramm A, et al. The number of identical kringle IV repeats in apolipoprotein(a) affects its processing and secretion by HepG2 cells. The Journal of Biological Chemistry. 1996;271(50):32403–32410. doi: 10.1074/jbc.271.50.32403. [DOI] [PubMed] [Google Scholar]
- 46.Boffelli D, Zajchowski DA, Yang Z, Lawn RM. Estrogen modulation of apolipoprotein(a) expression: identification of a regulatory element. The Journal of Biological Chemistry. 1999;274(22):15569–15574. doi: 10.1074/jbc.274.22.15569. [DOI] [PubMed] [Google Scholar]
- 47.Wade DP, Clarke JG, Lindahl GE, et al. 5′ control regions of the apolipoprotein(a) gene and members of the related plasminogen gene family. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(4):1369–1373. doi: 10.1073/pnas.90.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. The Journal of Clinical Investigation. 1992;90(1):52–60. doi: 10.1172/JCI115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen JC, Chiesa G, Hobbs HH. Sequence polymorphisms in the apolipoprotein(a) gene. Evidence for dissociation between apolipoprotein(a) size and plasma lipoprotein(a) levels. The Journal of Clinical Investigation. 1993;91(4):1630–1636. doi: 10.1172/JCI116370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hobbs HH, White AL. Lipoprotein(a): intrigues and insights. Current Opinion in Lipidology. 1999;10(3):225–236. doi: 10.1097/00041433-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 51.Puckey LH, Knight BL. Sequence and functional changes in a putative enhancer region upstream of the apolipoprotein(a) gene. Atherosclerosis. 2003;166(1):119–127. doi: 10.1016/s0021-9150(02)00315-5. [DOI] [PubMed] [Google Scholar]
- 52.Huby T, Afzal V, Doucet C, et al. Regulation of the expression of the apolipoprotein(a) gene: evidence for a regulatory role of the 5′ distal apolipoprotein(a) transcription control region enhancer in yeast artificial chromosome transgenic mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(9):1633–1639. doi: 10.1161/01.ATV.0000084637.01883.CA. [DOI] [PubMed] [Google Scholar]
- 53.Hofmann SL, Eaton DL, Brown MS, McConathy WJ, Goldstein JL, Hammer RE. Overexpression of human low density lipoprotein receptors leads to accelerated catabolism of Lp(a) lipoprotein in transgenic mice. The Journal of Clinical Investigation. 1990;85(5):1542–1547. doi: 10.1172/JCI114602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(2):522–528. doi: 10.1161/01.atv.20.2.522. [DOI] [PubMed] [Google Scholar]
- 55.Knight BL, Perombelon YFN, Soutar AK, Wade DP, Seed M. Catabolism of lipoprotein(a) in familial hypercholesterolaemic subjects. Atherosclerosis. 1991;87(2-3):227–237. doi: 10.1016/0021-9150(91)90025-x. [DOI] [PubMed] [Google Scholar]
- 56.Rader DJ, Mann WA, Cain W, et al. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. The Journal of Clinical Investigation. 1995;95(3):1403–1408. doi: 10.1172/JCI117794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Argraves KM, Kozarsky KF, Fallon JT, Harpel PC, Strickland DK. The atherogenic lipoprotein Lp(a) is internalized and degraded in a process mediated by the VLDL receptor. The Journal of Clinical Investigation. 1997;100(9):2170–2181. doi: 10.1172/JCI119753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edelstein C, Italia JA, Scanu AM. Polymorphonuclear cells isolated from human peripheral blood cleave lipoprotein(a) and apolipoprotein(a) at multiple interkringle sites via the enzyme elastase. Generation of mini-Lp(a) particles and apo(a) fragments. The Journal of Biological Chemistry. 1997;272(17):11079–11087. doi: 10.1074/jbc.272.17.11079. [DOI] [PubMed] [Google Scholar]
- 59.Kronenberg F, Trenkwalder E, Lingenhel A, et al. Renovascular arteriovenous differences in Lp(a) plasma concentrations suggest removal of Lp(a) from the renal circulation. Journal of Lipid Research. 1997;38(9):1755–1763. [PubMed] [Google Scholar]
- 60.Sechi LA, Zingaro L, De Carli S, et al. Increased serum lipoprotein(a) levels in patients with early renal failure. Annals of Internal Medicine. 1998;129(6):457–461. doi: 10.7326/0003-4819-129-6-199809150-00006. [DOI] [PubMed] [Google Scholar]
- 61.Frischmann ME, Kronenberg F, Trenkwalder E, et al. In vivo turnover study demonstrates diminished clearance of lipoprotein(a) in hemodialysis patients. Kidney International. 2007;71(10):1036–1043. doi: 10.1038/sj.ki.5002131. [DOI] [PubMed] [Google Scholar]
