

Abstract
We report here the genetic basis for a form of progressive hereditary spastic paraplegia (SPG43) previously described in two Malian sisters. Exome sequencing revealed a homozygous missense variant (c.187G>C; p.Ala63Pro) in C19orf12, a gene recently implicated in neurodegeneration with brain iron accumulation (NBIA). The same mutation was subsequently also found in a Brazilian family with features of NBIA, and we identified another NBIA patient with a three-nucleotide deletion (c.197_199del; p.Gly66del). Haplotype analysis revealed that the p.Ala63Pro mutations have a common origin, but MRI scans showed no brain iron deposition in the Malian SPG43 subjects. Heterologous expression of these SPG43 and NBIA variants resulted in similar alterations in the subcellular distribution of C19orf12. The SPG43 and NBIA variants reported here as well as the most common C19orf12 missense mutation reported in NBIA patients are found within a highly-conserved, extended hydrophobic domain in C19orf12, underscoring the functional importance of this domain.
Keywords: SPG43, NBIA, C19orf12, hereditary spastic paraplegia
Hereditary spastic paraplegias (HSPs) are heterogeneous neurological disorders (SPG1-57) characterized by progressive spasticity and weakness in the lower limbs [Harding, 1993]. The estimated prevalence is about 3-9/100,000 in most populations [Blackstone, et al., 2011; Erichsen, et al., 2009; Silva, et al., 1997]. All modes of inheritance have been described, but dominant HSPs are most common in North America and northern Europe, while recessive forms are predominant in North Africa, the Middle East, and the Mediterranean region [Erichsen, et al., 2009; Harding, 1993; Silva, et al., 1997]. Over 30 recessive HSPs have been described, and over 20 of the disease genes have been identified to date.
In HSPs, axons of the corticospinal tracts and posterior columns of the spinal cord are impaired in a length-dependent manner. The cellular pathophysiology of HSPs involves several functional areas, including organellar membrane shaping and traffic, mitochondrial function, myelination, and lipid and cholesterol metabolism [Blackstone, et al., 2011; Blackstone, 2012]. Some HSP proteins have been implicated in more than one of these areas. For example, SPG4, the most common form of HSP, is caused by mutations in SPAST (MIM# 604277), the gene encoding spastin, an ATPase involved in endoplasmic reticulum (ER) morphogenesis, endosomal trafficking, BMP signaling, cytokinesis, and cytoskeletal regulation [Hazan, et al., 1999].
We previously described a consanguineous Malian family with recessive HSP in which two sisters presented at ages 7 and 12 with gait difficulty, spasticity, and peripheral neuropathy, and shared a region of extended homozygosity on chromosome 19 [Meilleur, et al., 2010]. Five years after the initial examination, the older patient had severe atrophy and decreased sensation in the arms and legs, and reduced-to-absent reflexes, but no cognitive decline, facial and bulbar weakness, or vision loss. Brain MRI of one of the affected sisters showed no abnormalities, in particular no brain iron deposits (Fig. 1A). DNA from an affected sister was used to perform exome sequencing as previously described [Landouré, et al., 2012], and we identified a homozygous missense sequence variant in the coding region of the C19orf12 gene (NM_001031726.3; MIM# 614297) at position c.187G>C (Supp. Figure S1), predicting the amino acid substitution p.Ala63Pro (http://databases.lovd.nl/shared/variants/C19orf12). Sequencing of 298 Malian controls did not show the homozygous sequence variant. The sequence variant was also not seen in 951 samples in the ClinSeq® cohort [Biesecker, et al., 2009]. Interestingly, the variant was found in 3 of 3836 African-American alleles (but none of 8222 European-American alleles) in the NHLBI Exome Sequencing Project database (http://evs.gs.washington.edu/EVS/). We subsequently sequenced the full coding region of C19orf12 in 16 Australians, 46 French, 195 Americans, and 170 Japanese presenting with diverse HSP types, and none had the c.187G>C variant or any other detectable variant in the gene. Thus, C19orf12 mutation is likely a rare cause of autosomal recessive HSP.
Figure 1.
