

Abstract
Neurofibromatosis type 1 (NF1) is a common genetic disorder affecting 1 in 3500 individuals. Patients with NF1 are predisposed to debilitating skeletal manifestations, including osteopenia/osteoporosis and long bone pseudarthrosis (nonunion fracture). Hyperactivation of the Ras/mitogen-activated protein kinase (MAPK) pathway in NF1 is known to underlie aberrant proliferation and differentiation in cell lineages, including osteoclast progenitors and mesenchymal stem cells (MSCs) also known as osteoblast progenitors (pro-OBLs). Our current study demonstrates the hyper Ras/MAPK as a critical pathway underlying the pathogenesis of NF1-associated fracture repair deficits. Nf1-deficient pro-OBLs exhibit Ras/MAPK hyperactivation. Introduction of the NF1 GTPase activating-related domain (NF1 GAP-related domain) in vitro is sufficient to rescue hyper Ras activity and enhance osteoblast (OBL) differentiation in Nf1−/− pro-OBLs and NF1 human (h) MSCs cultured from NF1 patients with skeletal abnormalities, including pseudarthrosis or scoliosis. Pharmacologic inhibition of mitogen-activated protein kinase kinase (MEK) signaling with PD98059 partially rescues aberrant Erk activation while enhancing OBL differentiation and expression of OBL markers, osterix and osteocalcin, in Nf1-deficient murine pro-OBLs. Similarly, MEK inhibition enhances OBL differentiation of hMSCs. In addition, PD98059 rescues aberrant osteoclast maturation in Nf1 haploinsufficient bone marrow mononuclear cells (BMMNCs). Importantly, MEK inhibitor significantly improves fracture healing in an NF1 murine model, Col2.3Cre;Nf1flox/−. Collectively, these data indicate the Ras/MAPK cascade as a critical pathway in the pathogenesis of bone loss and pseudarthrosis related to NF1 mutations. These studies provide evidence for targeting the MAPK pathway to improve bone mass and treat pseudarthrosis in NF1.
INTRODUCTION
Mutations in NF1 cause neurofibromatosis type 1 (NF1), a common genetic disorder that affects 1 in 3500 individuals (1–3). Neurofibromin, the protein encoded by NF1, functions as a GTPase-activating protein (GAP) for Ras (4). The NF1 GAP-related domain (GRD) is critical for accelerating hydrolysis of active Ras-GTP to inactive Ras-GDP (4). Mutations in NF1 result in constitutive activation of Ras and its downstream signaling axis, which leads to a spectrum of clinical sequelae stemming from the aberrant growth and differentiation of multiple cell lineages (3,5–7). Individuals with NF1 are afflicted with a range of malignant and nonmalignant manifestations (5–14). Clinical studies have found that individuals with NF1 are at significant risk for both generalized (15–20) and focal skeletal abnormalities (19,21–23). The most debilitating of these bone anomalies is pseudarthrosis, which is a pathologic condition, in which spontaneous fracture occurs with a failure to unite across the callus. Often times, patients with pseudarthrosis require amputation of the affected limb following multiple failed orthopedic interventions in order to allow mobility (24). Interestingly, 50–90% of patients with pseudarthrosis have Nf1 mutations (25–28). Treatment options for NF1 patients with pseudarthrosis remain inadequate due to limited information on cellular/molecular mechanisms.
Independent groups have reported several murine models of NF1 with a spectrum of skeletal deficits, including diminished bone mass, tibial bowing and delayed fracture healing (29,30). Defective OBL differentiation has been considered to be the underlying cellular mechanism (29,30). We have previously reported that haploinsufficiency of Nf1 results in gain-in osteoclast differentiation and bone resorptive activity. These studies imply that gain-in osteoclast functions and impaired OBL differentiation may collaboratively impact skeletal manifestation in NF1 patients. Interestingly, a previous clinical study by Stevenson et al. showed that some NF1 individuals with pseudarthrosis exhibit NF1 nullizygosity in the microdissected pseudarthrosis tissue while being NF1 haploinsufficient in the peripheral blood (23). Based on these studies, we recently established two novel murine models, PeriCre;Nf1flox/− and Col2.3Cre;Nf1flox/−, in which MSCs, also known as pro-OBLs, and/or OBLs are nullizygous in Nf1 while being systemically heterozygous in cell types, including osteoclasts and their progenitors. These mice recapitulate multiple skeletal deficits of NF1 patients, including reduced bone mass and tibial fracture nonunion (31).
Increased activation of the Ras/ MAPK pathway in NF1 is known to underlie aberrant proliferation and differentiation in multiple cell types, including pro-OBLs (32) and osteoclasts (33). Pre-clinical studies have demonstrated protective effects of genetic and pharmacologic inhibition of the MAPK pathway in NF1-associated juvenile myelomonocytic leukemia murine models (34,35). Staser et al. reported that genetic disruption of both Erk1 and Erk2 in Nf1-deficient mice abates the development of myeloproliferative disease (34). Chang et al. showed a rescue in the myeloproliferative neoplasm phenotype by targeting the MAPK pathway using a mitogen-activated protein kinase kinase (MEK) inhibitor (35). Pharmacologic MEK inhibition has also been shown to reduce neurofibroma formation in mice and decrease aberrant cellular proliferation in human malignant peripheral nerve sheath tumors cell xenografts (36). Furthermore, studies have shown that deletion of Erk1 leads to increased bone formation (37–39) and inhibits osteoclastogenesis (40). We therefore reasoned that the combined anti-catabolic and pro-anabolic effects of pharmacologic MAPK inhibition may augment tibial fracture healing in two murine models of NF1 pseudarthrosis by both increasing bone formation and reducing bone erosive activity.
Here, we show that application of the MEK inhibitor, PD98059, significantly attenuates the capacity of Nf1+/− BMMNCs to form osteoclasts in tissue culture while promoting terminal OBL differentiation and expression of OBL differentiation markers in Nf1-deficient pro-OBLs. PD98059 treatment also increases OBL differentiation in human MSCs cultured from NF1 patients with tibial pseudarthrosis or scoliosis. More importantly, in vivo studies show that administration of PD98059 significantly improves tibial fracture healing in Col2.3Cre; Nf1flox/− and PeriCre;Nf1flox/− mice. Collectively, our results demonstrate that a hyper Ras/MAPK cascade contributes to bone loss and fracture nonunion in NF1. This suggests that targeting the MAPK pathway may be a viable therapeutic option for augmenting bone healing in NF1-associated pseudarthrosis and other osteopenic/osteoporotic diseases.
RESULTS
Increased Erk1/2 phosphorylation at the fracture sites of Col2.3Cre;Nf1flox/− mice
Previous in vitro studies have suggested that the MAPK pathway is critical for OBL differentiation and skeletal development (37,38). To examine the impact of neurofibromin in modulating Ras/MAPK signaling in vivo, Erk phosphorylation was evaluated in midsagittal histological sections from fractured tibia on day 28 by immunohistochemical staining (Fig. 1A). A significant increase in the percent p44/42 positive cells (p44/42 positive cells/total cells) is observed in the Nf1+/− and Col2.3Cre;Nf1flox/− tissue sections as compared to sections from WT mice (Fig. 1B).
Figure 1.

MAPK hyperactivation in Col2.3Cre+;Nf1flox/− tibiae. (A) Representative histological sections from the tibia fracture site of WT, Nf1+/−, and Col2.3Cre; Nf1flox/− mice are shown. Red arrows indicate cells that are p44/42 positive (brown). (B) Quantitative measurement of p44/42-positive cells (p44/42-positive cells/total cells) is shown. Data are represented as mean ± SEM from three individual experiments (*P < 0.05 for WT versus Nf1+/−mice, **P < 0.01 for Nf1+/− versus Col2.3Cre; Nf1flox/− mice, ***P < 0.001 for WT versus Col2.3Cre; Nf1flox/− mice).
