

Abstract
Classical molecular dynamics (MD) simulations based on atomistic models are increasingly used to study a wide range of biological systems. A prerequisite for meaningful results from such simulations is an accurate molecular mechanical force field. Most biomolecular simulations are currently based on the widely used AMBER and CHARMM force fields, which were parameterized and optimized to cover a small set of basic compounds corresponding to the natural amino acids and nucleic acid bases. Atomic models of additional compounds are commonly generated by analogy to the parameter set of a given force field. While this procedure yields models that are internally consistent, the accuracy of the resulting models can be limited. In this work, we propose a method, General Automated Atomic Model Parameterization (GAAMP), for generating automatically the parameters of atomic models of small molecules using the results from ab initio quantum mechanical (QM) calculations as target data. Force fields that were previously developed for a wide range of model compounds serve as initial guess, although any of the final parameter can be optimized. The electrostatic parameters (partial charges, polarizabilities and shielding) are optimized on the basis of QM electrostatic potential (ESP) and, if applicable, the interaction energies between the compound and water molecules. The soft dihedrals are automatically identified and parameterized by targeting QM dihedral scans as well as the energies of stable conformers. To validate the approach, the solvation free energy is calculated for more than 200 small molecules and MD simulations of 3 different proteins are carried out.
Introduction
Molecular dynamics simulations based on classical molecular mechanical (MM) force fields are increasingly used to provide atomic-level insights in studies of biological phenomena1-3. However, accurate force fields are needed to obtain meaningful results from MD simulations. The most widely used biomolecular force fields, such as CHARMM4-8, AMBER9, OPLS10, and GROMOS11, were optimized to model basic biological constituents, including proteins, nucleic acid and lipids. However, these force fields only cover a fairly restricted set of small organic compounds, and although models of additional compounds can be generated by analogy to the parameter set of a given force field, the accuracy of the resulting models can be limited. The challenges are even greater when compounds that have no close analogs within the popular biomolecular force fields are needed. This includes, for example, drug candidates, non-natural amino acids, and spectroscopic probes. The best way to address this issue is to have an objective algorithmic procedure to automatically parameterize an arbitrary molecule in a manner that is consistent with a given force field.
The first program able to model arbitrary organic compounds based on the atom types determined from local structure and pre-defined tabulated parameter sets was MacroModel12. While it addressed many of the issues arising when developing a general procedure, the models were not necessarily consistent with the most widely used force fields in biomolecular simulations. In this regard, a great leap forward was achieved by the general Amber force field (GAFF) presented by Wang et al.13, which automatically generates the parameters for arbitrary organic molecules consistent with the AMBER force field. In GAFF, atom types and internal parameters (bonds, angles, dihedrals and improper dihedrals) of a given compound are assigned from tabulated values according to an AMBER-consistent classification while atomic charges are fitted to match the results of quantum mechanical (QM) or semi-empirical calculations14. The program Antechamber15 in AmberTools was created to automatically parameterize small compounds in accord with GAFF. An independent effort to produce models of arbitrary small compound consistent with the OPLS force field was based on electrostatic partial charges determined using semi-empirical CM1 and CM3 calculations16. More recently, the CHARMM general force field (CGenFF) was introduced by Mackerell et al.17 to provide CHARMM-consistent force field parameters for small compounds and drug-like molecules. Two web portals, ParamChem (www.paramchem.org) and MATCH (http://brooks.chem.lsa.umich.edu/index.php?matchserver=submit), are available to automatically parameterize small compounds according to CGenFF17.
These computational tools represent important advances that greatly broaden the range of biomolecular systems that can be studied with simulations by enabling an objective and automatic parameterization of novel molecules. More importantly, most procedures above avoid the subjective manual adjustments of force field parameters, which ultimately undermine the predictive value of computations based on atomic models. Nevertheless, it is important to realize that despite the great advance that they represent, the accuracy of these MM models is not explicitly assessed during the automatic parameterization and may be limited. In particular, it is known that partial charges and dihedral parameters between molecules have limited transferabilities17,18, implying that any knowledge-based rule is necessarily an approximation to QM. Similarly, dihedral parameters are highly dependent on context and local non-bonded interactions and partial charges. For this reason, an automated method able to avoid tabulated values for these parameters is highly desirable.
Here we present an extension of these methods aiming at achieving an automatic parameterization for small molecules using ab initio QM results as the primary target data. Special efforts are made to optimize the electrostatic and dihedral parameters in a consistent manner. Atomic partial charges are optimized according to simultaneously best match the ESP from QM, as well as compound-water interactions with hydrogen-bonding donor or acceptor groups. ESP fitting has been used for the development of AMBER9,19 force fields and the fitting of water interactions has been used for the development of CHARMM6 force fields. Here, the two perspectives are combined to yield more robustly accurate models. Identifying automatically the dihedrals with low energy barriers that are most likely to undergo conformational change, the so-called “soft” dihedrals, the parameterization algorithm then proceeds from systematic one-dimensional (1D) dihedral scan and determination of conformer energies from QM. There have been a few attempts to parameterize MM models with QM target data recently; Ren et al. proposed a procedure to automatically generate a polarizable force field consistent with AMOEBA for small compounds20, and Wang et al. presented an iterative scheme to develop a polarizable model for water molecule targeting QM forces and energies of clusters of water molecules21. Nevertheless, to our knowledge, this is the first automatic parameterization tool relying on QM data that combines the information from ESP and water interactions together, and that detects, scans and optimizes all soft dihedral parameters. The methodology presented here has been implemented in a web server, General Automated Atomic Model Parameterization (GAAMP, http://gaamp.lcrc.anl.gov/). Although this server is not open to public yet since QM calculations are very expensive, we will release the source code for parameterization to public then one can use the code to do parameterization on local computers. A gateway for GAAMP will be set up based on XSEDE (www.xsede.org) for public access and the link will be announced on GAAMP website.
Parameterization Method
The functional form of the potential function used in the parameterization is compatible with the non-polarizable CHARMM force field1,
| (1) |
With some small modification, the force field can be optimized in a manner compatible with AMBER. The main difference is that the 1-4 non-bonded charge-charge interactions are scaled by 0.833 in AMBER, but are fully accounted for in CHARMM (the E14FAC parameter is 1.0). From this point on, three specific actions affect the final parameterization: (1) verification and adjustment of equilibrium bond length and angle parameters, (2) charge fitting using QM target data including ESP and specific interaction with water molecules (Fig. 1b), and (3) dihedral parameter fitting using QM target data (Fig. 1c). A detailed flowchart of the proposed scheme for automated parameter determination is depicted in Fig. 1. As input, the user must provide a structure file in the format of the protein data bank (pdb) or mol2. The initial input structure file must contain all atoms, including hydrogens, and ionizable groups must be correctly protonated. Since the initial structure is first refined by geometry optimization at the AM1 level, it is important that the bond length and angle in the initial structure be reasonably close to chemically realistic values. The refined atomic structure can be ran through the program Antechamber15 or CGenFF17 to generate initial topology and parameter files for the molecule in CHARMM format. A detailed flowchart is shown in Fig. 1.
Fig. 1.

a) This scheme shows the flowchart of GAAMP parameterization. b) This scheme shows the flowchart of electrostatic parameters optimization. c) This scheme shows the flowchart of dihedral parameters optimization.