- 62.Kostner KM, Banyai S, Banyai M, et al. Urinary apolipoprotein(a) excretion in patients with proteinuria. Annals of Medicine. 1998;30(5):497–502. doi: 10.3109/07853899809002492. [DOI] [PubMed] [Google Scholar]
- 63.Oida K, Takai H, Maeda H, et al. Apolipoprotein(a) is present in urine and its excretion is decreased in patients with renal failure. Clinical Chemistry. 1992;38(11):2244–2248. [PubMed] [Google Scholar]
- 64.Boffa MB, Marcovina SM, Koschinsky ML. Lipoprotein(a) as a risk factor for atherosclerosis and thrombosis: mechanistic insights from animal models. Clinical Biochemistry. 2004;37(5):333–343. doi: 10.1016/j.clinbiochem.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 65.Koschinsky ML, Marcovina SM. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Current Opinion in Lipidology. 2004;15(2):167–174. doi: 10.1097/00041433-200404000-00009. [DOI] [PubMed] [Google Scholar]
- 66.Kronenberg F, Kronenberg MF, Kiechl S, et al. Role of lipoprotein(a) and apolipoprotein(a) phenotype in atherogenesis: prospective results from the bruneck study. Circulation. 1999;100(11):1154–1160. doi: 10.1161/01.cir.100.11.1154. [DOI] [PubMed] [Google Scholar]
- 67.Danesh J, Erqou S. Risk factors: lipoprotein(a) and coronary disease-moving closer to causality. Nature Reviews. 2009;6(9):565–567. doi: 10.1038/nrcardio.2009.138. [DOI] [PubMed] [Google Scholar]
- 68.Tsimikas S, Witztum JL. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Current Opinion in Lipidology. 2008;19(4):369–377. doi: 10.1097/MOL.0b013e328308b622. [DOI] [PubMed] [Google Scholar]
- 69.Tsimikas S, Witztum JL, Miller ER, et al. High-dose atorvastatin reduces total plasma levels of oxidized phospholipids and immune complexes present on apolipoprotein B-100 in patients with acute coronary syndromes in the MIRACL trial. Circulation. 2004;110(11):1406–1412. doi: 10.1161/01.CIR.0000141728.23033.B5. [DOI] [PubMed] [Google Scholar]
- 70.Tsimikas S, Kiechl S, Willeit J, et al. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: five-year prospective results from the Bruneck study. Journal of the American College of Cardiology. 2006;47(11):2219–2228. doi: 10.1016/j.jacc.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 71.Tsimikas S, Brilakis ES, Miller ER, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. The New England Journal of Medicine. 2005;353(1):46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- 72.Tsimikas S, Bergmark C, Beyer RW, et al. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. Journal of the American College of Cardiology. 2003;41(3):360–370. doi: 10.1016/s0735-1097(02)02769-9. [DOI] [PubMed] [Google Scholar]
- 73.Tsimikas S, Lau HK, Han K, et al. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein(a): short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation. 2004;109(25):3164–3170. doi: 10.1161/01.CIR.0000130844.01174.55. [DOI] [PubMed] [Google Scholar]
- 74.Karabina S, Ninio E. Plasma PAF-acetylhydrolase: an unfulfilled promise? Biochimica et Biophysica Acta. 2006;1761(11):1351–1358. doi: 10.1016/j.bbalip.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 75.Morgan E, Boyle EM, Jr., Yun W, et al. Platelet-activating factor acetylhydrolase prevents myocardial ischemia-reperfusion injury. Circulation. 1999;100(supplement 19):II365–II368. doi: 10.1161/01.cir.100.suppl_2.ii-365. [DOI] [PubMed] [Google Scholar]
- 76.Quarck R, De Geest B, Stengel D, et al. Adenovirus-mediated gene transfer of human platelet-activating factor-acetylhydrolase prevents injury-induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;103(20):2495–2500. doi: 10.1161/01.cir.103.20.2495. [DOI] [PubMed] [Google Scholar]
- 77.Theilmeier G, De Geest B, van Veldhoven PP, et al. HDL-associated PAF-AH reduces endothelial adhesiveness in apoE(-/-) mice. The FASEB Journal. 2000;14(13):2032–2039. doi: 10.1096/fj.99-1029com. [DOI] [PubMed] [Google Scholar]
- 78.Noto H, Hara M, Karasawa K, et al. Human plasma platelet-activating factor acetylhydrolase binds to all the murine lipoproteins, conferring protection against oxidative stress. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(5):829–835. doi: 10.1161/01.ATV.0000067701.09398.18. [DOI] [PubMed] [Google Scholar]
- 79.Caslake MJ, Packard CJ, Suckling KE, Holmes SD, Chamberlain P, Macphee CH. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase: a potential new risk factor for coronary artery disease. Atherosclerosis. 2000;150(2):413–419. doi: 10.1016/s0021-9150(99)00406-2. [DOI] [PubMed] [Google Scholar]
