

Abstract
We have demonstrated that scar formation after myocardial infarction (MI) is associated with an endogenous pool of CD44posCD45neg multipotential mesenchymal stem cells (MSC). MSC differentiate into fibroblasts secreting collagen that forms a scar and mature into myofibroblasts that express alpha smooth muscle actin (α-SMA) that stabilizes the scar. In the aging mouse, cardiac repair after MI is associated with impaired differentiation of MSC; MSC derived from aged hearts form dysfunctional fibroblasts that deposit less collagen in response to transforming growth factor beta-1 (TGF-β1) and poorly mature into myofibroblasts. We found in vitro that the defect in myofibroblast maturation can be remedied by AICAR, which activates non-canonical TGF-β signaling through AMP-activated protein kinase (AMPK). In the present study, we injected aged mice with AICAR and subjected them to 1h occlusion of the left anterior descending artery (LAD) and then reperfusion for up to 30 days. AICAR-dependent AMPK signaling led to mobilization of an endogenous CD44posCD45neg MSC and its differentiation towards fibroblasts and myofibroblasts in the infarct. This was accompanied by enhanced collagen deposition and collagen fiber maturation in the scar. The AICAR-treated group has demonstrated reduced adverse remodeling as indicated by improved apical end diastolic dimension but no changes in ejection fraction and cardiac output were observed. We concluded that these data indicate the novel, previously not described role of AMPK in the post-MI scar formation. These findings can potentially lead to a new therapeutic strategy for prevention of adverse remodeling in the aging heart.
Keywords: aging, MSC, AMPK, myocardial infarction, scar formation, fibroblast
1. Introduction
The elderly population is increasing, in part due to improvements in medical care. Because the number of older patients suffering from cardiovascular disease has increased and because older patients often survive MI but subsequently demonstrate adverse remodeling, the frequency of congestive heart failure has strikingly risen [1].
Our studies in the aging mouse model demonstrate that cardiac repair after experimental MI is impaired as early as ten months of age, with progressive impairment extending through 24 months [2, 3]. The size of the induced MI is not larger; the major pathological feature is a poorly organized scar with reduced fibroblast and myofibroblast content. In the aged heart participating myofibroblasts/fibroblasts deposit substantially less collagen in the injured area when compared to young hearts [3], leading to adverse remodeling.
We have previously demonstrated that cardiac repair after MI is associated with an endogenous pool of CD44posCD45neg multipotential MSC that rapidly proliferates within the infarct a few days after MI [4]. These cells differentiate into collagen secreting fibroblasts that form a scar and later mature into myofibroblasts expressing α-SMA that contract the scar and these processes are mediated by TGF-β1 signaling [5, 6]. Effective collagen deposition within a scar is a very specific and targeted process that allows efficient healing after MI [7]. Impairment or delay of these reparative fibrotic responses is associated with increased dilative remodeling. By contrast, increased fibrosis in a non-reparative setting is found interstitially in the left ventricle in cardiac hypertrophy and failure and is associated with cardiac dysfunction [8]. Recently we reported that fibroblasts derived from the aged murine heart are defective; they matured poorly into myofibroblasts, displayed reduced motility towards TGF-β1, and did not effectively increase expression of collagen type I in response to TGF-β1, all as a result of reduced expression of the TGF-β receptor I (TβRI) [9]. Interestingly, in vitro, we were able to rescue the aged myofibroblast maturation through AICAR activation of AMPK and its downstream kinase p38MAPK (that is, a non-canonical TGF-β signaling pathway) [9]. The role of AMPK in ischemia has been extensively studied by many groups [10-12]. It has been demonstrated that AMPK deletion has a detrimental effect on the ischemic heart [13]; AMPK in the ischemic heart maintains cardiomyocyte metabolism by promoting fatty acid oxidation and glycolysis [11, 14]. However AMPK serves also as a key mediator in non-metabolic processes; AMPK (or its activator, AICAR) promotes differentiation of embryonic stem cells [15, 16], adipose tissue-derived MSC [17] and endothelial progenitors [18].
Based on our previous work showing impaired healing after MI in older mice [2, 3] and defects in MSC/fibroblasts/myofibroblasts derived from aged hearts, as well as in vitro rescue of the defective phenotype via an AMPK dependent pathway [9], we hypothesized that in vivo activation of AMPK might mobilize fibroblast precursors and enhance scar formation in the aged injured heart. Because the attenuated healing capacity after MI was observed in middle aged mice (as early as 14 months of age) [2], we used 14 month old mice for the proof of concept. Mice were injected with AICAR or saline for 7 days starting 2 days before ischemia reperfusion injury (MI), and then animals were followed for up to 30 days. Our data indicate that AICAR-dependent AMPK signaling leads to greater activation of endogenous CD44posCD45neg fibroblast precursors and enhances their differentiation towards fibroblasts and their maturation into myofibroblasts in the infarcted heart. These findings were accompanied by a reduced adverse remodeling in the AICAR-treated group (evaluated by echocardiography) with significantly improved post-ischemic systolic and diastolic function (evaluated by Doppler).
2. Material and methods
2.1. Reagents
AICAR: 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (Toronto Research Chemicals, Ontario, Canada)
2.2. Animals
14 month old male C57BL/6 mice were purchased from the National Institute of Aging and allowed one week to recover from shipping. Mice were anesthetized, intubated, and underwent thoracotomy. A ligature in a noose was placed around the LAD and the ends of the suture exteriorized to allow for closed chest induction of myocardial ischemia. Mice were allowed to recover for one week. Mice were then reanesthetized with 1% isoflurane and attached to an ECG board. The LAD was occluded for 1h with elevations of the ST segments monitored after which the tension on the suture was released and reperfusion occurred. This closed chest 1h ischemia followed by reperfusion reproducibly produces a modest infarction in mice [19]. Mice were then followed from 5 days to 4 weeks. Mice were injected with AICAR intraperitoneally (0.5mg/g of body weight) every day for 7 days starting 2 days before coronary occlusion. An equal volume of saline was used to inject control animals. Animals were used in agreement with the Baylor College of Medicine Animal Care and Research Advisory Committee guidelines.
2.3. Cell isolation
5 days after MI, hearts were excised, cut into 1 mm3 pieces and digested with Liberase TH (Roche Diagnostics, Indianapolis, IN, USA), incubated in a 37°C shaking water bath with regular trituration by pipet to obtain a single cell suspension. Afterwards, cells were centrifuged at 250×g for 5 min. The cell pellet was washed and then suspended in PBS [4]. The isolated cell population contains only non-myocytes.
