

Abstract
Mutations in bestrophin-1 (Best1) cause Best vitelliform macular dystrophy (BVMD), a dominantly inherited retinal degenerative disease. Best1 is a homo-oligomeric anion channel localized to the basolateral surface of retinal pigment epithelial (RPE) cells. A number of Best1 mutants mislocalize in Madin–Darby canine kidney (MDCK) cells. However, many proteins traffic differently in MDCK and RPE cells, and MDCK cells do not express endogenous Best1. Thus, effects of Best1 mutations on localization in MDCK cells may not translate to RPE cells. To determine whether BVMD causing mutations affect Best1 localization, we compared localization and oligomerization of Best1 with Best1 mutants V9M, W93C, and R218C. In MDCK cells, Best1 and Best1R218C were basolaterally localized. Best1W93C and Best1V9M accumulated in cells. In cultured fetal human retinal pigment epithelium cells (fhRPE) expressing endogenous Best1, Best1R218C and Best1W93C were basolateral. Best1V9M was intracellular. All three mutants exhibited similar fluorescence resonance energy transfer (FRET) efficiencies to, and co-immunoprecipitated with Best1, indicating unimpaired oligomerization. When human Best1 was expressed in RPE in mouse eyes it was basolaterally localized. However, Best1V9M accumulated in intracellular compartments in mouse RPE. Co-expression of Best1 and Best1W93C in MDCK cells resulted in basolateral localization of both Best1 and Best1W93C, but co-expression of Best1 with Best1V9M resulted in mislocalization of both proteins. We conclude that different mutations in Best1 cause differential effects on its localization and that this effect varies with the presence or absence of wild-type (WT) Best1. Furthermore, MDCK cells can substitute for RPE when examining the effects of BVMD causing mutations on Best1 localization if co-expressed with WT Best1.
INTRODUCTION
Mutations in the gene BEST1, encoding the protein human bestrophin-1 (hBest1), cause five clinically distinct forms of inherited retinal degeneration: Best vitelliform macular dystrophy (BVMD), adult-onset vitelliform macular dystrophy, autosomal recessive bestrophinopathy, autosomal dominant vitreoretinochoroidopathy, and retinitis pigmentosa (1–6). The most prevalent of these is BVMD, an autosomal-dominant macular degenerative disease that varies in age of onset and severity. Most patients will exhibit a vitelliform lesion in the macula that may progress through several stages to geographic atrophy and in some cases become neovascular. Some 7–9% of carriers of BVMD mutations will never experience vision loss, but all carriers have an aberrant electrooculogram with a normal electroretinogram (7,8).
Best1 is an integral membrane protein that, within the eye, is exclusively expressed in the retinal pigment epithelium (RPE), where it is localized to the basolateral plasma membrane of the cell (9). A great deal of data supports the hypothesis that Best1 is a homo-oligomeric anion channel. Numerous studies in heterologous expression systems (reviewed in 10 and 11) have since been complemented by the findings that Best1 functions in mouse cerebellar glia to carry a GABA conductance (12) and in mouse hippocampal astrocytes to carry a glutamate conductance (13). Similarly, we have shown that bestrophin-2, a close homolog of Best1, carries a bicarbonate conductance in colonic goblet cells (14). To date, every disease-causing mutation in BEST1 tested has resulted in a loss of anion channel activity, with those associated with BVMD dominantly inhibiting the conductance of wild-type (WT) Best1 (10,11). However, loss of anion channel activity is not the only consequence of BEST1 mutation.
Studies on Best1−/− and Best1 knock-in mice carrying the BVMD causing mutation W93C have shown that Best1 plays a critical role in regulating Ca2+ signaling in RPE cells (15,16). This finding is further supported by recent data using RPE cells generated from iPS cells of BVMD patients (17). The mechanism by which Best1 regulates Ca2+ signaling is not clear. While Best1 interacts physically and functionally with voltage dependent Ca2+ channels (15,18–21), there are data suggesting that a sub-population of Best1 may reside in the endoplasmic reticulum adjacent to the basolateral plasma membrane of the RPE where it may interact with STIM1 to regulate Ca2+ stores (17,22,23).
We have previously demonstrated that the BVMD causing mutants R218C and W93C correctly localize to the basolateral plasma membrane when expressed in the RPE of rat eyes via adenovirus mediated gene transfer (24). However, recent work by Milenkovic et al. (25) using stably transfected Madin–Darby canine kidney II (MDCK II) cells suggests that numerous other BVMD mutants exhibit defects in intracellular trafficking, suggesting that an inability to localize to the plasma membrane may underlie loss of channel activity, or alteration in Ca2+ store release for some mutants. However, the relevance of MDCK II cells to studies on RPE protein localization is questionable as many proteins exhibit different localizations in MDCK II and RPE cells (26) and, since BVMD is a dominantly inherited disease, both mutant and WT proteins will be typically expressed. In human iPS-derived RPE cells expressing both endogenous hBest1 and one of the two BVMD-associated hBest1 mutants, hBest1 is properly localized (17). MDCK II cells do not express endogenous Best1, and the effect of mutants on the localization of the WT protein has not been previously addressed in these cells. In this study, we examined the localization of hBest1 and three mutants, V9M, W93C and R218C in MDCK II cells and fetal human retinal pigment epithelium (fhRPE) cells, and for V9M mouse RPE in the eye. We found that the ability to localize correctly differs among the three mutants and can be influenced by the presence of WT hBest1 or, as is the case for V9M, cause mislocalization of WT hBest1. We conclude that some BVMD causing mutations in hBest1 cause mislocalization of hBest1 channels, preventing them from functioning at the plasma membrane and potentially interfering with other cellular processes such as Ca2+ signaling.
RESULTS
hBest1 mutants impair anion channel activity
To date, all disease-causing mutants of hBest1 tested have exhibited diminished anion channel activity (10,11). Mutations at positions W93 and R218 diminish or abolish hBest1 associated anion conductance (12,27). Mutations at amino acid V9 have been separately reported by five groups (2,3,28–30), making them among the more common mutations associated with BVMD (http://www-huge.uni-regensburg.de/BEST1_database). Mutations at position V9 have not previously been tested for anion channel activity. To address this, we performed whole-cell patch clamp of HEK293 cells heterologously expressing hBest1, hBest1V9M or hBest1 and hBest1V9M. As shown in Fig. 1, the V9M mutation significantly diminishes the hBest1 Cl− current. Furthermore, V9M appears to exert a dominant effect, causing a significantly diminished current even when co-expressed with WT hBest1 (Fig. 1).
Figure 1.