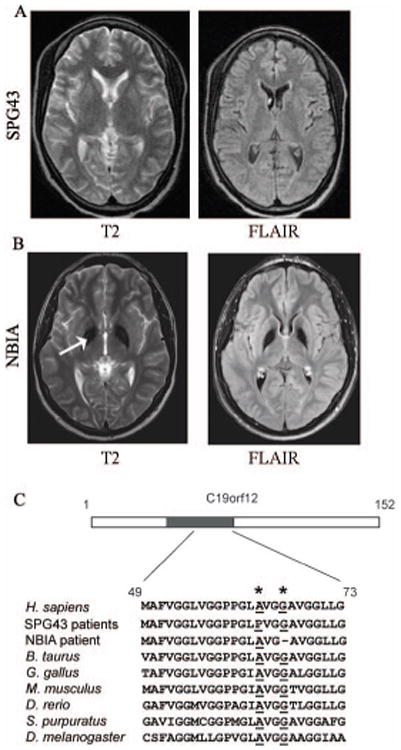
MRI and genetic characterization of SPG43 and NBIA. (A) Brain MRI images of a SPG43 subject with the homozygous C19orf12 mutation c.187G>C, p.Ala63Pro. (B) MRI images of a NBIA patient with the homozygous C19orf12 deletion c.197_199GGG, p.Gly66del. The white arrow indicates iron deposition. (C) Schematic diagram of C19orf12, with the hydrophobic sequence in blue, and a protein sequence alignment of C19orf12 in various species (amino acid numbers refer to the human sequence). The SPG43 and NBIA mutations cause amino acid changes at Ala63 and Gly66, respectively, both highly conserved residues (in red, asterisks above).
While this work was in progress, mutations in C19orf12 were reported in a series of patients with neurodegeneration with brain iron accumulation (NBIA) [Hartig, et al., 2011]. NBIA is a heterogeneous neurological disorder caused by mutations in a range of genes, some of which display allelic heterogeneity [Paisan-Ruiz, et al., 2009]. It is characterized primarily by extrapyramidal features, with spasticity and optic atrophy. Psychiatric symptoms and cognitive decline have been reported in some cases [Hartig, et al., 2011]. We evaluated an NBIA patient at the NIH Clinical Center under the auspices of the NIH Undiagnosed Diseases Program. The subject presented at age 4 with speech difficulty followed by progressive spasticity and impaired walking. On examination he had dysarthria, psychomotor slowness, brisk reflexes, mild weakness and atrophy in the distal extremities, and small, pale optic discs. Electromyography showed denervation changes over multiple body segments, worse distally, consistent with a motor neuropathy. Brain MRI imaging showed symmetrical excess iron deposition in the globus pallidus (Fig. 1B). Exome sequencing identified a previously reported in-frame deletion in the coding region of the C19orf12 gene (c.197_199del, p.Gly66del) [Deschauer et al., 2012].
We also evaluated a consanguineous Brazilian family in which two affected siblings presented with walking difficulties at ages 14 and 15. Their clinical examinations showed spasticity, distal wasting, weakness, reduced sensation, and visual loss with bilateral optic atrophy. They became wheelchair-bound in their mid-thirties. Electrodiagnostic studies showed an axonal sensory and motor neuropathy, and MRI scans showed evidence of brain iron deposits in the globus pallidus (Supp. Figure S2). The younger sibling had memory loss and depression, but the older sibling had no psychiatric features. Using the GEM.app software [Gonzalez, et al., 2013] to analyze exome sequencing data, we identified the same variant seen in the Malian family (Ala63>Pro). An analysis of the markers around the C19orf12 locus showed that the Malian and Brazilian families had a shared haplotype, indicating that the variants likely have a common origin (Supp. Table S1). The Ala63 and Gly66 residues are conserved across a wide range of species including mammals, fish, and insects, indicating functional importance (Fig. 1C).
Computational analyses (http://www.predictprotein.org/) predict that the Ala63 and Gly66 residues are within a membrane domain. The extended nature of this hydrophobic domain and the presence of single or paired Pro residues within the region in all known species further raises the possibility that this domain forms a hydrophobic hairpin, as has been described in many ER proteins mutated in other HSPs, including the three most common forms [Blackstone, 2012]. In any case, the p.Ala63Pro mutation is very likely to be disruptive. We found that, in cultured cells, recombinant N-terminally-tagged wild-type C19orf12 had a complex and variable distribution. In many cells, it co-localized closely with the ER marker calreticulin, while in some other cells C19orf12 appeared to localize more to mitochondria (Fig. 2A and Supp. Figure S3A). The p.Ala63Pro missense mutant showed a dramatically different, more generalized distribution throughout the cytoplasm in a majority of cells, similar to p.Gly66del and another commonly reported NBIA mutant (p.Gly69Arg) [Hartig, et al., 2011] (Fig. 2A-B and Supp. Figure S3B). However, known polymorphisms in healthy controls, p.Val52Ile and p.Leu55Phe, which are also in the predicted membrane domain, did not alter the subcellular localization (Fig. 2B). The position of the epitope tag did not appear to influence these findings, since expression of an N-terminally HA-tagged C19orf12 protein gave results similar to those of C-terminal Myc-tagged C19orf12 for both the wild-type and p.Ala63Pro mutant proteins (Fig. 2C-D).
