

Abstract
Facioscapulohumeral muscular dystrophy is a dominantly inherited myopathy associated with chromatin relaxation of the D4Z4 macrosatellite array on chromosome 4. DUX4 is encoded within each unit of the D4Z4 array where it is normally transcriptionally silenced and packaged as constitutive heterochromatin. Truncation of the array to less than 11 D4Z4 units (FSHD1) or mutations in SMCHD1 (FSHD2) results in chromatin relaxation and a small percentage of cultured myoblasts from these individuals exhibit infrequent bursts of DUX4 expression. There are no cellular or animal models to determine the trigger of the DUX4 producing transcriptional bursts and there has been a failure to date to detect the protein in significant numbers of cells from FSHD-affected individuals. Here, we demonstrate for the first time that myotubes generated from FSHD patients express sufficient amounts of DUX4 to undergo DUX4-dependent apoptosis. We show that activation of the Wnt/β-catenin signaling pathway suppresses DUX4 transcription in FSHD1 and FSHD2 myotubes and can rescue DUX4-mediated myotube apoptosis. In addition, reduction of mRNA transcripts from Wnt pathway genes β-catenin, Wnt3A and Wnt9B results in DUX4 activation. We propose that Wnt/β-catenin signaling is important for transcriptional repression of DUX4 and identify a novel group of therapeutic targets for the treatment of FSHD.
INTRODUCTION
FSHD is initially characterized by progressive weakening of select skeletal muscle groups in the face, trunk and lower extremities (1), but over time can affect nearly every muscle in the body. The disease is associated with the loss of markers of heterochromatin at the D4Z4 macrosatellite array on chromosome 4 (2) which can occur by array contraction dependent (FSHD1) or independent (FSHD2) mechanisms (3–5). The chromatin relaxation results in expression of the transcription factor double Homeobox protein 4 (DUX4) contained within each 3.3 kb D4Z4 unit. (6–9).
DUX4-induced toxicity has been demonstrated by overexpressing the gene in cultured cells and tissues (10–13) and produces cell death that is dependent on reactive oxygen species (10) and p53 activity (11,12). DUX4-induced cell death provides a convenient assay for the identification of compounds that directly interfere with the protein or the downstream apoptotic program. Molecular pathways that regulate DUX4 transcription would be missed with forced expression strategies and these are potentially important therapeutic targets. However, investigators have failed to detect significant levels of endogenously produced DUX4 protein in FSHD myoblasts with current estimates at 1 out of 1000 myoblasts cells (14) (Table 1). Here we describe and validate an approach to myoblast differentiation that increases the sensitivity for detecting DUX4 protein and produces DUX4-dependent toxicity in patient-derived muscle cell cultures. Thus, molecular pathways that activate or repress DUX4 transcription can be studied in addition to the downstream events in un-modified primary cells from FSHD-affected people.
Table 1.
Myoblast cells used in this study
| Biopsy code | D4Z4 numbera | Phenotype | DUX4b | >70% MIc | Deathd |
|---|---|---|---|---|---|
| 2082 | 8 | FSHD1 | *** | Y | Y |
| 2346 | 6 | FSHD1 | *** | N | N |
| 2391 | 5 | FSHD1 | N.D. | N | N |
| 2084 | 4 | FSHD1 | * | Y | Y |
| 2066 | 7 | FSHD1 | N.D. | Y | N |
| 2349 | 2 | FSHD1 | *** | Y | Y |
| 2088 | 9 | FSHD1 | * | Y | Y |
| 2062 | 15 | FSHD2 | ** | N | N |
| 1881 | 16 | FSHD2 | *** | Y | Y |
| 2305 | 12 | FSHD2 | *** | Y | Y |
| 2401 | 11 | Non-FSHD | N.D. | Y | N |
| 2081 | 74 | Non-FSHD | N.D. | Y | N |
| NR209 | 25 | Non-FSHD | N.D. | Y | N |
DUX4 was detected < 1/106 myoblast nuclei.
N.D., none detected.
aNumber of D4Z4 repeats on the permissive 4qA allele.
bDUX4 positive nuclei in myotubes. *<1%,**<10%, and ***>10%.
cMI; myotube index (% of total nuclei in MHC(+) cells at 48 h).
dMyotube death in cultures after 96 h of differentiation.
The Wnt/β-catenin signaling pathway has been implicated in FSHD pathology because of its role in muscle development, and facial muscle organization (15), and because people with mutations of the Wnt ligand, Norrin and the Wnt receptors frizzled-4, LRP5 and TSPAN12 have specific peripheral retinal vascular pathology in common with some patients with FSHD (16,17). Wnt genes encode a family of secreted proteins that play a role in many aspects of embryonic development and tissue homeostasis through the activation of receptor-mediated signaling pathways (18,19). The canonical Wnt signaling pathway involves Wnt-mediated stabilization of the transcriptional co-factor, β-catenin (20). In the absence of Wnt, β-catenin is phosphorylated by glycogen synthase 3-beta (GSK3β) and degraded. In the presence of Wnt, phosphorylation of β-catenin is prevented, allowing β-catenin to enter the nucleus and promote transcription of Wnt-target genes by binding to TCF/ LEF-1 transcription factors. Wnts can also initiate β-catenin independent pathways that oppose the effects of the canonical pathway (21). Understanding how these processes may regulate DUX4 has not been investigated, but it is clear that Wnt/β-catenin signaling plays important roles in muscle development (22,23) and postnatal muscle repair by facilitating myoblast differentiation and myotube fusion (24,25). Here we focus on the effect of Wnt/β-catenin signaling on DUX4 expression and for the first time show that activation of the Wnt/β-catenin pathway in FSHD myotubes results in reduced DUX4 expression levels and prevents DUX4-dependent myotube apoptosis. Reduction of transcripts that encode Wnt-pathway components results in DUX4 activation, consistent with a model where DUX4 transcription is under active Wnt-mediated suppression. Given that Wnt7a was recently shown to promote hypertrophy of myotubes in a mouse model of Duchenne Muscular Dystrophy, therapies that enhance or suppress components of the Wnt signaling pathway may be a practical strategy to block further muscle wasting in patients with FSHD (26–30).
