

Abstract
The total synthesis of 8-epi, 8,9-epi, and 9-epi-(−)-cephalimysin A is described. Our catalytic enantioselective synthesis takes advantage of a novel tandem photoisomerization/Stetter reaction. The approach provides rapid access to the desired spirofuranone lactam core in good yield and excellent enantioselectivity. A late stage oxidation strategy allows for flexible access to three of the four diastereomers of cephalimysin A. Access to the epimers provides further support for the correction of the initially proposed relative stereochemistry of cephalimysin A.
Introduction
In 2007 cephalimysin A (1) was isolated by Yamada and coworkers1 from the culture broth of Aspergillus fumigatus initially isolated from the marine fish Mugil cephalus. Cephalimysin A belongs to a family of natural products containing a unique 1-oxa-7-azaspiro[4.4]non-2-ene-4,6-dione core structure (Figure 1). This group of natural products is characterized by their highly substituted spirocyclic core and wide range of biological activities.2 Specifically, cephalimysin A has exhibited significant cytotoxicity against the murine P388 leukemia cell line (IC50 = 15.0 nM) and the human HL-60 leukemia cell line (IC50 = 9.5 nM).1 Several total syntheses of members of this family of natural products have been described in the literature.3 To date, there are no known syntheses of cephalimysin A (1).
Fig. 1.

Representative spirofuranone containing natural products.
Interestingly, the structure of cephalimysin A was initially proposed to possess a trans relationship between the C-8 methoxy group and C-9 alcohol (1a), a relationship unique within this family of natural products (Figure 1). In 2010 Yamada and co-workers published a report on the CD Cotton effect and the absolute stereochemistry of FD-838 (2a) and its seven stereoisomers.4 In their report the structure of cephalimysin A is corrected (see 1b, Figure 1). This correction of the structure is in agreement with observed 1H nmr splitting of the hydrogen at C-8 (vide infra). Intrigued by the densely functionalized stereochemically challenging core and the apparent disparity between it and its congeners, in 2008 we initiated a program directed at the total synthesis of cephalimysin A.
Recently, N-heterocyclic carbene organocatalyzed transformations have received increased attention in organic chemistry.5 Of these reactions the carbene catalyzed Stetter reaction6 has become an efficient method for the asymmetric formation of C-C bonds. Our group’s implementation of amino-indanol and pyrrolidine derived triazolium N-heterocyclic carbenes (NHCs)7 has been integral for the development of a variety of highly stereoselective intra-8 and intermolecular9 asymmetric Stetter reactions. In an effort to showcase the power of this newly developed stereoselective umpolung reactivity in the context of natural product synthesis, we became interested in the structurally unique and biologically active spirofuranone lactam containing natural products.
We recently disclosed the application of the intramolecular Stetter reaction for the asymmetric synthesis of spirocycle 5 in good yield and enantioselectivity (Figure 2).10 This process allowed rapid and selective access to the desired spirocyclic core of this family of natural products. Spirocycle 5 was further functionalized to an advanced intermediate but did not allow access to the target, FD-838 (2a). One of the difficulties in accessing the desired natural product was installation of the methyl group at C-3. We sought to overcome this problem by conducting the Stetter reaction with enal 6. Use of aldehyde 6 would not only circumvent the need for a late state installation of the methyl group but also produce the furanone oxidation state which is set for installation of the desired side chain at C-2.
Fig. 2.

Planned approach towards spirofuranone containing natural products.
Three potential complications were considered when proposing the desired Stetter reaction. First, the olefin geometry of the enal would be crucial to the success of the Stetter reaction with the (Z) isomer needed for the reaction to proceed. Second, we have previously shown that aldehydes bearing α substitution generally yield Stetter products with low enantioselectivity.11 Third, the possibility of carbene catalyzed redox5 pathways could inhibit the desired C-C bond formation. However, we believed that the potential utility of this kind of Stetter reaction warranted further investigation as synthesis of spirofuranone 7 could potentially allow access to a number of members of the spirofuranone lactam containing natural products.