Figure 2.
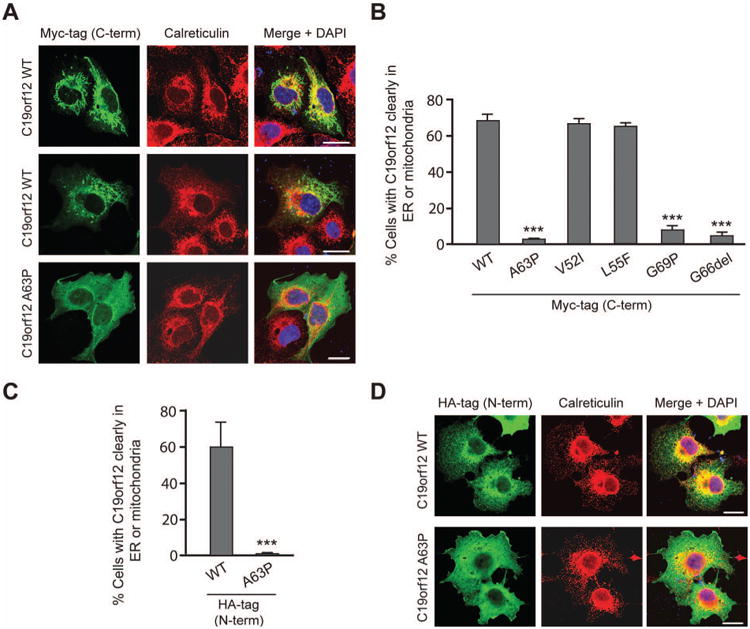
C19orf12 localizes to the ER, and the SPG43 and NBIA mutations alter its subcellular distribution. (A) COS7 cells expressing HA-tagged wild-type (WT) or p.Ala63Pro mutant C19orf12 were co-stained for endogenous calreticulin, an ER protein (red). (B) Quantification of the subcellular distribution of C19orf12 in cells expressing Myc-tagged, wild-type C19orf12 or C19orf12 containing the SPG43 mutation p.Ala63Pro, the NBIA-associated mutants p.Gly69Arg and p.Gly66del, or two known polymorphisms (p.Val52Ile and p.Leu55Phe). Cells with C19orf12 clearly at the ER or mitochondria were counted (n=3 trials, with >100 cells per trial). (C and D) Cells expressing HA-tagged wild-type or p.Ala63Pro mutant C19orf12 (green) were co-immunostained for calreticulin (red) and DAPI (blue) (D) and their distributions quantitated (C) as in panel B. Data represent the means ± SEM of three independent, blinded experiments. C-term, C-terminal; N-term, N-terminal. ***P<0.001. Bars, 20 μm.
A previous NBIA study localized C19orf12 to mitochondria [Hartig, et al., 2011]. Our studies of recombinant wild-type C19orf12 show a complex localization to a variety of organelles, primarily the ER but also the mitochondria (Fig. 2 and Supp. Fig. S2A). In future studies, it will be important to investigate the localization of the endogenous C19orf12 protein in more detail, with a particular focus on ER, mitochondria, and ER-mitochondrial contact sites.
SPG43 is an autosomal recessive spastic paraplegia with a presentation that includes distal amyotrophy, while NBIA is characterized by extrapyramidal and psychiatric features, optic atrophy, cognitive decline, and brain iron deposits in addition to spasticity. In this study, we show that C19orf12 mutation can present with or without typical features of NBIA, i.e., that it can cause spastic paraplegia with lower motor neuron features (SPG43) without vision loss and brain iron accumulation, as in the Malian family, or with vision loss and evidence of brain iron accumulation but without extrapyramidal features (dystonia and parkinsonism), as in the Brazilian family. These findings are an important extension of the phenotype previously reported with C19orf12 mutation [Hogarth et al., 2013] to include hereditary spastic paraplegia and formes frustres of NBIA.