2.4. Flow cytometry
Cells were incubated with antibodies to CD45-PE (553081, BD Pharmingen, San Diego, CA, USA) and CD44-biotin (731958, Beckman Coulter, Brea, CA, USA) or the appropriate isotype controls followed by streptavidin-PE-Cy5 (Beckman Coulter) and external antigens were analyzed. For internal antigens samples were fixed in 2% paraformaldehyde with 0.1% saponin, washed and then incubated with antibodies to collagen type I (600-401-103-05, Rockland Immunochemicals, Gilbertsville, PA), α-SMA-FITC (F3777, Sigma, St. Louis, MO, USA) or an IgG control. Then the secondary antibody conjugated to DyLight-488 (Jackson Immunoresearch, West Grove, PA, USA) was applied and cells were analyzed on a Cell Lab Quanta SC flow cytometer (Beckman Coulter) using the Quanta Analysis software or FlowJo (v.7.2.5, Tree Star Inc., Ashland, OR, USA). 50 nM calcein (Invitrogen, Carlsbad, CA, USA) was added to non-fixed samples to determine cell viability. n=4, 5 for saline and AICAR treated animals respectively.
2.5. Whole heart lysate
Hearts were snap frozen in liquid nitrogen then ground under liquid nitrogen to a fine powder. The tissue powder was resuspended in RIPA lysis buffer (Thermo Scientific, Rockford, IL, USA) supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific). Lysates were then frozen, thawed and centrifuged at 10,000 × g for 5 minutes. Supernatant was aliquoted and stored at −80°C until further analysis.
2.6. Western blot analysis
50 μg of protein were mixed with reducing Laemmli sample buffer and separated on 4%–15% Tris-HCl gels (Bio-Rad, Hercules, CA, USA). Proteins were then transferred to nitrocellulose membranes (Bio-Rad). The membranes were incubated with antibodies diluted according to the manufacturer's protocol. SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) was used to detect signals. Antibodies: anti-pAMPK (2535, Cell Signaling, Danvers, MA, USA) and AMPK (2603, Cell Signaling). Densitometry analysis was performed using ImageJ software (NIH, Bethesda, MD, USA). n=4.
2.7. Ultrasound procedure
2D echo guided M-mode images were acquired using a Vevo 770 RMV-707B 30-MHz probe (VisualSonics, Toronto, Canada). For the mitral inflow and aortic outflow Doppler measurements a Doppler signal processing workstation (DSPW, Indus Instruments, Houston, TX) was employed using a 10-MHz (1 mm diameter) pulsed Doppler probe as previously described [20]. Measurements of left ventricle function and structure were performed prior to any surgery (pre) and then 1, 2, 4 weeks after MI procedures. The following parameters were reported as indicators of function and remodeling: left ventricular internal diameter (diastole) LVID,d, ΔLVID,d = LVID,d(4week)- LVID,d(1week), E peak velocity, isovolumic contraction time (IVCT), isovolumic relaxation time (IVRT), peak aortic flow velocity (PAFV), mean aortic acceleration, left atrium (LA) dimensions and Tei index = (IVCT+IVRT)/ejection time, apical end diastolic and end systolic dimensions were performed as described [21]. n=9, 10 for saline and AICAR treated animals respectively except for apical diastolic and systolic dimensions where n=5 for both group.
2.8. Immunofluorescence staining
Tissue was fixed in zinc-Tris fixative (modified from Beckstead et al) [22], paraffin embedded and sectioned at a 5 μm thickness. Sections were deparaffinized, permeabilized in PBS containing 1% Triton X-100, blocked in 1% BSA and incubated with following antibodies overnight: anti-CD44-PE (731959, Beckman Coulter), anti-procollagen type I (sc-25973, Santa Cruz Biotechnology, Dallas, TX, USA), anti-αSMA-FITC (F3777, Sigma), anti-LOX (L4669, Sigma), anti-wheat germ agglutinin (W849, Invitrogen). Subsequently sections were washed and a secondary reagent was applied if applicable such as anti-goat or anti-rabbit IgG DyLight-488 conjugated (Jackson ImmunoResearch, West Grove, PA, USA) or anti-rabbit IgG DyLight-549 conjugated (Jackson ImmunoResearch). n=4, 5 for saline and AICAR treated animals respectively.
2.9. Immunohistochemistry (IHC)
IHC was performed on 5 μm paraffin heart sections using IHC kit (Vector Laboratories, Burlingame, CA, USA). Following antibodies were used: anti-pAMPK (2535, Cell Signaling), p-p38MAPK (4511, Cell Signaling and WT-1 (sc-192, Santa Cruz Biotechnology). Nuclei were counterstained with hematoxylin. n=4, 5 for saline and AICAR treated animals respectively.
2.10. Polarized light assessment of fibrillar collagen
The hearts were perfused first with cardioplegic solution and thereafter with a zinc buffer [22] for four hours before dehydration through alcohols to xylene followed by embedding in paraffin. Sections were cut at 5 μm, rehydrated and stained with picrosirius red (Sigma) [23] with a 0.01N HCl wash [24]. They were then dehydrated and mounted in Cytoseal-XYL (Richard-Allan Scientific, Kalamazoo, MI, USA). The sections were examined under brightfield illumination with an Olympus CKX41 or under an Olympus AX70 microscope equipped with filters for linear polarization [24]. The polarizer was rotated to achieve extinction with the analyzer, showing a black background and the birefringency of the picrosirius red-stained collagen. Images were obtained with 10× and 40× objective lenses, recorded with an Olympus Q-color 3 digital camera, and analyzed using ImageJ software. The RGB images were interrogated using the color threshold function for their hue spectrum (color histograms). We then converted the RGB image to an HSB (hue, saturation, brightness) stack and obtained histograms of the hue spectrum that were quantified for each hue after setting the spectral limits for red, orange, yellow, and green. n=4, 5 for saline and AICAR treated animals respectively.
2.11. Infarct size
Animals underwent 1 hour ischemia as described above. Twenty four hours later 1% Evans blue was perfused into the aorta and coronary arteries, and then animals were sacrificed. Hearts were sectioned into 1 mm sections and then incubated in 1.5% triphenyltetrazolium chloride (Sigma) for 30 minutes. Sections were placed in formalin and photographs were taken 24 hours later. Viable myocardium was red, the infarct site appears pale white and area at risk (AAR) was blue [25]. The area of infarction for each slice was determined by image analysis (ImageJ). Infarct size was determined by applying the following formulas: infarct weight = (A1 × Wt1) + (A2 × Wt2) + (A3 × Wt3) + (A4 × Wt4), where A is a percent area of infarction by planimetry and Wt is a weight of each section. Percentage of infarcted LV = (infarct weight/LV weight) × 100. AAR was determined using planimetry (Image J). n=4 for infarct size studies and n=3 for AAR evaluation.
2.11. Statistical analysis
Results are presented as mean ± SE. For statistical analysis an unpaired Student's t-test with Welch's correction or One Way ANOVA with Tukey's post-test was applied. Differences were considered statistically significant if p<0.05.