Results and discussion
Stetter reaction optimization
The synthesis of the desired Stetter reaction precursor bearing either a para-methoxybenzyl (PMB) or 2-(trimethylsilyl)-ethoxyethyl (SEM) protection group was carried out in two steps from known bromomaleimide 8 (Scheme 1).12 Mitsunobu protection13 of maleimide 8 affords the protected bromomaleimide 9a. Treatment of maleimide 9a with the potassium salt of dialdehyde 1014 in a mixture of acetonitrile and dimethylsulfoxide affords the desired aldehyde-tethered maleimide 11a in moderate yield. A similar sequence allows for the synthesis of the SEM protected maleimide 11b.15 However, in both cases the aldehydes 11a and 11b were formed exclusively as the undesired (E) isomer.
Scheme 1.
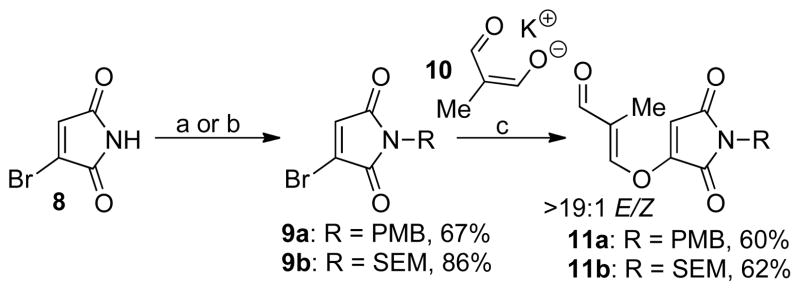
Synthesis of aldehydes 11a and 11b. Reagents and conditions: (a) DIAD, PPh3, para-methoxybenzyl alcohol, neopentyl alcohol, THF, −78 to 22 °C (67% of 9a); (b) SEMCl, DIPEA, CH2Cl2, −40 °C (86% of 9b); (c) 10, MeCN/DMSO, 22 °C (60% of 11a and 62% of 11b); DIAD = diisopropyl azodicarboxylate, SEMCl = 2-(trimethylsilyl)ethoxymethyl chloride, DIPEA = N,N-diisopropylethylamine, DMSO = dimethyl sulfoxide.
Although the undesired olefin isomer was obtained we believed that in situ isomerization of the olefin in the presence of the carbene catalyst could potentially allow for the desired Stetter reaction to proceed. Of the various methods for olefin isomerization, photoisomerization seemed to be the most attractive since it would not require the addition of any additional reagents that could interfere with the carbene catalyst. However, the stability and reactivity of the carbene upon exposure to high energy light (UV) radiation, typically necessary for isomerization, was not clear.16
We were encouraged to find that UV irradiation of a mixture of the aldehyde 11a in the presence of triazolium 13a and sodium acetate delivered the desired product, albeit in low yield (35%) and enantioselectivity (28%) (Table 1, entry 1). An investigation of amino-indanol derived precatalysts showed that trichlorophenyl triazolium 13b delivers the desired spirocycle with increased yield and enantioselectivity (entry 2). The bis(trifluromethyl)-phenyl catalyst17 13c delivers the product with further increase in yield (46%) (entry 3). Interestingly catalyst 13c yields the opposite major enantiomer of spirocycle 12a in a good 87% ee.18 Encouraged by these results, a series of phenyl-alanine derived catalysts were screened under the reaction conditions but no improvement of yield and enantioselectivity was observed (entries 4–6). It is worth noting that unlike catalyst 13c the phenyl-alanine derived bis(trifluromethyl)-phenyl catalyst 14c did not afford the opposite enantiomer of product 12a (entry 6).19 Having identified catalyst 13c as delivering the best selectivity a brief screen of solvents was conducted (entries 7–9). It was found that benzene affords the product in an acceptable 64% yield and excellent 94% ee.20 The same conditions could be applied to the SEM protected aldehyde 11b to afford the product with similar results (62% yield, 95% ee, entry 10).21,22 Importantly, the Stetter reaction could be carried out on 2.50 g scale with no change in the yield or enantioselectivity.23
Table 1.