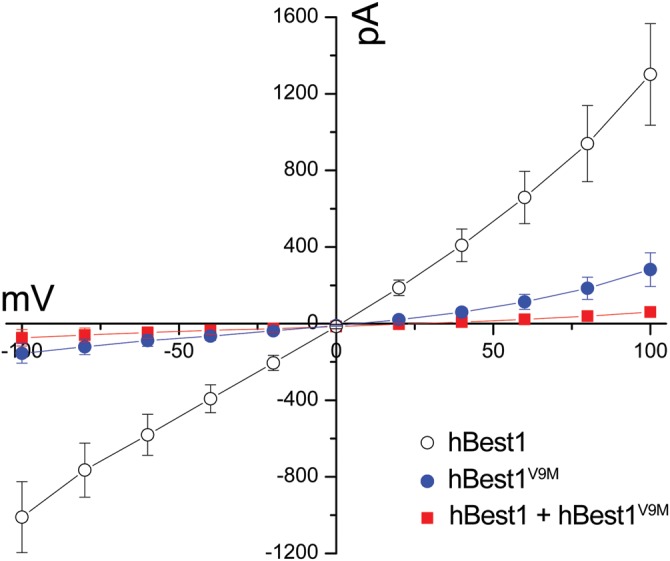
Anion permeability of hBest1 and hBest1V9M. Whole-cell patch clamp recordings were performed in HEK293 cells. Mean current–voltage relationships were obtained from cells transfected with hBest1 (n = 13), hBest1V9M (n = 9) or co-transfected with hBest1 and hBest1V9M (n = 10). hBest1V9M displayed a loss of Cl− conductance and, when co-expressed with WT hBest1, suppressed the Cl− conductance of WT hBest1 as well. Error bars indicate mean ± SD.
Recent studies by Davidson et al. (31) and Milenkovic et al. (25) suggest that mislocalization may underlie the loss of anion channel activity associated with some hBest1 mutants. In the latter study (25), a number of BVMD associated hBest1 mutants were expressed in MDCK II cells and found to accumulate in intracellular compartments. In contrast, we have previously demonstrated that the mutants hBest1W93C and hBest1R218C are, like WT hBest1, localized to the basolateral plasma membrane when expressed via adenovirus-mediated gene transfer in RPE cells in rat eyes (24). To determine whether MDCK II cells are a reasonable predictor of the localization of hBest1 and hBest1 mutants in RPE cells, we chose to compare the localization of the three mutants V9M, W93C and R218C hBest1 in MDCK II and fhRPE cells and, for hBest1V9M, in RPE in the murine eye.
Are hBest1 mutants mislocalized?
Expression of hBest1 and hBest1 mutants in MDCK II cells was accomplished using adenovirus-mediated gene transfer in polarized monolayers of MDCK II cells. Examination of the localization of hBest1 in MDCK II cells using confocal X-Y and X-Z scans demonstrated that hBest1 expressed in MDCK II cells was localized to the basolateral surface, similar to its localization in the RPE (Fig. 2A–C). The mutant hBest1R218C was also basolaterally polarized (Fig. 2D–F). These data are in agreement with the findings of Milenkovic et al. (25). Unlike R218C, however, both hBest1V9M (Fig. 2G–I) and hBest1W93C (Fig. 2J–L) accumulated in intracellular compartments.
Figure 2.
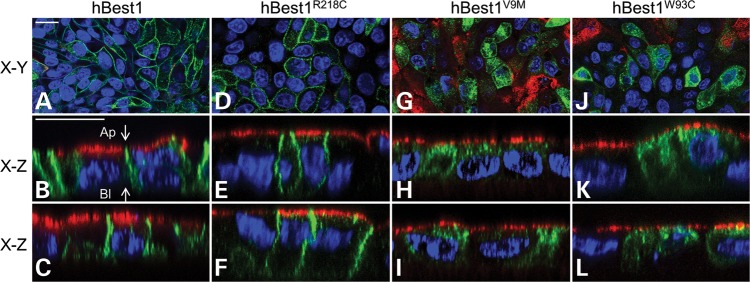
Localization of hBest1 and hBest1 mutants (R218C, V9M and W93C) in MDCK II cells as determined by confocal microscopy. Representative X–Y and X–Z scans of stained WT or mutant hBest1 (green) are shown. WT (A–C), R218C (D–F), V9M (G–I) or W93C (J–L) hBest1 were expressed via adenovirus-mediated gene transfer in polarized monolayers of MDCK II cells and stained for hBest1. The endogenous apical protein gp135 (red) and nuclei (blue) were stained for positional referencing. WT and R218C hBest1 localized to the basolateral plasma membrane, while V9M and W93C hBest1 were intracellular. Ap and Bl in (B) stand for apical and basolateral, respectively. Scale bars: 20 µm.
BVMD is a dominant disease. Thus, with few exceptions (32–34), affected individuals will carry both a WT and a mutant allele of the BEST1 gene. Therefore, it is possible that WT hBest1 can rescue the mislocalization of mutant hBest1. This possibility has not been previously tested. To examine the potential for rescue of mislocalization, we used fhRPE cells as a primary cell culture model. fhRPE cells express endogenous hBest1, though at low levels. As shown in Fig. 3K, it is difficult to detect endogenous hBest1 by western blot of a lysate derived from a single monolayer of fhRPE grown on a 1 cm diameter Millicell HA filter. However, when two or more monolayers are included in the lysate we can detect hBest1 by western blot. Immunofluorescence and confocal microcopy of fhRPE monolayers reveals that endogenous hBest1 is localized to the basolateral plasma membrane of the cells, consistent with findings in native RPE (9) (Fig. 3A and B). This did not change when hBest1 was substantially overexpressed (Fig. 3C and D), indicating that these cells have a high capacity to properly sort hBest1. This is a critical control, as we have shown in the past that sorting pathways can be saturated using viral overexpression (35).
Figure 3.
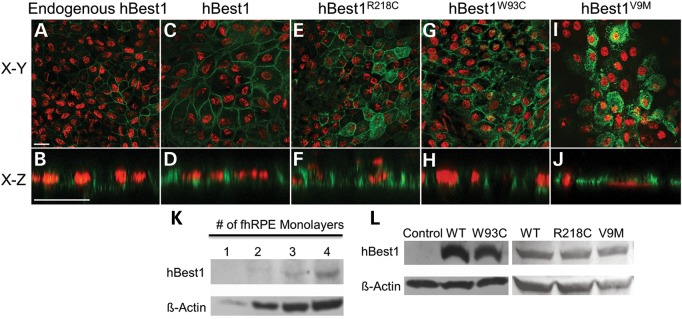
Localization of endogenous hBest1, overexpressed hBest1, and overexpressed hBest1 mutants (R218C, W93C, and V9M) in fhRPE cells. Representative X–Y and X–Z scans of hBest1 (green) localization are shown for endogenous hBest1 (A and B) or following adenovirus-mediated gene transfer, for overexpressed hBest1 (C and D), hBest1R218C(E and F), hBest1W93C(G and H), or hBest1V9M(I and J) in polarized monolayers of fhRPE cells. Nuclei (red) were used as a positional marker. Like endogenous hBest1, overexpressed hBest1, hBest1R218C and hBest1W93C localized to the basolateral plasma membrane, while hBest1V9M remained intracellular. Scale bars: 20 µm. (K) Western blotting of fhRPE monolayers revealed endogenous expression of hBest1 with β-actin as a loading control. (L) Western blotting of hBest1 and β-actin in fhRPE cells (control) or fhRPE cells overexpressing hBest1 or hBest1 mutants following adenovirus-mediated gene transfer demonstrates level of overexpression of hBest1 in infected cells.