- 80.Rosenson RS. Phospholipase A2 inhibition and atherosclerotic vascular disease: prospects for targeting secretory and lipoprotein-associated phospholipase A2 enzymes. Current Opinion in Lipidology. 2010;21(6):473–480. doi: 10.1097/MOL.0b013e32833eb581. [DOI] [PubMed] [Google Scholar]
- 81.Mannheim D, Herrmann J, Versari D, et al. Enhanced expression of Lp-PLA2 and lysophosphatidylcholine in symptomatic carotid atherosclerotic plaques. Stroke. 2008;39(5):1448–1455. doi: 10.1161/STROKEAHA.107.503193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blankenberg S, Stengel D, Rupprecht HJ, et al. Plasma PAF-acetylhydrolase in patients with coronary artery disease: results of a cross-sectional analysis. Journal of Lipid Research. 2003;44(7):1381–1386. doi: 10.1194/jlr.M300086-JLR200. [DOI] [PubMed] [Google Scholar]
- 83.Pedersen TR. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) The Lancet. 1994;344(8934):1383–1389. [PubMed] [Google Scholar]
- 84.Cai A, Zheng D, Qiu R, Mai W, Zhou Y. Lipoprotein-associated phospholipase A2 (Lp-PLA(2)): a novel and promising biomarker for cardiovascular risks assessment. Disease Markers. 2013;34(5):323–331. doi: 10.3233/DMA-130976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nambi V, Ballantyne CM. Lipoprotein-associated phospholipase A2: pathogenic mechanisms and clinical utility for predicting cardiovascular events. Current Atherosclerosis Reports. 2006;8(5):374–381. doi: 10.1007/s11883-006-0034-8. [DOI] [PubMed] [Google Scholar]
- 86.Tsironis LD, Katsouras CS, Lourida ES, et al. Reduced PAF-acetylhydrolase activity associated with Lp(a) in patients with coronary artery disease. Atherosclerosis. 2004;177(1):193–201. doi: 10.1016/j.atherosclerosis.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 87.Tsimikas S, Tsironis LD, Tselepis AD. New insights into the role of lipoprotein(a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(10):2094–2099. doi: 10.1161/01.ATV.0000280571.28102.d4. [DOI] [PubMed] [Google Scholar]
- 88.Hervio L, Girard-Globa A, Durlach V, Anglés-Cano E. The antifibrinolytic effect of lipoprotein(a) in heterozygous subjects is modulated by the relative concentration of each of the apolipoprotein(a) isoforms and their affinity for fibrin. European Journal of Clinical Investigation. 1996;26(5):411–417. doi: 10.1046/j.1365-2362.1996.156288.x. [DOI] [PubMed] [Google Scholar]
- 89.Erqou S, Thompson A, Di Angelantonio E, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58, 000 participants. Journal of the American College of Cardiology. 2010;55(19):2160–2167. doi: 10.1016/j.jacc.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 90.Danik JS, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. Journal of the American Medical Association. 2006;296(11):1363–1370. doi: 10.1001/jama.296.11.1363. [DOI] [PubMed] [Google Scholar]
- 91.Rifai N, Ma J, Sacks FM, et al. Apolipoprotein(a) size and lipoprotein(a) concentration and future risk of angina pectoris with evidence of severe coronary atherosclerosis in men: the physicians’ health study. Clinical Chemistry. 2004;50(8):1364–1371. doi: 10.1373/clinchem.2003.030031. [DOI] [PubMed] [Google Scholar]
- 92.Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. Journal of the American Medical Association. 2009;301(22):2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 93.Chretien J-P, Coresh J, Berthier-Schaad Y, et al. Three single-nucleotide polymorphisms in LPA account for most of the increase in lipoprotein(a) level elevation in African Americans compared with European Americans. Journal of Medical Genetics. 2006;43(12):917–923. doi: 10.1136/jmg.2006.042119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lanktree MB, Anand SS, Yusuf S, Hegele RA. Comprehensive analysis of genomic variation in the LPA locus and its relationship to plasma lipoprotein(a) in South Asians, Chinese, and European Caucasians. Circulation. 2010;3(1):39–46. doi: 10.1161/CIRCGENETICS.109.907642. [DOI] [PubMed] [Google Scholar]