RESULTS
Use of media supplemented with knock-out serum replacer improves differentiation of human myoblasts
Modeling FSHD has been challenging because DUX4 is only present in the primate lineage and expression is sporadic and infrequent in myoblasts derived from human muscle biopsies. Furthermore, growth and differentiation of FSHD-biopsy derived myoblasts appears to occur normally without obvious differences when comparing growth and differentiation characteristics to those of myoblasts derived from unaffected controls (31). Differentiation of cultured human myoblasts is initiated by mitogen depletion, usually accomplished by reducing serum concentrations in the medium (32). We found that growth factor reduction without serum starvation by using medium containing 20% knock out serum replacer (KOSR) improved myoblast differentiation and reproducibly generated large myotubes from multiple patient biopsies(33) (Fig. 1A–C). Applying this differentiation protocol to FSHD-muscle-derived myoblasts resulted in an increase in the number of DUX4(+) myotube nuclei when cells differentiated with KOSR were compared with the same cells differentiated using conventional conditions (HS/ITS) (Fig. 1A and B). Since the percentage of DUX4(+) nuclei varied between different cell lines, we investigated DUX4 immunoreactivity in myotubes generated from a number of FSHD-derived muscle biopsies (Supplementary Material, Table S1) and chose myoblast cell lines that formed myotubes with the highest percentage of DUX4(+) nuclei for subsequent studies. These included FSHD1 (2349), which contained two D4Z4 units, and cells from people carrying SMCHD1 mutations (34) FSHD2(2305) with 12 units and FSHD2(1881) with 16 units.
Figure 1.

Culture medium supplemented with KOSR improves myoblast differentiation and produces myotubes that express DUX4. (A) Immunofluorescent microscopy of primary human FSHD myoblasts (2082) differentiated using HS/ITS or KOSR and stained with an antibody specific for the C-terminus of DUX4 (red) and MHC (green). Nuclei are counterstained with DAPI (blue). Scale bar = 50 μm. (B) High magnification images of insets numbered in (A). Scale bar = 10 μm. (C) Scatter plot displaying the number of nuclei per cluster and demonstrating that myotubes differentiated in medium supplemented with KOSR contain large clusters that have more nuclei. *P < 0.05 by Mann–Whitney test for non-parametric distribution. (D) Fold change in the percentage of total nuclei positive for DUX4 immunofluorescence within MHC(+) myotubes for two non-FSHD and three FSHD-derived myoblasts when comparing HS/ITS or KOSR differentiation conditions. Results are pooled between the three cell lines (FSHD1(2082), FSHD2(2305) and FSHD2(1881). P < 0.05 based on Student's t-test. (E) Percentage of DUX4(+) nuclear clusters as a fraction of total number of nuclei in myotubes. (F) FSHD myoblasts were fused with mouse C2C12 myoblasts which do not carry the DUX4 gene. Mouse nuclei are distinguished by pronounced pericentric heterochromatin staining with DAPI and several examples are circled in yellow. DUX4(+) mouse nuclei (circled in yellow and immunostained red for DUX4) are indicated. (G) Quantitative RT-PCR of DUX4-activated genes, MBD3L2 and CCNA1 in three FSHD-derived cell lines [FSHD1(2082), FSHD2(2305), FSHD2(1881)]. Data is displayed as the change in expression level when myoblasts are differentiated in KOSR compared with HS/ITS. *P < 0.05 by Student's t-test.
Regardless of the differentiation protocol, DUX4 was exclusively expressed in MHC(+) myotubes. To account for variable myotube formation between different cell cultures, the percentage of DUX4(+) nuclei is expressed as a fraction of nuclei incorporated into MHC(+) myotubes. Using this metric DUX4 protein was detected in 0.5–2.0% of myotubes when myoblasts were differentiated using conventional HS/ITS conditions and 3.0–16.5% when differentiated using KOSR (Fig. 1D). Because the increased percentage of DUX4(+) nuclei could be attributed to abundant transcription in a sentinel nucleus with distribution of DUX4 protein to multiple adjacent nuclei (35) sharing a common cytoplasm, we quantified sentinel transcription events by counting DUX4(+) clusters in myotubes and divided by the total number of nuclei in myotubes. When FSHD-myoblasts were differentiated in medium supplemented with HS/ITS or KOSR, no difference in the number of sentinel events was observed (Fig. 1E) suggesting that the apparent increase in prevalence of DUX4 protein seen in FSHD-myotubes produced by KOSR-mediated differentiation is due to distribution of DUX4 to more nuclei present in clusters rather than an increase in the number of nuclei undergoing active DUX4 transcription. Consistent with this hypothesis, we observed human DUX4 protein in mouse nuclei when FSHD myoblasts were fused with mouse C2C12 myoblasts that do not contain the DUX4 gene (Fig. 1F). The consequence of more nuclei being exposed to DUX4 produced from sentinel activation events is an amplification of expression of downstream targets of the DUX4 transcription factor as demonstrated by quantifying the change in MBD3L2, and CCNA1 transcript levels that occurs with myoblast differentiation (36) (Fig. 1G).
Myotubes from FSHD-derived biopsies undergo apoptosis
The DUX4 signal amplification occurring by exposing more nuclei to the transcription factor also produced a cell-culture phenotype. FSHD myoblasts induced to differentiate in KOSR-containing medium developed cytopathic lesions that appeared as retracted myotubes surrounding a lawn of myoblasts (Fig. 2A and B) and the lesions progressed to complete loss of myotubes in the cultures after 72–96 h of differentiation. The myotubes adjacent to lesions incorporated DAPI indicating a loss in cell membrane integrity and DAPI incorporation was only present in FSHD-myoblast cultures differentiated in KOSR (Supplementary Material, Fig. S1). DUX4 protein was consistently detected within 500 μm of retracted myotubes and infrequently detected outside of the lesions suggesting the cytopathology was directly related to DUX4 exposure (Fig. 2C). Cytopathic lesions were present in differentiated cultures of myoblasts from most of our FSHD-derived samples, including cells from people with either FSHD1 or FSHD2 (Supplementary Material, Table S1).
Figure 2.