Verify equilibrium bond and angle parameters in GAFF
For some specific molecules, the equilibrium bond lengths or angles from GAFF or CGenFF may be inaccurate. For this reason, the bond lengths and angles of the molecule in the structure optimized at HF/6-31G* or higher level are compared with the values observed in the structure optimized using the MM force field. If the deviations are too large, e.g., 0.05 Å for bond length and 8° for angle, then the internal equilibrium values of the force field are substituted by the values obtained in the optimized QM geometry.
Charge fitting in non-polarizable model
The MM charges of a molecule of interest are fitted to best-reproduce target data obtained by QM calculations. As is customarily done, the numerical problem is cast as the optimization of an objective function constructed to account for all the target data19. The target data includes the ESP calculated from a QM method at a large number of points disposed around the molecule (illustrated in Fig. 2, left). In addition, the target data also includes the interaction energy (Eint) and associated inter-molecular distances (Rint) with explicit water molecules6 if the molecule has hydrogen bonding donors or acceptors (illustrated in Fig. 2, right). Lastly, the objective function also includes weak restraints to prevent unphysical values of the MM charges, which is particularly important in the case of buried atoms. With these elements, the objective function used in the optimization procedure of the MM charges is written as the sum of three terms: the objective function for the electrostatic potential (Eq. 3), the objective function for compound-water interactions (Eq. 4) and the restraints on reference charges (Eq. 5).
| (2) |
The contribution to the objective function from the electrostatic potential is,
| (3) |
where ngrid is the number of grid points at which the ESP are calculated, and and are the values of the ESP calculated at the i-th point from QM and MM, respectively. This part essentially follows the procedure used in the development of the AMBER force field.19 In the present implementation, the points where the ESP is evaluated are organized into five layers of grids that are 1.4, 1.6, 1.8, 2.0, and 2.2 times the van der Waals radii. 5To remain consistent with the standard approaches used for the non-polarizable force fields CHARMM and AMBER, the QM ESP calculations are carried out at the HF/6-31G* level.
Fig. 2.

This figure illustrates the strategy of GAAMP used for charge fitting which combines ESP fitting (shown in the left plot) and compound-water interaction fitting (shown in the right plot).
The contribution to the objective function from compound-water interactions is,
| (4) |
where wEint and wRint are the weights set for Eint and Rint respectively. The standard output of the program reports on how the various target data ϕiQM, EintQM, and RintQM are reproduced by MM model. Accounting explicitly for the interactions with water molecules follows the procedure commonly used in the development of CHARMM force field6. Following the protocol recommended by MacKerell et al.,6 the QM calculations are performed at the HF/6-31G* level without basis set superposition error (BSSE) correction. The QM interaction energy Eint is kept unchanged for charged molecules, while it is scaled by 1.16 for neutral molecule. The QM optimal distance, Rint is shifted by −0.20 for neutral molecule. For charged compounds, a shift of −0.05 Å has been used in this work considering the average Rint from MM is often slightly smaller than the value in QM for a set of ion-water interactions22. Different value for such a shift (e.g., −0.1 in ref23 and −0.2 Å in ref17) have been previously suggested. Determining an optimal shift should be done in future work. Partial charges are then re-optimized, now targeting simultaneously the ESP and the compound-water interactions (Eint and Rint). The geometry of the molecule is taken from the optimized QM structure and kept rigid during the calculation of Eint and Rint in MM. Only relatively strong hydrogen bonds (Eint < −2kcal/mol) are included in the target data.
Lastly, the objective function also includes weak restraints preventing the fitted MM charges from deviating too far from reference values. The latter are taken as the AM1-BCC14 charges assigned by Antechamber. This contribution to the objective function is written as,
| (5) |
where wCG is the weight set for charge restraint, f(qi, qi0) = 0 if ∣qi – qi0∣<=0.02 , otherwise, f(qi, qi0) = (∣qi – qi0∣–0.02)2). This form allows the MM charges to deviate slightly from the reference values without penalty. It should be noted that the present choice of restraint and reference values differs from the original RESP procedure19, where the MM charges were weakly restrained to zero.
Electrostatic parameter fitting for the Drude model
The method described above can easily be generalized with minor modifications to automatically generate the electrostatic parameters of a polarizable model based on the classical Drude oscillators24-34. In the Drude polarizable force field24,25, a charged auxiliary particle attached to an atom via a harmonic spring is introduced to mimic the electronic response and account for induced polarization effects. As such, all the Drude particles can be treated as part of the MM force field and minimizing the energy over the position of the Drude particles recovers the familiar induced polarization self-consistent field (SCF) treatment. The fitting procedure for the electrostatic parameters of a model is essentially the same, except that more QM data are needed to evaluate how a polarizable molecule responds to an applied external electric field. As described in Anisimove et al.25, this is accomplished by placing a test charge of +0.5e at various positions around the molecule and re-calculating the perturbed ESP from QM to determine the polarizability of the different atoms in the molecule. The same situation with a test charge is reproduced in the MM model during the charge fitting procedure. In addition, the potential function comprises a few contributions that are specific to the Drude model, including anisotropic polarization and screened induced dipole interactions26. Lastly, as in the case of the non-polarizable force field, some restraints are introduce to prevent large unphysical deviations of all the parameters. The objective function is written as,
| (6) |
The contribution from ESP is,
| (7) |
where ϕi and ϕij, p represents the unperturbed and perturbed ESP respectively. There are npert configurations used to calculate perturbed ESP and ngrid grid points where ESP are calculated. The contribution from the restraint on target charges assigned by antechamber (AM1-BCC14) is,
| (8) |
where f(qi, qi0) = 0 if ∣qi – qi0∣<=0.03 , otherwise, f(qi, qi0) = (∣qi – qi0∣–0.03)2). Generally, smaller weight (wCG) on the charge restraint than that in non-polarizable model should be used since AM1-BCC14 charges were specifically parameterized for non-polarizable model. The restraint on the atomic polarizabilities takes the form,
| (9) |
where represents the default polarizability and wα represents the weight of the restraint on target polarizabilities. Atomic polarizabilities from Miller35 scaled by a factor of 0.7 serve as the target value, . The restraint on the shielding parameters takes the form,
| (10) |
where represents the default “Thole” parameters that controls the electrostatic screening of induced dipole interactions within 1-2 and 1-3 pairs. The fitted parameters are restrained to avoid large deviations from the original values . In current development of Drude force field in CHARMM, a starting value of 1.3 is used as for . However, some molecules can be unstable with this value, especially those with hetero-cycles bonded with atoms with high electronegativity. A smaller value, e.g., 0.2 was used for such cases. It is possible that such instabilities may be circumvented with alternative treatments of the 1-2, 1-3, and 1-4 intramolecular non-bonded interactions within the MM model in the future. The anisotropic contribution is an energy term introduced to improve the induced polarization in response to applied electric field in the case of specific groups such as the backbone carbonyl in proteins26. The restraint on the anisotropic term is written as,
| (11) |
f(K11,i, K110)=0 if ∣K11,i – K110∣<=200 kcal/Å2 where K110 is set 50 kcal/Å2, otherwise, f(K11,i, K110)=(∣K11,i – K110∣–200)2. F(K22,i, K220) and f(K33,i, K330) have the same form as f (K11,i, K110). Such restraints are necessary to make sure that the effective spring constant for the Drude particle will not be too small (which may allow the Drude go far away from nucleus and lead to issues of instabilities) or too large (which would require an inefficiently small integration time step). The definition of χ2wat_int is same as that in non-polarizable model. In practice, the starting topology and parameter file will be automatically generated by introducing the entries of default polarizability, anisotropy and shielding parameters into the GAFF topology and parameter files generated by Antechamber.