Optimization of photoinduced isomerization asymmetric Stetter reaction.a

| |||||
|---|---|---|---|---|---|
| Entry | Aldehyde | Cat. | Solvent | Yield (%)b | ee (%)c |
| 1 | 11a | 13a | CHCl3 | 35 | 28 |
| 2 | 11a | 13b | CHCl3 | 40 | 60 |
| 3 | 11a | 13c | CHCl3 | 47 | −87 |
| 4 | 11a | 14a | CHCl3 | 37 | 50 |
| 5 | 11a | 14b | CHCl3 | 20 | 72 |
| 6 | 11a | 14c | CHCl3 | 32 | 44 |
| 7 | 11a | 13c | DCE | 40 | −92 |
| 8 | 11a | 13c | PhMe | 43 | −94 |
| 9 | 11a | 13c | PhH | 58 | −94 |
| 10d | 11b | ent-13c | PhH | 62e | 95 |
Reagents and conditions unless otherwise noted: triazolium salt (30 mol %), sodium acetate (1.0 equiv.), benzene (0.05 M), 45 °C, UV irradiation (300 < λ < 400), 24 h.
Isolated yield of spirocycle 12a or 12b.
Enantiomeric excess was determined by HPLC analysis on a chiral stationary phase.
20 mol % triazolium salt, 48 h.
2.50 g scale.
Retrosynthetic analysis
Having established an efficient enantioselective synthesis of key spirocycle 12b we aimed to develop a strategy for the synthesis of cephalimysin A, due to both the ambiguity surrounding the C-8 stereochemistry and its high biological activity. Indeed, at the time the program was initiated we were under the impression that the stereochemistry at C-8 was as initially proposed. However, we were leery of this assignment based on the known stereochemistry of all other members of this family of natural products. With this in mind we knew our approach must be flexible in installing the methoxy group at C-8 and would ideally do so at a late stage. We hoped that this strategy would allow for further confirmation of relative stereochemistry of cephalimysin A.
Our retrosynthetic plan is outlined in Scheme 2. We envisioned a late stage oxidation of spirocycle 17 to afford the natural product 1. The enone 17 would arise from oxidation and elimination of the benzylidene lactam 16. We would arrive at benzylidene lactam 16 via a Barbier coupling between spirocyclic succinimide 15 and benzyl bromide followed by oxidation of the enol ether to the desired furanone. In turn succinimde 15 would be delivered via conjugate addition followed by enolate trapping of the spirocyclic furanone 12b, which is easily accessed on gram scale from previously described asymmetric Stetter reaction. We believed that this route should allow for late stage functionalization of the C-8 position with the possibility of accessing multiple diastereomers of cephalimysin A for comparison to the natural isolate.
Scheme 2.

Retrosynthetic analysis of cephalimysin A.
Synthesis
Our synthesis of key enone 17 began with conjugate addition of Grignard 18 in the presence of copper(I) bromide to furanone 12b followed by in situ enolate trapping with tert-butyldimethylsilyl chloride (TBSCl) to afford silylenol ether 19 in 74% yield as a 10:1 mixture of diastereomers. Under our previously developed conditions,10,24 treatment of succinimide 19 with benzyl bromide and samarium diiodide followed by mild acidic work-up gives the benzylidene lactam 20 in good yield as a 5:1 mixture of E/Z olefins. Oxidation of the silylenol ether with DDQ25 followed by deprotection of the SEM group affords the unprotected lactam 16. Treatment of benzylidene lactam 16 with dimethyldioxirane (DMDO) generates the diol 21 as an inconsequential mixture of diastereomers.26 Surprisingly, exposure of diol 21 to Martin-sulfurane (2.2 equiv.) mediates both oxidation of the secondary alcohol as well as elimination of the tertiary alcohol to yield the desired enone 17. Although unexpected, oxidation of activated secondary alcohols by Martin-sulfurane has been previously reported.27 With the desired enone in hand we were set to explore our planned late stage oxidation of enone 17 to afford cephalimysin A.
Initial attempts at oxidation of the enone 17 with electrophilic oxidants such as m-chloroperoxybenzoic acid (m-CPBA), DMDO, trifluoroperoxyacetic acid, and osmium tetroxide led to either epoxidation of the distal alkene with no observed oxidation of the desired olefin or no reaction. Nucleophilic epoxidation conditions generally led to decomposition of the molecule again failing to deliver any of the desired oxidized product. Having found the selective electrophilic epoxidation of the enamide double bond to be problematic we turned our attention to an alternate strategy to functionalize the lactam.