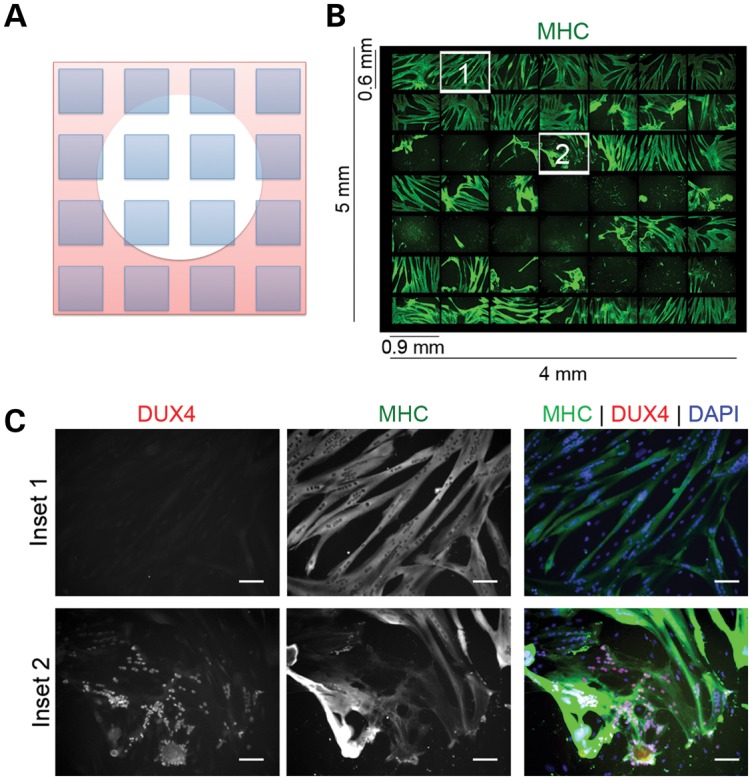
Myoblasts from FSHD-muscle biopsies die when efficiently differentiated to myotubes. (A) Drawing of photo arrangement shown in (B) showing a cytopathic lesion represented as a white circle. The microscope was programmed to take pictures (shown as blue squares) spanning the lesion. (B) Immunofluorescent microscopy of a 49 images spanning a 4 × 5 mm lesion from an FSHD myotube culture differentiated in medium supplemented with KOSR for 48 h. Green = MHC. (C) Numbered fields from (B) shown at increased magnification to demonstrate that nuclei adjacent to the lesion (Inset #2) are DUX4(+), whereas nuclei outside of the lesion are DUX4(−) (Inset #1). Red = antibody to C-terminus of DUX4. Green = antibody to MHC. Scale Bar = 50 μm.
FSHD-myotube apoptosis is due to DUX4 expression
DUX4(+) nuclei had condensed chromatin and contracted myotubes contained clusters of TUNEL(+) pyknotic nuclei that stained for activated caspase-3 suggesting an apoptotic mechanism (Supplementary Material, Fig. S2). To determine whether apoptosis could be prevented, the myotubes were transfected with siRNA targeting DUX4 (36). The siRNAs efficiently reduced transcript and protein levels, and reduced expression of the DUX4-activated genes MBD3L2 and CCNA1 by over 80% (Fig. 3A–C). After 96 h of differentiation, FSHD-derived cell cultures had widespread loss of myotubes, that was reversed by transfection with an siRNA targeting DUX4 (36) (Fig. 4A). Because toxicity was myotube specific, myoblasts transfected with DUX4 siRNA produced myotubes with a higher myotube index (Fig. 4B), lower numbers of pyknotic nuclear clusters (Fig. 4C), lower DNA content in the medium and fewer myotubes containing activated caspase-3 (data not shown). Similar results were achieved by transfection of cells with an siRNA targeting p53 transcripts supporting previous studies performed in mouse cells and tissues (Fig. 4D) (37).
Figure 3.

Optimization of DUX4 knockdown. (A) FSHD myoblasts were transfected with two siRNAs targeting DUX4 (DUX4-1 and DUX4-4). Twenty-four hours later, the medium was changed to KOSR differentiation medium and 48 h later harvested and assayed for DUX4 transcripts by RT-PCR. (B) Cells prepared as in (A), were assayed by qRT-PCR for expression of the two DUX4-activated genes, MBD3L2 and CCNA1. (C) Cells prepared as in (A) were fixed and stained for DUX4 (red) and MHC (green) and counterstained with DAPI (blue). Scale Bar = 50 μm.
Figure 4.

Myotube death is DUX4 dependent. (A) FSHD myoblasts were transfected with non-targeting (SCR) or DUX4-targeting siRNAs and differentiated into myotubes. Ninety-six hours after the initiation of differentiation, dishes were stained with Coomassie blue, and imaged at low magnification (×10) to demonstrate the widespread loss of myotubes in the culture. Scale bar = 0.5 mm. (B) FSHD myoblasts were transfected with non-targeting or DUX4-targeting siRNAs and differentiated into myotubes. Ninety-six hours post-differentiation, dishes were stained with antibodies to MHC and the myotube fusion index was calculated. P < 0.05 by Student's t-test. (C) Average number of contracted MHC(+) myotubes containing clusters of pyknotic nuclei per well of a 384-well dish 96 h following transfection of the DUX4-targeting siRNA. P < 0.05 by Student's t-test. (D) FSHD1(2349) myoblasts were transfected with non-targeting, DUX4-targeting and p53-targeting siRNA and stained for MHC 96 h post-differentiation. Scale bar = 50 μm. (E) FSHD1(2349) myoblasts cultured in 384-well dishes were treated with non-targeting, DUX4-targeting or FRG1-targeting siRNAs and assayed for myotube viability by staining with an anti-MHC antibody (green) and counterstained with DAPI (blue). Scale bar = 50 μm.
FSHD related gene 1 (FRG1) has also been implicated in FSHD pathology due to its location adjacent to the D4Z4 array on chromosome 4 (38), and the existence of transgenic mice that exhibit myopathology when the gene is overexpressed in muscle tissue (39). However, reduction of FRG1 transcripts did not ameliorate myotube toxicity (Fig. 4E and Supplementary Material, Fig. S3) suggesting its effects, if any, are secondary to DUX4. These experiments demonstrate that DUX4 is principally responsible for the apoptosis seen in patient samples.
This cell-culture platform allowed us to quickly test several chemicals that might abrogate DUX4 toxicity and thus be candidates for FSHD therapies. Oxidative stress has been a frequently sited feature of FSHD pathology because of a screen that showed increased expression of genes in oxidative response pathways (40), and because dysregulation of oxidative response pathways was also identified in mouse myoblasts overexpressing DUX4 (10). We treated FSHD myotubes with the antioxidants β-mercaptoethanol and ascorbic acid but observed DUX4-dependent myotube apoptosis despite treatments at concentrations that rescued mouse myoblasts under conditions of DUX4 overexpression (10) (Supplementary Material, Fig. S4A). Since KOSR contains selenium, which may have antioxidant properties, we differentiated myotubes in medium containing lipid-rich albumin fraction, Albumax—the protein and lipid constituent of KOSR (41). Myotubes from FSHD patients underwent apoptosis at the same rate in either KOSR or Albumax containing medium with or without β-mercaptoethanol, thus the pro-survival effects of antioxidants may not suffice to rescue the phenotype in myotubes.