Dihedral parameter fitting
Once the electrostatic parameters are determined, it is necessary to obtain accurate parameters for the dihedral angles. Of particular importance for the force field are those dihedrals with small energy barriers because they largely control the accessible rotameric states and the overall flexibility of the molecule. Once such “soft” dihedrals have been identified, the parameterization uses information from QM calculations as target data for both a series of 1D dihedral energy profile as well as the energy of conformers. An important first step concerns the automatic identification of all the soft dihedrals within a molecule. There are several possible ways to carry out this task. The simple protocol that is adopted in the current algorithm consists in constructing a list of all dihedrals in the molecule, and then excluding those involved in cycles. Also excluded are the dihedral associated with the trivial rotation of methyl groups considering that the QM energy profile of such dihedrals generally can be reproduced reasonably by the GAFF or CGenFF, which also decreases the overall computational cost.
The second step consists in determining all the putatively stable local minima based on isomerization of the pre-identified soft dihedrals in the following way. All possible combinations of soft dihedrals are enumerated and local geometry optimizations are carried out for all putative conformers. This is followed by a clustering of the dihedral value to detect redundancies and obtain an estimate of all possible minima for each soft dihedral. As a first pass, this initial task relies on the dihedral potential from the MM force field obtained directly from GAFF or CGenFF. If the number of soft dihedrals is too large, the initial configurations for geometry optimization can be randomly generated and special care is taken to make sure all soft dihedrals are sampled thoroughly. Once this is done, an optimal structure is selected for each soft dihedral to carry out a 1D dihedral scan at the QM level. During this 1D scan, all other soft dihedrals are kept fixed at their local minima to avoid abrupt changes of configurations in the molecule, which also allow us to fit soft dihedrals independently. The configuration used to carry out the QM scan along a given dihedral angle must be selected carefully to decrease the possibility of non-bonded steric clashes that would obscure the data. For this purpose, corresponding 1D scans are first carried out using the MM force field and only the configuration producing the lowest torsion energy barrier is retained. The 1D scans from QM determine all local minima of each soft dihedral. Once this is done, the information is then used to carry out a first optimization of the dihedral parameters for all the soft dihedrals in which they are all considered separately. The objective function is,
| (12) |
where wi represents the weight set for configurations in 1D torsion scan, with n=1,2,3,4,6; σn=0/π, kn and E0 can be determined efficiently by solving a set of linear equations or other quasi-Newton method like L-BFGS36,37. The implementation of L-BFGS in NLopt38 is adopted. Eothers is defined as the total MM energy without the contribution from the dihedral energy of the soft dihedral selected for parameter fitting. Configurations with very high energies, e.g., 20 kcal/mol higher than the lowest energy along the 1D scan are not included in the optimization. During optimization, the force constants kn are constrained to remain positive for the sake of simplicity.
The above optimization procedure based on the 1D scans leads to improved dihedral parameters, but it is not guarantied to yield accurate energy ranking of the accessible conformers of the molecule. To this end, an additional step is taken in which the dihedral parameters are fitted again, this time using simultaneously the information from the 1D scans and the conformer energies, with
| (13) |
and
| (14) |
where Erot0 is a constant to be fitted and . The weights of conformers in this form are chosen to enhance the contribution from configurations with low MM and QM energies. Optimization of the parameters using the conformer energies as target data is helpful to obtain a final model able to accurately reproduce the relative energies of the accessible conformations with lowest energies. A maximum of 200 conformers are selected based on the MM energy for further geometry optimization using QM. For very large molecules (more than 8 soft dihedrals), it becomes very challenging to select meaningful conformers among numerous possible conformers. In this case, the energies of conformers are not included in our target data and dihedral parameter fitting only relies on 1D dihedral scans.
Parameterization of unnatural amino acids side chains
With minor adjustments, the current algorithm can be used to parameterize any amino acids, including unnatural amino acids (UAAs), in a manner that is consistent with the backbone from the rest of the MM force field. Here the procedure was used to produce UAAs models consistent with the backbone of the CHARMM force field, although models consistent with the AMBER force field could be produced in a similar fashion. First, the program determines the partial charges of the side-chain compound (the side-chain plus one hydrogen atom) using the procedure of charge fitting described above under the constraint that the charge of the hydrogen atom added is fixed at zero. As a second step, the program generates CHARMM format topology, parameter and coordinate files for the full molecule, comprising the side-chain molecule and the backbone of an alanine dipeptide. As a third step, the program identifies the soft dihedrals within the side-chain and the parameters are optimized according to the procedure described above. During the side-chain dihedral fitting, the backbone atoms are fixed with the ϕ and ψ backbone dihedrals in an α=helical conformation (−60 and −45 for ϕ and ψ). For the sake of simplicity, only 1D dihedral scans from QM are used for dihedral parameter fitting to avoid considering the multiple conformers of the dipeptide. The parameters of the resulting model has the Param27 CHARMM (CHARMM27) force field6,7 for the backbone, and the current optimization for the side-chain.
Computational details
The library of NLOpt38 was used for parameter optimizations. L-BFGS36,37 algorithm was used for charge and dihedral parameter optimization as well as the molecular geometry optimization without constraints. Augmented Lagrangian algorithm39,40 conjugated with L-BFGS36,37 was used for the geometry optimization with constraints on selected soft dihedrals. Numerical gradients by central differences are used for L-BFGS optimizer. Our programs were written in C++, bash shell script and Python.
Solvation free energy calculations
The absolute solvation free energy of small compounds was calculated and decomposed into three components (repulsive, dispersive and charge term) following a FEP simulation protocol developed in our group41-43. Replica exchange method42,44 was used to enhance the sampling to get better convergence. We recently ported the implementation of FEP/REMD42 in CHARMM into NAMD45. The simulations of non-polarizable models were performed in NAMD and the simulations of polarizable model were performed in CHARMM1. In non-polarizable models, the compound was solvated in a cubic water box of TIP3P water molecules46 with dimension ~20 Å and periodic boundary condition (PBC) was imposed. Long-range electrostatic interactions were computed using particle mesh Ewald summation47,48 with a Ewald splitting parameter 0.34 Å−1, a grid spacing of ~0.6 Å, and a sixth-order interpolation of the charge to the grid. Non-bonded van der Waals interactions were smoothly switched to zero between 10 and 12 Å. The isothermal-isobaric ensemble was simulated using Langevin thermostat49 and Langevin piston50. The SETTLE algorithm51 was used to keep TIP3P water molecules rigid and RATTLE algorithm52 was used to fix the length of those bonds connecting heavy atoms and hydrogen atoms in the compound. The multiple time step, RESPA algorithm53 implemented in NAMD45, was used for 4fs integration time step for non-bonded interactions and 2fs time step for bonded interactions. For each value of the thermodynamic coupling parameter, λ, equilibrium properties were averaged over a 500 ps molecular dynamics simulation after an initial equilibration of 300 ps. Exchanges of neighboring replica were attempted every 200 fs. Weighted histogram analysis method (WHAM)54 was used in data processing. A long-range correction for Lennard-Jones interactions55 beyond the cutoff was added to the calculated solvation free energy in post-analysis. MD simulations of the polarizable Drude models were carried out according to a similar approach, with a few differences: the CHARMM program was used with the SWM4 water model27, and the VV2 integrator56 and Nosé-Hoover thermostat were used with no multiple time step algorithm to sample isothermal-isobaric ensemble28.