Re-expression of the NF1 GRD enhances Nf1−/− OBL differentiation in vitro
It has previously been shown that Nf1+/−OBLs exhibit Ras hyper activation (32). Using Raf-pull down assay, we demonstrate a gene dose-dependent increase of Ras-GTP activity in Nf1+/− and Nf1−/− pro-OBLs when compared withe WT pro-OBLs (Fig. 2A). In addition, pErk1/2 is elevated in Nf1+/− and Nf1−/− pro-OBLs in comparison to WT pro-OBLs at both the basal levels and following recombinant human platelet-derived growth factor-BB (PDGF) stimulation. Furthermore, PD98059 inhibits Erk phosphorylation in WT, Nf1+/−, and Nf1−/− pro-OBLs (Fig. 2B). To evaluate whether the elevated Ras activity and Erk1/2 phosphorylation are responsible for impaired OBL differentiation in Nf1−/− pro-OBLs, we re-expressed the NF1 GRD in Nf1−/− pro-OBLs. OBL differentiation assays reveal a substantial restoration of alkaline phosphatase (ALP) expression in Nf1−/− pro-OBLs transduced with NF1 GRD as compared to Nf1−/− pro-OBLs transduced with the MSCV-pac control vector (Fig. 3A). Ras activity, both at the baseline and after PDGF stimulation, was reduced after re-expression of NF1 GRD in Nf1−/−pro-OBLs in comparison to MSCV-pac transduced Nf1−/−pro-OBLs, which demonstrate persistently elevated Ras-GTP levels (Fig. 3B).
Figure 2.

PD98059 inhibits hyperactive Ras/MAPK signaling in Nf1−/− pro-OBLs. (A) Ras activity in WT, Nf1+/−, Nf1−/− pro-OBLs was measured at indicated time following stimulation with PDGF (20 ng/ml). A representative immunoblot is shown. (B) Phosphorylation of Erk1/2 (p44/42) was measured in WT, Nf1+/−, Nf1−/− pro-OBLs in response to PDGF stimulation in the presence or absence of PD98059 (100 nm). Data represent one of the four independent results.
Figure 3.
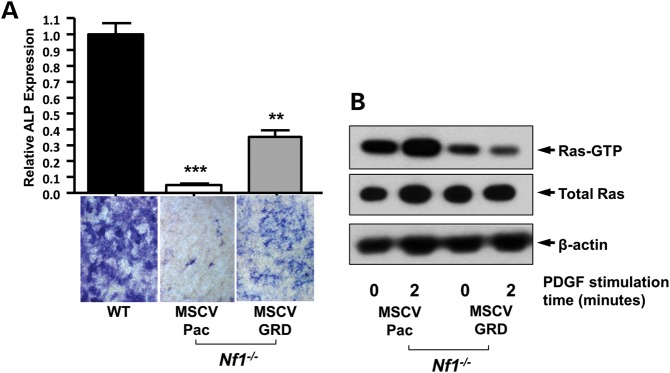
Expression of NF1 GRD promotes OBL differentiation in Nf1−/− pro-OBL cultures. (A) OBL differentiation was evaluated by ALP staining after 5 days of culture in OBL differentiation medium using WT and Nf1−/− pro-OBLs transduced with MSCV-pac control or MSCV-NF1 GRD-pac. Representative high power fields (20×) of OBL cultures were used to quantify OBL differentiation by measuring ALP expression relative to WT controls (**P < 0.001 for NF1 GRD Nf1−/− pro-OBLs versus WT MSCV-pac, and MSCV-pac Nf1−/− pro-OBLs, ***P < 0.001 for MSCV-pac Nf1−/− pro-OBLs versus MSCV-WT, and NF1 GRD Nf1−/− pro-OBLs). (B) Ras activity in Nf1−/− pro-OBLs transduced with either MSCV-pac or NF1 GRD was measured at indicated times following PDGF (20 ng/ml) stimulation.
Pharmacologic MEK inhibition enhances OBL differentiation and increases osteocalcin and osterix : mRNA expression in Nf1−/− pro-OBLs
We next examined whether PD98059 could improve OBL differentiation of WT, Nf1+/− and Nf1−/− pro-OBLs. A gene dosage-dependent defect in OBL differentiation is observed in Nf1+/− and Nf1−/− pro-OBLs as determined by ALP expression (Fig. 4A). To verify the impact of PD98059 on OBL differentiation, we assessed the expression of OBL differentiation markers, osterix and osteocalcin. Osterix and osteocalcin mRNA expression was dramatically diminished in Nf1−/− pro-OBLs versus WT pro-OBLs at a basal level (Fig. 4B and C). After PD98059 treatment, Nf1−/− pro-OBLs demonstrate a significant increase in mRNA expression of both osterix and osteocalcin in comparison to WT pro-OBLs (Fig. 4B and C). Restoration of osterix and osteocalcin expression in Nf1−/− pro-OBLs following PD98059 treatment is consistent with the increase in ALP expression in PD98059 treated Nf1-deficient pro-OBLs, suggesting the important role of MAPK pathway in impaired Nf1-deficient OBL differentiation.
Figure 4.

Pharmacologic MEK inhibition improves OBL differentiation and enhances mRNA expression of OBL differentiation markers. (A) WT, Nf1+/− and Nf1−/− pro-OBLs were cultured in OBL differentiating medium in the presence or absence of PD98059 (100 nm). OBL differentiation was measured by quantifying ALP expression from representative photomicrographs (20×). Data reflect three independent experiments (*P < 0.05 for Nf1−/− pro-OBLs treated with PD98059 versus WT pro-OBLs, **P < 0.01 for Nf1+/− pro-OBLs treated with vehicle versus PD98059 treated Nf1+/− pro-OBLs and WT pro-OBLs, ***P < 0.001 for Nf1−/− pro-OBLs treated with vehicle versus PD98059). (B) Osterix mRNA expression in untreated versus PD98059 (100 nm) treated WT and Nf1−/− pro-OBLs was determined by a real-time PCR. Osterix expression, relative to untreated WT, was quantified in three individual experiments (***P < 0.001 for untreated WT versus untreated Nf1−/− pro-OBLs and untreated Nf1−/− pro-OBLs versus PD98059 treated Nf1−/− pro-OBLs). (C) Osteocalcin mRNA expression in untreated versus PD98059 treated WT and Nf1−/− pro-OBLs was determined by a real-time PCR in five individual experiments. Osteocalcin expression was measured relative to untreated WT (***P < 0.001 for untreated WT versus untreated Nf1−/− pro-OBLs and untreated Nf1−/− pro-OBLs versus PD98059 treated Nf1−/− pro-OBLs).
PD98059 attenuates the aberrant frequency of osteoclast progenitors and osteoclast maturation of Nf1+/−BMMNCs
Colony-forming unit-macrophage (CFU-M) and osteoclast maturation assays were performed in WT and Nf1+/−BMMNCs to evaluate the consequence of Nf1 disruption in macrophage and osteoclast differentiation. Consistent with our previous study (33), in comparison to WT, Nf1+/−BMMNCs exhibit significantly elevated numbers of osteoclast progenitors (CFU-M) in methylcellulose colony-forming assays (Fig. 5A) as well as an increased capacity for osteoclast maturation (Fig. 5B). Importantly, pharmacologic inhibition of MEK by PD98059 reduces CFU-M formation and osteoclast maturation in vitro in Nf1+/−BMMNCs.