We next examined the localization of hBest1 mutants in fhRPE cells (Fig. 3E–J). As was observed in MDCK II cells, hBest1R218C was basolaterally polarized (Fig. 3E and F) and hBest1V9M was predominantly in intracellular compartments (Fig. 3I and J). Interestingly, hBest1W93C, which was mislocalized in MDCK II cells, was basolaterally polarized in fhRPE (Fig. 3G and H). Western blot of fhRPE lysates indicated that all the three mutants were overexpressed (Fig. 3L), but none appeared to be expressed in quantities substantially greater than WT hBest1. This was confirmed by densitometric analysis, which found that the ratio of hBest1 to β-actin for the mutants did not exceed the ratio of WT to β-actin in any given experiment by >22%.
Do WT and Best1 mutants interact to form oligomers?
When we previously expressed the hBest1W93C mutant in the RPE in rat eyes, it was found to localize to the basolateral plasma membrane (24). Since Best1 forms oligomers of unknown stoichiometry (27,36,37), the most plausible explanation for the proper localization of hBest1W93C in rat RPE and fhRPE cells is that endogenous Best1 has the capacity to rescue mislocalization of at least some Best1 mutants. For hBest1V9M, there are several possibilities that could explain the lack of rescue in fhRPE cells. One possibility is that V9 is involved in the formation of Best1 oligomers and that the mutation V9M prevents oligomer formation. To test this, we co-expressed c-myc tagged WT hBest1 together with mutant hBest1 fused to yellow fluorescence protein (YFP) in MDCK II cells and performed reciprocal immunoprecipitation of the two tagged forms of hBest1 using antibodies specific for the tags. As shown in Fig. 4, both hBest1-c-myc and mutant hBest1-YFP have the ability to immunoprecipitate the other tagged form of hBest1, indicating formation of oligomers. The results did not differ significantly when R218C, W93C or V9M were expressed together with hBest1.
Figure 4.

Reciprocal co-immunoprecipitation of hBest1 and mutant hBest1 in MDCK II cells. MDCK II cells were co-transfected with hBest1-c-myc and an YFP-tagged hBest1 mutant (R218C, W93C and V9M). Co-expressing cells were lysed and hBest1 or mutant hBest1 immunoprecipitated using anti-c-myc or anti-YFP antibodies, respectively, and western blotted using the opposite antibody. Control lanes were loaded with immunoprecipitates prepared from untransfected MDCK II cells. Lysate lanes were loaded using lysates from co-transfected cells.
To confirm that hBest1V9M and other mutants interact with WT hBest1 to form oligomers rather than aggregates resulting from overexpression of the protein, we co-expressed hBest1 fused to cyan fluorescent protein (CFP) with WT or mutant hBest1 fused to YFP in MDCK II cells. Performing live-cell fluorescence resonance energy transfer (FRET) using confocal microscopy in X–Y sections of the basal plasma membrane or the lateral borders near the basal membrane revealed that hBest1-CFP and hBest1-YFP exhibit plasma membrane FRET (Fig. 5A and B). As a positive control for FRET, we transfected cells with CFP fused to YFP. For a negative control, we expressed hBest1-CFP and YFP. FRET was determined by the increase in CFP (donor) fluorescence following photobleaching of the acceptor YFP as indicated in the Methods section. FRET efficiencies for controls were 45.12 ± 3.32% for CFP–YFP (mean ± SD, n = 26) and 0.94 ± 1.36% for hBest1-CFP co-expressed with YFP (mean ± SD, n = 25). As shown in Fig. 5, the FRET efficiency of the hBest1-CFP/hBest1-YFP combination was 13.27 ± 2.13% (mean ± SD, n = 23), which was significantly different (P < 0.001) from either the negative control (P < 0.001) or the positive control (P < 0.001). When hBest1-CFP was co-expressed with hBest1 mutants (R218C, W93C, or V9M) fused to YFP (Fig. 5C), the FRET efficiencies were similar to those obtained using WT hBest1-YFP [13.25 ± 4.09%, 14.65 ± 2.70%, and 13.50 ± 2.70% for R218C, V9M and W93C, respectively (mean ± SD, n = 24, 23, and 21)]. hBest1R218C-YFP and hBest1W93C-YFP exhibited FRET at the plasma membrane with hBest1-CFP (Supplementary Material, Fig. S1). In contrast, hBest1V9M-YFP and hBest1-CFP exhibited FRET in intracellular compartments (Supplementary Material, Fig. S1). We conclude from these data that none of the mutations tested block the interaction of hBest1 monomers to form oligomers.
Figure 5.
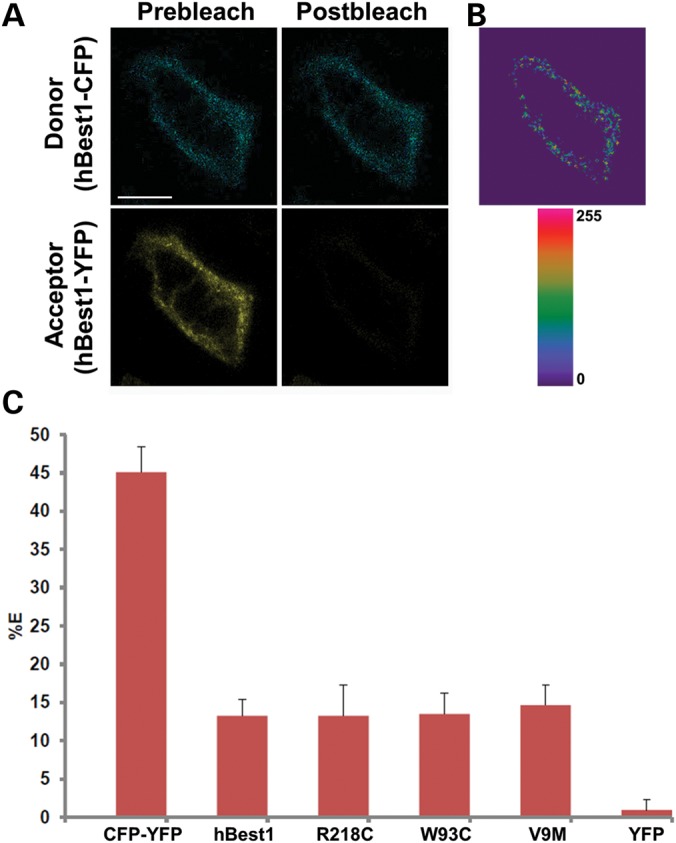
Live-cell, confocal FRET acceptor photobleaching of hBest1-CFP and hBest1-YFP or mutant (R218C, W93C and V9M) hBest1-YFP in MDCK II cells. (A) Representative X–Y scan of hBest1-CFP (blue, donor) and hBest1-YFP (yellow, acceptor) co-expressed in confluent MDCK II cells using adenovirus-mediated gene transfer. Live-cell acceptor photobleaching was performed by bleaching the acceptor, generating the resultant image in (B), which highlights regions in the plasma membrane where donor intensity increased. (C) FRET efficiencies (%E's) were determined for hBest1-CFP paired with hBest1-YFP (n = 23) or hBest1V9M-YFP (n = 23) via adenovirus-mediated gene transfer or hBest1W93C-YFP (n = 21) or hBest1R218C-YFP (n = 24) hBest1 via transfection. MDCK II cells were transfected with a CFP–YFP fusion protein (n = 26) as a positive control and hBest1-CFP and YFP (n = 25) as a negative control. Both hBest1 and mutant hBest1 had %E's significantly different (P < 0.001) than the negative and positive controls. Scale bar: 10 µm. Error bars indicate ± SD.