We predicted that halo etherification may allow for a selective functionalization of the desired alkene due to the reversible nature of halonium formation.28 In this case halonium opening would now be the product determining step. We postulated that the enamide alkene should be more prone to halonium opening due to donation of the nitrogen lone pair into the σ* orbital of the halonium ion. Indeed, treatment of spirocycle 17 with N-iodosuccinimide (NIS) in methanol affords the desired iodoether 22a as a 7:1 mixture of diastereomers.29 Notably, oxidation of the distal alkene was not observed under these conditions. Dehalogenative hydroxylation of the alkyl iodide 22a using modified Nakamura conditions30 affords the desired diastereomeric alcohols 1c and 1a as a 1.5:1 mixture in 65% combined yield. The relative stereochemistry of the products was determined by comparison of the 1H nmr to the natural product cephalimysin A,1 and to the three diastereomers of related natural product FD-8384 vide infra.
Compound 1c was determined to contain a cis relationship between C-9 alcohol and C-8 methoxy group based on the 12.6 Hz coupling observed for the C-9 proton. This rather large coupling constant is indicative of an intramolecular hydrogen bond only accessible via a cis relationship and is consistent with other members of this family of natural products (Figure 2). However, the 1H and 13C nmr signals of 1c did not match that of the natural isolate. Thus we determine we have accessed 8,9-epi-cephalimysin A (1c). Spirocycle 1a was determined to possess a trans relationship between C-9 alcohol and C-8 methoxy group based on the much smaller 4.1 Hz coupling observed for the C-9 proton. This relatively small coupling is consistent with a lack of intramolecular hydrogen bonding and therefore a trans relationship. The stereochemistry at C-8 should be conserved during the hydroxylation and the two resulting alcohols should be epimeric at C-9; thus the stereochemistry of 1a is as shown. This structure matches the originally proposed structure of cephalimysin A, but the 1H and 13C nmr do not match that of the natural isolate.
Having accessed two diastereomers of the natural cephalimysin A we sought to synthesize the correct diastereomer. We concluded that inversion of the diastereoselectivity of the haloetherification should allow access to the desired natural product. We were delighted to find that addition of trifluoroacetic acid to our previously developed conditions for haloetherification results in a reversal in diastereoselectivity to give 22b as the major diastereomer in a 4:1 ratio. We propose two possible explanations for this observed reversal in diastereoselectivity. First, addition of acid may prevent the Lewis basic carbonyl of the furanone from stabilizing the iodonium from the top face of the molecule. Second, acidic reaction conditions may disrupt hydrogen bonding between the nucleophile and the substrate. In particular hydrogen bonding between the furanone oxygen and methanol in a six-membered ring transition state would deliver the nucleophile from the bottom face of the molecule. Interruption of this hydrogen bonding interaction could lead to the observed diastereoselectivity.
With the desired diastereomer 22b we were poised to execute the dehalogenative hydroxylation. However exposure of 22b to same conditions described previously failed to deliver the desired product; the minor diastereomer 22a was consumed but the major diastereomer 22b failed to react. We circumvented this problem by carrying out a two step process with TEMPO31 as the terminal oxidant to form the desired oxygenated species. Subsequent cleavage of the N-O bond with zinc metal in the presence of acetic acid delivered 1d. A single diastereomer was isolated and the 3.6 Hz coupling observed for the proton at C-9 indicated a trans relationship between the alcohol and methoxy group. Therefore 9-epi-cephalimysin A (1d) represents the third diastereomer of cephalimysin A (1b) synthesized from common spirocycle 17.
Our ability to rapidly access three diastereomers of cephalimysin A (1a, c–d) has allowed the comparison of the spectroscopic information of naturally isolated cephalimysin A to our synthetic samples. This data (vide supra) strongly suggests that the originally proposed structure of cephalimysin A (1a) is in error and that the revised structure as first reported by Yamada4 (1b) possesses the correct relative stereochemistry.
Conclusions
In conclusion, we have a described an efficient method for the enantioselective synthesis of the core of spirocyclic furanone containing natural products via a novel photoisomerization intramolecular asymmetric Stetter reaction. This approach allows for a rapid entry to the cephalimysin A framework. Our late stage oxidation strategy has allowed for the synthesis of three diastereomers of cephalimysin A (1a, c–d). Access to these epimers give further credence for the correction of the structure of cephalimysin A (1b) and will allow for further biological testing of these unnatural stereoisomers.
Supplementary Material
Fig. 3.

Comparison of C-9 1H NMR data of synthetic compounds 1a and 1c to natural isolates cephalimysin A–D (1b, 2b–2d).
Scheme 3.