3. Results
3.1. In vivo AICAR treatment activates AMPK in cells populating the infarcted area
Mice were injected daily with AICAR or saline for 7 days. Because we hypothesized that AICAR treatment would mobilize endogenous progenitors, AICAR injections started 2 days before occlusion. 5 days after the MI, hearts were excised and analyzed. Activation of AMPK was assessed by the level of Thr172 phosphorylation. Thr172 is located within the activation loop of the AMPK catalytic subunit α [26]. AMPK in the whole heart lysate was activated in response to AICAR treatment, as we found a 2 fold increase in AMPK phosphorylation in the AICAR-treated group but the difference was not statistically significant (p=0.16) (Fig. 1A). However, immunohistochemical analysis of non-myocyte cells populating the infarcted area revealed a statistically significant increase of phosphorylated AMPK in the AICAR-treated group compared to saline-treated mice (Fig. 1B). In other cell type (cardiomyocytes) AICAR treatment has been shown to increase AMPK phosphorylation for up to 24 hours in [27] and in our experiment hearts were excised 24 hours after the last AICAR or saline injection.
Figure 1.
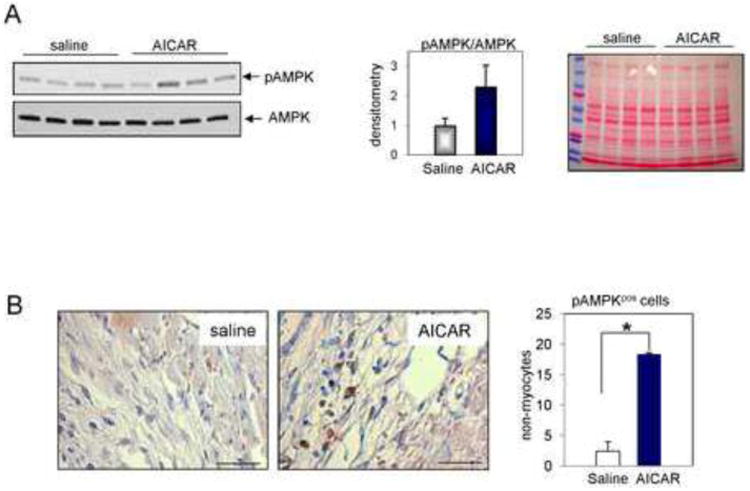
AICAR-dependent activation of AMPK. A. Phosphorylation of AMPK was doubled in hearts isolated from AICAR-treated mice. Animals were treated with AICAR for 7 days. Hearts were isolated 24 h after last AICAR injection. The middle panel shows densitometry results and the right panel shows Ponceau staining on the gel. n=4. B. Non-myocytes populating the infarcted area express activated (phosphorylated) AMPK 5 days post occlusion in AICAR-treated animals as evaluated by IHC staining. Cells were counterstained with hematoxylin. Scale bar = 50 μm. n=4, 5 for saline and AICAR treated animals respectively. * denotes p<0.05. Student's t-test with Welch's correction was used for analysis.
3.2. AICAR mobilizes a pool of endogenous fibroblast precursors
We recently reported that in the young infarcted heart 2 days after injury the number of CD44posCD45neg mesenchymal precursors was elevated. The number of these cells peaks at 3 days post-MI and accounts for ∼5% of all viable non-myocyte cells [4]. To see if AMPK activation has an effect on these cells, infarcted mice were injected daily with AICAR or saline 2 days before occlusion and then throughout the reperfusion, which lasted 5 days. Then, non-myocyte cells were isolated and analyzed by flow cytometry. Our data indicated that AICAR treatment mobilized CD44posCD45neg cells in the ischemic heart (Fig. 2A, left and right panels). The number of CD44posCD45neg cells was doubled in AICAR-stimulated hearts compared to saline-treated controls. The number of CD44posCD45negcollagenpos fibroblasts (Fig. 2B) was increased in the AICAR-treated heart by ∼3 fold when compared to saline-treated controls and the immunofluorescence staining revealed an accumulation of CD44posprocollagen type Ipos cells in the infarct of AICAR-treated hearts as compared to controls (Fig. 2C). Note that almost all cells in the infarcted area in the AICAR-treated heart are double positive. The density of cells expressing procollagen in non-infarcted areas was not affected by AICAR treatment (Fig. 2D).
Figure 2.
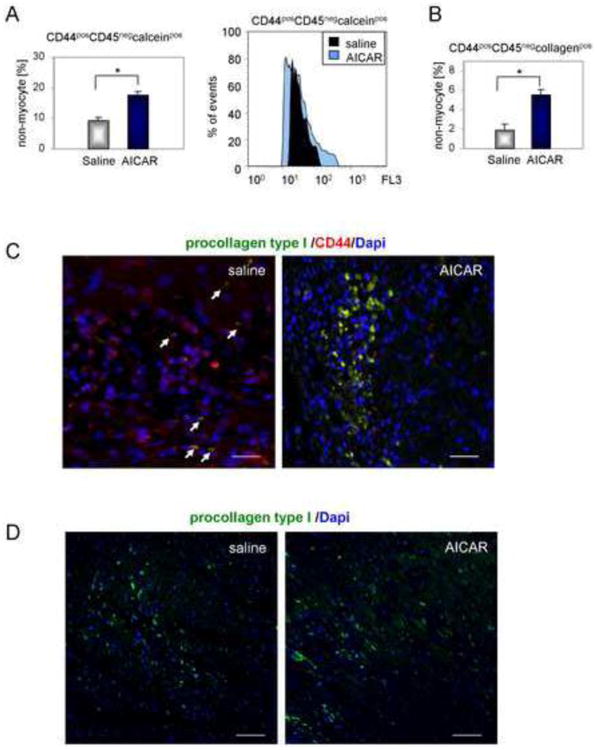
AICAR activates primitive endogenous precursors in the ischemic heart. Cells were isolated 5 days after occlusion. Daily AICAR or saline injections were given for 7 days, starting 2 days before occlusion. A. Cardiac CD44posCD45neg viable cell number is elevated by AICAR treatment. Right panel shows representative overlaid histograms. B. AMPK activation enhances fibroblast differentiation as the number of CD44posCD45negcollagen type Ipos cells was increased in hearts isolated from AICAR-treated animals. C. Immunofluorescence staining showing increased accumulation of CD44posprocollagen type Ipos cells in the infarcted area of AICAR-treated mouse. Arrows indicate double stained cells. Scale bar = 50μm. D. AICAR treatment did not affect the number of procollagen type Ipos cells in non-infarcted areas. Scale bar = 200μm. n=4, 5 for saline and AICAR treated animals respectively. * denotes p<0.05. Student's t-test with Welch's correction was used for analysis.
Although the evidence suggests that fibroblasts of hematopoietic origin are not critical to repair after MI [28, 29], AMPK may affect transendothelial migration [30, 31]. For this reason we evaluated the number of cardiac CD45pos cells or their differentiation into fibroblasts and myofibroblasts in AICAR treated animals. We found no significant difference (Supplemental Figure 1), suggesting that endogenous precursors but not blood-derived cells participate in AICAR-dependent enhanced repair after MI.
