

Abstract
An enantioselective rhodium (I) catalyzed [2+2+2] cycloaddition with a cleavable tether has been developed. The reaction proceeds with a variety of alkyne substrates in good yield and high enantioselectivity. Upon reduction of the vinylogous amide in high diastereoselectivity (>19:1) and cleavage of the tether, N-methylpiperidine products with functional group handles can be accessed.
Keywords: Asymmetric synthesis, Heterocyclic compd, Cycloaddition react
Due to their prevalence in drug targets and natural products, the asymmetric synthesis of nitrogen containing heterocycles is an important focus of the synthetic community. Our lab has a longstanding interest in the catalytic asymmetric synthesis of such moieties (Scheme 1). In 2006, our lab reported the rhodium (I) catalyzed asymmetric [2+2+2] cycloaddition between alkenylisocyanates and alkynes. This catalytic, asymmetric method allows facile access to indolizidines and quinolizidines, important scaffolds in natural products and pharmaceutical targets, in good yields with high enantioselectivities.[1,2] Extension of this methodology to the synthesis of monocyclic nitrogen containing heterocycles would be useful, as piperidines are present in numerous compounds with interesting biological activities,[3] such as alkaloid 241D,[4] isosolenopsin A[5] and palinavir[6] (Figure 1). Recently, several new methods have been reported for the synthesis of poly-substituted piperidines,[7,8] highlighted by Bergman and Ellman’s recent contribution.[9] Catalytic asymmetric approaches to polysubstituted piperidines, however, remain scarce with the notable exception of the powerful aza-Diels-Alder reaction.[10] Complementary approaches to piperidines relying on the union of two or more fragments with concomitant control of stereochemistry in the process would be of significant value.[11,12] Herein, we report a partial solution to this problem relying on an asymmetric rhodium catalyzed cycloaddition of an alkyne, alkene and isocyanate, bringing three components together wherein two of the three are attached by a removal linker.
Scheme 1.

Previous and Envisioned [2+2+2] cycloadditions.
Figure 1.

Piperidine containing compounds.
We sought to develop a catalytic asymmetric method to access piperidine scaffolds utilizing the rhodium (I) catalyzed [2+2+2] cycloaddition. While the fully intermolecular reaction faces several challenges, such as competitive insertion of the alkene component over insertion of a second alkyne to form a pyridone and regioselectivity of π component insertion, the use of a cleavable tether in the isocyanate backbone provides a solution to these obstacles (Scheme 1).[13–15] Products of net intermolecular [2+2+2] cycloaddition would be accessed after cleavage of the tether, allowing for the synthesis of substituted piperidine scaffolds in a catalytic asymmetric fashion. In this communication, we report the use of a cleavable tether in the rhodium catalyzed [2+2+2] cycloaddition between oxygen-linked alkenyl isocyanates and alkynes to access piperidine scaffolds after cleavage of the tether. The products are obtained in high enantioselectivity and yield. Differentially substituted piperidines with functional group handles for further manipulation can be accessed in a short sequence, in which the stereocenter introduced in a catalytic asymmetric fashion controls the diastereoselectivity of two more stereocenters.
Our investigations began with the oxygen-linked alkenyl isocyanate shown to participate in the rhodium (I) catalyzed [2+2+2] cycloaddition (Table 1).[1f] As with previous rhodium (I) catalyzed [2+2+2] cycloadditions, [Rh(C2H4)2Cl]2 proved to be the most efficient precatalyst.[16,17] A variety of TADDOL based phosphoramidite ligands provided the vinylogous amide. However, poor product selectivity (Table 1, Entry 1) and low yield (Table 1, Entries 2, 3) are observed. BINOL based phosphoramidite ligands such as Guiphos B1 provided vinylogous amide with low enantioselectivity (Table 1, Entry 4). The recently developed electron withdrawing phosphoramidite, CKphos, proved to be the best ligand (Table 1, entry 5).[18] Using CKphos, vinylogous amide was obtained in 77% yield and 94% ee. As expected with CKphos, product selectivity favored 3 over 4 by >19:1.[19]
Table 1.
 | ||||
|---|---|---|---|---|
| Entry | Ligand | 3:4b | Yield 3 [%] | ee 3c [%] |
| 1 | T1 | 2.7:1 | 53 | 88 |
| 2 | T2 | >19:1 | 20 | 91 |
| 3 | T3 | >19:1 | 33 | 93 |
| 4 | B1 | 1:1 | 30 | 40 |
| 5 | CKphos | >19:1 | 77 | 94 |
Reaction conditions: 1.6 equiv 1a, 1.0 equiv 2, 0.05 equiv [Rh(C2H4)2Cl]2, 0.10 equiv ligand, reaction concentration: 0.05M.