Ab initio calculations
All the ab initio calculations were performed with the program Gaussian 0957. AM1 was used for the pre-optimization for the initial structure (step 2 in Fig. 1b) before calling Antechamber to generate the initial force field (step 3 in Fig. 1b). HF/6-31G* was used for geometry optimization (step 5 in Fig. 1b) as well as ESP calculation (step 7 in Fig. 1b) in the non-polarizable force field parameterization. B3LYP/aug-cc-pVDZ was used in the unperturbed and perturbed ESP calculation in the Drude force field parameterization25. The interactions between the molecule to be parameterized and water were calculated at the HF/6-31G* level without BSSE (step 9 in Fig. 1b), following the recommended prescription6. Calculation at the HF/6-31G* or MP2/6-31G* level were used to perform the 1D dihedral scan (step 13 in Fig. 1c), and the geometry optimization of the various conformer states (step 15 in Fig. 1c). 6-31+G* basis set was used for anions. Ultimately, the choice of basis set and QM level depends on the size of the molecule and the accuracy desired. Any reasonable combinations of theory level and basis set can be applied with the current parameterization procedure.
Result and discussion
Electrostatic parameters
Coulomb interactions play an important role in intra- and inter-molecular interactions. As a consequence, carefully optimized partial charges are essential for an accurate MM force field. There is a wide variety of methods to determine partial charges58. The electrostatic potential (ESP) on the surface of a given molecule, calculated using QM or semi-empirical methods as illustrated in the left plot of Fig. 2, serves as the target data for the charge fitting in AMBER force field development9,19. Methods based on ESP fitting are easy to implement and carry the important advantage that the resulting charges are not coupled with Lennard-Jones parameters during fitting. Alternatively, matching the interactions between the compound and water molecule calculated by QM as illustrated in the right plot of Fig. 2 is the common approach used in the development of the CHARMM6 force field. For polar molecules, the hydrogen bonds between the compound and water molecules are very important, and including these interactions directly in the optimization can help to generate models that are more accurate.
Combining ESP and compound-water interactions together in the charge optimization makes it possible to take advantage of both perspectives. Because the strength of the hydrogen bonding interaction in a MM model is primarily determined by the charges of a small number of atoms close to the hydrogen bond donor/acceptor in the compound, a reasonable assumption is that a model based on ESP partial charges can be improved if QM data of compound-water interactions is included in the target data during parameter optimization. First, charges are optimized based on QM ESP data (step 8 in Fig. 1b). Then, the charges are further optimized with the QM data of compound-water interactions together with QM ESP (step 10 in Fig. 1b). For the sake of internal consistency, the compound-water interactions within the MM force field should preferably be evaluated using the geometry of the compound energy-minimized within the MM force field. However, as the initial MM model is either incomplete or perhaps grossly inaccurate, it is not possible to rely on the optimized geometry based on the initial MM model. To circumvent this problem, an iterative procedure is used to optimize all the parameters of the force field. First, a cycle of charge optimization (steps 8-10 in Fig. 1b) is carried out using the fixed QM geometry of the compound. Then, using the resulting charges, a first optimization of the dihedral parameters is carried out using the 1D dihedral scan profiles from QM (step 14 in Fig 1c). Once this done, the charges of the model are re-optimized (step 10 in Fig. 1b) but this time using the energy-minimized geometry of the compound based on the MM force field. Using this new set of partial charges, the dihedral parameters are then re-optimized once more using the 1D dihedral scan profiles from QM (step 14 in Fig 1c). Finally, this is followed by a global optimization targeting both the 1D scan and conformer energies from QM (step 16 in Fig 1c). This iterative procedure, where charges and dihedral parameters are completely re-optimized twice, helps increase the stability of the optimization and the accuracy of the final model.
Dihedral parameter
Dihedral parameters often correspond to some of the softest degrees of freedom in a molecule and an accurate parameterization is critical to sample correct configurations in simulations. With the increase of the number of soft dihedrals, the number of accessible configurations increases exponentially. Both GAFF13 and CGenFF17 use lookup tables to assign dihedral parameters for given dihedral types. However, this method does not always give reasonable parameters, especially when the assigned partial charges in the compound to be parameterized are significantly changed from those charges assigned in the analog used in the development of the force field.
The results of two small compounds are presented to demonstrate the performance of the algorithm of dihedral parameter fitting. The 1D dihedral energy profiles for butyric acid methyl ester are shown in Fig. 3. GAFF/AM1-BCC works reasonably for this molecule. The 1D dihedral energy profiles calculated by the parameters fitted by GAAMP perfectly match QM results as shown in Fig. 3b. Fig. 3c shows that the QM conformer energies also can be reproduced reasonably well.
Fig. 3.

a) A snapshot of butyric acid methyl ester is shown. ϕ1, ϕ2 and ϕ3 are corresponding to dihedrals, 1-2-3-4, 2-3-4-6 and 3-4-6-7 respectively. b) The comparison of QM, GAAMP and GAFF/AM1-BCC dihedral energy profiles for three dihedrals. c) The energies of eight conformers calculated in QM and MM are compared.
The results for a slightly larger compound, N-phenylbenzamide, are shown in Fig. 4. The model with the optimized GAAMP dihedral parameters can reproduce the 1D dihedral energy profiles from QM reasonably well. In contrast, the torsion potential from GAFF/AM1-BCC encounters some difficulties with this molecule. For instance, the energy profile along the ϕ1 dihedral, particularly the energy basin around 180°, is not described accurately. Moreover, the energy profile of the ϕ3 dihedral significantly deviates from the QM result. The origin of these inaccuracies seems due to the improper parameters for four dihedrals in GAFF, “X-C-CA-X 3.625 2 180.0”. Although HF/6-31G* was used for the 1D dihedral scan in the present example, it would be straightforward to generate QM target data using other affordable high-level QM methods and larger basis sets.
Fig. 4.

a) A snapshot of N-phenylbenzamide is shown and three soft dihedrals are highlighted with red eclipses. ϕ1, ϕ2 and ϕ3 are the three dihedrals highlighted from left to right in the snapshot. b) The comparison of QM, GAAMP and GAFF/AM1-BCC dihedral energy profiles for three dihedrals.
As an illustrative example of a large molecule, the present procedure was used to parameterize Imatinib (or Gleevec), a commercial drug used in the treatment of certain cancers59. As shown in Fig. 5, this molecule contains 69 atoms and 8 soft dihedrals. The 1D dihedral energy profiles for the fitted parameters and GAFF/AM1-BCC are compared with QM in Fig. 5. GAFF/AM1-BCC does not perform well for ϕ4 and ϕ6, although the dihedral energy profiles for other dihedrals are reproduced correctly. The dihedral ϕ6 in Imatinib is similar to ϕ3 in N-phenylbenzamide studied above. The deviation also comes from improper dihedral parameters in GAFF, X-C-CA-X. In contrast, the optimized dihedral parameters from GAAMP can reproduce QM energy profiles reasonably well for all dihedrals. This parameterization took ~40 hours on 12 cores of Intel Xeon 2.67GHz using only the 1D dihedral scan QM profiles at the HF/6-31G* level (no conformer energies fitting). Starting from the optimized structure in QM, the optimized structure using the GAAMP optimized parameters deviates from the initial structure by 0.33 Å.
Fig. 5.

a) A snapshot of Imatinib is shown and ϕ1, ϕ2,…,ϕ8 are the eight dihedrals highlighted from left to right in the snapshot. b) The comparison of QM, GAAMP and GAFF/AM1-BCC dihedral energy profiles for eight dihedrals.