P53 is a component of the final common pathway of DUX4-mediated apoptosis (37,42), we therefore tested the effects of a small library of anti-apoptotic reagents for their ability to prevent DUX4-mediated myotube death. Treatment of FSHD-myotubes with the p53 inhibitor pifithrin-α (37) or arsenic trioxide, a chemotherapeutic agent that prevents p53 activation by disrupting PML nuclear bodies, efficiently prevented DUX4-mediated myotube death (Supplementary Material, Fig. S4B).
Wnt/β-catenin signaling suppresses DUX4 expression and prevents FSHD-associated myotube apoptosis
The Wnt/β-catenin signaling pathway has been implicated in FSHD pathology, so we tested whether Wnt-pathway activation might affect DUX4 expression. Treatment of primary human cells [normal(NR201) and FSHD2(2305)] with an inhibitor of glycogen synthase kinase 3β (GSKi) to activate Wnt signaling, reduced the characteristic increase in DUX4 expression observed during myotube formation (43) (Fig. 5A). FSHD myoblasts differentiated in the presence of GSKi showed a >50% reduction in the number of DUX4(+) nuclei and the number was further reduced to 2% of total nuclei by adding recombinant Wnt3A (Fig. 5B and C) which prevented DUX4-mediated apoptosis (Fig. 5D and E). The results could not be explained by changes in myotube formation or nuclear clustering, since the myotube index did not change significantly between conditions, and myogenin levels showed only a slight increase when Wnt signaling was activated in non-FSHD myotubes (Supplementary Material, Fig. S5). Since Wnt activated DUX4 suppression by treatment with GSKi occurred in both FSHD1 and FSHD2 derived cells (Fig. 5F), we hypothesized that the effect was independent of chromatin structure at D4Z4. To test whether transcriptional suppression occurs in the absence of chromatin, we determined whether treatment with GSKi could repress the activity of a single-repeat DUX4 promoter controlling expression of firefly luciferase (D4Z4(FFL)). Treatment of C2C12 myotubes carrying D4Z4(FFL) with GSKi prevented the characteristic increase in luciferase expression noted during myotube formation (Fig. 5G).
Figure 5.

Activation of the Wnt/β-catenin signaling reduces DUX4 expression in FSHD myotubes. (A) RT-PCR of non-FSHD and FSHD-derived cells cultured in myoblast proliferation or differentiation medium in the absence or presence of GSK3β inhibitor (GSKi). Transcription of the Wnt target gene, AXIN2, was monitored as a control for activation of Wnt/β-catenin signaling. (B) Primary FSHD(2349) myoblasts were differentiated in medium containing GSKi or 250 ng/ml recombinant Wnt3A (rWnt3A) or relevant vehicle controls (DMSO for GSKi or 0.1% CHAPS for rhWnt3A). Myotubes were fixed at 48 h to quantify DUX4 protein expression. (C) Immunofluorescent microscopy images of cells prepared in (B). Scale Bar = 50 μm. Myotube index for the image displayed in each condition is displayed in white text in the lower left corner of the panel (MI). (D) Primary FSHD(2349) myoblasts were cultured as in (B) and fixed at 96 h to measure myotube loss. Photos from the 96 h timepoint stained with MHC (green) are shown. The percentage of DUX4(+)/MHC(+) nuclei from a 48 h timepoint of the matching experiment are shown in white text in each quadrant. Scale bar = 200 μm. (E) Myotube index of experiments shown in (D). P < 0.05 by Student's t-test. (F) FSHD2 myotubes were treated with GSKi to confirm that the effect of Wnts on DUX4 protein levels was independent of D4Z4 chromatin structure. DUX4(+) nuclei were quantified. P < 0.05 by Student's t-test. (G) C2C12 myoblasts infected with a lentivirus encoding D4Z4(FFL) were cultured in the presence of absence of GSKi, and Luciferase activity was measured. P < 0.05 by Student's t-test.
Reduction of Wnt pathway signaling mediators results in increased DUX4 expression in FSHD-derived cells
To determine which components of the canonical Wnt pathway are involved in DUX4 suppression, Wnt signaling activity was reduced by knockdown of the genes downstream of GSK3β. β-catenin and Wnt3A siRNA-mediated transcript reduction resulted in a 3-fold and 4-fold increase in the percentage of DUX4(+) nuclei, respectively (Fig. 6A), suggesting that activity from the canonical pathway is the main source of DUX4 suppression. However, inhibition of the acyltransferase, PORCN, which is required for Wnt post-translational modification and concomitant secretion of all Wnt proteins, only resulted in a 20% increase in DUX4(+) nuclei, suggesting that other Wnt pathways may have opposing effects on the Wnt/β-catenin pathway (Fig. 6B). Consistent with this observation, we found that FSHD myotubes treated with rWnt3A in the presence or absence of IWP2 resulted in an 80% decrease in DUX4(+) nuclei indicating that when the repressive effects of Wnt3A on DUX4 are isolated by inhibiting the action of other Wnt molecules the suppression of DUX4 production is most apparent (Fig. 6C). To determine if other Wnt-related genes are involved in the suppression of DUX4 expression in non-diseased cells, we used siRNAs to reduce transcript levels of a small panel of Wnt signaling regulators in a non-FSHD myotube. DUX4(+) nuclei were only observed after reduction of Wnt9B transcripts. (Fig. 6D). Reduction of Wnt9B transcripts in the FSHD(2349) cell line resulted in a 10-fold increase in the number of DUX4(+) nuclei (Fig. 6E) demonstrating that Wnt/β-catenin signaling contributes to DUX4 suppression from hypomethylated D4Z4 arrays that are kept in equilibrium with transcriptionally silenced counterparts in cultures of FSHD1 cells (Fig. 6F).
Figure 6.