Synthesis of spirocyclic enone 17. Reagents and conditions: (a) 18, CuBr•SMe2, THF, −78 °C, then TBSCl, HMPA, −78 °C to 0 °C (74%, 10:1 dr); (b) SmI2, benzyl bromide, THF, 22 °C then 0.05 M HCl (aq) (77%, 5:1 E/Z); (c) DDQ, H2O/CH2Cl2, 22 °C (68%); (d) i) TFA, CH2Cl2, 0 °C to 22 °C; ii) ethylene diamine, 10 M NaOH, MeOH, 23 °C (65%); (e) DMDO, acetone, −40 °C (70%); (f) Martin-sulfurane (2.2 equiv.), CH2Cl2, 23 °C (67%); TBSCl = tertbutyldimethylsilyl chloride, HMPA = hexamethylphosphoramide, TFA = trifluoroacetic acid, DMDO = dimethyldioxirane.
Scheme 4.

Synthesis of 8-epi-cephalimysin A (1a) and 8,9-epi-ephalimysin A (1c). Reagents and conditions: (a) NIS, MeOH, 0 °C (80%, 7:1 dr); (b) ((CH3)3Si)3SiH, AIBN, air, toluene 80 °C (65%, 1.5:1 dr); NIS = N-iodosuccinimide, AIBN = azobisisobutyronitrile.
Scheme 5.

Synthesis of 9-epi-cephalimysin A (1d). Reagents and conditions: (a) NIS, TFA, MeOH, (71%, 4:1 dr); (b) i) TEMPO, tributyltin hydride, toluene, 70 °C; ii) Zn, AcOH/THF/H2O, 70 °C (38%); TEMPO = 2,2,6,6-tetramethyl-1-piperidinyloxy free radical.
Acknowledgments
We thank NIGMS for generous support of this research (GM72586). S. P. L. thanks Sanofi-Aventis (Organic Division Fellowship) for funding. T.R. thanks Amgen and Roche for unrestricted support. We thank Donald Gauthier and Greg Hughes (Merck) for a generous gift of aminoindanol and Kevin M. Oberg and Derek M. Dalton (CSU) for crystal structure analysis. We thank Professor T. Yamada for generously supplying a sample of cephalimysin A for spectroscopic comparison.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Yamada T, Imai E, Nakatuji K, Numata A, Tanaka R. Tetrahedron. 2007;48:6294. [Google Scholar]
- 2.(a) Weber HP, Petcher TJ, Bloch P, Tamm C. Helv Chim Acta. 1976;59:137. doi: 10.1002/hlca.19760590115. [DOI] [PubMed] [Google Scholar]; (b) Mizoue K, Okazaki T, Hanada K, Amamoto T, Yamagashi M, Omura S. 216607 Eur Pat Appl EP. ; 205278. JP Appl. 1987; Chem Abstr. 1987;107:132627x. [Google Scholar]; (c) Ando O, Satake H, Nakajima M, Sato A, Nakamura T, Kinoshita T, Furuya K, Haneishi T. J Antibiot. 1991;44:382. doi: 10.7164/antibiotics.44.382. [DOI] [PubMed] [Google Scholar]; (d) Wenke J, Anke H, Sterner O. Biosci Biotech Biochem. 1993;57:961. [Google Scholar]; (e) Komagata D, Fujita S, Yamashita N, Saito S, Morino T. J Antibiot. 1996;49:958. doi: 10.7164/antibiotics.49.958. [DOI] [PubMed] [Google Scholar]; (f) Asami Y, Kakeya H, Onose R, Yoshida A, Matsuzaki H, Osada H. Org Lett. 2002;4:2845. doi: 10.1021/ol020104+. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hayashi Y, Shoji M, Yamaguchi J, Sato K, Yamaguchi S, Mukaiyama T, Sakai K, Asami Y, Kakeya H, Osada H. J Am Chem Soc. 2002;124:12078. doi: 10.1021/ja0276826. [DOI] [PubMed] [Google Scholar]; (b) Hayashi Y, Shoji M, Yamaguchi S, Mukaiyama T, Yamaguchi J, Kakeya H, Osada H. Org Lett. 2003;5:2287. doi: 10.1021/ol034630s. [DOI] [PubMed] [Google Scholar]; (c) Aoki S, Oi T, Shimizu K, Shiraki R, Takao K, Tadano K. Bull Chem Soc Jpn. 2004;77:1703. [Google Scholar]; (d) Aoki S, Oi T, Shimizu K, Shiraki R, Takao K, Tadano K. Heterocycles. 2004;62:161. [Google Scholar]; (e) Hayashi Y, Shoji M, Mukaiyama T, Gotoh H, Yamaguchi S, Nakata M, Kakeya H, Osada H. J Org Chem. 2005;70:5643. doi: 10.1021/jo050664x. [DOI] [PubMed] [Google Scholar]; (f) Hayashi Y, Sankar K, Ishikawa H, Nozawa Y, Mizoue K, Kakeya H. Bioorg Med Chem Lett. 2009;19:3863. doi: 10.1016/j.bmcl.2009.03.154. [DOI] [PubMed] [Google Scholar]