Dihedral parameters are coupled to the underlying non-bonded parameters. For this reason, it can be very challenging to automatically fit dihedral parameters when 1-4 bonded pair of atoms carry large partial charges. Hydrazine is used as an example to demonstrate this issue in Fig. 6. The partial charges on hydrogen atoms 3, 4, 5 and 6 are 0.379 e. The energy barriers between local minima cannot be captured correctly neither by GAAMP nor GAFF although the positions of the local minima are closely reproduced. Coulomb interactions in the MM force field are very strong and the model cannot reproduce the QM dihedral energy profile, even when trying to adjust the dihedral parameters. In this case, scaling down the non-bonded interactions between two atoms with short distance might be helpful, showing that the electrostatic parameters cannot always be determined without considering the internal energy of the molecules.
Fig. 6.

a) A snapshot of hydrazine is shown. b) The comparison of QM, GAAMP and GAFF/AM1-BCC dihedral energy profiles for dihedrals 3-1-2-5.
Solvation free energies of amino acid side-chain analogs
Examining the hydration free energies of amino acid side-chain analogs is of interest as it reflects the accuracy of protein force field60. To assess the performance of GAAMP, the solvation free energies of 15 neutral amino acid side-chain analogs was calculated and compared with GAFF and other force fields in literature60. The results are given in Table 1. For small non-polar molecules, e.g., alkanes like Ala, Val, Leu and Ile, the results from GAAMP are almost the same as the values using GAFF. This is expected since the Lennard-Jones parameters from GAFF are used, and the electrostatic contribution is minor in these molecules. For other molecules with hydrogen donors/acceptors, such as Ser, Thr and Hid, noticeable improvements are observed in terms of solvation free energies when using GAAMP. Other than GAFF, CHARMM Param27 (CHARMM27) was also used to provide the initial parameters. The results of the optimized parameters, CHARMM27-GAAMP, lead to reasonably good solvation free energies compared with GAFF/AM1-BCC, although the results are a little better for the original CHARMM27. Most errors in CHARMM27-GAAMP come from polar molecules, Gln, Hid and Hie. Based on the average unsigned error (AUE), the three best models are, CHARMM27, OPLS and GAFF-GAAMP, in this order. A systematic shift of ~−0.4 kcal/mol in solvation free energies were found in CHARMM27 compared with CHARMM22. Possible discrepancies may be attributed to different TIP3P models between Shirts’60 and this work. Unlike the TIP3P model used by Shirts, Lennard-Jones parameters on the hydrogen atoms were added in the TIP3P model used in CHARMM, which makes the solvation free energies in present work more negative. Differences in the free energy schemes may also be the cause of these small discrepancies.
Table 1.
The solvation free energies of 15 amino acid side-chain analogs calculated with GAFF/AM1-BCC, GAFF-GAAMP, CHARMM27 and CHARMM27-GAAMP, other force fields (AMBER, CHARMM22 and OPLS-AA)60 as well as experiment data are shown. Unit is in kcal/mol.
| Mol | GAFF/ AM1-BCC |
GAFF- GAAMP |
CHARMM27 | CHARMM27- GAAMP |
AMBER | CHARMM22 | OPLS-AA | exp |
|---|---|---|---|---|---|---|---|---|
| Ala | 2.49 | 2.51 | 2.31 | 2.33 | 2.57 | 2.44 | 2.31 | 1.94 |
| Val | 2.42 | 2.36 | 1.98 | 2.06 | 2.69 | 2.52 | 2.59 | 1.99 |
| Leu | 2.42 | 2.28 | 2.37 | 2.40 | 2.72 | 2.94 | 2.69 | 2.28 |
| Ile | 2.43 | 2.35 | 2.04 | 2.13 | 2.84 | 2.67 | 2.73 | 2.15 |
| Ser | −3.60 | −3.74 | −4.96 | −3.48 | −4.37 | −4.59 | −4.36 | −5.06 |
| Thr | −3.62 | −3.88 | −4.86 | −3.53 | −3.83 | −4.22 | −4.11 | −4.88 |
| Phe | −1.29 | −0.87 | −0.53 | −1.14 | 0.10 | 0.09 | −0.54 | −0.76 |
| Tyr | −5.86 | −6.17 | −5.16 | −5.82 | −4.23 | −4.46 | −5.25 | −6.11 |
| Cys | −0.45 | −0.06 | −0.52 | −1.30 | 0.11 | 0.02 | −1.59 | −1.24 |
| Met | 0.04 | 0.19 | 0.27 | −1.00 | 0.91 | 1.08 | −1.27 | −1.48 |
| Asn | −8.99 | −7.96 | −8.15 | −7.66 | −7.80 | −7.89 | −8.53 | −9.68 |
| Gln | −9.03 | −7.01 | −7.82 | −6.77 | −7.69 | −7.51 | −8.4 | −9.38 |
| Trp | −7.34 | −5.72 | −4.57 | −5.43 | −4.88 | −3.57 | −4.44 | −5.88 |
| Hid | −8.01 | −9.47 | −10.44 | −8.89 | −8.43 | −10 | −8.87 | −10.27 |
| Hie | −8.46 | −9.03 | −10.77 | −8.11 | −8.98 | −10.27 | −9.05 | −10.27 |
| AUE | 0.92 | 0.85 | 0.63 | 0.89 | 1.22 | 1.06 | 0.75 |
Solvation free energies of 217 compounds in the non-polarizable models
To further test the current procedure, we parameterized 217 small neutral compounds and calculated the solvation free energies. For those molecules without hydrogen bonding donor/acceptor, the partial charges were fitted only on the basis of ESP data. The calculated values for 98 compounds without hydrogen-bonding donor/acceptor using GAFF/AM1-BCC and GAAMP fitted parameters are compared with experimental values in Fig. 7a and 7b. Higher correlation coefficient and smaller AUE can be achieved using our RESP fitting compared with using GAFF/AM1-BCC. For the remaining 119 molecules having H-bond donor/acceptor, the partial charges were fitted with RESP and RESP combined with molecule-water interactions. The solvation free energies calculated with three sets of parameters, GAFF/AM1-BCC, GAAMP/RESP and GAAMP (RESP combined with molecule-water interactions), are compared with experimental values in Fig. 8a, 8b and 8c respectively.
Fig. 7.

The calculated solvation free energies for 98 compounds without hydrogen-bond donor/acceptor are compared with experimental value. a) Using GAFF/AM1-BCC. The AUE is 0.74 kcal/mol. b) Using GAAMP/RESP charges. The AUE is 0.58 kcal/mol.
Fig. 8.


The calculated solvation free energies for 119 compounds with hydrogen-bond donor/acceptor are compared with experimental value. a) Using GAFF/AM1-BCC. The AUE is 0.94 kcal/mol. b) Using GAAMP/RESP charges. The AUE is 1.35 kcal/mol. c) Using GAAMP charges (by fitting both RESP and compound-water interactions). The AUE is 1.00 kcal/mol.
It is important to note that models based on RESP alone do not lead to good correlation between calculated and experimental solvation free energies for the compounds including hydrogen-bond donor/acceptor, although such method works reasonable well in deriving partial charges for the compounds without hydrogen-bond donor/acceptor. For the partial charges derived with the original RESP19, the same behavior is observed and a low correlation coefficient with experimental data (0.60) is found for the solvation free energy of the compounds with hydrogen-bond donor/acceptor by analyzing the data in literature43. The results in this work suggest that including compound-water interactions as target data can substantially improve the quality of partial charges derived from RESP when a compound has hydrogen-bond donor/acceptor.