Does WT Best1 rescue mislocalization of Best1 mutants?
Another potential explanation for the failure to rescue hBest1V9M trafficking could rest in differences in RPE cells in culture and the eye. Many plasma membrane proteins traffic differently in native RPE cells, cultured RPE cells and MDCK II cells (26). To test this hypothesis, we chose to express hBest1V9M in mouse RPE in the eye via adenovirus-mediated gene transfer. For this experiment to be a valid test of the effect of WT Best1 on the localization of hBest1V9M, it was critical to first determine whether hBest1 could interact with mouse Best1 (mBest1). To test this, we repeated the experiments performed to examine hBest1-hBest1 interaction, but this time to test mBest1-hBest1 interaction. As observed for hBest1-hBest1 interaction, hBest1 and mBest1-green fluorescent protein (GFP) were capable of reciprocal co-immunoprecipitation as was hBest1V9M and mBest1-GFP (Supplementary Material, Fig. S2).
To confirm that hBest1 and hBest1V9M interact with mBest1 to form oligomers rather than aggregates resulting from overexpression of the protein, we co-expressed mBest1-CFP with WT or V9M hBest1-YFP in MDCK II cells. When we performed live-cell FRET, mBest1-CFP and hBest1-YFP were found to exhibit FRET at the plasma membrane (Supplementary Material, Fig. S3A and B). FRET efficiencies for controls were 45.12 ± 3.32% for CFP-YFP (mean ± SD, n = 26) and 1.70 ± 2.72% for mBest1-CFP/YFP (mean ± SD, n = 23). The FRET efficiency of the mBest1-CFP/hBest1-YFP combination was 13.07 ± 2.01% (mean ± SD, n = 24), which was significantly different (P < 0.001) from either the negative control (P < 0.001) or the positive control (P < 0.001), but similar to the efficiency determined for hBest1-CFP/hBest1-YFP interaction (Supplementary Material, Fig. S3). When mBest1-CFP was co-expressed with hBest1V9M fused to YFP, the FRET efficiency was 13.88 ± 2.35% (mean ± SD, n = 24) (Supplementary Material, Fig. S3C), similar to that obtained using the mBest1-CFP/hBest1-YFP combination or hBest1-CFP as the donor. We conclude from these data that mBest1 and hBest1 can form oligomers, and that the V9M mutation does not block the interaction of mBest1 and hBest1 monomers to form oligomers.
To determine whether endogenous mBest1 could rescue the mislocalization of hBest1V9M, we expressed hBest1V9M in RPE in the mouse eye using adenovirus-mediated gene transfer, a technique that we have previously used to study the localization of plasma membrane proteins, including hBest1 (24). As shown in Fig. 6, hBest1 expressed in the mouse eye was localized to the basolateral plasma membrane of the RPE (Fig. 6A–C). In contrast, hBest1V9M was clearly localized to intracellular compartments (Fig. 6D–F), indicating that mBest1 either does not rescue the mislocalization of hBest1V9M or is not present in sufficient quantities to exert a discernible rescue effect.
Figure 6.
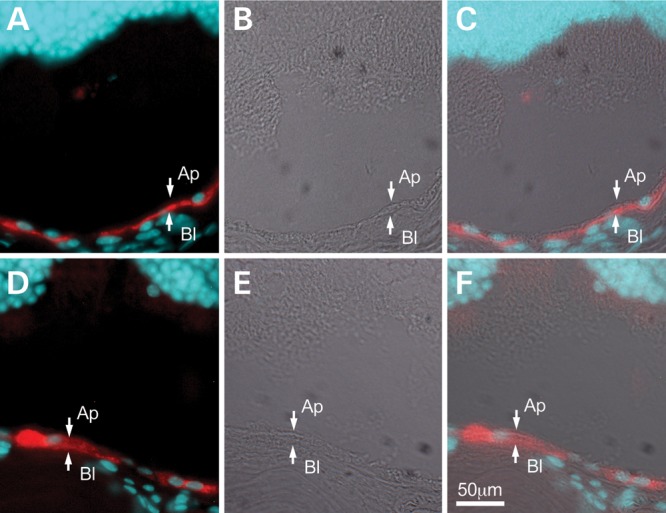
Localization of hBest1 and hBest1V9M in the mouse eye. hBest1 or hBest1V9M were expressed in the RPE of mouse eyes via adenovirus-mediated gene transfer. hBest1 (A, red) was concentrated along the basal surface of the RPE. hBest1V9M (D, red) was observed to be intracellular. Nuclei were stained with DAPI (blue) in (A) and (D) and differential interference contrast (DIC) images were provided in (B) and (E) for positional referencing. Images in (C) and (F) are merged from fluorescent and DIC images. Scale bar: 50 µm.
We next tested the ability of WT hBest1 to rescue the mislocalization of hBest1V9M in MDCK II cells where we could experimentally control the level of expression of hBest1. To do this, polarized MDCK II monolayers were induced to co-express hBest1-CFP and hBest1-YFP, or hBest1-CFP and hBest1V9M-YFP using adenovirus-mediated gene transfer. Both hBest1-CFP and hBest1-YFP were co-localized to the basolateral plasma membrane (Fig. 7A). In contrast, both hBest1-CFP and hBest1V9M-YFP were co-localized to intracellular compartments (Fig. 7B), indicating that hBest1 cannot rescue the mislocalization of hBest1V9M, and that the V9M mutation dominantly causes mislocalization of WT hBest1.
Figure 7.