- 4.Yamada T, Kitada H, Kajimoto T, Numata A, Tanaka R. J Org Chem. 2010;75:4146. doi: 10.1021/jo100496f. [DOI] [PubMed] [Google Scholar]
- 5.For reviews see: Marion N, Díez-González S, Nolan SP. Angew Chem Int, Ed. 2007;46:2988. doi: 10.1002/anie.200603380.Enders D, Niemeir O, Henseler A. Chem Rev. 2007;107:5606. doi: 10.1021/cr068372z.Moore JL, Rovis T. Top Curr Chem. 2010;291:77. doi: 10.1007/978-3-642-02815-1_18.Vora HU, Rovis T. Aldrichimica Acta. 2011;44:3.
- 6.(a) Stetter H, Schreckenberg M. Angew Chem, Int Ed Engl. 1973;12:81. [Google Scholar]; (b) Stetter H. Angew Chem, Int Ed Engl. 1976;15:639. [Google Scholar]; (c) Stetter H, Kuhlmann H. Org React. 1991;40:407. [Google Scholar]; (d) Christmann M. Angew Chem, Int Ed. 2005;44:2632. doi: 10.1002/anie.200500761. [DOI] [PubMed] [Google Scholar]; (e) Read de Alaniz J, Rovis T. Chem Lett. 2008;37:2. [Google Scholar]
- 7.Kerr MS, Read de Alaniz J, Rovis T. J Am Chem Soc. 2002;124:10298. doi: 10.1021/ja027411v.Read de Alaniz J, Kerr MS, Moore JL, Rovis T. J Org Chem. 2008;73:2033. doi: 10.1021/jo702313f.For synthesis of these triazolium salts, see: Kerr MS, Read de Alaniz J, Rovis T. J Org Chem. 2005;70:5725. doi: 10.1021/jo050645n.Vora HU, Lathrop SP, Reynolds NT, Kerr MS, Read de Alaniz J. Org Synth. 2010;87:350.
- 8.For a review of the asymmetric intramolecular Stetter reaction, see: Read de Alaniz J, Rovis T. Syn Lett. 2009:1189. doi: 10.1055/s-0029-1216654.
- 9.(a) Liu Q, Perreault S, Rovis T. J Am Chem Soc. 2008;130:14066. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu Q, Rovis T. Org Lett. 2009;11:2856. doi: 10.1021/ol901081a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) DiRocco DA, Oberg KM, Dalton DM, Rovis T. J Am Chem Soc. 2009;131:10872. doi: 10.1021/ja904375q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) DiRocco DA, Rovis T. J Am Chem Soc. 2011;133:10402. doi: 10.1021/ja203810b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) DiRocco DA, Noey EL, Houk KN, Rovis T. Angew Chem, Int Ed. 2012;51:2391. doi: 10.1002/anie.201107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orellana A, Rovis T. Chem Commun. 2008:730. doi: 10.1039/b716445a. [DOI] [PubMed] [Google Scholar]
- 11.Reynolds NT, Rovis T. Tetrahedron. 2005;61:6368. [Google Scholar]
- 12.Tedaldi LM, Smith MEB, Nathani RI, Baker JR. Chem Commun. 2009:6583. doi: 10.1039/b915136b. [DOI] [PubMed] [Google Scholar]
- 13.Walker MA. J Org Chem. 1995;60:5352. [Google Scholar]
- 14.(a) Ullrich FW, Rotscheidt K, Breitmaier E. Chem Ber. 1986;119:1737. [Google Scholar]; (b) O’Sullivan PT, Buhr W, Fuhry MAM, Harrison JR, Davies JE, Feeder N, Marshall DR, Burton JW, Holmes AB. J Am Chem Soc. 2004;126:2194. doi: 10.1021/ja038353w. [DOI] [PubMed] [Google Scholar]
- 15.For a similar synthesis of9b, see: Coleman RS, Walczak MC, Campbell EL. J Am Chem Soc. 2005;127:16038. doi: 10.1021/ja056217g.