Figure 5.

Inhibition of MAPK pathway attenuates frequency of osteoclast progenitors and osteoclast maturation of Nf1+/− BMMNCs. (A) CFU-M formation was measured in WT and Nf1+/−BMMNCs following M-CSF (30 ng/ml) stimulation in the presence of escalating dose of PD98059. Data represent the mean ± SEM of three individual experiments (**P < 0.01 for untreated and PD98059 (100 nm) treated WT versus untreated and PD98059 (100 nm) treated Nf1+/−CFU-M, *P < 0.05 for PD98059 treated (250 nM, 500 nM) WT versus PD98059-treated (250, 500 nm) Nf1+/−CFU-M. (B) WT and Nf1+/−BMMNCs were cultured for 7 days and stained with TRACP to identify mature osteoclasts. (a) Data represent the mean ± SEM of % TRACP-positive area in response to varying concentrations of PD98059 of three independent experiments (**P < 0.01 for untreated WT versus untreated Nf1+/−osteoclasts). (b) Representative photographs (20×) of untreated and PD98059 (100 nM) treated WT and Nf1+/−osteoclasts are shown.
MEK inhibition improves fracture healing in Col2.3Cre;Nf1flox/− and PeriCre;Nf1flox/−mice
Given the in vitro data of the multifaceted abilities of PD98059 in the restoration of Nf1-deficient OBL differentiation while attenuating the Nf1+/− osteoclastogenic capacity, we sought to further test whether pharmacologic MEK inhibition could improve the fracture healing in a mouse model of NF1-associated pseudarthrosis. PD98059 was administered to Col2.3Cre;Nf1flox/− mice following tibial fracture via a micro-osmotic pump (contralateral to fractured tibia), which maintains a constant release of the compound (0.25 μL/h at 10 mg/kg/day) for a period of 28 days (41). Tibiae fracture healing was monitored weekly by radiographs and quantified at the end of 4 weeks by histologic analysis of the fracture site. Radiographic quantification of the number of healed fractures, as defined by complete cortical bridging across the fracture site, has been shown to be 80% in WT mice infused with a vehicle solution (31). More importantly, radiograph analysis reveals a 3-fold increase in the percentage of healed fractures in Col2.3Cre;Nf1flox/− mice receiving PD98059 (66.7%) in comparison to Col2.3Cre;Nf1flox/− mice infused with a vehicle solution (22%) (Fig. 6A, Table 1). No changes are detected via radiography on Days 7 and 14 in both vehicle and PD98059 groups, whereas two mice in the PD98059 group have complete fracture healing at Day 21.
Figure 6.

MEK inhibition promotes tibial fracture-healing in Col2.3Cre; Nf1flox/−mice in vivo. Tibial fractures were generated for WT and Col2.3Cre;Nf1flox/− mice. Osmotic pumps containing either vehicle control (PBS) or PD98059 were implanted subcutaneously in the contralateral dorsal flank of Col2.3Cre;Nf1flox/− mice. WT mice received vehicle control (PBS) implants. (A) Showing are representative radiographs acquired at 0, 14, 21, 28 days post-fracture in WT mice with PBS vehicle and Col2.3Cre;Nf1flox/− mice with or without PD98059 (10 mg/kg/day). (B, a) Representative photomicrographs (20×) of MacNeal's/Von Kossa counter stain shows accumulation of cartilage and infiltration of marrow cells filling the fracture area in the control pump group. (B, b) Representative photomicrographs (20×) of TRACP stains are shown. Red area represents TRACP-positive-staining osteoclasts. (C) TRACP-positive osteoclasts to the bone surface ratio (Oc.N/BS) was enumerated as shown (***P < 0.01). (D) The ratio of bone volume/tissue volume was quantified to calculate percent tissue mineralization (***P < 0.01). (E) The number of OBLs per bone surface (Ob.N/BS) was quantified as shown (**P < 0.01).). Data represent mean ± SEM from five mice per treatment group.
Table 1.
MEK inhibition improves tibial fracture-healing in Col2.3Cre; Nf1flox/− mice in vivo
| Treatment | Vehicle pump | PD98059 pump |
|---|---|---|
| Healed | 2 | 12 |
| Fractured | 9 | 18 |
| % Healed | 22% | 66.7% |
The percentage of healed fractures after 28 days was determined in mice receiving the vehicle versus PD98059 treatment. Healed fractures were defined radiographically by complete periosteal bridging across the fracture site.
The cellular and structural changes within the fracture sites are histologically evaluated by MacNeal/von Kossa counter stain (Fig. 6B, a) and tartrate-resistant acid phosphatase (TRACP) stain (Fig. 6B, b). PD98059-treated Col2.3Cre;Nf1flox/− mice exhibit a significant reduction in TRACP-positive osteoclasts when compared with mice receiving vehicle (Fig. 6C). In contrast, a significant increase in the trabecular bone area accompanied with augmentation of tissue mineralization (Fig. 6D) and increase in the number of OBLs (Fig. 6E) is observed in Col2.3Cre;Nf1flox/− mice that received PD98059 compared with vehicle controls. These results indicate that inhibition of the MAPK pathway significantly improves fracture healing in Col2.3Cre;Nf1flox/− mice by augmenting OBL bone formation while inhibiting excessive osteoclast bone resorption.
To verify our findings, we repeated the experiments in a second NF1 murine model, PeriCre;Nf1flox/−, which have been shown to develop similar skeletal phenotypes as the Col2.3Cre;Nf1flox/− model (31). Administration of PD98059 (10 mg/kg/day) induces comparable improvements in fracture healing outcome in the PeriCre;Nf1flox/− mouse model (Supplementary Material, Fig. S1A) as evidenced by an increase in the percentage of healed fractures (Supplementary Material, Fig. S1B), reduction in osteoclast number and an enhancement in callus mineralization (Supplementary Material, Fig. S1C).
Reintroduction of the NF1 GRD and PD98059 rescues OBL differentiation in NF1 hMSCs
To determine whether the Ras/MAPK signaling pathway is also impactful in the human system, we generated hMSCs from bone marrow of NF1 patients with pseudarthrosis or scoliosis (obtained during surgical procedures). We first examined the neurofibromin expression in NF1 hMSCs. As shown in Supplementary Material, Figure S2, neurofibromin levels are reduced in NF1 hMSCs compared with control MSCs derived from donor bone marrow. We next introduced NF1 GRD or MSCV-pac into hMSCs cultures established from NF1 patients and evaluated OBL differentiation of hMSCs via an ALP expression assay. Consistent with murine Nf1-deficient pro-OBLs, reduced ALP expression is observed in NF1 hMSCs compared with control MSCs. Transduction of NF1 GRD into NF1 hMSCs substantially restores ALP expression when compared with the control vector (Fig. 7A). To test whether MEK inhibition restores OBL differentiation in NF1 hMSC cultures, as seen in the murine system, PD98059 was applied to hMSC cultures containing OBL differentiation media. Analogous to the results achieved in murine-derived Nf1 nullizygous pro-OBLs, ALP activity is significantly enhanced in NF1 hMSC cultures following PD98059 treatment in comparison to vehicle controls (Fig. 7B). These data indicate that the Ras/MAPK pathway plays an important role in the impaired Nf1−/− OBL differentiation.
Figure 7.