Recent studies have demonstrated that epicardium can be activated after MI and that epicardial-derived cells can populate the damaged area [32]. We analyzed hearts excised 5 days post-occlusion for the presence of Wilm's Tumor 1 antigen (WT1), an epicardial marker. We found that only a small percentage of cells expressing WT1 were present in the epicardium, the subepicardium and the infarcted area; however, the number of WT1pos cells within the infarcted area was increased in AICAR-treated hearts (Supplemental Figure 2). As TGF-β1 stimulates epicardial-to-mesenchymal transition (EMT) [33] and AICAR augments TGFβ1 signaling [9], the activated TGFβ1/AMPK pathway may account for the elevated EMT in AICAR-treated hearts. Because of the small portion of WT1pos cells in the infracted area they might be contributing to a minor extent to fibroblast formation in the aging heart as suggested by others [32].
3.3. Improved scar formation in the AICAR-treated post-ischemic heart
The collagen content of the infarct scar was examined using picrosirius red staining. Brightfield images show the greater collagen content in the scar of AICAR-treated animals compared to that of the saline control (Fig.3). Quantified collagen content in the scar area was computed using ImageJ and plotted in a graph shown on right panel of Fig.3.
Figure 3.
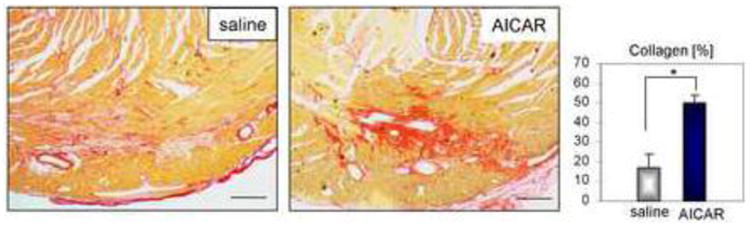
AICAR advances collagen deposition in the scar of the aged ischemic heart. A. Representative image of picrosirius red staining of collagen network 5 days after occlusion. Right panel illustrates enhanced collagen deposition in the scar from AICAR-treated animals computed by ImageJ. Scale bar = 200μm. n=4, 5 for saline and AICAR treated animals respectively. * denotes p<0.05. Student's t-test with Welch's correction was used for analysis.
To characterize the collagen structure further, we viewed the picrosirius red stained sections under polarized light (see Materials and methods). This reveals the thickness of the collagen fibrils, with the thinnest ones appearing green, intermediates yellow to orange, and the thickest ones red [34]. The images show not only the increased collagen content in the scar (Fig. 4A, B), but also that the fibrils are thicker in the AICAR-treated animals than in the saline controls (Fig. 4B). The shift in the spectrum of the collagen birefringency can be seen in the color histograms (Fig. 4B) and this is quantified in the graph (Fig. 4B right panel). There are more small (green) fibrils in the saline control, and more thick (red) fibrils in the AICAR-treated animals, indicating that AICAR-treated mice make a better scar.
Figure 4.
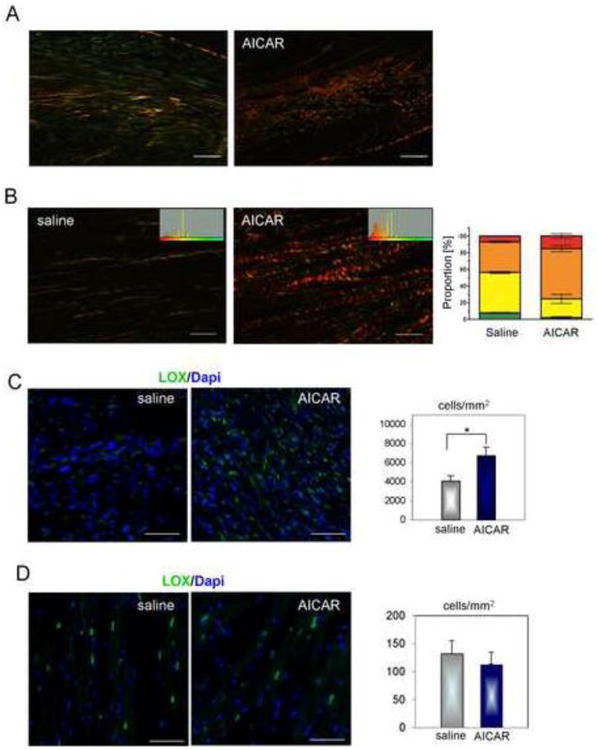
AICAR improves scar formation in the aged ischemic heart via enhanced collagen fibril maturation. Representative images 5 days post occlusion are shown. A. Lower magnification image shows mature collagen fibrils in AICAR-treated mice. Scale bar = 200 μm. B. Higher magnification images were analyzed for the degree of collagen fiber maturation. Green, yellow, orange and red correspond to fibril maturation where green corresponds to the thinnest and least mature and red the most mature. Changes in color where recorded in a histogram (inside window) and used to calculate proportion of each fiber type in the scar of saline and AICAR-treated mice (right panel graph). Scale bar = 50 μm. C. Representative images of LOX immunofluorescence staining in the infarcted area (left panel). Right panel shows LOX expression evaluated as a number of positive cells per area within infarct. D. Representative images of LOX immunofluorescence staining in the non-infarcted area (left panel). Right panel shows LOX expression evaluated as a number of positive cells per area Scale bar = 50 μm. n=4, 5 for saline and AICAR treated animals respectively. * denotes p<0.05. Student's t-test with Welch's correction was used for analysis.
To examine one possible reason for the increased collagen fibril thickness, we investigated the ability of cells in the infarct to cross-link collagen by production of lysyl oxidase (LOX) [35]. It can be seen in Fig. 4C that there are many more cells that are LOXpos in the AICAR-treated scar. As expected, the number of LOX-expressing cells in non-infarcted areas was much lower and was not significantly different between saline and AICAR-treated hearts (Fig. 4D).
3.4. Increased myofibroblast content in AICAR-treated post-ischemic heart
Scar density and mechanical effectiveness is also influenced by scar contraction. We have previously shown that the in vitro rescue of defective aged fibroblast maturation occurred via a non-canonical TGFβ/AMPK pathway [9] that involves activation of p38MAPK. Our current in vivo data support these findings. The number of CD44posα-SMApos myofibroblasts was increased in AICAR-treated hearts by 11 fold when compared to the saline treated group (Fig. 5A) as evaluated by flow cytometry. Immunofluorescence analysis of the infarct in heart isolated from animals treated with AICAR also indicated that the damaged area was more densely populated with CD44posα-SMApos myofibroblasts (Fig. 5B) and with cells expressing phosphorylated/activated p-p38MAPK (Fig. 5C) suggesting an enhanced myofibroblast maturation due to in vivo AMPK pathway activation. α-SMA expression in cells residing in areas outside of the infarct was mostly associated with blood vessels regardless of treatment (Fig. 5D).