Effect of hBest1 on localization of hBest1V9M, or hBest1W93C in MDCK II cells. Polarized monolayers of MDCK II cells were made to co-express hBest1-CFP and hBest1-YFP, hBest1V9M-YFP, or hBest1W93C via adenovirus-mediated gene transfer. Gp135 (cyan) was used as an apical protein marker for positional referencing. Representative X–Y and X–Z scans are shown for each co-localization experiment. (A) Both hBest1-CFP (red) and hBest1-YFP (green) co-localized to the basolateral plasma membrane. (B) Both hBest1-CFP (red) and hBest1V9M-YFP (green) co-localized in intracellular compartments. (C) hBest1-YFP (green) was co-expressed with untagged hBest1W93C and cells were stained for hBest1 (red). hBest1 staining was in the basolateral plasma membrane, indicating that the presence of WT hBest1 rescued the mislocalization of hBest1W93C. Scale bars: 20 µm.
We repeated this experiment for hBest1-CFP and untagged hBest1W93C, finding that co-expression of equal amounts of these proteins results in basolateral localization of hBest1, demonstrating that hBest1 rescues hBest1W93C mislocalization in MDCK II cells (Fig. 7C). Western blotting of lysates from MDCK II cells co-expressing hBest1-YFP and untagged hBest1W93C demonstrates staining of two distinct bands corresponding to untagged and tagged hBest1, confirming that both WT and W93C hBest1 were expressed (Supplementary Material, Fig. S4). Although western blotting suggests that both WT hBest1 and hBest1W93C were successfully co-expressed (Supplementary Material, Fig. S4), we employed an untagged form of hBest1W93C. Therefore, we cannot exclude the possibility that the antibody is detecting the WT hBest1-YFP protein and not the hBest1W93C mutant. However, these results are consistent with our initial FRET studies in which hBest1-CFP and hBest1W93C-YFP were co-expressed in MDCK II cells following transfection. In those experiments, FRET was concentrated at the periphery of cells, consistent with a plasma membrane localization of hBest1W93C (Supplementary Material, Fig. S1).
DISCUSSION
Best1 is an integral membrane protein that is localized to the basolateral plasma membrane of RPE cells (9). Mutations in Best1 cause five clinically distinct retinal degenerative diseases, with BVMD by far the most common (1–8). In this study, we sought to further our understanding of how mutations in hBest1 cause BVMD. Prior studies have suggested that disease causing mutations may lead to mislocalization of hBest1 in BVMD (25). Those studies performed in MDCK II cells conflict with our previously published data (24) and that of Singh et al. (17), in which the hBest1 mutants W93C, R218C, N296H, and A146K expressed either endogenously or following virus mediated gene transfer were found to be properly localized in RPE cells. With the goal of resolving the differences between data obtained from MDCK II and RPE cells, which have often been conflicting with regard to polarized plasma membrane proteins (26), we examined three hBest1 mutants, W93C, R218C, and V9M in MDCK II and fhRPE cells. Our data demonstrate that BVMD causing mutations in BEST1 do in some instances result in mislocalization of the protein. Our data also demonstrate that, for a subset of the mutations that cause mislocalization, the presence of WT Best1 can rescue the mislocalization caused by the mutation (e.g. W93C) or, as is the case for hBest1V9M, the mutant protein can prevent the proper localization of WT hBest1.
To date, all studies examining the anion channel function of Best1 have observed that disease causing mutations in the protein result in diminished anion channel activity (10,11). This has been previously demonstrated for mutants at position R218 as well as for the mutation W93C (12,27). Here, we show that the V9M mutation also results in diminished anion channel activity and, like previously described BVMD mutants, dominantly impairs the hBest1 associated anion conductance. These three mutants, however, differed dramatically in their localizations when expressed in MDCK II and fhRPE cells. Like hBest1, hBest1R218C was localized to the basolateral plasma membrane of MDCK II and fhRPE cells, suggesting that the mutation impairs channel activity. hBest1W93C and hBest1V9M, however, were localized to intracellular compartments when expressed in MDCK II cells, similar to many of the mutants studied by Milenkovic et al. (25). However, when expressed in fhRPE cells, which produce endogenous hBest1, hBest1W93C exhibited a basolateral localization similar to that of WT hBest1 and hBest1R218C. Studies on the closely related bestrophin-2 have shown that W93 is likely a part of the channel pore (38,39). More likely than not, this mutation renders hBest1 inactive. In contrast, hBest1V9M remained in intracellular compartments, suggesting that the diminished anion channel activity due to this mutation may be due to lack of the protein in the plasma membrane rather than a functional impairment. Importantly, hBest1V9M also prevented proper localization of WT hBest1, suggesting a mechanism for dominant inhibition of hBest1 channel activity.
Based on our data, we conclude that the underlying pathogenic mechanism that causes BVMD can vary from mutant to mutant, but that loss of anion channel activity is always the result. Some mutations likely interfere with the structure of the channel (e.g. W93C), while others, such as V9M, prevent its proper delivery to the plasma membrane. hBest1W93C is an enigma in this regard. When co-expressed with WT hBest1, as would be the case in most individuals with BVMD, it appears to properly traffic to the basolateral plasma membrane. However, an individual homozygous for Best1W93C has been well studied (34). This individual had classical BVMD and did not have a worse disease phenotype than his heterozygous progeny. Our laboratory examined the eyes of this individual postmortem and concluded that hBest1W93C was expressed and that it was not correctly localized (34). In that report, we could not be certain whether the mislocalization was due to the advanced state of the donor's disease and the effects on the RPE or whether it was due to the mutation. Based on the data in Fig. 2, we can now conclude that hBest1 in this individual should have been mislocalized due to the absence of WT hBest1. This mislocalization did not alter the disease phenotype in comparison to that of his heterozygous progeny (34) who, based on our previous work (24) and our data in Fig. 3 and 7, and Supplementary Material, Fig. S1, should have a normal localization for hBest1. Our laboratory has also generated a mouse model carrying the W93C mutation (16). We have observed no significant difference in the phenotype of heterozygous and homozygous mice. Unfortunately, lack of high fidelity anti-mBest1 antibodies has hindered our efforts at examining the localization of mBest1W93Cin these mice.
In summary, we have shown that single amino acid mutations in hBest1 associated with BVMD can differentially affect the localization of the protein, and that these effects can, in some instances, be rescued by interaction with WT hBest1. Like mutations in rhodopsin that cause retinitis pigmentosa (40,41), it appears that these mutations can be segregated into a group that cause mislocalization/misfolding and a separate group that directly inactivate channel activity. In neither case does the disease phenotype change. Understanding which mutations affect trafficking in the presence of WT hBest1 will be critical to efforts aimed at developing gene therapy approaches to the treatment of BVMD. Understanding how the anion channel function and localization of hBest1 influences its ability to regulate Ca2+ signaling should also be the subject of future studies.