- 95.Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. The New England Journal of Medicine. 2009;361(26):2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 96.Erqou S, Thompson A, Di Angelantonio E, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58, 000 participants. Journal of the American College of Cardiology. 2010;55(19):2160–2167. doi: 10.1016/j.jacc.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 97.Li Y, Luke MM, Shiffman D, Devlin JJ. Genetic variants in the apolipoprotein(a) gene and coronary heart disease. Circulation. 2011;4(5):565–573. doi: 10.1161/CIRCGENETICS.111.959601. [DOI] [PubMed] [Google Scholar]
- 98.Luke MM, Kane JP, Liu DM, et al. A polymorphism in the protease-like domain of apolipoprotein(a) is associated with severe coronary artery disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(9):2030–2036. doi: 10.1161/ATVBAHA.107.141291. [DOI] [PubMed] [Google Scholar]
- 99.Shiffman D, O'Meara ES, Bare LA, et al. Association of gene variants with incident myocardial infarction in the Cardiovascular Health Study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(1):173–179. doi: 10.1161/ATVBAHA.107.153981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hopewell JC, Clarke R, Parish S, et al. Lipoprotein(a) genetic variants associated with coronary and peripheral vascular disease but not with stroke risk in the heart protection study. Circulation. 2011;4(1):68–73. doi: 10.1161/CIRCGENETICS.110.958371. [DOI] [PubMed] [Google Scholar]
- 101.Shiffman D, Louie JZ, Rowland CM, Malloy MJ, Kane JP, Devlin JJ. Single variants can explain the association between coronary heart disease and haplotypes in the apolipoprotein(a) locus. Atherosclerosis. 2010;212(1):193–196. doi: 10.1016/j.atherosclerosis.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 102.Carlson LA, Hamsten A, Asplund A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidaemic subjects treated with nicotinic acid. Journal of Internal Medicine. 1989;226(4):271–276. doi: 10.1111/j.1365-2796.1989.tb01393.x. [DOI] [PubMed] [Google Scholar]
- 103.Vigna GB, Donegà P, Zanca R, et al. Simvastatin, transdermal patch, and oral estrogen-progestogen preparation in early-postmenopausal hypercholesterolemic women: a randomized, placebo-controlled clinical trial. Metabolism. 2002;51(11):1463–1470. doi: 10.1053/meta.2002.35584. [DOI] [PubMed] [Google Scholar]
- 104.Taskinen M, Puolakka J, Pyörälä T, et al. Hormone replacement therapy lowers plasma Lp(a) concentrations: comparison of cyclic transdermal and continuous estrogen-progestin regimens. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(10):1215–1221. doi: 10.1161/01.atv.16.10.1215. [DOI] [PubMed] [Google Scholar]
- 105.Godsland IF. Effects of postmenopausal hormone replacement therapy on lipid, lipoprotein, and apolipoprotein (a) concentrations: analysis of studies published from 1974–2000. Fertility and Sterility. 2001;75(5):898–915. doi: 10.1016/s0015-0282(01)01699-5. [DOI] [PubMed] [Google Scholar]
- 106.Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210(2):353–361. doi: 10.1016/j.atherosclerosis.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 107.Shlipak MG, Simon JA, Vittinghoff E, et al. Estrogen and progestin, lipoprotein(a) and the risk of recurrent coronary heart disease events after menopause. Journal of the American Medical Association. 2000;283(14):1845–1852. doi: 10.1001/jama.283.14.1845. [DOI] [PubMed] [Google Scholar]
- 108.Lippi G, Targher G. Optimal therapy for reduction of lipoprotein(a) Journal of Clinical Pharmacy and Therapeutics. 2012;37(1):1–3. doi: 10.1111/j.1365-2710.2011.01244.x. [DOI] [PubMed] [Google Scholar]
- 109.Ramharack R, Barkalow D, Spahr MA. Dominant negative effect of TGF-β1 and TNF-α on basal and IL-6- induced lipoprotein(a) and apolipoprotein(a) mRNA expression in primary monkey hepatocyte cultures. Arteriosclerosis, Thrombosis, and Vascular Biology. 1998;18(6):984–990. doi: 10.1161/01.atv.18.6.984. [DOI] [PubMed] [Google Scholar]
- 110.Schultz O, Oberhauser F, Saech J, et al. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (A) levels in human subjects with rheumatoid diseases. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0014328.e14328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand A, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. Journal of the American College of Cardiology. 2012;59(25):2344–2353. doi: 10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 112.Lippi G, Franchini M, Targher G. Platelets and lipoprotein(a) in retinal vein occlusion: mutual targets for aspirin therapy. Thrombosis and Haemostasis. 2007;97(6):1059–1060. [PubMed] [Google Scholar]