The differences in phenotype could be due to different genetic or environmental modifiers in the Malian and Brazilian families. With the shared haplotype around the locus, the p.Ala63Pro mutation likely has a common origin and other variations within the gene are unlikely, but there could be different modifiers elsewhere in the genome. Alternatively the phenotypic differences could be stochastic. It is unlikely that they are due to a difference in the evaluations, since both families had thorough neurological exams and MRI brain scans. The presence of the heterozygous p.Ala63Pro mutation in about 1 in 1300 African-American alleles indicates that it may be a rare, previously unidentified cause of spastic paraplegia or NBIA in homozygotes in this population.
One of the pathogenic themes of recessive HSP mutations is abnormality of intracellular membrane trafficking, a process important for the maintenance of long corticospinal axons [Blackstone, 2012; Reid and Rugarli, 2010]. C19orf12 is predicted to be a membrane-bound protein [Hartig, et al., 2011], with a long hydrophobic domain extending from approximately amino acid residues 42 to 75. As with the mutations described in this study, mutations in most patients with NBIA cluster within the predicted helical membrane domain, highlighting its potentially important functional role. The mutations identified here and the p.Gly69Arg mutation described in other patients with NBIA, but not known polymorphisms in the predicted membrane domain, disrupt the subcellular distribution of C19orf12, indicating that the altered localization of the disease-causing mutations may contribute to the pathologic mechanism.
Although the cellular functions of C19orf12 remain unclear, an interesting parallel is seen with the clinical spectrum of FA2H (MIM# 611026) mutations. This gene is mutated in SPG35 as well as NBIA and leukodystrophy [Kruer, et al., 2010]. The FA2H gene product functions in fatty acid synthesis; the C19orf12 mutation characterized here alters sub-cellular localization of C19orf12 to the ER and may affect similar pathways. Furthermore, the phospholipase A2 gene PLA2G6 (MIM# 603604) is mutated in autosomal recessive NBIA, and this enzyme is involved in the metabolism of complex lipids [Lamari et al., 2013]. Though patients with NBIA present with features not seen in the SPG43 siblings, some overlapping findings are present, indicating that mutations in C19orf12 can also present across a neurological disease spectrum. Further functional and localization studies of C19orf12 should shed light on any common cellular mechanisms by which the mutations result in common clinical manifestations, and other genetic or environmental factors that account for the phenotypic differences.
Supplementary Material
Acknowledgments
We are grateful to the subjects and their families for participating in this study. We thank Dr. Elena Rugarli (University of Cologne) and the UDP bioinformatics team for assistance with bioinformatics, the NINDS DNA sequencing facility for DNA sequencing, Ms. Taryn Edsall (University of Michigan) for assistance with patient DNA sequencing, Dr. Fanny Mochel (Hôpital de La Salpêtrière) for her input in the manuscript, and Dr. Dramane Coulibaly of the Department of Neurology, Hôpital de Fann, Dakar, Sénégal for organizing the brain MRI for the Malian patient. We also thank the DNA and cell bank of UMR975 INSERM/UPMC and the SPATAX international network for providing patient DNA. This work was supported by the Intramural Research Programs of NINDS (1ZIA NS002974-13) and NHGRI, NIH grants R01NS072248 and R01NS054132 to S.Z., l'Agence Nationale pour la Recherche (SPAX, to A.D.), and grants from the VERUM Foundation and Association Strümpell-Lorrain to G.S. and A.D.
Grant Sponsor: NIH/NINDS intramural research funds (1ZIA NS002974-13: Studies of hereditary neurological disease: Disease gene identification).
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
Disclosure statement: The authors report no conflict of interest related to this study.