Knockdown of endogenous Wnt/β-catenin signaling pathway components activates DUX4. (A) Quantification of DUX4(+) nuclei from myotubes transfected with siRNAs targeting CTNNB1 (the gene encoding β-catenin) (siCTNNB1) or Wnt3A (siWnt3A.) P < 0.05 by Student's t-test (B) FSHD1(2349) myoblasts were differentiated in medium supplemented with KOSR and containing 100 nmol IWP2 or vehicle control. The fold change of DUX4(+) nuclei is shown. (C) FSHD1(2349) myoblasts were differentiated in medium containing rWnt3A with or without IWP2. DUX4 was quantified by immunohistochemstriy. P < 0.05 by ANOVA. (D) Non-FSHD(2401) myoblasts were seeded onto wells of at 384-well dish and transfected with siRNAs to the genes shown on the X-axis (si[GENE]). Wells were stained for DUX4 and inspected for DUX4 expression. Two DUX4(+) nuclear clusters were observed when Wnt9B was knocked down. P < 0.05 by ANOVA. (E) Knockdown of Wnt9B (siWnt9B) results in a 10-fold increase in DUX4(+) nuclei than siSCR controls. P < 0.05 by Student's t-test. (F) Model depicted two states of a truncated D4Z4 array existing in equilibrium in a myotube culture. On the left of the equation, silenced heterochromatinized arrays are shown coated in white circles. When the array reaches a chromatin conformation amenable for DUX4 transcription (right of the equation), the canonical Wnt pathway serves to repress DUX4 transcription.
DISCUSSION
This is the first report of DUX4 protein in FSHD-cells at significant levels and sufficient quantities to an intrinsic viability defect in myotubes generated from FSHD-muscle biopsies. Cell death is directly associated with and dependent upon DUX4, and independent of the FSHD-candidate gene FRG1. The muscle-specific activation of DUX4 may explain why no defects were previously identified when cells were cultured in HS/ITS, since nuclear clustering in the larger myotubes produced by KOSR-mediated differentiation allows DUX4 to enter more nuclei and amplify its effect on transcriptional targets. Preferential myotube expression may also explain why DUX4 levels reported here are different from those previously reported (<0.01%) (36), since the myotube index was not taken into account. In addition, variations in myotube fusion can account for differences observed in secondary marker expression since less nuclei are exposed to translated DUX4 protein. In primary myotubes, the apoptotic mechanisms appear to be similar to those described in models in which DUX4 is artificially overexpressed in myoblasts (10,37,44).
By improving myotube differentiation, more nuclei become exposed to the DUX4 protein made from relatively infrequent bursts of expression. Because of this signal amplification, we observed the consequence of DUX4 transcriptional activation which is apoptosis in primary FSHD myotubes. Others have described a more subtle ‘atrophic’ phenotype of cultured FSHD myotubes that might also be evidence of the activity of DUX4 (44,45); however, the differentiation conditions may have produced DUX4 levels that were perhaps insufficient to induce myotube apoptosis. The extensive myotube depletion in our cultures provided an endpoint for performing genetic knockdown experiments that confirmed that the pathology was due to DUX4 expression and suggests that the DUX4-induced genetic program initiates intrinsic apoptosis in the muscles of FSHD-affected people in addition to the potential of eliciting an immune response proposed previously (36). While it is possible that apoptosis occurs due to disruption of a specific muscle cell transcriptional program by inappropriately expressing germline-specific genes, we are also intrigued by the possibility that specific targets of DUX4 may be principally responsible for the apoptosis observed here and the use of this system to identify DUX4 target genes with potent apoptosis-inducing activities is an active area of investigation.
The observation that Wnt/β-catenin signaling actively suppresses DUX4 is a novel finding that may provide a framework for connecting the muscular and non-muscular features of FSHD. For example, mutations in Wnt signaling receptors Frizzled 4, LRP5 and TSPAN12 that disrupt Wnt/β-catenin signaling in the retinal vasculature can cause the clinically indistinguishable developmental retinal exudative retinopathies, including Norrie Disease and familial exudative vitreoretinopathy (FEVR), while the mutations of the Norrin gene have been reported to cause Coats disease (46). Furthermore, the finding that extracellular signaling can influence the expression of DUX4 in myotubes suggests that sporadic DUX4 expression in the nuclei of cultured myotubes, and the focal pattern of affected muscle observed using MRI (47,48) may involve a stimulus that results in escape from the effects of a repressive pathway.
The mechanism of DUX4 regulation by Wnt/β-catenin is likely independent of direct binding of β-catenin at D4Z4. An extensive search revealed that sequences within D4Z4 do not contain TCF/LEF binding sites or other known Wnt responsive elements. Thus, Wnt/β-catenin signaling likely alters the levels of myotube specific transcription factors that bind the DUX4 promoter, or activates transcription of genes involved in direct suppression of DUX4 expression. Furthermore, Wnt/β-catenin signaling may alter properties of skeletal muscle that can not be measured by simply measuring the myotube index, such as muscle fiber type. These are topics of active investigation because of the possibility that these pathways may be specific targets for DUX4 suppressive therapies. Given the recent success in treating a mouse model of muscular dystrophy with Wnt7a (30), treatment of FSHD with Wnt agonists may be a feasible goal and the results presented here suggest that it should reduce DUX4-mediated pathology in patients.
MATERIALS AND METHODS
Ethics statement
Control and FSHD cell lines were obtained from patients using consents approved by the IRB at the University of Rochester (approval numbers 12146 and 22880). Study subjects gave written informed consent for the use of their cells in these studies.
Cell lines and cell-culture reagents
Primary human myoblasts were obtained by written informed consent through the Fields Center at the University of Rochester cell bank (http://www.urmc.rochester.edu/fields-center/protocols/myoblast-cell-cultures.cfm). Myoblasts were grown in F10 medium (Invitrogen, Carlsbad, CA, USA; www.invitrogen.com) supplemented with 20% fetal bovine serum [FBS; Thermo Scientific (Hyclone)], and 50 U/50 μg penicillin/streptomycin (Pen-Strep; Invitrogen), 10 ng bFGF (Invitrogen) and 1 μm dexamethasone (Sigma-Aldrich, St Louis, MI, USA, www.sigmaldrich.com) (9). Differentiation was induced using F10 medium supplemented with 1% equine serum and ITS supplement (insulin 0.1%, 0.000067% sodium selenite, 0.055% transferrin; Invitrogen), or in DMEM:F12 (1:1) with 3.151 g/l glucose, 10 mm non-essential amino acids (Invitrogen), 100 mm sodium pyruvate, 20% knockout serum replacer (Invitrogen, # 10828010). Albumax (Invitrogen #11020) was used at a final concentration of 10 mg/ml in DMEM:F12. Pharmacological inhibitor of glycogen synthase kinase-3-β (GSKi) was used at a final concentration of 3 μm (CHIR99021, StemGent; Cambridge. MA, USA). Recombinant human Wnt3A was used at a concentration of 250 ng/ml, and confirmed to be bioactive prior to use in our assays. IWP2 was used at a final concentration of 100 nmol (Sigma-Aldrich).