Determined by 1H NMR after purification.
ee’s were determined by HPLC analysis on a chiral stationary phase.
![[c]](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/108c/3820010/74fd3546cae5/nihms515945f4.jpg)
With optimal conditions in hand, the alkyne scope was explored (Table 2). Aryl alkynes with electron donating and electron withdrawing groups participate in the reaction with moderate to high yield and high enantioselectivity (3a–3j). Substitution at the ortho-and meta- positions (3f–3j) is tolerated without decrease in yield or enantioselectivity. Heteroaromatic alkynes and enynes are also competent substrates in the reaction, providing 3k and 3l with high enantioselectivity. In all cases, product selectivity is >19:1 favoring vinylogous amide.
Table 2.
 |
Reaction conditions: 1.6 equiv 1, 1.0 equiv 2.
Yields for isolated vinylogous amide product. In all cases product selectivity is >19:1 (3:4). ee’s were determined by HPLC analysis on a chiral stationary phase.
Slow addition of 2 was required.
3n could be obtained in 16% yield with the use of B1 as ligand.
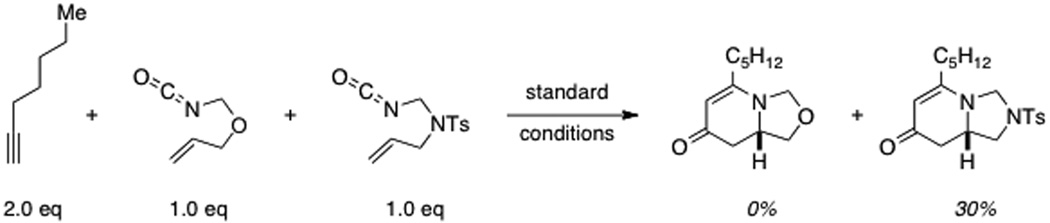
Alkyl and internal alkynes do not undergo the desired reaction with oxygen-linked alkenyl isocyanates under the standard conditions.[20] Excess alkyne (5.0 equivalents) or prolonged reaction times (48 h) do not lead to cycloadduct formation. However, in the case of 1-heptyne, we found that with slow addition of the isocyanate, 3m could be isolated in modest yield. Interestingly, isocyanates with a N-Ts linker provide the desired products with both aromatic and alkyl alkynes (Table 2). Importantly, the reaction also tolerates Cbz and Boc protecting groups on nitrogen (6p–6r, Table 2).
Vinylogous amide products 8 containing a tetrasubstituted carbon could be obtained when alkenyl isocyanate 7 was used in the reaction (Table 3).[1c] These reactions proceed in slightly lower yield and enantioselectivity. A variety of alkynes are tolerated, including aryl alkynes with electron donating or withdrawing substituents and enynes. Substrates bearing a homologous tether afford vinylogous amides 10 with a 6,6-bicyclic ring system (Table 4).
Table 3.
 |
Reaction conditions: 1.6 equiv 1, 1.0 equiv 7.
See Table 2.
Table 4.
 |
Reaction conditions: 1.6 equiv 1, 1.0 equiv 9.
See Table 2.
We then turned our attention to cleavage of the tether. Unfortunately, a one step cleavage of the tether proved problematic.[21] We found that reduction of the vinylogous amide allows cleavage of the aminal. 5% Palladium on carbon under a hydrogen atmosphere affords bicyclic aminals with high diastereoselectivity (>19:1, Table 5).[1b]
Table 5.
Access to Trisubstituted Piperidines.[a]
 |
Reaction conditions: 5 mol% Pd/C in EtOAc for 24–36h at 23 °C; 2.0 equiv NaCNBH3 in MeOH/AcOH for 24h at 23 °C.
A screen of a variety of conditions to cleave the aminal revealed reductive amination as an effective method to provide the N-methylpiperidinol products. Thus, treatment of the aminal with sodium cyanoborohydride in a mixture of methanol and acetic acid (3:1) at ambient temperature provides the desired product.[22] Using vinylogous amide 3l in this two-step procedure affords piperidinol 12l with alkyl substitution. This presents a solution to the incorporation of alkyl alkynes in the reported [2+2+2] cycloaddition.
An X-ray crystal structure was obtained of compound 13a. The protons of the three tertiary carbons are all on the same face of the piperidinol ring, confirming the stereochemistry of the reduction of the vinyologous amide with Pd/C.[23]
In conclusion, we present a route to access piperidinol scaffolds based on the rhodium (I) catalyzed asymmetric [2+2+2] cycloaddition between alkynes and an oxygen-linked alkenyl isocyanate. The cycloaddition proceeds with good yield and high enantioselectivity for a variety of substrates. The stereocenter introduced in a catalytic, asymmetric fashion is then used to control diastereoselectivity in a subsequent hydrogenation to afford diastereoselectivities of >19:1. Piperidinol scaffolds with functional group handles for further manipulation can then be accessed following reductive amination.