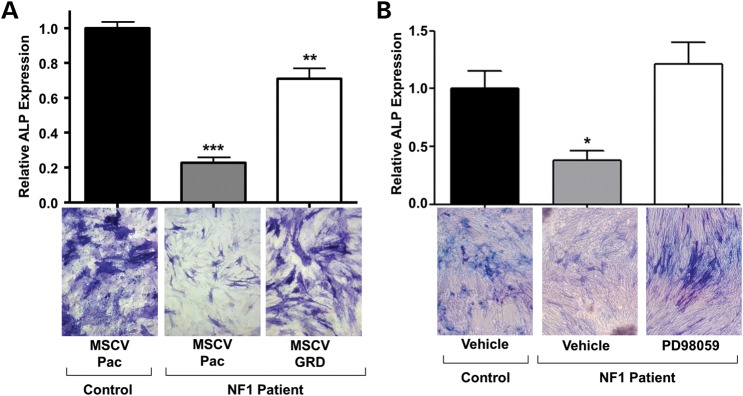
Inhibition of Ras/MAPK signaling enhances OBL differentiation in NF1 hMSCs. (A) Reintroduction of the NF1 GRD into NF1 patient hMSCs restores OBL differentiation. ALP expression, relative to WT control, is shown (***P < 0.001 for MSCV-pac control hMSCs versus MSCV-pac NF1 hMSCs, **P < 0.01 for MSCV-pac NF1 hMSCs versus NF1 GRD NF1 hMSCs,). (B) Pharmacologic MEK inhibition (100 nm PD98059) enhances OBL differentiation in NF1 patient hMSCs (*P < 0.05 for vehicle-treated NF1 hMSCs versus control hMSCs, and NF1 hMSCs vehicle versus PD98059 -reated NF1 hMSCs. Data are representative of three NF1 and two control samples.
DISCUSSION
Pseudarthrosis is a debilitating consequence of NF1 with limited successful intervention. In the current study, we demonstrate that the activated Ras/MAPK pathway plays a prominent role in the pathogenesis of fracture nonunion in NF1. Targeting with a MEK inhibitor dramatically increases OBL differentiation and inhibits osteoclast maturation in vitro, which enhances the fracture healing in vivo in Nf1 murine models.
Ras proteins regulate the proliferation, survival, differentiation and cytoskeletal functions of many cell lineages, including cells that are central to bone formation and remodeling (42–48). The biochemical output of Ras proteins is tightly regulated by neurofibromin, which functions as a negative regulator of the cycle between an active Ras-GTP and an inactive Ras-GDP state (43,44). Ras activation regulates OBL differentiation (42,45,46). In previous studies, we and others have shown that haploinsufficiency of Nf1 results in a reduction of the maturation of OBLs and a modest, but not statistically significant, decline in skeletal formation (32,49). In the present study, we show that deletion of Nf1 in pro-OBLs leads to a reduction in OBL differentiation as evidenced by decreased ALP expression and reduced mRNA expression of OBL specific markers, osterix and osteocalcin. Transduction of the full-length NF1 GRD restores OBL differentiation in both Nf1-deficient pro-OBLs and NF1 patient hMSCs. Our study is consistent with a previous study that demonstrates an increase in OBL differentiation and mineralization following adenovirus vector-mediated dominant negative Ras (Ras (DN)) expression in a wild-type osteoblastic cell line (39).
Ras-GTP recruits the serine-threonine kinase, Raf-1, to the plasma membrane, which in turn activates a series of downstream effectors such as Erks (p44/p42 MAP kinase) (47,50). The activation of these molecules is important in the extracellular stimuli induced mitogenic response, including proliferation and differentiation of OBLs (39,51,52). Kono et al. established the Erk pathway as a negative regulator of matrix mineralization both in vitro and in vivo (39). Highuchi et al. have shown that the MAPK pathway negatively regulates the OBL differentiation and the expression of osteocalcin (53). Consistent with the findings of these studies, we now show that inhibition of hyperactive MAPK signaling by application of an MEK inhibitor (PD98059) partially restores Nf1-deficient OBL differentiation evidenced by increased ALP expression and mRNA expression of OBL differentiation markers, osterix and osteoclacin. More importantly, PD98059 treatment restores the OBL differentiation of hMSCs derived from NF1 patients.
Osteoclasts are specialized cells derived from the myeloid monocyte–macrophage lineage that successively adhere and remove bone tissue by resorbing the mineralized matrix. Many skeletal diseases, in particular diseases with decreased bone mineral density, occur as a consequence of a skeletal imbalance that favors bone resorption over bone deposition (54). We have previously shown that Nf1+/− myeloid cells are hypersensitive to M-CSF as evidenced by an increase in CFU-M and CFU-GM (33). In addition, using a genetic mouse model, we have recently reported that Erk1 plays a critical role in regulating osteoclast differentiation and bone resorptive activity using a genetic murine model (40). In the present study, we show that pharmacologic inhibition of the MAPK pathway using the MEK inhibitor, PD98059, inhibits aberrant osteoclast differentiation and maturation. In addition, compared with WT, Nf1+/− osteoclasts are hypersensitive to MEK inhibition, which demonstrates the biological relevance of this pathway in mediating Nf1+/− osteoclast gain-in-functions.
Schindeler et al. previously showed that the combination of bisphosphonates with recombinant bone morphogenetic protein 2 (BMP-2) promotes heterotypic bone formation (55) and tibial fracture repair (56) in Nf1+/− mice. This combinatorial approach provides insight for targeting both the anabolic and catabolic pathways of bone remodeling. Kolanczyk et al. reported that treatment with lovastatin improved bone healing defects in a Nf1Prx−/− cortical bone injury model (57). Wang et al. reported that locally applied lovastatin improved tibial fracture healing in a Col2.3Cre+;Nf1flox/floxmouse model (29). By indirectly inhibiting the prenylation of G-proteins including Ras, lovastatin has generated interest for its potential to modulate NF1 molecular defects, although nonspecifically. In the present study, we use a MEK inhibitor to target downstream effectors of Ras to restore the aberrant functions of Nf1 mutant OBLs and osteoclasts. By attenuating hyperactive Ras/MAPK signaling in Nf1-deficient OBLs and osteoclasts, PD98059 exhibited potent anabolic and anti-catabolic effects on bone remodeling within the fracture site, significantly improving tibial fracture healing in two previously established murine models of pseudarthrosis associated with NF1.
In summary, our current study demonstrates the pivotal role Ras cascade likely plays in both OBLs and osteoclasts. We show that hyperactive MAPK signaling is responsible, at least in part, for the defective OBL differentiation/bone formation and increased osteoclast maturation in vivo in NF1. This study provides pre-clinical evidence for targeting MEK components of the Ras/MAPK pathway, which may have potential therapeutic value to improve fracture healing in NF1 patients. However, PD98059 has been shown to have side effects on different cell lineages (58,59), which requires further testing of clinical-grade MAPK inhibitors to determine whether Ras-MAPK inhibition may be a viable therapeutic strategy to increase bone mass and decrease bone resorption in NF1 patients with long bone healing defects.