Figure 5.
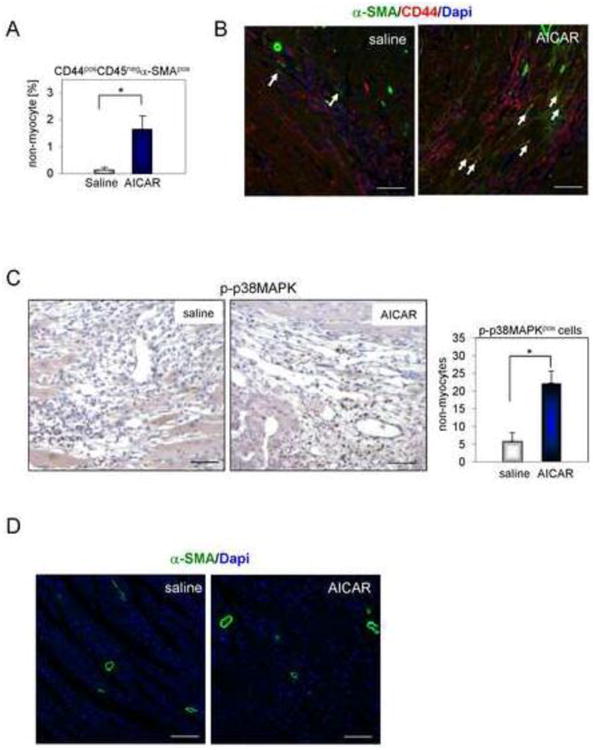
AMPK activation promotes myofibroblast maturation in the aged heart. A. Increased number of CD44posCD45neg non-myocytes expressing α-SMA in the hearts from AICAR-treated animals. B. Immunofluorescence staining of CD44posα-SMApos cells in the infarcted area. Arrows indicate double stained cells. Scale bar = 50 μm. C. Representative images of phosphorylated p38MAPK (p-p38MAPK) IHC staining in the infarcted area of saline and AICAR-treated hearts. Cells were counterstained with hematoxylin. Number of p-p38MAPKpos cells per total number of cells populating infarcted area was plotted in a graph shown on a right panel. Scale bar = 50 μm. * denotes p<0.05. Student's t-test with Welch's correction was used for statistical analysis. D. Immunofluorescence staining of α-SMApos cells in the non-infarcted area. Scale bar = 100 μm. n=4, 5 for saline and AICAR treated animals respectively.
3.5. Effect of in vivo AMPK activation on post-ischemic heart function
Next, we evaluated the role of AMPK activation in post-MI heart function. Again, mice were injected with AICAR or saline for 7 days (as above), but followed for 4 weeks. Functional studies were performed before any manipulation (pre) to establish the baseline and then after MI.
The left ventricle (LV) internal dimensions were not significantly different between saline and AICAR at one week post infarct (Supplemental Table 1A). Over the next three weeks, the saline treated mice increased their left ventricular internal diameter in diastole (LVID,d) while the AICAR treated had no change in dimension (Fig. 6A, Supplemental Table 1A). We have reported previously that the aged heart displayed adverse remodeling after MI [3]. These findings suggested that the AICAR treatment prevented the post infarction adverse remodeling in older mice. Consistent with the effects on remodeling, AICAR preserved diastolic and end systolic dimensions at the apical (site of infarct) post infarction (Fig. 6B) whereas diastolic dimension was significantly increased in saline treated group (end systolic dimension at the apex appeared to be increased in saline treated animals as well but the statistical significance was p=0.16).
Figure 6.
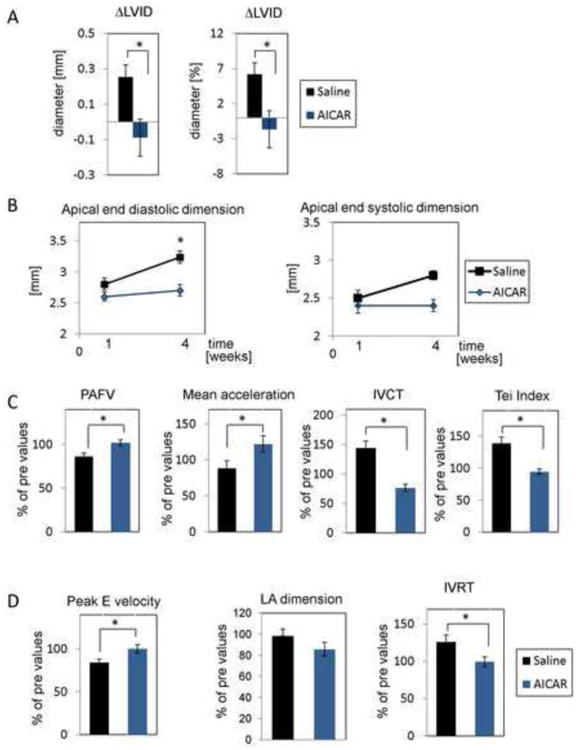
The effect of AMPK activation on the aged murine heart. Daily AICAR or saline injections were given for 7 days starting 2 days before occlusion. A. AICAR-dependent AMPK activation prevents adverse remodeling after MI. Parameters are the difference between 1 and 4 weeks after MI in saline and AICAR treated mice (left graph change in mm, right graph change in %). n=9, 10 for saline and AICAR treated animals respectively. B. Apical end diastolic (left panel) and end systolic (right panel) dimensions in AICAR and saline treated mice. n=5 for both treatment groups. One –way ANOVA with Tukey's post-test correction was used to evaluate statistical differences. C. Systolic and D. diastolic dysfunctions were prevented by AICAR treatment. These data represent changes in the heart function at four weeks post-MI compared to pre values. For A, C and D Student's t-test was used for statistical analysis. * denotes p<0.05.
Doppler parameters were used to reflect global cardiac function. In the older saline treated mice, there were decrements in the systolic and diastolic function after 4 weeks post occlusion (Fig. 6C, D, and Supplemental Table 1B). Peak Aortic Flow Velocity (PAFV) and mean acceleration decreased (Fig. 6C left and middle panel) about 10% at four week post infarct. For diastolic function Peak E decreased and isovolumic relaxation time (IVRT) (Fig. 6D left and right panel) was prolonged at four week, consistent with worsened filling. By contrast, the AICAR treated mice show preserved Peak E velocity, PAFV and Tei Index at 4 weeks post infarct (Fig. 6C, D). To be certain the indices of left ventricular filling in the AICAR-treated mice did not result from paradoxical increases in atrial filling pressure, we measured left atrial (LA) dimensions in both control and AICAR-treated mice (Figure 6D middle panel). There were no significant changes seen in the left atrial dimensions in either group over the time course of these experiments. Thus, the differences seen are compatible with increased diastolic dysfunction in the control group and preserved diastolic function of the AICAR-treated group. Representative 2D M-Mode echo images from saline and AICAR-treated mice are shown in Supplemental Figure 3. The left panel shows images acquired 1 week post-MI and the right panel represents images obtained 4 weeks post-MI. No dilation was observed in AICAR-treated hearts.