Immunofluorescent microscopy
Immunofluorescent staining for DUX4 was performed using the E5-5 rabbit monoclonal antibody to the C-terminus of DUX4 (49). Staining was performed by fixing the cells in 4% paraformaldehyde for 10 min at room temperature, washing with PBS, permeablizing with 0.5% TritonX-100 in PBS for 10 min at room temperature. The cells were then washed again in PBS and incubated overnight in a 1:10 dilution of the E5-5 antibody. The following day, the cells were washed 3–5 min in PBS and stained using a fluorescent conjugated goat-anti rabbit secondary antibody for 1–2 h at room temperature. Following a final wash, the cells were counterstained with 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich) and visualized by microscopy using uv illumination. Where applicable, the cultures were co-stained with other antibodies including the myosin heavy chain (1:1000 dilution MF20 clone; R&D systems), anti-caspase-3 (Cell Signaling Technology, Danvers, MA, USA; www.cellsignal.com).
Image quantification
To quantify DUX4 expression, the Nikon Ti Eclipse microscope was programmed using the Nikon Elements software to take 36 pictures at random on a 35 mm culture dish, or to scan an entire well of a 384-well dish. These images were then manually inspected and DUX4 (+) nuclei counted. To determine the myotube index, the Nikon Elements software was used to create binary masks of the DAPI and MHC channels for each image. This technique created a border around each nucleus or cluster of nuclei which could then be used to measure the sum pixel intensity from the DAPI channel. The average sum pixel intensity for single nuclei from each image was determined, and used to calculate the number of nuclei within clusters. By intersecting the binary images from the DAPI and MHC channels, we could exclude the nuclei that were outside of MHC(+) myotubes, and create a myotube fusion index. The myotube fusion index was compared with manual counts to confirm accuracy of our calculations.
DNA cloning
The D4Z4 regulatory region controlling the expression of DUX4 (43) was subcloned upstream of the open-reading frame of the pGL3-basic vector (pGL3) to make D4Z4(FFL). The D4Z4(FFL) was excised using XbaI/SpeI restriction enzymes and ligated into the SpeI site of a third-generation lentiviral vector multiple cloning site (pRRL-sinc-PPT-MCS; gift from Dr Grant Trobridge).
Luciferase assays
Cells transduced with D4Z4(FFL) lentivirus vector were seeded in a 96-well dish. Thirty minutes prior to the assay, Hoescht Dye was added to the cell-culture medium to a final concentration of 5 μm, and 30 min later the plate was analyzed for DNA content by measuring fluorescence at 355 nm using a multi-well plate reader (Envision; Perkin-Elmer). An equal volume of Steady-Glo (Promega) was added to each well, and transferred to an opaque dish. Luminescence was read using the plate reader, and normalized to DNA content for each well.
Cell viability assays
Hoescht Dye was added to cells in culture for 30 min at a final concentration of 5 μm. Following detatchment with trypsin a 50 μl aliquot of the cells was assayed for fluorescence at 355 nm to give an absorbance value for DNA content. An equal volume of Cell Titre Glo (Promega) was then added to the aliquot, and luminescence was measured. The luminescent value was normalized to the absorbance value for DNA content.
Semi-quantitative PCR
RNA was isolated by Trizol extraction according to the manufacturer’s protocol (Invitrogen), except that the final ethanol precipitate was resuspended 1× DNase I buffer and 5 U DNase I for 15 min and the RNA was extracted with phenol/chloroform and precipitated with ethanol using RNase-free glycogen (New England Biolabs) as a carrier. One microgram of RNA was converted to DNA by reverse transcription using Oligo dT primers and the Superscript III first-strand cDNA synthesis kit. The reverse transcription reaction was incubated first at 65°for 5 min, followed by 45°for 45 min, 50°for 15 min, 55°for 15 min and 75°for 15 min in a total of 50 μl. PCR for DUX4 was performed using the PCRX enhancer system for GC rich sequences (Invitrogen) and Taq polymerase (New England Biolabs). Cycling conditions: 35 cycles of 94°C × 30 s, 55°C × 30 s, 68°C × 120 s. To control for genomic DNA contamination, a no-RT control was always included for each PCR sample. Primer sequences:
587: GCTGGAAGCACCCCTCAGCGAGGAA;
588: GAATTCCATGGCGTCGTTTACTTTGACCAACAAGAA;
GAPDHF: ATCTTCCAGGAGCGAGATCC;
GAPDHR: ACCACTGACACGTTGGCAGT
siRNA transfections
siRNA for DUX4 was custom synthesized using previously published sequences (49) (Thermo Scientifc). Five microliters of 20 μm stock was resuspended in 250 μl optimem (Invitrogen) and incubated for 15 min with 6 μl Lipofectamine RNAiMax (Invitrogen). The mixture was added to myoblasts that had been seeded 24 h prior at 50 000 cells/cm2. Twenty-four hours later, the medium was changed to KOSR differentiation medium and RNA from the cells was assayed by PCR and cells fixed in formaldehyde were assessed by immunofluorescent microscopy for DUX4 reduction. Conditions were scaled down to be performed in 384-well dishes. In these experiments, 1.8 μl of Lipofectamine RNAiMax was added to 100 μl optimem. One microliter of siRNA was added to 19 μl of liposome-containing optimem. Five microliters of the siRNA mixture was used per well of a 384-well dish. siRNA sequences: siDUX4-1: 5′-r(GAUGAUUAGUUCAGAGAUA)d(TT)-3′. siDUX4-2: 5′-r(GCGCAACCUCUCCUAGAAA)d(TT)-3′). For a non-targeting control, we used the non-targeting control siRNA #1 (Thermo Scientific). Silencer Select siRNAs to β-catenin, p53, FRG1, Wnt9B and a corresponding scrambled control were purchased from Invitrogen (#s437, #s606, #s5367, #s14901 and Silencer Select Negative Control 1, respectively).
Western blot
Western blots were performed using standard procedures as described previously (34). Mouse anti-myogenin clone F5D was obtained from the Developmental Studies Hybridoma Bank (http://dshb.biology.uiowa.edu) and used at a 1:1000 dilution. Rat anti-tubulin (AbCam) was used at a final dilution of 1:5000. Blots were imaged using a VersaDoc digital imaging system (BioRad).