MATERIALS AND METHODS
Animals
Nf1+/− mice were obtained from Dr Tyler Jacks at the Massachusetts Institute of Technology (Cambridge, MA, USA) (60). Nf1flox/flox mice were provided by Dr Luis Parada at the University of Texas Southwestern Medical Center (8). Col2.3Cre transgenic mice were generated as described elsewhere (61). Conditional bi-allelic deletion of Nf1 in pre-OBLs was achieved by genetic intercross of Nf1flox/flox mice with Col2.3Cre+ mice, placing the Cre gene under the control of the 2.3 kb proximal fragment of the α1(I)-collagen promoter, which is specifically expressed in OBLs (62,63). Col2.3Cre+;Nf1flox/− mice (Nf1 null in OBLs, systemically Nf1 heterozygous) were generated by intercrossing Col2.3Cre+;Nf1flox/floxmice with Nf1+/− mice as described previously (31). PeriCre+mice were intercrossed with mice bearing conditional genetic targeting of the Nf1 allele (Nf1flox/flox) to generate PeriCre+;Nf1flox/floxmice (Nf1 null in pro-OBLs and OBLs, systemically WT). Similarly, PeriCre+;Nf1flox/− mice (Nf1 null in pro-OBLs and OBLs, systemically Nf1 heterozygous) were generated by intercrossing PeriCre+;Nf1flox/floxmice with Nf1+/− mice (31). Mice were born at the expected Mendelian ratio. All studies were approved by the Institutional Animal Care and Use Committee. Chemicals were purchased from Sigma (St. Louis, MO, USA) unless otherwise stated.
Pro-OBL cell culture
Pro-OBLs were generated from WT, Nf1+/−, and PeriCre+;Nf1flox/− mice as previously described (32). Briefly, BMMNCs were separated by low-density gradient centrifugation from bone marrow of 6 to 8-week-old mice. BMMNCs were then resuspended and cultured in mouse MesenCult basal medium containing MesenCult Supplement (Stem Cell Technologies Inc., CA, USA) at a concentration of 2 × 106 cells/ml in a 10 cm tissue culture plate. Once the cultures reached 80–90% confluency, cells were trypsinized and replated at a density of 5 × 105 cells/75 cm2. Pro-OBLs at passage 5–10 were used for the following experiments.
OBL differentiation assays
Pro-OBLs were cultured in osteogenic differentiation medium, consisting of MSC complete medium supplemented with 10−8 m dexamethasone, 50 ng/ml ascorbic acid and 10 mm β-galactophosphate. Pro-OBLs underwent differentiation for 5 days in the presence and absence of PD98059 (100 nm) (64,65). OBL differentiation cultures were processed for ALP expression using an ALP staining kit (32,66). Cells were then evaluated microscopically and OBL differentiation was quantified by measuring the intensity of ALP expression determined by ImageJ software (32).
Clonogenic progenitor assays
CFU-M of BMMNCs was assayed, as described previously (33). Briefly, 2.5 × 104 WT and Nf1+/− BMMNCs were seeded onto a 35 mm gridded dish containing methylcellulose supplemented with murine recombinant M-CSF (30 ng/ml) and murine recombinant RANKL (20 ng/ml) with or without PD98059 (100 nm) for 7 days at 37°C in a 5% CO2 incubator. Colonies were scored using an inverted light microscope. M-CSF and RANKL cytokines were purchased from PeproTech (Rocky Hills, NC, USA).
Generation of murine osteoclasts and TRACP staining
Mouse osteoclasts were obtained in vitro using BMMNCs, as described previously (40). Briefly, BMMNCs from 8-week-old WT and Nf1+/− mice were isolated by long bone marrow flush and Ficoll density gradient centrifugation (2400g) and cultured in alpha-modified minimum essential medium (α-MEM) supplemented with 10% fetal bovine serum (FBS), 30 ng/ml M-CSF and 20 ng/ml RANKL for 3 days with or without MEK inhibitor (PD98059). On day four, the cell culture medium was changed to α-MEM supplemented with 10% FBS, M-CSF (30 ng/ml) and 60 ng/ml RANKL for another 3 days with or without PD98059. Osteoclasts were identified by fixation of adherent cells with a solution containing 25 ml citrate solution, 65 ml acetone and 8 ml 3.7% formaldehyde, followed by TRACP stain using a commercially available kit. Osteoclasts were visualized with a Nikon TE2000-S microscope (Nikon Inc., Melville, NY, USA), and images were taken by a QImaging camera and QCapture-Pro software (Fryer Company Inc., Cincinnati, OH, USA). Multinucleated TRACP-positive-staining cells containing more than three nuclei were scored as mature osteoclasts. The area of multinucleated, TRACP-positive osteoclasts was normalized to the bone surface area and calculated (osteoclast area % per field) using ImageJ software (32).
Human MSC culture and OBL differentiation assays
Human samples used in this study were approved by the Indiana University School of Medicine and University of Utah Institutional Review Boards. Human MSCs (hMSCs) were cultured from bone marrow aspirates collected from discarded bone and marrow tissues of healthy controls and NF1 patients undergoing surgical procedures for pseudarthrosis or scoliosis. Cells were cultured using a MesenCult basal medium containing MesenCult Supplement (Stem Cell Technologies Inc., CA, USA) and the medium was changed once a week. Passages 5–10 were used for experiments. The hMSCs underwent OBL differentiation in the medium supplemented with ascorbic acid (50 ng/ml), β-glycerolphosphotase (10 mm) and dexamethasone (10−8 m) for 5 days with or without PD98059 (100 nm). Cells were stained with an ALP staining kit and microscopically examined to quantify OBL differentiation using the Image J software (32). Two normal controls and three NF1 patient samples were used to conduct the experiments.
Generation of recombinant retroviral plasmids and retroviral transduction of pro-OBLs and hMSCs
Previously developed recombinant retrovirus constructs were used in these studies (67). A virus expressing the full-length NF1 GTPase activating-related domain (NF1 GRD) pac (MSCV-NF1 GRD-pac) under transcriptional control of the myeloproliferative sarcoma retrovirus promoter and a virus expressing the selective marker gene alone (MSCV-pac) under transcriptional control of a phosphoglycerate kinase promoter were employed in this study via a viral transduction methodology described elsewhere (32). Briefly, pro-OBLs and hMSCs were cultured in MesenCult media and transduced with either MSCV-NF1 GRD-pac or MSCV-pac followed by puromycin-resistant selection (5 µg/ml) beginning 72 h following transduction.
Tibial fracture, radiography and osmotic pump implantation
A single tibial fracture was induced as described previously (31). To stabilize the bone post-fracture and permit load-bearing, an intramedullary rod (a 30-G needle) was surgically inserted via the proximal end of the tibia into the medullary canal to achieve internal fixation. A fracture was generated between the middle 1/3 and distal 1/3 of the tibia by dropping a 200 g weight from a height of 30 cm. To ensure standardization of fractures, radiographs were obtained following the fracture using a pixRay-100 X-ray scanner (Bioptics, Pheonix, AZ, USA). The mice were allowed to recover on a heated pad and then placed in recovery cages. Radiography was performed weekly to monitor fracture repair, defined by complete cortical bridging across the fracture site, for a duration of 4 weeks, time course determined by the previous study (31). In the event where internal fixation failed (due to pin slippage, bending or breakage), the affected mouse was euthanized and excluded from subsequent analysis.
For osmotic pump implantation, immediately following fracture, Col2.3Cre;Nf1flox/− and PeriCre;Nf1flox/− mice were implanted with a subcutaneous Alzet 1007D micro-osmotic pump (Durect, Alzet, Cupertino, CA, USA) under the contralateral dorsal skin under avertin anesthesia. To obtain a constant release, PD98059 (10 mg/kg/day) (68,69) or vehicle (PBS) was released over 28 days (31) at a rate of 0.25 μl/h.