It is important to emphasize that these experiments were done with the intention of evaluating the cell biology and pathophysiology of cardiac repair in the aging animal and the potential remedial effects of stimulating non-canonical TGFβ signaling. In previous experiments, we have described that myocardial infarction in older animals has a very high mortality rate [2]; therefore, we elected to make small infarcts to allow us sufficient numbers of experimental survivors in each group in a highly labile model. The infarct size in both groups was the same modest in size (Supplemental Figure 4B, discussed below) and, indeed, there was no difference in the ejection fraction or cardiac output (Supplemental Table 1A) in the two groups at four weeks. Thus, both groups were able to compensate for the injury despite their age. Nevertheless, utilizing standard evaluators of systolic and diastolic function, AICAR prevented the functional decrements seen in the saline treated mice at four weeks, suggesting that, even in a compensated infarcted heart, there is potential value in intervention to prevent adverse remodeling. It is possible that, over time, the regional dysfunction and the inherent decrease in ventricular efficiency from adverse remodeling might result in progression of heart failure even with a small infarct. It is to be noted that, in aging patients, congestive heart failure frequently appears at time remote from the initial infarction which apparent recovery has occurred (reviewed in [36]).
To further evaluate adverse remodeling, heart sections (4 weeks post-MI) were stained with picrosirius red (Fig.7A). Because at this time point analyzed sections contain not only collagen that forms scar but also collagen contributing to interstitial fibrosis in contrast to the results presented in Fig. 3 showing exclusively scar-forming collagen, we have chosen 3 sections per each heart separated by a 50-60 μm distance with the highest collagen content for analysis. We found that saline treated mice had a 3.5 fold increase in collagen content when compared to AICAR-treated mice and that the difference was statistically significant (Fig. 7A right panel). Cardiomyocyte hypertrophy (as another useful tool to analyze adverse remodeling) was evaluated by the mean cross-sectional area of left ventricular cardiomyocytes in the infarct border zone and non-infarcted myocardium 4 weeks post-MI (Fig. 7B). We observed that cardiomyocyte size in saline-treated hearts when compared to AICAR-treated mice was increased by 15.5% in the remote myocardium suggesting that AICAR treatment may reduce cardiomyocyte hypertrophy as reported by others [37] however the observed difference was not statistically significant.
Figure 7.
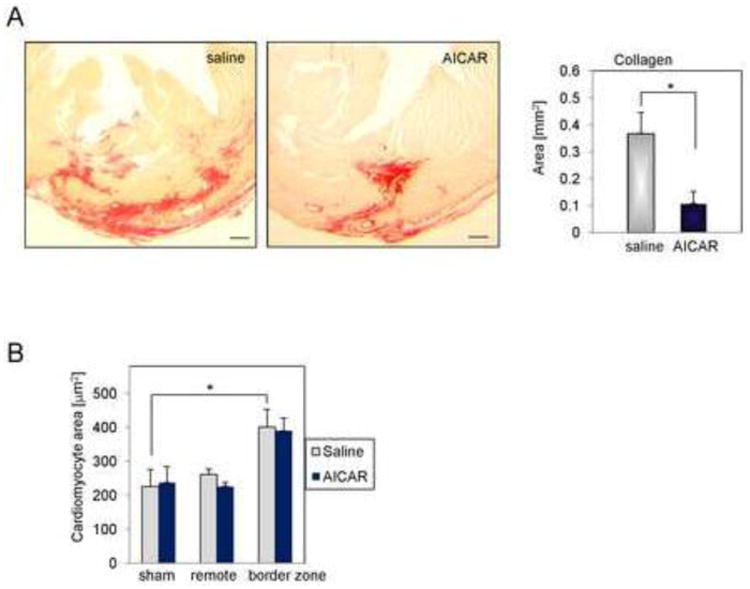
AICAR treatment reduces post-MI adverse remodeling. A. Representative images of picrosirius red staining of collagens in heart sections 4 weeks post-MI. Right panel illustrates elevated collagen deposition per area as computed by ImageJ. Scale bar = 200μm. * denotes p<0.05. Student's t-test with Welch's correction was used for analysis. B. Cardiomyocyte hypertrophy was evaluated by wheat germ agglutinin staining. Cell area was computed by analyzing cross-sectioned area of the heart using ImageJ software. At least 100 cells per heart section and 2 sections per mouse were analyzed. n=4, 5 for saline and AICAR treated animals respectively. 3 sham animals were used per treatment group. * denotes p<0.05. One –way ANOVA with Tukey's post-test correction was used to evaluate statistical differences.
Heart weight to body weight ratios were not changed by AICAR treatment as shown in Supplemental Figure 4A and importantly, AICAR-treatment did not affect the infarct size at 24 hours reperfusion (Supplemental Figure 4B left panel) nor was the area at risk compared to infarct size statistically different in both groups (Supplemental Figure 4B right panel). Total survival rate was the same for both groups (95.1%). The animals that died did so after implantation of a ligature before saline or AICAR was injected (see Material and methods section). 2/41 of saline-treated and 2/41 of AICAR-treated animals died after occlusion.
4. Discussion
Here we present new data showing that enhancement of AMPK activation improves post-MI cardiac function in the aged heart via promoting scar formation (see schema Fig. 8). This effect was accomplished by counteracting the defective responses to TGF-β by cardiac (myo)fibroblast. Not only did the fibroblasts deposit more collagen in the scar (Fig.3) which was comprised of more mature fibrils (Fig. 4A, B), but they also expressed more α-SMA (Fig. 5A, B) with which they contract the scar.
Figure 8.
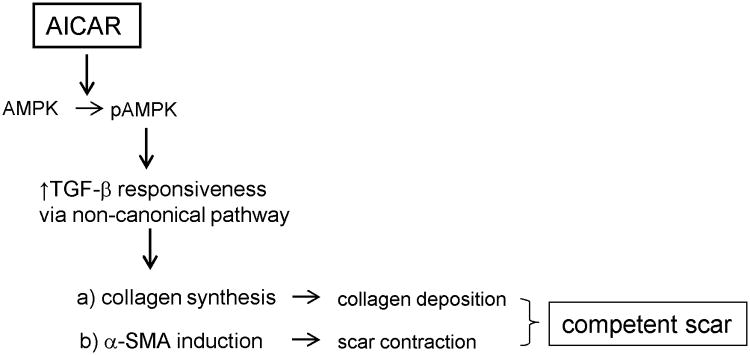
A proposed overview of AMPK-dependent regulation of post-MI scar formation in the aged heart. AICAR treatment in the aged heart activates AMPK. AMPK phosphorylation increases TGF-β responsiveness via activation of a non-canonical TGF-β pathway. Enhanced TGF-β signaling results in increased collagen and α-SMA expressions. Elevated collagen deposition and scar contraction leads to the formation of a competent scar that improves postishemic heart function and prevents adverse remodeling.