Statistics
For experiments in which two means were being compared, statistical significance was determined using a two-tailed Student's t-test. When more than two means were being compared, a one-way analysis of variance was performed. When counts were being compared, for example when counting DUX4(+) nuclei, a Fisher exact test was used. When comparing the distribution of nuclei in clusters (Fig. 1C), statistical significance was determined using the Mann–Whitney test. Statistical data was processed using Graphpad software (Prism; www.graphpad.com).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the NIH (NIH P01NS069539 and NIH UL1RR024160), The Friends of FSH Research, and The Fields Center for FSHD and Neuromuscular Research.
Supplementary Material
REFERENCES
- 1.Tawil R. Facioscapulohumeral muscular dystrophy. Neurotherapeutics. 2008;5:601–606. doi: 10.1016/j.nurt.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Overveld P.G., Lemmers R.J., Sandkuijl L.A., Enthoven L., Winokur S.T., Bakels F., Padberg G.W., van Ommen G.J., Frants R.R., van der Maarel S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003;35:315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 3.de Greef J.C., Lemmers R.J., Camano P., Day J.W., Sacconi S., Dunand M., van Engelen B.G., Kiuru-Enari S., Padberg G.W., Rosa A.L., et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–1554. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Greef J.C., Lemmers R.J., van Engelen B.G., Sacconi S., Venance S.L., Frants R.R., Tawil R., van der Maarel S.M. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum. Mutat. 2009;30:1449–1459. doi: 10.1002/humu.21091. [DOI] [PubMed] [Google Scholar]
- 5.de Greef J.C., Wohlgemuth M., Chan O.A., Hansson K.B., Smeets D., Frants R.R., Weemaes C.M., Padberg G.W., van der Maarel S.M. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology. 2007;69:1018–1026. doi: 10.1212/01.wnl.0000271391.44352.fe. [DOI] [PubMed] [Google Scholar]
- 6.Dixit M., Ansseau E., Tassin A., Winokur S., Shi R., Qian H., Sauvage S., Matteotti C., van Acker A.M., Leo O., et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl Acad. Sci. USA. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabriels J., Beckers M.C., Ding H., De Vriese A., Plaisance S., van der Maarel S.M., Padberg G.W., Frants R.R., Hewitt J.E., Collen D., et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/s0378-1119(99)00267-x. [DOI] [PubMed] [Google Scholar]
- 8.Lemmers R.J., van der Vliet P.J., Klooster R., Sacconi S., Camano P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W., et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snider L., Asawachaicharn A., Tyler A.E., Geng L.N., Petek L.M., Maves L., Miller D.G., Lemmers R.J., Winokur S.T., Tawil R., et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Gen. 2009;18:2414–2430. doi: 10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosnakovski D., Xu Z., Gang E.J., Galindo C.L., Liu M., Simsek T., Garner H.R., Agha-Mohammadi S., Tassin A., Coppee F., et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace L.M., Garwick S.E., Mei W., Belayew A., Coppee F., Ladner K.J., Guttridge D., Yang J., Harper S.Q. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 2010;69:540–552. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallace L.M., Liu J., Domire J.S., Garwick-Coppens S.E., Guckes S.M., Mendell J.R., Flanigan K.M., Harper S.Q. RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol. Ther. 2012;20:1417–1423. doi: 10.1038/mt.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wuebbles R.D., Long S.W., Hanel M.L., Jones P.L. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int. J. Clin. Exp. Pathol. 2010;3:386–400. [PMC free article] [PubMed] [Google Scholar]
- 14.Snider L., Geng L.N., Lemmers R.J., Kyba M., Ware C.B., Nelson A.M., Tawil R., Filippova G.N., van der Maarel S.M., Tapscott S.J., et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tajbakhsh S., Borello U., Vivarelli E., Kelly R., Papkoff J., Duprez D., Buckingham M., Cossu G. Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development. 1998;125:4155–4162. doi: 10.1242/dev.125.21.4155. [DOI] [PubMed] [Google Scholar]
- 16.Robitaille J., MacDonald M.L., Kaykas A., Sheldahl L.C., Zeisler J., Dube M.P., Zhang L.H., Singaraja R.R., Guernsey D.L., Zheng B., et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 2002;32:326–330. doi: 10.1038/ng957. [DOI] [PubMed] [Google Scholar]
- 17.Fitzsimons R.B. Retinal vascular disease and the pathogenesis of facioscapulohumeral muscular dystrophy. A signalling message from Wnt? Neuromuscul. Disord. 2011;21:263–271. doi: 10.1016/j.nmd.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Sethi J.K., Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem. J. 2010;427:1–17. doi: 10.1042/BJ20091866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nusse R., Fuerer C., Ching W., Harnish K., Logan C., Zeng A., ten Berge D., Kalani Y. Wnt signaling and stem cell control. Cold Spring Harb. Symp. Quant. Biol. 2008;73:59–66. doi: 10.1101/sqb.2008.73.035. [DOI] [PubMed] [Google Scholar]
- 20.Katoh M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007;13:4042–4045. doi: 10.1158/1078-0432.CCR-06-2316. [DOI] [PubMed] [Google Scholar]
- 21.Vladar E.K., Antic D., Axelrod J.D. Planar cell polarity signaling: the developing cell's compass. Cold Spring Harb. Perspect. Biol. 2009;1:a002964. doi: 10.1101/cshperspect.a002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polesskaya A., Seale P., Rudnicki M.A. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell. 2003;113:841–852. doi: 10.1016/s0092-8674(03)00437-9. [DOI] [PubMed] [Google Scholar]
- 23.Cossu G., Borello U. Wnt signaling and the activation of myogenesis in mammals. EMBO J. 1999;18:6867–6872. doi: 10.1093/emboj/18.24.6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Velden J.L., Langen R.C., Kelders M.C., Wouters E.F., Janssen-Heininger Y.M., Schols A.M. Inhibition of glycogen synthase kinase-3beta activity is sufficient to stimulate myogenic differentiation. Am. J. Physiol. Cell Physiol., 2006;290:C453–C462. doi: 10.1152/ajpcell.00068.2005. [DOI] [PubMed] [Google Scholar]