Histomorphometric measurements
After euthanasia, tibiae were dissected from the mice and fixed in 10% neutral buffered formalin for 48 h, dehydrated in graded ethanols, and embedded undecalcified in methylmethacrylate. Midsagittal sections (4 µm thick) were cut from the tibiae using a motorized microtome equipped with a tungsten-carbide knife (Leica Inc, Deerfield, IL, USA). The sections were processed with TRACP and MacNeal and/or von Kossa stain for the identification and enumeration of osteoclasts and OBLs, respectively (31). In addition, immunohistochemical staining with an anti-phosphoErk1/2 antibody (Cell Signaling, Danvers, MA, USA) was performed in post-fractured, control tibiae to determine the activity of Ras/MAPK signaling. The sections were imaged at 20× magnification on a Leitz DMRXE microscope (Leica Mikroskopie und System GmbH, Wetzlar, Germany) equipped with a SPOT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI, USA).
Western blot and Ras activation assays
Murine pro-OBLs were deprived of serum and growth factors for 24 h. For the stimulation only group, cells were stimulated with PDGF (20 ng/ml) for 2 min. For the inhibitor group, cells were pre-inhibited with PD98059 (100 nm) for 30 min, followed by stimulation with PDGF (20 ng/ml). Cells were lysed in nonionic lysis buffer (20 mm Tris–Cl, 137 mm NaCl, 1 mm EGTA (ethylene-glycol-tetra-acetic acid), 1% Triton X-100, 10% glycerol, 1.5 mm MgCl2) containing protease inhibitors (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The protein concentration of the lysates was normalized using the bicinchoninic acid assay (BCA assay) kit (Pierce, Rockford, IL, USA). Proteins were separated by 12.5% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to high-quality polyvinylidene difluoride membranes (Roche Diagnostics, Indianapolis, IN, USA) in a Tris (20 mm), glycine (150 mm) and methanol (20%) buffer at 250 mV for 2 h at 4°C. After blocking in 5% nonfat dry milk in phosphate buffered saline tween-20 (PBST), the membranes were incubated with primary antibodies at 1:1000 dilution (phospho-ERK, total-ERK, β-Actin; Cell Signaling) in 5% milk in PBST overnight at 4°C. Following overnight exposure, the membranes were washed three times with PBST and incubated with secondary antibodies (anti-rabbit, anti-mouse, GE Healthcare UK Limited, UK) conjugated with horseradish peroxidase at 1:5000 dilution in 5% milk in PBST for 1 h at room temperature. Membranes were washed again in PBST three times at room temperature. Membranes were exposed to a supersignal west pico chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA) and protein bands were visualized on an X-ray film (GeneMate, Kaysville, UT, USA).
To measure Ras activity, pro-OBLs were deprived of serum and growth factors for 24 h and then stimulated with PDGF (20 ng/ml) for 2 min. Cells were lysed in nonionic lysis buffer (20 mm Tris–Cl, 137 mm NaCl, 1 mm EGTA, 1% Triton X-100, 10% glycerol, 1.5 mm MgCl2) containing protease inhibitors (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The protein concentration of the lysates was normalized using a BCA assay kit (Pierce, Rockford, IL , USA). Ras activity was determined using a Ras activation assay kit (Millipore Corporation, Temecula, CA, USA) (67). Briefly, GTP-bound Ras levels were determined by incubating cell lysates with Raf-1 Ras-binding domain conjugated to agarose beads followed by an immunoblot using an anti-Ras antibody (Millipore Corporation). Total Ras was also measured (Cell Signaling). Image J was used to quantitate densitometry for all protein bands.
Real time reverse-transcriptase polymerase chain reaction
To evaluate the expression of OBL differentiation markers in pro-OBLs, a quantitative real-time reverse-transcriptase polymerase chain reaction (qRT-PCR) was performed. Briefly, total RNA was extracted from differentiated WT and Nf1−/− pro-OBLs in the presence or absence of PD98059 (100 nm) using the RNeasy mini kit (Qiagen, Valencia, CA, USA). Complementary DNA (cDNA) was synthesized according to the protocol and using reagents from the high-capacity cDNA reverse transcription kit (Life Technologies, Grand Island, NY, USA). A qRT-PCR was performed (100 ng RNA was used for each reaction) using the Taqman Fast universal PCR Master mix (2X) and Taqman murine primers Sp7 (osterix), BGLAP (osteocalcin), and Gapdh (Life Technologies).
Statistical analysis
Two-tailed Student's t-test and analysis of variance were used to evaluate statistical difference. P-values <0.05 were considered significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement: None declared.
FUNDING
This work was supported in part by the Department of Defense (NF100087, NF043032, NF073112 to F.C.Y.) and March of Dimes (YF08-246 to F.C.Y.). R.S. was supported by a Howard Hughes Medical Institute Medical Research Fellowship. S.D.R. was supported in part by a Children's Tumor Foundation (CTF) Young Investigator Award (2011-01-010) and an Indiana CTSI Career Development Award (5TL1RR025759-03). D.A.S. was supported by the Department of Defense (NF100057 to D.A.S.) and Thrasher Research Fund.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Heather Daniel for administrative support. We thank Heather Hanson, Austin Stevens and the orthopedic NF team at Shriner's Hopsital for Children, Salt Lake City (Drs J.C. Carey, J. D’ Astous, K. Carroll, J. Roach, T. Hennessey, A. Stotts, L. Randall, M. Pond, M. Woiczik and S. Santora) for help with participant recruitment and sample collection.
REFERENCES
- 1.Le L.Q., Parada L.F. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–4616. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutmann D.H. The neurofibromatoses: when less is more. Hum. Mol. Genet. 2001;10:747–755. doi: 10.1093/hmg/10.7.747. [DOI] [PubMed] [Google Scholar]
- 3.Theos A., Korf B.R. Pathophysiology of neurofibromatosis type 1. Ann. Intern. Med. 2006;144:842–849. doi: 10.7326/0003-4819-144-11-200606060-00010. [DOI] [PubMed] [Google Scholar]
- 4.Xu G.F., O'Connell P., Viskochil D., Cawthon R., Robertson M., Culver M., Dunn D., Stevens J., Gesteland R., White R., et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 5.Atit R.P., Crowe M.J., Greenhalgh D.G., Wenstrup R.J., Ratner N. The Nf1 tumor suppressor regulates mouse skin wound healing, fibroblast proliferation, and collagen deposited by fibroblasts. J. Invest. Dermatol. 1999;112:835–842. doi: 10.1046/j.1523-1747.1999.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atit R.P., Mitchell K., Nguyen L., Warshawsky D., Ratner N. The neurofibromatosis type 1 (Nf1) tumor suppressor is a modifier of carcinogen-induced pigmentation and papilloma formation in C57BL/6 mice. J. Invest. Dermatol. 2000;114:1093–1100. doi: 10.1046/j.1523-1747.2000.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rutkowski J.L., Wu K., Gutmann D.H., Boyer P.J., Legius E. Genetic and cellular defects contributing to benign tumor formation in neurofibromatosis type 1. Hum. Mol. Genet. 2000;9:1059–1066. doi: 10.1093/hmg/9.7.1059. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y., Ghosh P., Charnay P., Burns D.K., Parada L.F. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ajuebor M.N., Hogaboam C.M., Kunkel S.L., Proudfoot A.E., Wallace J.L. The chemokine RANTES is a crucial mediator of the progression from acute to chronic colitis in the rat. J. Immunol. 2001;166:552–558. doi: 10.4049/jimmunol.166.1.552. [DOI] [PubMed] [Google Scholar]