AMPK activity is stimulated via various pathways involving stress (ischemic, oxidative, hypoglycemic), exercise, hormones (adiponectin, leptin) and pharmacological agents (AICAR, metformin, A769662) [38]. AMPK's role in post-ischemic heart protection is quite complex. AMPK is activated due to an increased AMP:ATP ratio, which occurs during an ischemic event. In cardiomyocytes, AMPK switches metabolism to preserve energy [10] and reduces cardiac hypertrophy by targeting protein synthesis [39]. It has been demonstrated that reduced AMPK expression in cardiomyocytes is damaging to the ischemic heart because of the reduction of glucose uptake and ATP homeostasis, and increased necrosis and apoptosis after injury [10]. AMPK cardioprotective properties are not only associated with cardiomyocytes since it has been demonstrated that AMPK also promotes differentiation of endothelial progenitor cells and vasculogenesis [18], decreases recruitment of monocytes to endothelium and therefore lowers inflammation [31] and as we demonstrated targets fibroblast progenitors.
As the aging heart displays difficulties in healing after MI, it has been reported that the infarct size is actually reduced with aging [2]. We evaluated the infarct size of AICAR and saline-treated animals. We found that infarct size in aged animals was relatively small (∼12%) (as previously reported [2]) and AICAR treatment did not alter infarct size or area at risk (Supplemental Figure 4B). Despite the fact that ischemia itself markedly activates AMPK, it has been demonstrated in isolated reperfused hearts that AICAR reduces infarct size in young animals [40-42]. In this study the role of AICAR does not appear to relate to myocardial infarction size; moreover, there is no apparent AICAR effect seen in the first week of infarction (AICAR treatment ceased 5 days post-MI). Thus, the AICAR effect appears to relate to the facilitation of an effective mechanical scar formation through action on non-myocyte fibroblast precursors.
The heart responds to injury by activating repair mechanisms [43]. In one aspect of this repair, the number of MSC/fibroblast precursors increases and within a few days after MI these cells differentiate into fibroblasts and mature into myofibroblasts [4]. We have shown in Fig. 1B, 2C, 4C, and 5B a significant number of non-myocytes populating the infarcted area 5 days post-occlusion-reperfusion and indicated that these cells differentiate into (myo)fibroblasts. Post-occlusion CD44posCD45neg infiltrates in the infarcted area has been reported by Carlson et al. [4] that peak at day 3 post-MI and then differentiate into CD44posfibroblast and CD44posmyofibroblast by day 5 and 7 post-occlusion reperfusion respectively [4]. Although it has been shown that neutrophils and macrophages also infiltrate infarcted area, in the murine heart it is a robust but transient event and by day 5 post-occlusion (the time frame we mostly studied) the content of inflammatory cells most likely was reduced as indicated by the timecourse described by Dewald et al [44].
We recently reported that aged cardiac MSC were characterized by defective fibroblast differentiation and function [9], linked to reduced TβRI expression in MSC and consequent impairment of the TGF-β canonical pathway and downstream induction of α-SMA in myofibroblasts. The MSC defect could be reduced by stimulation of a non-canonical pathway [9] and led to the present study. In our in vivo study, AICAR treatment resulted in doubling the number of fibroblast precursors (CD44posCD45neg) in the ischemic area (Fig. 2A). Differentiation of precursors into fibroblasts and myofibroblasts in a setting of post-MI recovery in the aged heart has been shown to be reduced compared to what we observed in the young hearts [3]. At 5 days post-MI (peak for fibroblast differentiation) the CD44posCD45neg fibroblast and myofibroblast content in saline-treated mice was equal or lower to what we observed in sham-operated young animals [4]. However AICAR treatment increased the number of these cells to the level observed in young animals 5-7 days post-MI (compare Carlson et al. [4] and Fig. 2B, 5A), suggesting enhanced differentiation.
TGF-β signaling is crucial for scar contraction and collagen deposition [6, 43, 45-48]. In the aged murine heart, we found that collagen content in the cardiac scar is markedly attenuated in aged mouse infarcts, resulting in formation of a scar that contains loose connective tissue [3]. However, cells within the infarct site in hearts treated with AICAR expressed more LOX, an enzyme essential for cross-linking of collagens (Fig. 5C). It has been demonstrated that both LOX transcript level and enzymatic activity and α-SMA expression are controlled by TGF-β [6, 49]. Thus AICAR activation of a non-canonical TGF-β/AMPK pathway may promote transcriptional regulation of other TGF-β-sensitive genes. As we have demonstrated in vivo increased phosphorylation of p38MAPK in AICAR-treated hearts that were specifically targeted to the infarcted area (Fig. 5C), we further confirmed the importance of AMPK → p38MAPK → SMA pathway that was previously characterized in vitro [9]. The functional association between AMPK and p38MAPK activities has been characterized by others in cardiomyocytes [50]. Furthermore in cardiomyocytes, the AMPK/p38MAPK axis depends on TGF-β signaling similar to what we described previously in vitro in myofibroblasts [9].
AICAR's cardioprotective role (other than reported in this manuscript), has been described by others. It has been demonstrated that AICAR reduces migration and proliferation of vascular smooth muscle cells following balloon injury [51], inhibits Na+/H+ exchanger during reperfusion [52] and may inhibit ceramide accumulation thus preventing fatty acid induced apoptosis [53]. AICAR also reduces obesity related inflammation, improves insulin sensitivity [54] and increases lipolysis [55] that is also beneficial for the heart. Although AICAR has targets other than AMPK, the use of other activators also has been problematic due to specificity issues [56, 57]. We recognize that this study has limitations and we intend to follow up with more detailed analyses using different AMPK activators and different times of application such as during occlusion or reperfusion. However, AICAR preconditioning prior to ischemia (similar to what we reported) has been shown to be beneficial and prevented post-ischemic leukocyte adhesion [58]. The purpose of the current study was to establish if AICAR mobilizes endogenous cardiac CD44pos precursors and improves scar formation. Our data suggest that AICAR targets CD44pos fibroblasts precursors and enhances their differentiation. It has been demonstrated that the CD44 transmembrane protein is essential for ischemia-dependent AMPK activation [59], suggesting that perhaps two signaling pathways (ischemia and activation by pharmacological agent) are necessary to achieve optimal level of AMPK activity in the older heart especially that reduced AMPK activation was observed in aged heart [60] and cardiac fibroblasts [9].
In a recently reported large clinical trial, investigators found that AICAR therapy resulted only in mild side effects such as increased uric acid and hyperuricemia [61] however AICAR application did not reduce mortality, stroke and left ventricular dysfunction after coronary artery bypass graft surgery. Our data presented here indicate that different applications for AICAR therapy such as improvement of post-MI scar formation may prove more beneficial.