- 25.Vyas D.R., Spangenburg E.E., Abraha T.W., Childs T.E., Booth F.W. GSK-3beta negatively regulates skeletal myotube hypertrophy. Am. J. Physiol. Cell Physiol. 2002;283:C545–C551. doi: 10.1152/ajpcell.00049.2002. [DOI] [PubMed] [Google Scholar]
- 26.Le Grand F., Jones A.E., Seale V., Scime A., Rudnicki M.A. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009;4:535–547. doi: 10.1016/j.stem.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seale P., Polesskaya A., Rudnicki M.A. Adult stem cell specification by Wnt signaling in muscle regeneration. Cell Cycle. 2003;2: 418–419. [PubMed] [Google Scholar]
- 28.von Maltzahn J., Bentzinger C.F., Rudnicki M.A. Wnt7a-Fzd7 signalling directly activates the Akt/mTOR anabolic growth pathway in skeletal muscle. Nat. Cell. Biol. 2011;14:186–191. doi: 10.1038/ncb2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Maltzahn J., Chang N.C., Bentzinger C.F., Rudnicki M.A. Wnt signaling in myogenesis. Trends Cell Biol. 2012;22:602–609. doi: 10.1016/j.tcb.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Maltzahn J., Renaud J.M., Parise G., Rudnicki M.A. Wnt7a treatment ameliorates muscular dystrophy. Proc. Natl Acad. Sci. USA. 2012;109:20614–20619. doi: 10.1073/pnas.1215765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Homma S., Chen J.C., Rahimov F., Beermann M.L., Hanger K., Bibat G.M., Wagner K.R., Kunkel L.M., Emerson C.P., Jr., Miller J.B. A unique library of myogenic cells from facioscapulohumeral muscular dystrophy subjects and unaffected relatives: family, disease and cell function. Eur. J. Hum. Genet. 2012;20:404–410. doi: 10.1038/ejhg.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blau H.M., Webster C. Isolation and characterization of human muscle cells. Proc. Natl Acad. Sci. USA. 1981;78:5623–5627. doi: 10.1073/pnas.78.9.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semsarian C., Sutrave P., Richmond D.R., Graham R.M. Insulin-like growth factor (IGF-I) induces myotube hypertrophy associated with an increase in anaerobic glycolysis in a clonal skeletal-muscle cell model. Biochem. J. 1999;339:443–451. [PMC free article] [PubMed] [Google Scholar]
- 34.Lemmers R.J.L.F., Tawil R., Petek L.M., Balog J., Block G.J., Santen G.W.E., Amell A.M., van der Vliet P.J., Almomani R., Straasheijm K.R., et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2 Nat. Genetics. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tassin A., Laoudj-Chenivesse D., Vanderplanck C., Barro M., Charron S., Ansseau E., Chen Y.W., Mercier J., Coppee F., Belayew A. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J Cell. Mol. Med. 2012;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng L.N., Yao Z., Snider L., Fong A.P., Cech J.N., Young J.M., van der Maarel S.M., Ruzzo W.L., Gentleman R.C., Tawil R., et al. DUX4 Activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallace L.M., Garwick S.E., Mei W., Belayew A., Coppee F., Ladner K.J., Guttridge D., Yang J., Harper S.Q. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 2011;69:540–552. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Deutekom J.C., Lemmers R.J., Grewal P.K., van Geel M., Romberg S., Dauwerse H.G., Wright T.J., Padberg G.W., Hofker M.H., Hewitt J.E., et al. Identification of the first gene (FRG1) from the FSHD region on human chromosome 4q35. Hum. Mol. Gen. 1996;5:581–590. doi: 10.1093/hmg/5.5.581. [DOI] [PubMed] [Google Scholar]
- 39.Gabellini D., D'Antona G., Moggio M., Prelle A., Zecca C., Adami R., Angeletti B., Ciscato P., Pellegrino M.A., Bottinelli R., et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–977. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- 40.Winokur S.T., Barrett K., Martin J.H., Forrester J.R., Simon M., Tawil R., Chung S.A., Masny P.S., Figlewicz D.A. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul. Disord. 2003;13: 322–333. doi: 10.1016/s0960-8966(02)00284-5. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Gonzalo F.R., Izpisua Belmonte J.C. Albumin-associated lipids regulate human embryonic stem cell self-renewal. PloS One. 2008;3:e1384. doi: 10.1371/journal.pone.0001384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kowaljow V., Marcowycz A., Ansseau E., Conde C.B., Sauvage S., Matteotti C., Arias C., Corona E.D., Nunez N.G., Leo O., et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 43.Block G.J., Petek L.M., Narayanan D., Amell A.M., Moore J.M., Rabaia N.A., Tyler A., van der Maarel S.M., Tawil R., Filippova G.N., et al. Asymmetric bidirectional transcription from the FSHD-causing D4Z4 array modulates DUX4 production. PloS One. 2012;7:e35532. doi: 10.1371/journal.pone.0035532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanderplanck C., Ansseau E., Charron S., Stricwant N., Tassin A., Laoudj-Chenivesse D., Wilton S.D., Coppee F., Belayew A. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PloS One. 2011;6:e26820. doi: 10.1371/journal.pone.0026820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barro M., Carnac G., Flavier S., Mercier J., Vassetzky Y., Laoudj-Chenivesse D. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J. Cell. Mol. Med. 2010;14:275–289. doi: 10.1111/j.1582-4934.2008.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Black G.C., Perveen R., Bonshek R., Cahill M., Clayton-Smith J., Lloyd I.C., McLeod D. Coats’ disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum. Mol. Genet. 1999;8:2031–2035. doi: 10.1093/hmg/8.11.2031. [DOI] [PubMed] [Google Scholar]
- 47.Kan H.E., Scheenen T.W., Wohlgemuth M., Klomp D.W., van Loosbroek-Wagenmans I., Padberg G.W., Heerschap A. Quantitative MR imaging of individual muscle involvement in facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2009;19: 357–362. doi: 10.1016/j.nmd.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 48.Friedman S.D., Poliachik S.L., Carter G.T., Budech C.B., Bird T.D., Shaw D.W. The magnetic resonance imaging spectrum of facioscapulohumeral muscular dystrophy. Muscle Nerve. 2012;45:500–506. doi: 10.1002/mus.22342. [DOI] [PubMed] [Google Scholar]
- 49.Geng L.N., Tyler A.E., Tapscott S.J. Immunodetection of human double homeobox 4. Hybridoma (Larchmt) 2011;30:125–130. doi: 10.1089/hyb.2010.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.