- 10.Bajenaru M.L., Hernandez M.R., Perry A., Zhu Y., Parada L.F., Garbow J.R., Gutmann D.H. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–8577. [PubMed] [Google Scholar]
- 11.Ingram D.A., Yang F.C., Travers J.B., Wenning M.J., Hiatt K., New S., Hood A., Shannon K., Williams D.A., Clapp D.W. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J. Exp. Med. 2000;191:181–188. doi: 10.1084/jem.191.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang F.C., Ingram D.A., Chen S., Hingtgen C.M., Ratner N., Monk K.R., Clegg T., White H., Mead L., Wenning M.J., et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/- mast cells. J. Clin. Invest. 2003;112:1851–1861. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johannessen C.M., Reczek E.E., James M.F., Brems H., Legius E., Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl Acad. Sci. USA. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cichowski K., Jacks T. NF1 Tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 15.Kuorilehto T., Kinnunen P., Nissinen M., Alanne M., Leskela H.V., Lehenkari P., Peltonen J. Vasculopathy in two cases of NF1-related congenital pseudarthrosis. Pathol. Res. Pract. 2006;202:687–690. doi: 10.1016/j.prp.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Kuorilehto T., Nissinen M., Koivunen J., Benson M.D., Peltonen J. NF1 Tumor suppressor protein and mRNA in skeletal tissues of developing and adult normal mouse and NF1-deficient embryos. J. Bone Miner. Res. 2004;19:983–989. doi: 10.1359/JBMR.040130. [DOI] [PubMed] [Google Scholar]
- 17.Kuorilehto T., Poyhonen M., Bloigu R., Heikkinen J., Vaananen K., Peltonen J. Decreased bone mineral density and content in neurofibromatosis type 1: lowest local values are located in the load-carrying parts of the body. Osteoporos. Int. 2005;16:928–936. doi: 10.1007/s00198-004-1801-4. [DOI] [PubMed] [Google Scholar]
- 18.Lammert M., Kappler M., Mautner V.F., Lammert K., Storkel S., Friedman J.M., Atkins D. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos. Int. 2005;16:1161–1166. doi: 10.1007/s00198-005-1940-2. [DOI] [PubMed] [Google Scholar]
- 19.Jacques C., Dietemann J.L. Imaging features of neurofibromatosis type 1. J. Neuroradiol. 2005;32:180–197. doi: 10.1016/s0150-9861(05)83136-0. [DOI] [PubMed] [Google Scholar]
- 20.Illes T., Halmai V., de Jonge T., Dubousset J. Decreased bone mineral density in neurofibromatosis-1 patients with spinal deformities. Osteoporos. Int. 2001;12:823–827. doi: 10.1007/s001980170032. [DOI] [PubMed] [Google Scholar]
- 21.Heuze Y., Piot B., Mercier J. [Difficult surgical management of facial neurofibromatosis type I or von Recklinghausen disease in children] Rev. Stomatol. Chir. Maxillofac. 2002;103:105–113. [PubMed] [Google Scholar]
- 22.Stevenson D.A., Moyer-Mileur L.J., Murray M., Slater H., Sheng X., Carey J.C., Dube B., Viskochil D.H. Bone mineral density in children and adolescents with neurofibromatosis type 1. J. Pediatr. 2007;150:83–88. doi: 10.1016/j.jpeds.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevenson D.A., Zhou H., Ashrafi S., Messiaen L.M., Carey J.C., D'Astous J.L., Santora S.D., Viskochil D.H. Double inactivation of NF1 in tibial pseudarthrosis. Am. J. Hum. Genet. 2006;79:143–148. doi: 10.1086/504441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevenson D.A., Birch P.H., Friedman J.M., Viskochil D.H., Balestrazzi P., Boni S., Buske A., Korf B.R., Niimura M., Pivnick E.K., et al. Descriptive analysis of tibial pseudarthrosis in patients with neurofibromatosis 1. Am. J. Med. Genet. 1999;84:413–419. doi: 10.1002/(sici)1096-8628(19990611)84:5<413::aid-ajmg5>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 25.Gilbert A., Brockman R. Congenital pseudarthrosis of the tibia. Long-term followup of 29 cases treated by microvascular bone transfer. Clin. Orthop. Relat. Res. 1995;314:37–44. [PubMed] [Google Scholar]
- 26.Ippolito E., Corsi A., Grill F., Wientroub S., Bianco P. Pathology of bone lesions associated with congenital pseudarthrosis of the leg. J. Pediatr. Orthop. B. 2000;9:3–10. doi: 10.1097/01202412-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Morrissy R.T., Riseborough E.J., Hall J.E. Congenital pseudarthrosis of the tibia. J. Bone. Joint Surg. Br. 1981;63-B:367–375. doi: 10.1302/0301-620X.63B3.6790551. [DOI] [PubMed] [Google Scholar]
- 28.Sofield H.A. Congenital pseudarthrosis of the tibia. Clin. Orthop. Relat. Res. 1971;76:33–42. doi: 10.1097/00003086-197105000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Wang W., Nyman J.S., Moss H.E., Gutierrez G., Mundy G.R., Yang X., Elefteriou F. Local low-dose lovastatin delivery improves the bone-healing defect caused by Nf1 loss of function in osteoblasts. J. Bone Miner. Res. 2010;25:1658–1667. doi: 10.1002/jbmr.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schindeler A., Morse A., Harry L., Godfrey C., Mikulec K., McDonald M., Gasser J.A., Little D.G. Models of tibial fracture healing in normal and Nf1-deficient mice. J. Orthop. Res. 2008;26:1053–1060. doi: 10.1002/jor.20628. [DOI] [PubMed] [Google Scholar]
- 31.Wu X., Chen S., He Y., Rhodes S.D., Mohammad K.S., Li X., Yang X., Jiang L., Nalepa G., Snider P., et al. The haploinsufficient hematopoietic microenvironment is critical to the pathological fracture repair in murine models of neurofibromatosis type 1. PLoS One. 2011;6:e24917. doi: 10.1371/journal.pone.0024917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X., Estwick S.A., Chen S., Yu M., Ming W., Nebesio T.D., Li Y., Yuan J., Kapur R., Ingram D., et al. Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum. Mol. Genet. 2006;15:2837–2845. doi: 10.1093/hmg/ddl208. [DOI] [PubMed] [Google Scholar]
- 33.Yang F.C., Chen S., Robling A.G., Yu X., Nebesio T.D., Yan J., Morgan T., Li X., Yuan J., Hock J., et al. Hyperactivation of p21ras and PI3 K cooperate to alter murine and human neurofibromatosis type 1-haploinsufficient osteoclast functions. J. Clin. Invest. 2006;116:2880–2891. doi: 10.1172/JCI29092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staser K., Park S.J., Rhodes S.D., Zeng Y., He Y.Z., Shew M.A., Gehlhausen J.R., Cerabona D., Menon K., Chen S., et al. Normal hematopoiesis and neurofibromin-deficient myeloproliferative disease require Erk. J. Clin. Invest. 2013;123:329–334. doi: 10.1172/JCI66167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang T., Krisman K., Theobald E.H., Xu J., Akutagawa J., Lauchle J.O., Kogan S., Braun B.S., Shannon K. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J. Clin. Invest. 2013;123:335–339. doi: 10.1172/JCI63193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jessen W.J., Miller S.J., Jousma E., Wu J., Rizvi T.A., Brundage M.E., Eaves D., Widemann B., Kim M.O., Dombi E., et al. MEK Inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Invest. 2013;123:340–347. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mimori K., Komaki M., Iwasaki K., Ishikawa I. Extracellular signal-regulated kinase 1/2 is involved in ascorbic acid-induced osteoblastic differentiation in periodontal ligament cells. J. Periodontol. 2007;78:328–334. doi: 10.1902/jop.2007.060223. [DOI] [PubMed] [Google Scholar]