- 16.For a recent example of dual NHC and photoredox catalysis using much lower energy light, see: DiRocco DA, Rovis T. J Am Chem Soc. 2012;134:8094. doi: 10.1021/ja3030164.
- 17.Takikawa H, Suzuki K. Org Lett. 2007;9:2713. doi: 10.1021/ol070929p. [DOI] [PubMed] [Google Scholar]
- 18.The inversion in the sense of absolute stereochemistry as a function of solvent polarity has previously been observed ( Liu Q, Rovis T. Org Proc Res Dev. 2007;11:598. doi: 10.1021/op600278f. To our knowledge, inversion as a function of arene electronics has not been observed.
- 19.Addition of photosensitizers such as benzophenone or acetone did not result in increase in product formation.
- 20.For further reaction optimization including the screening of alternate bases, see: Supporting Information.
- 21.Conducting the reaction in the absence of UV radiation at 60 °C only returned starting material ruling out the possibility of carbene catalyzed or thermal enal isomerization.
- 22.Absolute stereochemistry was determined by X-ray crystallography of a related compound. See Supporting Information for details.
- 23.The moderate yield of the Stetter reaction is attributed to the relative instability of the starting aldehydes11aand11b. Storage of the aldehydes on the bench top open to the air leads to significant decomposition after a few days indicating its sensitive nature. The presumed mode of decomposition is through conjugate addition to the enal followed by elimination of 3-hydroxymaleimide. This process is presumably exacerbated under the reaction conditions via conjugate addition by either the base or NHC followed by elimination.
- 24.Farcas S, Namy JL. Tetrahedron Lett. 2001;42:879. [Google Scholar]
- 25.For similar oxidations of silyenolethers with DDQ, see: Fleming I, Paterson I. Synthesis. 1979:736.Fevig TL, Elliott RL, Curran DP. J Am Chem Soc. 1988;110:5064.Kanazawa A, Delair P, Pourashraf M, Greene AE. J Chem Soc, Perkin Trans 1. 1997:1911.Sha CK, Santhosh KC, Lih SH. J Org Chem. 1998;63:2699. doi: 10.1021/jo9723570.
- 26.Koseki Y, Kusano S, Ichi D, Yoshida K, Nagasaka T. Tetrahedron. 2000;56:8855. Hayashi has utilized a similar DMDO oxidation in his syntheses of various spirofuranone containing natural products, see refs: 3b–c. [Google Scholar]
- 27.Wensley AM, Hardy AO, Gonsalves KM, Koviach JL. Tetrahedron Lett. 2007;48:2431. [Google Scholar]
- 28.Brown RS. Acc Chem Res. 1997;30:131. [Google Scholar]
- 29.Diastereomers are presumed to be a mixture of trans isomers based on high trans selectivity observed in similar systems, see: Matos MN, Afonso CAM, Batey RA. Tetrahedron. 2005;61:1221.
- 30.(a) Nakamura E, Inubushi T, Aoki S, Machii D. J Am Chem Soc. 1991;113:8980. [Google Scholar]; (b) Moutel S, Prandi J. Tetrahedron Lett. 1994;35:8163. [Google Scholar]; (c) Nakamura E, Sato K, Imanishi Y. Synlett. 1995:525. [Google Scholar]; (d) Sawamura M, Kawaguchi Y, Nakamura E. Synlett. 1997:801. [Google Scholar]; (e) Mayer S, Prandi J. Tetrahedron Lett. 1998;54:8753. [Google Scholar]
- 31.(a) Barrett AGM, Bezuidenhoudt BCB, Melcher LM. J Org Chem. 1990;55:5196. [Google Scholar]; (b) Boger DL, McKie JA. J Org Chem. 1995;60:1271. [Google Scholar]; (c) Castagner B, Leighton JL. Tetrahedron. 2007;63:5895. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.