MATERIALS AND METHODS
Cell culture
fhRPE were grown on 1.0 cm Millicell HA filters (Millipore, Billerica, MA, USA) in a 95% air 5%/CO2 environment at 37°C according to the method of Hu and Bok (42). MDCK II cells (American Type Culture Collection, Manassas, VA, USA) were maintained in a 95% air 5%/CO2 environment at 37°C. Cells were grown in Dulbecco's Modification Eagle's Medium (Cellgro, Manassas, VA, USA) supplemented with 10% fetal bovine serum and penicillin/streptomycin solution (Cellgro, Manassas, VA, USA). For studies using cells where hBest1 was expressed via adenovirus-mediated gene transfer, MDCK II cells were plated at confluence on 1.0 cm2 Transwell filters (Corning, Tewksbury, MA, USA) or 35 mm coverslips in glass, bottom-well dishes (MatTek Corporation, Ashland, MA, USA) and maintained for 5 days prior to infections with replication defective adenovirus vectors at an MOI of 30. That the cells had become polarized was ensured by staining for the apical marker protein Gp135 in immunofluorescence experiments and by measurement of transepithelial resistance, only using monolayers with a transepithelial electrical resistance exceeding120 Ω × cm2. fhRPE cells were maintained for >2 months in culture on Millicell HA filters and transduced with adenovirus vectors at an MOI of 3. Only monolayers with a transepithelial electrical resistance exceeding 400 Ω × cm2 were used. For transfection studies, MDCK II cells were plated on 6 cm plates or 35 mm coverslips in glass, bottom-well dishes. At ∼80% confluence, the cells were transfected using Lipofectamine 2000 (Invitrogen, Grand Island, NY, USA) and allowed to grow to 100% confluence prior to use.
Plasmid constructs, site-directed mutagenesis and adenoviral production
To generate mBest1 tagged with CFP or YFP, mBest1 was excised from a pEGFP-mBest1 plasmid using BglII and BamHI and ligated into the corresponding restriction sites in pECFP-N1 and pEYFP-N1 vectors (Clontech, Mountain View, CA, USA). Similarly, CFP- and YFP-tagged hBest1 were generated by excising hBest1 from pEGFP-hBest1 using the restriction enzymes NheI and AgeI and ligated into the corresponding restriction sites in pECFP-N1 and pEYFP-N1. hBest1V9M tagged with YFP was generated by site-directed mutagenesis of pEYFP-hBest1 using a kit according to the manufacturer's instructions (Agilent Technologies, Santa Clara, CA, USA). hBest1 tagged with the 6 × c-myc epitope at the C-terminus in pRK5 was kindly provided by Dr Jeremy Nathans (John Hopkins University, Baltimore, MD, USA).
Replication defective adenovirus vectors carrying hBest1, hBest1R218C and hBest1W93C were previously described (9,24). A replication defective adenovirus vector carrying hBest1V9M was generated as described previously using the method of Hardy et al. (43). These vectors were amplified, purified and titrated as previously described (9,24). Additional replication deficient adenovirus vectors carrying CFP- or YFP-tagged hBest1 were generated, amplified, purified and titrated by the Gene Transfer Vector Core at the University of Iowa using the viral shuttle vector pacAd5CMVK-NpA (provided by the Gene Transfer Vector Core, University of Iowa, Iowa City, IA). Tagged hBest1 was inserted into the shuttle vector using the KpnI and XhoI restriction sites. hBest1V9M tagged with YFP was generated in pacAd5CMVK-hBest1-YFP and hBest1V9M tagged with c-myc in pRK5 were generated using site-directed mutagenesis as described above.
Immunofluorescence
To study localization and co-localization of WT and mutant hBest1, hBest1 was expressed in confluent monolayers of MDCK II cells via adenovirus-mediated gene transfer. Twenty-four to forty-eight hours later, transwells were immersed in ice-cold methanol for 10 min, washed using phosphate-buffered saline containing 0.13 mm CaCl2 and 1.0 MgCl2, and blocked in the same buffer containing 3% bovine serum albumin. Cells were then stained for hBest1 using the previously described rabbit, polyclonal anti-hBest1 antibody Pab125 (9), gp135 using the mouse, monoclonal antibody 3F4 (generous gift of Dr George Ojakian, SUNY Health Science Center at Brooklyn) and/or nuclei using 4′,6-diamidino-2-phenylindole (DAPI). Images were obtained using a Leica SP5 confocal microscope with a ×40 oil immersion objective.
Immunoprecipitation and western blotting
To assess physical interaction between mBest1 and hBest1, MDCK II cells grown on 6 cm plates were co-transfected with pAdlox-mBest1-GFP and pAdlox-hBest1 or pAdlox-hBest1V9M. Forty-eight hours later, cells were lysed in 1% Triton X-100, 20 mm Tris–HCl, pH 8.0, 150 mm NaCl, 5 mm EDTA containing a protease inhibitor cocktail (Millipore) for 1 h at 4°C. After centrifugation, 25% of the volume of each lysate was removed and mixed with 4× Laemmli sample buffer. The remaining lysate was immunoprecipitated with the rabbit, polyclonal anti-mBest1 antibody Pab-003 or with Pab125, a rabbit, polyclonal anti-hBest1 antibody as described previously (9,44). Lysates and immunoprecipitates were resolved via sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (GE Healthcare, Waukesha, WI, USA) overnight. Blots were incubated with the mouse, monoclonal anti-GFP antibody JL-8 (Clontech) or the previously described mouse, monoclonal anti-hBest1 antibody E6-6 (9). Following incubation with an anti-mouse, alkaline-phosphatase conjugated secondary antibody (Rockland, Gilbertsville, PA, USA), blots were developed using the substrates nitro-blue tetrazolium chloride and 5-bromo-4-chloro-3′-indolyphosphate P-toluidine salt (Promega, Madison, WI). For co-immunoprecipitation experiments between WT and mutant hBest1, rabbit polyclonal antibodies were used to immunoprecipitate c-myc (Invitrogen) or YFP (Clontech). Western blotting was performed using a mouse, monoclonal antibody specific to c-myc (9E10; Invitrogen) or YFP (JL-8; Clontech). Western blots of hBest1 in fhRPE cells were performed using the mouse, monoclonal antibody E6-6 and with a mouse, monoclonal anti-β-actin (AC-74; Sigma-Aldrich, St Louis, MO, USA) antibody. For non-immunoprecipitation studies, western blotting of hBest1 in MDCK II cells was performed using the mouse, monoclonal antibody E6-6.