To gain more insights about how including compound-water interactions improves the fitted charges in solvation free energy calculations, the AUE of the solvation free energies have been compared within the compounds with same functional groups for GAAMP/RESP and GAAMP models. In several categories, such as aliphatic amines, aromatic amines, esters, ethers and nitro compounds, the AUE using GAAMP models are 0.5~1.1 kcal/mol smaller compared with the AUE using GAAMP/RESP models. On the other hand, the AUE using GAAMP models are 0.4~0.7 kcal/mol larger compared with the AUE using GAAMP/RESP models for amides, carboxylic acids and ketones. More information is provided in supplemental material.
The data used in both Fig. 7 and 8 can be plotted together as shown in Fig. 9. The parameter sets fitted by GAAMP lead to comparable correlation coefficient between the calculated and experimental solvation free energies for 217 small compounds compared with GAFF/AM1-BCC. The average unassigned error using the parameters from GAFF/AM1-BCC and GAAMP are 0.85 and 0.81 kcal/mol respectively.
Fig. 9.

The calculated solvation free energies for 217 compounds, including both polar and non-polar molecules, are compared with experimental value. a) Using GAFF/AM1-BCC. The AUE is 0.85 kcal/mol. b) Using GAAMP parameters. The AUE is 0.81 kcal/mol.
Solvation free energies of 217 compounds in the Drude polarizable models
The method for automated parameterization is general and also applicable for polarizable model based on classic Drude oscillator24,25. A polarizable force field is expected to be more accurate since more details are added to account for induced electronic polarization effect24. However, this is only true if all the parameters in the Drude model have been optimized carefully. During the parameterization, we rely on the bond, angle, improper dihedral and Lennard-Jones parameters from GAFF, which may limit the accuracy of the Drude models generated here. Ultimately, we would need to generate a basis set of Lennard-Jones parameters suitable for the Drude models. Here some preliminary results on the calculations of solvation free energies of 217 small compounds using the automatically generated Drude models are reported in Fig. 10. The correlation coefficient is 0.87, which is comparable with the value using GAFF/AM1-BCC report by Shivakumar43. Although the polarizable models do not yield a significant improvement over the non-polarizable models in the present case, they may be more accurate with further refinement of the Lennard-Jones parameters of the different atom types. Ren et al. calculated the solvation free energy for 25 small compounds parameterized automatically within the AMOEBA polarizable force field20. For these 25 compounds, they reported an AUE of 0.65 kcal/mol. This is slightly smaller than the AUE reported in this work for 217 compounds, which is 0.81 and 0.92 kcal/mol with non-polarizable and Drude models respectively. However, the set of compounds considered in the present work is considerably larger and more diverse than theirs.
Fig. 10.
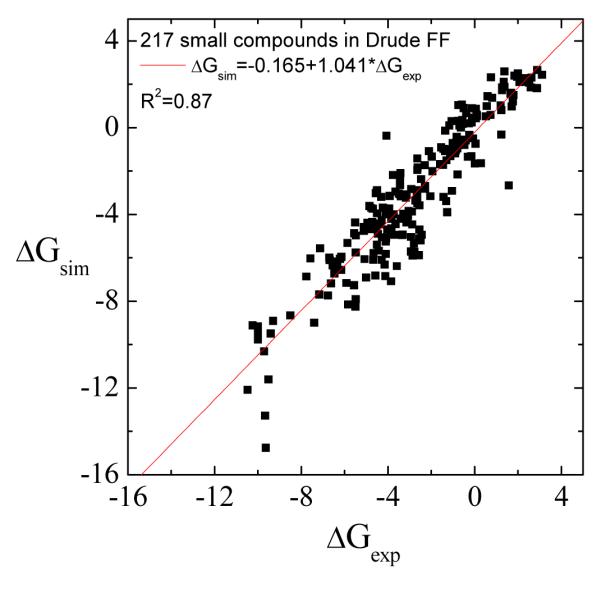
The calculated solvation free energies using GAAMP Drude model for 217 compounds are compared with experimental value. The AUE is 0.92 kcal/mol.
Parameterization of unnatural amino acid side-chains
Site directed incorporation of unnatural amino acids (UAAs) by exploiting the so-called nonsense suppression approach is a powerful experimental technique that considerably expands the chemical space of available perturbations for biochemical and biophysical studies61-63. In principle, simulation studies of any of these chemically modified systems could be carried out to complement the experimental information. However, the implication is that accurate MM models will be needed for an ever-growing number of possible UAAs. To test how the amino acid parameters obtained by GAAMP perform, we re-parameterize de novo all amino acids except glycine and proline to be consistent with the CHARMM27 force field. Three proteins with diverse topology, shown in Fig. 11, are used to compare the resulting FF (denoted as GAAMP) with CHARMM27: 1ctf (mixed α-helices and β-sheets), 1mjc (all β-sheets) and 1r69 (all α-helices). Four independent 100 ns MD simulations were conducted starting from the crystal structure of each protein. These three proteins are stable in 100 ns simulations both in CHARMM27 and GAAMP with conformational fluctuations. The simulations may suggest that the parameters of amino acids generated by GAAMP are consistent with existing CHARMM27. The automated algorithm is expected to serve as an efficient method to parameterize UAAs. As an example, MM models were generated for a set of 17 UAAs, which are commonly used in studies of membrane proteins. The residue topology and parameter files are provided in Supplementary Information.
Fig. 11.

The snapshots of three proteins with diverse structures are shown at the left of this figure. Their PDB IDs are 1ctf, 1mjc and 1r69 respectively. The plots are generated by PyMol65. The traces of RMSD for all Cα atoms for four trajectories of 100 ns MD simulations are compared between using CHARMM 27 (middle) and using GAAMP FF (right) for 1ctf, 1mjc and 1r69 respectively.
Database of force fields for small molecules
Currently there is no efficient way to retrieve the existing FF generated previously for an arbitrary molecule. Searching through literatures for molecule FF parameters is time consuming and difficult due to the lack of complete information. To solve this problem, we compile our parameterized molecule into a database, which allow users to search and download previously generated FF conveniently at our website, http://gaamp.lcrc.anl.gov/mol-search.html. All molecules parameterized by the web server including QM data used during parameterization will be added into the FF database and users can search and download any FF freely. Future users could choose to parameterize their molecules using our code locally and upload their parameterized molecules to our database if desired. This could be a convenient way to share FF with the community. Nonetheless, quality control could become an issue if there exist several variants for the same molecule using different initial configurations or different QM methods/basis sets during parameterization.
Limitation of the present method and possible improvements
GAAMP targets ab initio calculations, which could be extremely expensive depending on the size of the molecule or the level of the QM methods used. Limited by available computing resource, the present method may be only applicable to a molecule with less than 100 atoms. For larger molecules, one may need to consider smaller fragments, parameterize them separately, then join them together. Proper fragments also need to be selected for fitting the dihedral parameters at the junction. Currently, these operation must be carried out manually to generate the FF for the whole large molecule. There are a number of empirical parameters, e.g., the weights in charge fitting and dihedral fitting, which could affect the behavior and performance of the current method. Different value leads to slightly different models. More work is under way to tune the present method with the aim of accurately reproducing experimental data, including liquid densities, heat of evaporation, and solvation free energies, etc.