Experimental Section
Standard [2+2+2] Conditions
In a glove box, a round bottom flask was charged with chlorobisethylene rhodium (I) dimer (0.005 mmol) and CKphos (0.01 mmol). The flask was equipped with a reflux condensor and septum. Outside the glove box, toluene (1 mL) was added, and the mixture was stirred for 15 min. after which time alkenyl isocyanate (0.10 mmol) and alkyne (0.16 mmol) in toluene (1 mL) were added dropwise. The reaction mixture was heated to reflux and stirred for 16 h. Upon completion of the reaction, the flask was cooled to 23 °C, solvent removed via rotary evaporation, and the crude material was subjected to column chromatography (EtOAc to 20:1 EtOAc:MeOH).
Supplementary Material
Acknowledgments
We thank NIGMS (GM80442) for generous support and Roche and Amgen for unrestricted support. We thank Johnson Matthey for a generous loan of Rh salts.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:2782–2783. doi: 10.1021/ja057803c. [DOI] [PubMed] [Google Scholar]; b) Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:12370–12371. doi: 10.1021/ja064868m. [DOI] [PubMed] [Google Scholar]; c) Lee EE, Rovis T. Org. Lett. 2008;10:1231–1234. doi: 10.1021/ol800086s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yu RT, Rovis T. J. Am. Chem. Soc. 2008;130:3262–3263. doi: 10.1021/ja710065h. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Friedman RK, Rovis T. J. Am. Chem. Soc. 2009;131:10775–10782. doi: 10.1021/ja903899c. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Dalton DM, Oberg KM, Yu RT, Lee EE, Perreault S, Oinen ME, Pease ML, Malik G, Rovis T. J. Am. Chem. Soc. 2009;131:15717–15728. doi: 10.1021/ja905065j. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Oinen ME, Yu RT, Rovis T. Org. Lett. 2009;11:4934–4937. doi: 10.1021/ol9020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Michael JP. Nat. Prod. Rep. 2004;21:625–649. doi: 10.1039/b310689f. [DOI] [PubMed] [Google Scholar]; b) Michael JP. Nat. Prod. Rep. 2008;25:139–165. doi: 10.1039/b612166g. [DOI] [PubMed] [Google Scholar]
- 3.a) Strunz GM, Findlay JA. In: The Alkaloids. Brossi A, editor. Vol. 26. New York: Academic Press; 1985. p. 89. [Google Scholar]; b) O’Hagan D. Nat. Prod. Rep. 2000;17:435. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]; c) Michael JP. Nat. Prod. Rep. 2008;25:166–187. doi: 10.1039/b612168n. [DOI] [PubMed] [Google Scholar]
- 4.a) Edwards MW, Daly JW. J. Nat Prod. 1988;51:1188–1197. doi: 10.1021/np50060a023. [DOI] [PubMed] [Google Scholar]; b) Edwards MW, Garaffo HM, Daly JW. Synthesis. 1994:1167–1170. [Google Scholar]
- 5.Jones TH, Blum MS, Fales HM. Tetrahedron. 1982;38:1949–1958. [Google Scholar]
- 6.a) Anderson PC, Soucy F, Yoakim C, Lavallee P, Beaulieu PL. 5614533 A 19970325. US Patent. 1997; b) Lamarre D, Croteau G, Wardrop E, Bourgon L, Thibeault D, Clouette C, Vaillancourt M, Cohen E, Pargellis C, Yoakim C, Anderson PC. Antimicrob. Agents Chemother. 1997;41:965–971. doi: 10.1128/aac.41.5.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For select recent examples of poly-substituted piperidine syntheses in which two or more components are united to form the azacycle, see: Peltier HM, Ellman JA. J. Org. Chem. 2005;70:7342–7345. doi: 10.1021/jo051020s. Kobayashi T, Hasegawa F, Tanaka K, Katsumura S. Org. Lett. 2006;8:3813–3816. doi: 10.1021/ol0614065. Takahashi M, Micalizio GC. J. Am. Chem. Soc. 2007;129:7514–7516. doi: 10.1021/ja071974v. Wang Y, Zhu S, Ma D. Org. Lett. 2011;13:1602–1605. doi: 10.1021/ol200004s. Kim H, Rhee YH. J. Am. Chem. Soc. 2012;134:4011–4014. doi: 10.1021/ja2116298. Duttwyler S, Lu C, Rheingold AL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2012;134:4064–4067. doi: 10.1021/ja2119833. Ho KYT, Aïssa C. Chem. Eur. J. 2012;18:3486–3489. doi: 10.1002/chem.201200167. Ishida N, Yuhki T, Murakami M. Org. Lett. 2012;14:3898. doi: 10.1021/ol3016447. Kumar P, Louie J. Org. Lett. 2012;14:2026–2029. doi: 10.1021/ol300534j.