- 38.Ge C., Xiao G., Jiang D., Franceschi R.T. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J. Cell Biol. 2007;176:709–718. doi: 10.1083/jcb.200610046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kono S.J., Oshima Y., Hoshi K., Bonewald L.F., Oda H., Nakamura K., Kawaguchi H., Tanaka S. Erk pathways negatively regulate matrix mineralization. Bone. 2007;40:68–74. doi: 10.1016/j.bone.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 40.He Y., Staser K., Rhodes S.D., Liu Y., Wu X., Park S.J., Yuan J., Yang X., Li X., Jiang L., et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS One. 2011;6:e24780. doi: 10.1371/journal.pone.0024780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ingram D.A., Hiatt K., King A.J., Fisher L., Shivakumar R., Derstine C., Wenning M.J., Diaz B., Travers J.B., Hood A., et al. Hyperactivation of p21(ras) and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J. Exp. Med. 2001;194:57–69. doi: 10.1084/jem.194.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satoh T., Nakafuku M., Miyajima A., Kaziro Y. Involvement of ras p21 protein in signal-transduction pathways from interleukin 2, interleukin 3, and granulocyte/macrophage colony-stimulating factor, but not from interleukin 4. Proc. Natl Acad. Sci. USA. 1991;88:3314–3318. doi: 10.1073/pnas.88.8.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bourne H.R., Sanders D.A., McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 44.Hall A. Signal transduction through small GTPases—a tale of two GAPs. Cell. 1992;69:389–391. doi: 10.1016/0092-8674(92)90441-e. [DOI] [PubMed] [Google Scholar]
- 45.Hill C.S., Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 46.Satoh T., Uehara Y., Kaziro Y. Inhibition of interleukin 3 and granulocyte-macrophage colony-stimulating factor stimulated increase of active ras.GTP by herbimycin A, a specific inhibitor of tyrosine kinases. J. Biol. Chem. 1992;267:2537–2541. [PubMed] [Google Scholar]
- 47.Stokoe D., Macdonald S.G., Cadwallader K., Symons M., Hancock J.F. Activation of Raf as a result of recruitment to the plasma membrane. Science. 1994;264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- 48.Hall A. The cellular functions of small GTP-binding proteins. Science. 1990;249:635–640. doi: 10.1126/science.2116664. [DOI] [PubMed] [Google Scholar]
- 49.Yu X., Chen S., Potter O.L., Murthy S.M., Li J., Pulcini J.M., Ohashi N., Winata T., Everett E.T., Ingram D., et al. Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning. Bone. 2005;36:793–802. doi: 10.1016/j.bone.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 50.Leevers S.J., Paterson H.F., Marshall C.J. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 51.Hata K., Ikebe K., Wada M., Nokubi T. Osteoblast response to titanium regulates transcriptional activity of Runx2 through MAPK pathway. J. Biomed. Mater. Res. A. 2006;81:446–452. doi: 10.1002/jbm.a.31086. [DOI] [PubMed] [Google Scholar]
- 52.Hotchin N.A., Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J. Cell Biol. 1995;131:1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Higuchi C., Myoui A., Hashimoto N., Kuriyama K., Yoshioka K., Yoshikawa H., Itoh K. Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J. Bone Miner. Res. 2002;17:1785–1794. doi: 10.1359/jbmr.2002.17.10.1785. [DOI] [PubMed] [Google Scholar]
- 54.Raisz L.G. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J. Clin. Invest. 2005;115:3318–3325. doi: 10.1172/JCI27071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schindeler A., Ramachandran M., Godfrey C., Morse A., McDonald M., Mikulec K., Little D.G. Modeling bone morphogenetic protein and bisphosphonate combination therapy in wild-type and Nf1 haploinsufficient mice. J. Orthop. Res. 2008;26:65–74. doi: 10.1002/jor.20481. [DOI] [PubMed] [Google Scholar]
- 56.Schindeler A., Birke O., Yu N.Y., Morse A., Ruys A., Baldock P.A., Little D.G. Distal tibial fracture repair in a neurofibromatosis type 1-deficient mouse treated with recombinant bone morphogenetic protein and a bisphosphonate. J. Bone Joint Surg. Br. 2011;93:1134–1139. doi: 10.1302/0301-620X.93B8.25940. [DOI] [PubMed] [Google Scholar]
- 57.Kolanczyk M., Kuhnisch J., Kossler N., Osswald M., Stumpp S., Thurisch B., Kornak U., Mundlos S. Modelling neurofibromatosis type 1 tibial dysplasia and its treatment with lovastatin. BMC Med. 2008;6:21. doi: 10.1186/1741-7015-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gould M.C., Stephano J.L. Inactivation of Ca(2+) action potential channels by the MEK inhibitor PD98059. Exp. Cell Res. 2000;260:175–179. doi: 10.1006/excr.2000.5014. [DOI] [PubMed] [Google Scholar]
- 59.Sjogreen B., Wiklund P., Ekstrom P.A. Mitogen activated protein kinase inhibition by PD98059 blocks nerve growth factor stimulated axonal outgrowth from adult mouse dorsal root ganglia in vitro. Neuroscience. 2000;100:407–416. doi: 10.1016/s0306-4522(00)00278-5. [DOI] [PubMed] [Google Scholar]
- 60.Jacks T., Shih T.S., Schmitt E.M., Bronson R.T., Bernards A., Weinberg R.A. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 61.Liu F., Woitge H.W., Braut A., Kronenberg M.S., Lichtler A.C., Mina M., Kream B.E. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int. J. Dev. Biol. 2004;48:645–653. doi: 10.1387/ijdb.041816fl. [DOI] [PubMed] [Google Scholar]
- 62.Dacquin R., Starbuck M., Schinke T., Karsenty G. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev. Dyn. 2002;224:245–251. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- 63.Elefteriou F., Benson M.D., Sowa H., Starbuck M., Liu X., Ron D., Parada L.F., Karsenty G. ATF4 Mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006;4:441–451. doi: 10.1016/j.cmet.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salvarezza S.B., Lopez H.S., Masco D.H. The same cellular signaling pathways mediate survival in sensory neurons that switch their trophic requirements during development. J. Neurochem. 2003;85:1347–1358. doi: 10.1046/j.1471-4159.2003.01771.x. [DOI] [PubMed] [Google Scholar]
- 65.Watson K., Fan G.H. Macrophage inflammatory protein 2 inhibits beta-amyloid peptide (1-42)-mediated hippocampal neuronal apoptosis through activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling pathways. Mol. Pharmacol. 2005;67:757–765. doi: 10.1124/mol.104.004812. [DOI] [PubMed] [Google Scholar]
- 66.Meirelles Lda S., Nardi N.B. Murine marrow-derived mesenchymal stem cell: isolation, in vitro expansion, and characterization. Br. J. Haematol. 2003;123:702–711. doi: 10.1046/j.1365-2141.2003.04669.x. [DOI] [PubMed] [Google Scholar]
- 67.Hiatt K.K., Ingram D.A., Zhang Y., Bollag G., Clapp D.W. Neurofibromin GTPase-activating protein-related domains restore normal growth in Nf1−/− cells. J. Biol. Chem. 2001;276:7240–7245. doi: 10.1074/jbc.M009202200. [DOI] [PubMed] [Google Scholar]
- 68.Cheng J., Ye H., Liu Z., Xu C., Zhang Z., Liu Y., Sun Y. Platelet-derived growth factor-BB accelerates prostate cancer growth by promoting the proliferation of mesenchymal stem cells. J. Cell Biochem. 2013;114:1510–1518. doi: 10.1002/jcb.24492. [DOI] [PubMed] [Google Scholar]
- 69.Bergfeld S.A., DeClerck Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010;29:249–261. doi: 10.1007/s10555-010-9222-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.