More fundamentally, the accuracy of the methods for charge fitting and dihedral parameter fitting is unknown when applied for large molecules. Both RESP19 and compound-water interactions6 for charge fitting have only been extensively tested with relatively small molecules (e.g., smaller than 40 atoms). The dihedral fitting also replies on QM calculations in vacuum. However, the intra-molecular charge-charge interactions within a large compound could be substantially screened out if the environment is taken into account. Consequently, considering QM calculations with implicit solvent might be necessary. Alternatively, breaking a large compound into several small fragments for separate parameterizations could partially avoid having the torsional energy component to compensate for long-range electrostatics contributions.
The geometry optimizations of QM have been performed in vacuum in present work. Recently, MacKerell et al.64 reported that the bond length in charged alkyl-phosphate in the optimized structure with QM can deviate the X-ray experimental value by as much as 0.1~0.2 Å. As pointed out by one anonymous reviewer, a QM geometry optimization with a continuum solvent method prior to the QM energy evaluations with desired method could help when large deviations are observed between QM optimized structure and experimental value, such as bond length and angle.
Most of the present tests were carried out using GAFF to provide the initial parameters for GAAMP. Only charge and dihedral parameters are currently optimized, while the remaining parameters are essentially unchanged. Equivalently, the optimization could rely on CGenFF. For this reason, the quality of the resulting models relies on the accuracy of the initial force field. For those molecules inherently not supported by GAFF13 or CGenFF, including metal complexes, inorganic compounds, or unstable species such as radicals, one needs to manually prepare a reasonable initial FF, then use GAAMP to optimize charge and dihedral parameters. The whole parameterization could be done automatically for those special cases mentioned above if the process of fitting bonded parameters (including bond, angle, improper dihedral, dihedral) is incorporated into GAAMP.
Conclusion
A fully general and automatic method to parameterize non-polarizable or Drude polarizable atomic models of small molecules based on QM target data was implemented. The parameterization can start with GAFF or CGenFF as initial model, then verifies bond and angle parameters followed by charge and dihedral parameter fitting. Both ESP and the compound-water interactions from QM are used as target data in the optimization of electrostatic parameters. The dihedral parameters are optimized on the basis of 1D dihedral scans and the energy of conformers from QM.
The method of automated parameterization was applied to develop non-polarizable FF for small compounds including the analogs of the side-chain of neutral amino acid as well as 217 small molecules with diverse functional groups. The algorithm for dihedral fitting was shown to work well for small molecules. The solvation free energies of those small molecules parameterized with GAAMP show noticeable improvement over GAFF/AM1-BCC and GAFF/RESP43. The possibilities for further improvement were discussed. We also extended the method to automated UAA parameterization to be consistent with the backbone from CHARMM 27. The parameters of side-chain are taken from GAFF and GAAMP charge and dihedral parameters. MD simulations with side-chain parameterized according to the present procedure showed the native structures for three proteins with diverse structures are stable. Finally, the method was used to parameterize a set of 17 UAAs. Lastly, the method was also applied to parameterize Drude polarizable models. The preliminary results for solvation free energy calculations of 217 small molecules are promising compared with the results of using GAFF/AM1-BCC in literature43. More work to improve Drude models is under way. A database featuring searching and downloading force field for small molecule was also presented as a convenient platform for searching and sharing force fields.
Supplementary Material
Acknowledgement
We thank Drs. Alexander D. MacKerell Jr., Christopher N. Rowley, Janamejaya Chowdhary, James Gumbart, Haibo Yu, Yen-lin Lin and Yilin Meng for valuable discussions. We thank Allen Zhu for preparing the molecule structures for 17 UAAs. We are grateful to two referees for their insightful comments and suggestions. This work was supported by NIH/NIGMS through grant U54-GM087519 and was carried out in the context of the Membrane Protein Structural Dynamics Consortium. The computations were made possible by the resources provided by the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory through NIH Grant S10 RR029030-0.
Footnotes
Supporting Information The tabulated solvation free energies of 119 compounds, which were used in Fig. 8, with GAFF/AM1-BCC, GAAMP/RESP and GAAMP models are attached. The coordinates, topologies and parameters files in CHARMM format are provided for these 119 compounds. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002;9(9):646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- (4).Foloppe N, MacKerell AD. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21(2):86–104. [Google Scholar]
- (5).MacKerell AD, Banavali NK. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000;21(2):105–120. [Google Scholar]
- (6).MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- (7).Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- (8).Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21(12):1049–1074. [Google Scholar]
- (10).Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105(28):6474–6487. [Google Scholar]
- (11).Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004;25(13):1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- (12).Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel - an Integrated Software System for Modeling Organic and Bioorganic Molecules Using Molecular Mechanics. J. Comput. Chem. 1990;11(4):440–467. [Google Scholar]
- (13).Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- (14).Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000;21(2):132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- (15).Wang JM, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006;25(2):247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- (16).Udier-Blagovic M, De Tirado PM, Pearlman SA, Jorgensen WL. Accuracy of free energies of hydration using CM1 and CM3 atomic charges. J. Comput. Chem. 2004;25(11):1322–1332. doi: 10.1002/jcc.20059. [DOI] [PubMed] [Google Scholar]
- (17).Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mackerell AD. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004;25(13):1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- (19).Bayly CI, Cieplak P, Cornell WD, Kollman PA. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges - the Resp Model. J. Phys. Chem. 1993;97(40):10269–10280. [Google Scholar]
- (20).Wu JC, Chattree G, Ren PY. Automation of AMOEBA polarizable force field parameterization for small molecules. Theor. Chem. Acc. 2012;131(3):1138. doi: 10.1007/s00214-012-1138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang LP, Chen JH, Van Voorhis T. Systematic Parametrization of Polarizable Force Fields from Quantum Chemistry Data. J. Chem. Theory Comput. 2013;9(1):452–460. doi: 10.1021/ct300826t. [DOI] [PubMed] [Google Scholar]
- (22).Jorgensen WL, Tiradorives J. The Opls Potential Functions for Proteins - Energy Minimizations for Crystals of Cyclic-Peptides and Crambin. J. Am. Chem. Soc. 1988;110(6):1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- (23).MacKerell AD. Atomistic Models and Force Fields. In: Becker OM, MacKerell AD, Roux B, Watanabe M, editors. Computational Biochemistry and Biophysics. first edition CRC Press; 2001. [Google Scholar]