- 8.For recent syntheses of substituted 4-piperidinols, see: Davis FA, Rao A, Carroll PJ. Org. Lett. 2003;5:3855–3857. doi: 10.1021/ol035390j. Gnamm C, Krauter CM, Brödner K, Helmchen G. Chem. Eur. J. 2009;15:2050–2054. doi: 10.1002/chem.200802525. Cui L, Li C, Zhang L. Angew. Chem. Int. Ed. 2010;49:9178–9181. doi: 10.1002/anie.201004712. Coombs TC, Lushington GH, Douglas J, Aubé J. Angew. Chem. Int. Ed. 2011;50:2734–2737. doi: 10.1002/anie.201007133.
- 9.Duttwyler S, Chen S, Takase MK, Wiberg KB, Bergman RG, Ellman JA. Science. 2013;339:678–682. doi: 10.1126/science.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For an excellent recent review, see: Masson G, Lalli C, Benohoud M, Dagousset G. Chem. Soc. Rev. 2013;42:902–923. doi: 10.1039/c2cs35370a.
- 11.Catalytic asymmetric methods are poorly represented in the above list, outside of the aza-Diels-Alder reaction. Stereocontrol via a chiral auxiliary has been explored by Micalizio (2 steps) and Zhang (4 steps) (refs. 7c and 8c respectively). The two catalytic asymmetric examples are Ma’s prolinol catalyzed coupling of aldehydes and allylic amines bearing a nitro group on the olefin and Helmchen’s Ir catalyzed allylic amination reaction which provides trisubstituted piperidinols in 8 steps from allylic carbonates; see refs. 7d and 8b.
- 12.For reviews of piperidine synthesis, see: Baliah V, Jeyaraman R, Chandrasekaran L. Chem. Rev. 1983;83:379–423. Sardina FJ, Rapoport H. Chem. Rev. 1996;96:1825–1872. doi: 10.1021/cr9300348. Weintraub PM, Sabol JS, Kane JM, Borcherding DR. Tetrahedron. 2003;59:2953–2989. Buffat MGP. Tetrahedron. 2004;60:1701–1729. Bull JA, Mousseau JJ, Pelletier G, Charette AB. Chem. Rev. 2012;112:2642–2713. doi: 10.1021/cr200251d.
- 13.Use of alkenes with a tethered coordinating group such as a thioether, pyridine, amine, or second alkene did not provide the desired product.
- 14.For reviews on the use of cleavable tethers, see: Fensterbank L, Malacria M, Sieburth S. Synthesis. 1997:813–854. Gauthier D, Jr, Zandi KS, Shea KJ. Tetrahedron. 1998;54:2289–2338. Bracegirdle S, Anderson EA. Chem. Soc. Rev. 2010;39:4114–4129. doi: 10.1039/c0cs00007h.
- 15.Murakami has reported a fully intermolecular asymmetric [2+2+2] cycloaddition between an allene and two equivalents of isocyanate catalyzed by Ni; see: Miura T, Morimoto M, Murakami M. J. Am. Chem. Soc. 2010;132:15836–15838. doi: 10.1021/ja105541r.
- 16.Chlorocyclooctadienerhodium (I) dimer is also a competent catalyst, but provided slightly lower yield (60%, 92% ee) of the desired vinylogous amide product with CKphos.
- 17.Rh(PPh3)3Cl, [Rh(cod)2]BF4, Ni(cod)2, Pd(PPh3)4, or [IRcod)Cl]2 did not provide any observed product.
- 18.(a) Dalton DM, Rappé AK, Rovis T. Chem. Sci. 2013 doi: 10.1039/C3SC50271F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Perreault S, Rovis T. Synthesis. 2013:719–728. doi: 10.1055/s-0032-1316786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dalton DM, Rovis T. submitted. [Google Scholar]
- 19.Attempts to access 4 as the major product resulted in either no reaction or decomposition of 2.
-
20.The reasons behind this observation are unclear. We note, however, that the presence of 2 does not prevent other cycloadditions from occurring with heptyne as per the following experiment:
- 21.Acidic conditions led to a mixture of unidentified byproducts. Also, variation of the heteroatom tether did not alleviate the problems with one-pot cleavage.
- 22.Direct reductive amination of the vinylogous amide leads toobserved formation of the desired product, albeit in poor diasteroselectivity.
- 23.See supporting information for X-ray structure and data.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.