References
- Biesecker LG, Mullikin JC, Facio FM, Turner C, Cherukuri PF, Blakesley RW, Bouffard GG, Chines PS, Cruz P, Hansen NF, Teer JK, Maskeri B, et al. The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19(9):1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C, O'Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12(1):31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschauer M, Gaul C, Behrmann C, Prokisch H, Zierz S, Haack TB. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J Neurol. 2012;259(11):2434–2439. doi: 10.1007/s00415-012-6521-7. [DOI] [PubMed] [Google Scholar]
- Erichsen AK, Koht J, Stray-Pedersen A, Abdelnoor M, Tallaksen CM. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain. 2009;132(Pt 6):1577–1588. doi: 10.1093/brain/awp056. [DOI] [PubMed] [Google Scholar]
- Gonzalez MA, Acosta Lebrigio RF, Van Booven D, Ulloa RH, Powell E, Speziani F, Tekin T, Schüle R, Züchner S. GEnomes Management Application (GEM.app): A New Software Tool for Large-Scale Collaborative Genome Analysis. Hum Mut. 2013;34(6):842–846. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelly C, Zhu PP, Leonardis L, Papic L, Zidar J, Schabhuttl M, Strohmaier H, Weis J, Strom TM, Baets J, Willems J, De Jonghe P, et al. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am J Hum Genet. 2011;88(1):99–105. doi: 10.1016/j.ajhg.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AE. Hereditary spastic paraplegias. Semin Neurol. 1993;13(4):333–336. doi: 10.1055/s-2008-1041143. [DOI] [PubMed] [Google Scholar]
- Hartig MB, Iuso A, Haack T, Kmiec T, Jurkiewicz E, Heim K, Roeber S, Tarabin V, Dusi S, Krajewska-Walasek M, Jozwiak S, Hempel M, et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet. 2011;89(4):543–550. doi: 10.1016/j.ajhg.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, Davoine CS, Cruaud C, Durr A, Wincker P, Brottier P, Cattolico L, et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet. 1999;23(3):296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- Hogarth P, Gregory A, Kruer MC, Sanford L, Wagoner W, Natowicz MR, Egel RT, Subramony SH, Goldman JG, Berry-Kravis E, Foulds NC, Hammans SR, et al. New NBIA subtype: genetic, clinical, pathologic, and radiographic features of MPAN. Neurology. 2013;80(3):268–275. doi: 10.1212/WNL.0b013e31827e07be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruer MC, Paisan-Ruiz C, Boddaert N, Yoon MY, Hama H, Gregory A, Malandrini A, Woltjer RL, Munnich A, Gobin S, Polster BJ, Palmeri S, et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA) Ann Neurol. 2010;68(5):611–618. doi: 10.1002/ana.22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamari F, Mochel F, Sedel F, Saudubray Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: toward a new category of inherited metabolic diseases. J Inherit Metab Dis. 2013;36(3):411–425. doi: 10.1007/s10545-012-9509-7. [DOI] [PubMed] [Google Scholar]
- Landouré G, Knight MA, Stanescu H, Taye AA, Shi Y, Diallo O, Johnson JO, Hernandez D, Traynor BJ, Biesecker LG, Elkahloun A, Rinaldi C, et al. A candidate gene for autoimmune myasthenia gravis. Neurology. 2012;79(4):342–347. doi: 10.1212/WNL.0b013e318260cbd0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilleur KG, Traoré M, Sangaré M, Britton A, Landouré G, Coulibaly S, Niaré B, Mochel F, La Pean A, Rafferty I, Watts C, Shriner D, et al. Hereditary spastic paraplegia and amyotrophy associated with a novel locus on chromosome 19. Neurogenetics. 2010;11(3):313–318. doi: 10.1007/s10048-009-0230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65(1):19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid E, Rugarli EI. The Online Metabolic and Molecular Bases of Inherited Diseases. 2010 http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease/b/abstract/part28/ch228.1.
- Silva MC, Coutinho P, Pinheiro CD, Neves JM, Serrano P. Hereditary ataxias and spastic paraplegias: methodological aspects of a prevalence study in Portugal. J Clin Epidemiol. 1997;50(12):1377–1384. doi: 10.1016/s0895-4356(97)00202-3. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, Martin E, Ouvrard-Hernandez AM, Tessa A, Bouslam N, Lossos A, Charles P, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007;39(3):366–372. doi: 10.1038/ng1980. [DOI] [PubMed] [Google Scholar]
- Zhao X, Alvarado D, Rainier S, Lemons R, Hedera P, Weber CH, Tukel T, Apak M, Heiman-Patterson T, Ming L, Bui M, Fink JK. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29(3):326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- Züchner S, Wang G, Tran-Viet KN, Nance MA, Gaskell PC, Vance JM, Ashley-Koch AE, Pericak-Vance MA. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet. 2006;79(2):365–369. doi: 10.1086/505361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.