- (24).Lopes PEM, Roux B, MacKerell AD. Molecular modeling and dynamics studies with explicit inclusion of electronic polarizability: theory and applications. Theor. Chem. Acc. 2009;124(1-2):11–28. doi: 10.1007/s00214-009-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Anisimov VM, Lamoureux G, Vorobyov IV, Huang N, Roux B, MacKerell AD. Determination of electrostatic parameters for a polarizable force field based on the classical Drude oscillator. J. Chem. Theory Comput. 2005;1(1):153–168. doi: 10.1021/ct049930p. [DOI] [PubMed] [Google Scholar]
- (26).Harder E, Anisimov VM, Vorobyov IV, Lopes PEM, Noskov SY, MacKerell AD, Roux B. Atomic level anisotropy in the electrostatic modeling of lone pairs for a polarizable force field based on the classical Drude oscillator. J. Chem. Theory Comput. 2006;2(6):1587–1597. doi: 10.1021/ct600180x. [DOI] [PubMed] [Google Scholar]
- (27).Lamoureux G, Harder E, Vorobyov IV, Roux B, MacKerell AD. A polarizable model of water for molecular dynamics simulations of biomolecules. Chem. Phys. Lett. 2006;418(1-3):245–249. [Google Scholar]
- (28).Lamoureux G, Roux B. Modeling induced polarization with classical Drude oscillators: Theory and molecular dynamics simulation algorithm. J. Chem. Phys. 2003;119(6):3025–3039. [Google Scholar]
- (29).Lamoureux G, MacKerell AD, Roux B. A simple polarizable model of water based on classical Drude oscillators. J. Chem. Phys. 2003;119(10):5185–5197. [Google Scholar]
- (30).Lamoureux G, Roux B. Absolute hydration free energy scale for alkali and halide ions established from simulations with a polarizable force field. J. Phys. Chem. B. 2006;110(7):3308–3322. doi: 10.1021/jp056043p. [DOI] [PubMed] [Google Scholar]
- (31).Anisimov VM, Vorobyov IV, Roux B, MacKerell AD. Polarizable empirical force field for the primary and secondary alcohol series based on the classical drude model. J. Chem. Theory Comput. 2007;3(6):1927–1946. doi: 10.1021/ct700100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lopes PEM, Lamoureux G, Roux B, MacKerell AD. Polarizable empirical force field for aromatic compounds based on the classical drude oscillator. J. Phys. Chem. B. 2007;111(11):2873–2885. doi: 10.1021/jp0663614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yu H, Mazzanti CL, Whitfield TW, Koeppe RE, Andersen OS, Roux B. A Combined Experimental and Theoretical Study of Ion Solvation in Liquid N-Methylacetamide. J. Am. Chem. Soc. 2010;132(31):10847–10856. doi: 10.1021/ja103270w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yu HB, Whitfield TW, Harder E, Lamoureux G, Vorobyov I, Anisimov VM, MacKerell AD, Roux B. Simulating Monovalent and Divalent Ions in Aqueous Solution Using a Drude Polarizable Force Field. J. Chem. Theory Comput. 2010;6(3):774–786. doi: 10.1021/ct900576a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Miller KJ. Additivity Methods in Molecular Polarizability. J. Am. Chem. Soc. 1990;112(23):8533–8542. [Google Scholar]
- (36).Nocedal J. Updating Quasi-Newton Matrices with Limited Storage. Math. Comput. 1980;35(151):773–782. [Google Scholar]
- (37).Liu DC, Nocedal J. On the Limited Memory Bfgs Method for Large-Scale Optimization. Math. Program. 1989;45(3):503–528. [Google Scholar]
- (38).Johnson SG. [accessed August 08, 2011];The NLopt nonlinear-optimization package. http://ab-initio.mit.edu/nlopt.
- (39).Conn AR, Gould NIM, Toint PL. A Globally Convergent Augmented Lagrangian Algorithm for Optimization with General Constraints and Simple Bounds. SIAM J. Numer. Anal. 1991;28(2):545–572. [Google Scholar]
- (40).Birgin EG, Martinez JM. Improving ultimate convergence of an augmented Lagrangian method. Optim. Method. Softw. 2008;23(2):177–195. [Google Scholar]
- (41).Deng YQ, Roux B. Hydration of amino acid side chains: Nonpolar and electrostatic contributions calculated from staged molecular dynamics free energy simulations with explicit water molecules. J. Phys. Chem. B. 2004;108(42):16567–16576. [Google Scholar]
- (42).Jiang W, Hodoscek M, Roux B. Computation of Absolute Hydration and Binding Free Energy with Free Energy Perturbation Distributed Replica-Exchange Molecular Dynamics. J. Chem. Theory Comput. 2009;5(10):2583–2588. doi: 10.1021/ct900223z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shivakumar D, Deng YQ, Roux B. Computations of Absolute Solvation Free Energies of Small Molecules Using Explicit and Implicit Solvent Model. J. Chem. Theory Comput. 2009;5(4):919–930. doi: 10.1021/ct800445x. [DOI] [PubMed] [Google Scholar]
- (44).Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314(1-2):141–151. [Google Scholar]
- (45).Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79(2):926–935. [Google Scholar]
- (47).Darden T, York D, Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993;98(12):10089–10092. [Google Scholar]
- (48).Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995;103(19):8577–8593. [Google Scholar]
- (49).Kubo R, Toda M, Hashitsume N. Statistical Physics II: Nonequilibrium Statistical Mechanics. 2 ed Springer; New York: 1991. [Google Scholar]
- (50).Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J. Chem. Phys. 1995;103(11):4613–4621. [Google Scholar]
- (51).Miyamoto S, Kollman PA. Settle - an Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992;13(8):952–962. [Google Scholar]
- (52).Andersen HC. Rattle - a Velocity Version of the Shake Algorithm for Molecular-Dynamics Calculations. J. Comput. Phys. 1983;52(1):24–34. [Google Scholar]
- (53).Tuckerman M, Berne BJ, Martyna GJ. Reversible Multiple Time Scale Molecular-Dynamics. J. Chem. Phys. 1992;97(3):1990–2001. [Google Scholar]
- (54).Roux B. The Calculation of the Potential of Mean Force Using Computer-Simulations. Comput. Phys. Commun. 1995;91(1-3):275–282. [Google Scholar]
- (55).Shirts MR, Mobley DL, Chodera JD, Pande VS. Accurate and efficient corrections for missing dispersion interactions in molecular Simulations. J. Phys. Chem. B. 2007;111(45):13052–13063. doi: 10.1021/jp0735987. [DOI] [PubMed] [Google Scholar]
- (56).Martyna GJ, Tuckerman ME, Tobias DJ, Klein ML. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996;87(5):1117–1157. [Google Scholar]
- (57).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JJA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc.; Wallingford CT: 2009. Gaussian 09, Revision A.02. [Google Scholar]
- (58).Martin F, Zipse H. Charge distribution in the water molecule - A comparison of methods. J. Comput. Chem. 2005;26(1):97–105. doi: 10.1002/jcc.20157. [DOI] [PubMed] [Google Scholar]
- (59).Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CDM, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002;347(7):472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- (60).Shirts MR, Pitera JW, Swope WC, Pande VS. Extremely precise free energy calculations of amino acid side chain analogs: Comparison of common molecular mechanics force fields for proteins. J. Chem. Phys. 2003;119(11):5740–5761. [Google Scholar]
- (61).Pless SA, Galpin JD, Niciforovic AP, Ahern CA. Contributions of counter-charge in a potassium channel voltage-sensor domain. Nat. Chem. Biol. 2011;7(9):617–623. doi: 10.1038/nchembio.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Lacroix JJ, Pless SA, Maragliano L, Campos FV, Galpin JD, Ahern CA, Roux B, Bezanilla F. Intermediate state trapping of a voltage sensor. J Gen Physiol. 2012;140(6):635–652. doi: 10.1085/jgp.201210827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Pless SA, Ahern CA. Unnatural Amino Acids as Probes of Ligand-Receptor Interactions and Their Conformational Consequences. Annu. Rev. Pharmacol. Toxicol. 2013;53:211–229. doi: 10.1146/annurev-pharmtox-011112-140343. [DOI] [PubMed] [Google Scholar]
- (64).Mallajosyula SS, Guvench O, Hatcher E, MacKerell AD. CHARMM Additive All-Atom Force Field for Phosphate and Sulfate Linked to Carbohydrates. J. Chem. Theory Comput. 2012;8(2):759–776. doi: 10.1021/ct200792v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).The PyMOL Molecular Graphics System. Version 1.3r1 Schrodinger, LLC; New York: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.