FRET
Forty-eight hours after transfection or transduction, MDCK II cells grown on 35 mm coverslips in glass, bottom-well dishes were placed on the stage of a Leica SP5 confocal microscope and imaged using a ×40 oil immersion objective. The FRET donor (CFP) was excited at 458 nm and emission was collected from 465 to 505 nm. The acceptor (YFP) was excited at 514 nm and emissions collected from 525 to 600 nm. FRET acceptor photobleaching was performed using the 514 nm laser at 100% power. FRET efficiency (%E) was calculated as follows: using ImageJ software (NIH), thresholds were determined for background fluorescence and saturation of images from cells expressing a positive FRET control (CFP tagged to YFP, a generous gift from Dr Robert Tarran, University of North Carolina, Chapel Hill, NC, USA), and all images adjusted to the same threshold values. Donor fluorescence values were then determined for all images before and after acceptor photobleaching and %E was calculated as:
Subretinal injections
Subretinal injections were performed using replication defective adenovirus vectors carrying hBest1 or hBest1V9M on Balb/c mice between 2 and 4 months of age, as described previously (24) with the following modifications: in brief, mice were anesthetized with avertin (250 mg/kg body weight), and their eyes dilated with phenylephrine (2.5%) and atropine (1%) drops. Prior to surgery, a topical anesthetic (1% proparacaine) was also applied to the eye. Under a stereomicroscope, a custom-made 32-gauge cannula was inserted through an incision made 1 mm posterior to the limbus. With a modified syringe (Hamilton, Reno, NV, USA) attached to a foot activated pump, a 2-µl volume of vector diluted to 1.25 × 107 particle/µl in Hanks' balanced salt solution was injected. Ten days after injection, mice were sacrificed by CO2 asphyxiation and their eyes enucleated and embedded in Tissue-Tek® O.C.T.™ Cryosections were then stained for hBest1 and nuclei using the mouse, monoclonal antibody E6-6 and DAPI, respectively.
Patch clamp
WT or mutant hBest1 and pEGFP (Invitrogen) were transfected into HEK293 cells (5:1 ratio, 2 μg total DNA per 3.5 cm plate), using a blend of lipids (Fugene-6; Roche Molecular Biochemicals, Indianapolis, IN, USA). Single cells identified by GFP fluorescence were used for whole-cell patch clamp experiments within 72 h. Transfected HEK293 cells were recorded using a conventional whole-cell patch-clamp technique with the amplifier (EPC-7, HEKA, Bellmore, NY, USA). Fire-polished borosilicate glass patch pipettes were 3–5 MΩ. Experiments were conducted at room temperature (20–24°C). Since the liquid junction potentials were small (<2 mV), no correction was made. The high Ca2+ intracellular solution contained (mm): 146 CsCl, 2 MgCl2, 5 Ca2+-EGTA (free Ca2+∼20 μm), 10 HEPES, 10 sucrose, pH 7.3, adjusted with N-Methyl-d-glucamine. The standard extracellular solution contained (mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 15 glucose, 10 HEPES, pH 7.4 with NaOH. This combination of intracellular and extracellular solutions set Erev for Cl− currents to zero, while cation currents carried by Na+ or Cs+ had very positive or negative Erev, respectively. Osmolarity was adjusted with sucrose to 303 mOsm for all solutions.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by grants from the NIH (EY13160 to A.D.M., EY014465 to L.Y.M.), the Macular Vision Research Foundation and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology at the University of Arizona.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Dr Benjamin Bakall for his work on the early phases of this study.
REFERENCES
- 1.Petrukhin K., Koisti M.J., Bakall B., Li W., Xie G., Marknell T., Sandgren O., Forsman K., Holmgren G., Andreasson S., et al. Identification of the gene responsible for best macular dystrophy. Nat. Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 2.Marquardt A., Stöhr H., Passmore L.A., Krämer F., Rivera A., Weber B.H. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best's disease) Hum. Mol. Genet. 1998;7:1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- 3.Krämer F., White K., Pauleikhoff D., Gehrig A., Passmore L., Rivera A., Rudolph G., Kellner U., Andrassi M., Lorenz B., et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur. J. Hum. Genet. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- 4.Burgess R., Millar I.D., Leroy B.P., Urquhart J.E., Fearon I.M., De Baere E., Brown P.D., Robson A.G., Wright G.A., Kestelyn P., et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008;82:19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yardley J., Leroy B.P., Hart-Holden N., Lafaut B.A., Loeys B., Messiaen L.M., Perveen R., Reddy M.A., Bhattacharya S.S., Traboulsi E., et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Invest. Ophthalmol. Vis. Sci. 2004;45:3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- 6.Davidson A.E., Millar I.D., Urquhart J.E., Burgess-Mullan R., Shweikh Y., Parry N., O'Sullivan J., Maher G.J., McKibbin M., Downes S.M., et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am. J. Hum. Genet. 2009;85:581–592. doi: 10.1016/j.ajhg.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marmorstein A.D., Cross H.E., Peachey N.S. Functional roles of bestrophins in ocular epithelia. Prog. Retin. Eye Res. 2009;28:206–226. doi: 10.1016/j.preteyeres.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boon C.J., Klevering B.J., Leroy B.P., Hoyng C.B., Keunen J.E., den Hollander A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009;28:187–205. doi: 10.1016/j.preteyeres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Marmorstein A.D., Marmorstein L.Y., Rayborn M., Wang X., Hollyfield J.G., Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl Acad. Sci. USA. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartzell H.C., Qu Z., Yu K., Xiao Q., Chien L.T. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol. Rev. 2008;88:639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- 11.Xiao Q., Hartzell H.C., Yu K. Bestrophins and retinopathies. Pflugers Arch. 2010;460:559–569. doi: 10.1007/s00424-010-0821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S., Yoon B.E., Berglund K., Oh S.J., Park H., Shin H.S., Augustine G.J., Lee C.J. Channel-mediated tonic GABA release from glia. Science. 2010;330:790–796. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- 13.Woo D.H., Han K.S., Shim J.W., Yoon B.E., Kim E., Bae J.Y., Oh S.J., Hwang E.M., Marmorstein A.D., Bae Y.C., et al. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell. 2012;151:25–40. doi: 10.1016/j.cell.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Yu K., Lujan R., Marmorstein A., Gabriel S., Hartzell H.C. Bestrophin-2 mediates bicarbonate transport by goblet cells in mouse colon. J. Clin. Invest. 2010;120:1722–1735. doi: 10.1172/JCI41129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marmorstein L.Y., Wu J., McLaughlin P., Yocom J., Karl M.O., Neussert R., Wimmers S., Stanton J.B., Gregg R.G., Strauss O., et al. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1) J. Gen. Physiol. 2006;127:577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y., Stanton J.B., Wu J., Yu K., Hartzell H.C., Peachey N.S., Marmorstein L.Y., Marmorstein A.D. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum. Mol. Genet. 2010;19:1108–1118. doi: 10.1093/hmg/ddp583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh R., Shen W., Kuai D., Martin J.M., Guo X., Smith M.A., Perez E.T., Phillips M.J., Simonett J.M., Wallace K.A., et al. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum. Mol. Genet. 2013;22:593–607. doi: 10.1093/hmg/dds469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reichhart N., Milenkovic V.M., Halsband C.A., Cordeiro S., Strauss O. Effect of bestrophin-1 on L-type Ca2+ channel activity depends on the Ca2+ channel beta-subunit. Exp. Eye Res. 2010;91:630–639. doi: 10.1016/j.exer.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Milenkovic V.M., Krejcova S., Reichhart N., Wagner A., Strauss O. Interaction of bestrophin-1 and Ca2+ channel ß-subunits: identification of new binding domains on the bestrophin-1 C-terminus. PLoS One. 2011;6:e19364. doi: 10.1371/journal.pone.0019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu K., Xiao Q., Cui G., Lee A., Hartzell H.C. The Best disease-linked Cl- channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains. J. Neurosci. 2008;28:5660–5670. doi: 10.1523/JNEUROSCI.0065-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenthal R., Bakall B., Kinnick T., Peachey N., Wimmers S., Wadelius C., Marmorstein A., Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2006;20:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- 22.Neussert R., Müller C., Milenkovic V.M., Strauss O. The presence of bestrophin-1 modulates the Ca2+ recruitment from Ca2+ stores in the ER. Pflugers Arch. 2010;460:163–175. doi: 10.1007/s00424-010-0840-2. [DOI] [PubMed] [Google Scholar]
- 23.Gómez N.M., Tamm E.R., Strauβ O. Role of bestrophin-1 in store-operated calcium entry in retinal pigment epithelium. Pflugers Arch. 2013;465:481–495. doi: 10.1007/s00424-012-1181-0. [DOI] [PubMed] [Google Scholar]
- 24.Marmorstein A.D., Stanton J.B., Yocom J., Bakall B., Schiavone M.T., Wadelius C., Marmorstein L.Y., Peachey N.S. A model of best vitelliform macular dystrophy in rats. Invest. Ophthalmol. Vis. Sci. 2004;45:3733–3739. doi: 10.1167/iovs.04-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milenkovic V.M., Röhrl E., Weber B.H., Strauss O. Disease-associated missense mutations in bestrophin-1 affect cellular trafficking and anion conductance. J. Cell Sci. 2011;124:2988–2996. doi: 10.1242/jcs.085878. [DOI] [PubMed] [Google Scholar]
- 26.Marmorstein A.D. The polarity of the retinal pigment epithelium. Traffic. 2001;2:867–872. doi: 10.1034/j.1600-0854.2001.21202.x. [DOI] [PubMed] [Google Scholar]
- 27.Sun H., Tsunenari T., Yau K.W., Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl Acad. Sci. USA. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponjavic V., Eksandh L., Andréasson S., Sjöström K., Bakall B., Ingvast S., Wadelius C., Ehinger B. Clinical expression of Best's vitelliform macular dystrophy in Swedish families with mutations in the bestrophin gene. Ophthalmic Genet. 1999;20:251–257. doi: 10.1076/opge.20.4.251.2270. [DOI] [PubMed] [Google Scholar]
- 29.Querques G., Zerbib J., Santacroce R., Margaglione M., Delphin N., Rozet J.M., Kaplan J., Martinelli D., Delle Noci N., Soubrane G., et al. Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Mol. Vis. 2009;15:2960–2972. [PMC free article] [PubMed] [Google Scholar]
- 30.Cohn A.C., Turnbull C., Ruddle J.B., Guymer R.H., Kearns L.S., Staffieri S., Daggett H.T., Hewitt A.W., Mackey D.A. Best's macular dystrophy in Australia: phenotypic profile and identification of novel BEST1 mutations. Eye (Lond.) 2011;25:208–217. doi: 10.1038/eye.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davidson A.E., Millar I.D., Burgess-Mullan R., Maher G.J., Urquhart J.E., Brown P.D., Black G.C., Manson F.D. Functional characterization of bestrophin-1 missense mutations associated with autosomal recessive bestrophinopathy. Invest. Ophthalmol. Vis. Sci. 2011;52:3730–3736. doi: 10.1167/iovs.10-6707. [DOI] [PubMed] [Google Scholar]
- 32.Iannaccone A., Kerr N.C., Kinnick T.R., Calzada J.I., Stone E.M. Autosomal recessive best vitelliform macular dystrophy: report of a family and management of early-onset neovascular complications. Arch. Ophthalmol. 2011;129:211–217. doi: 10.1001/archophthalmol.2010.367. [DOI] [PubMed] [Google Scholar]
- 33.Schatz P., Klar J., Andréasson S., Ponjavic V., Dahl N. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006;27:51–56. doi: 10.1080/13816810600677990. [DOI] [PubMed] [Google Scholar]
- 34.Bakall B., Radu R.A., Stanton J.B., Burke J.M., McKay B.S., Wadelius C., Mullins R.F., Stone E.M., Travis G.H., Marmorstein A.D. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2) Exp. Eye Res. 2007;85:34–43. doi: 10.1016/j.exer.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 35.Marmorstein A.D., Csaky K.G., Baffi J., Lam L., Rahaal F., Rodriguez-Boulan E. Saturation of, and competition for entry into, the apical secretory pathway. Proc. Natl Acad. Sci. USA. 2000;97:3248–3253. doi: 10.1073/pnas.070049497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanton J.B., Goldberg A.F., Hoppe G., Marmorstein L.Y., Marmorstein A.D. Hydrodynamic properties of porcine bestrophin-1 in Triton X-100. Biochim. Biophys. Acta. 2006;1758:241–247. doi: 10.1016/j.bbamem.2006.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu Z., Cheng W., Cui Y., Cui Y., Zheng J. Human disease-causing mutations disrupt an N–C-terminal interaction and channel function of bestrophin-1. J. Biol. Chem. 2009;284:16473–16481. doi: 10.1074/jbc.M109.002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qu Z., Hartzell C. Determinants of anion permeation in the second transmembrane domain of the mouse bestrophin-2 chloride channel. J. Gen. Physiol. 2004;124:371–382. doi: 10.1085/jgp.200409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu Z., Chien L.T., Cui Y., Hartzell H.C. The anion-selective pore of the bestrophins, a family of chloride channels associated with retinal degeneration. J. Neurosci. 2006;26:5411–5419. doi: 10.1523/JNEUROSCI.5500-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iannaccone A., Man D., Waseem N., Jennings B.J., Ganapathiraju M., Gallaher K., Reese E., Bhattacharya S.S., Klein-Seetharaman J. Retinitis pigmentosa associated with rhodopsin mutations: Correlation between phenotypic variability and molecular effects. Vision Res. 2006;46:4556–4567. doi: 10.1016/j.visres.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 41.Mendes H.F., van der Spuy J., Chapple J.P., Cheetham M.E. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol. Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Hu J., Bok D. A cell culture medium that supports the differentiation of human retinal pigment epithelium into functionally polarized monolayers. Mol. Vis. 2001;7:14–19. [PubMed] [Google Scholar]
- 43.Hardy S., Kitamura M., Harris-Stansil T., Dai Y., Phipps M.L. Construction of adenovirus vectors through Cre-lox recombination. J. Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bakall B., Marmorstein L.Y., Hoppe G., Peachey N.S., Wadelius C., Marmorstein A.D. Expression and localization of bestrophin during normal mouse development. Invest. Ophthalmol. Vis. Sci. 2003;44:3622–3628. doi: 10.1167/iovs.03-0030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.