5. Conclusions
In summary, we present new evidence that short-term AICAR-dependent AMPK activation in the aged ischemic heart improves scar formation and thereby results in better systolic and diastolic function and prevents adverse post-ischemic remodeling. We attribute the beneficial role of AICAR treatment to increased differentiation of MSC into fibroblasts and their subsequent maturation into functional myofibroblasts. After further study, these findings may suggest a potential mechanism by which AMPK activation in the aged ischemic heart may be developed into a therapeutic strategy for older patients suffering from MI.
Supplementary Material
Highlights.
In the old murine heart AICAR activates AMPK in cells populating the infarct area
Activated AMPK mobilizes a pool of endogenous CD44posCD45neg fibroblast precursors
AMPK activation increases (myo)fibroblast number in the infarcted area
Activated AMPK enhances collagen maturation and improves scar formation
AICAR improves post-ischemic heart function and prevents adverse remodeling
Acknowledgments
We would like to thank Dorellyn B. Lee, Hao Zhang, Jennifer Pocius and Thuy Pham for excellent technical assistance.
Sources of Funding: This work was supported by the Huffington Center on Aging grant (KC), NIH grant R01HL089792 (MLE), the Medallion Foundation grant (JT) and the Hankamer Foundation.
Glossary
- MSC
mesenchymal stem cells
- TGF-β
transforming growth factor-β
- α-SMA
α-smooth muscle actin
- AMPK
AMP activated protein kinase
- p38MAPK
p38 mitogen-activated protein kinase
- LVID
left ventricular internal diameter
- LVPW
left ventricular posterior wall thickness
- RWT
relative wall thickness
- IVCT
isovolumic contraction time
- IVRT
isovolumic relaxation time
- PAFV
peak aortic flow velocity
- LAD
left anterior descending coronary artery
- LOX
lysyl oxidase
Footnotes
Disclosures: none declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart Disease and Stroke Statistics--2012 Update: A Report From the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gould KE, Taffet GE, Michael LH, Christie RM, Konkol DL, Pocius JS, et al. Heart failure and greater infarct expansion in middle-aged mice: a relevant model for postinfarction failure. Am J Physiol Heart Circ Physiol. 2002;282:H615–21. doi: 10.1152/ajpheart.00206.2001. [DOI] [PubMed] [Google Scholar]
- 3.Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–92. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlson S, Trial J, Soeller C, Entman ML. Cardiac mesenchymal stem cells contribute to scar formation after myocardial infarction. Cardiovasc Res. 2011;91:99–107. doi: 10.1093/cvr/cvr061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. J Invest Dermatol. 1999;112:49–57. doi: 10.1046/j.1523-1747.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 6.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res. 1999;44:232–41. doi: 10.1016/s0008-6363(99)00212-6. [DOI] [PubMed] [Google Scholar]
- 8.Crawford JR, Haudek SB, Cieslik KA, Trial J, Entman ML. Origin of Developmental Precursors Dictates the Pathophysiologic Role of Cardiac Fibroblasts. J Cardiovasc Transl Res. 2012;5:749–59. doi: 10.1007/s12265-012-9402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cieslik KA, Trial J, Entman ML. Defective myofibroblast formation from mesenchymal stem cells in the aging murine heart rescue by activation of the AMPK pathway. Am J Pathol. 2011;179:1792–806. doi: 10.1016/j.ajpath.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kudo N, Gillespie JG, Kung L, Witters LA, Schulz R, Clanachan AS, et al. Characterization of 5′AMP-activated protein kinase activity in the heart and its role in inhibiting acetyl-CoA carboxylase during reperfusion following ischemia. Biochim Biophys Acta. 1996;1301:67–75. doi: 10.1016/0005-2760(96)00013-6. [DOI] [PubMed] [Google Scholar]
- 12.Carvajal K, Zarrinpashneh E, Szarszoi O, Joubert F, Athea Y, Mateo P, et al. Dual cardiac contractile effects of the alpha2-AMPK deletion in low-flow ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2007;292:H3136–47. doi: 10.1152/ajpheart.00683.2006. [DOI] [PubMed] [Google Scholar]
- 13.Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, et al. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am J Physiol Heart Circ Physiol. 2006;291:H2875–83. doi: 10.1152/ajpheart.01032.2005. [DOI] [PubMed] [Google Scholar]
- 14.Russell RR, 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999;277:H643–9. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- 15.Dzeja PP, Chung S, Faustino RS, Behfar A, Terzic A. Developmental enhancement of adenylate kinase-AMPK metabolic signaling axis supports stem cell cardiac differentiation. PLoS One. 2011;6:e19300. doi: 10.1371/journal.pone.0019300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padmasekar M, Sharifpanah F, Finkensieper A, Wartenberg M, Sauer H. Stimulation of Cardiomyogenesis of Embryonic Stem Cells by Nitric Oxide Downstream of AMP-Activated Protein Kinase and mTOR Signaling Pathways. Stem Cells Dev. 2011;20:2163–75. doi: 10.1089/scd.2010.0581. [DOI] [PubMed] [Google Scholar]
- 17.Kim EK, Lim S, Park JM, Seo JK, Kim JH, Kim KT, et al. Human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by AMP-activated protein kinase. J Cell Physiol. 2012;227:1680–7. doi: 10.1002/jcp.22892. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Han Y, Pang W, Li C, Xie X, Shyy JY, et al. AMP-activated protein kinase promotes the differentiation of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1789–95. doi: 10.1161/ATVBAHA.108.172452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, et al. A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol. 2000;278:H1049–55. doi: 10.1152/ajpheart.2000.278.4.H1049. [DOI] [PubMed] [Google Scholar]
- 20.Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. 2011;50:248–56. doi: 10.1016/j.yjmcc.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinones MA, Waggoner AD, Reduto LA, Nelson JG, Young JB, Winters WL, Jr, et al. A new, simplified and accurate method for determining ejection fraction with two-dimensional echocardiography. Circulation. 1981;64:744–53. doi: 10.1161/01.cir.64.4.744. [DOI] [PubMed] [Google Scholar]
- 22.Beckstead JH. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues. J Histochem Cytochem. 1994;42:1127–34. doi: 10.1177/42.8.8027531. [DOI] [PubMed] [Google Scholar]
- 23.Constantine VS, Mowry RW. Selective staining of human dermal collagen. II. The use of picrosirius red F3BA with polarization microscopy. J Invest Dermatol. 1968;50:419–23. doi: 10.1038/jid.1968.68. [DOI] [PubMed] [Google Scholar]
- 24.Junqueira LC, Cossermelli W, Brentani R. Differential staining of collagens type I, II and III by Sirius Red and polarization microscopy. Arch Histol Jpn. 1978;41:267–74. doi: 10.1679/aohc1950.41.267. [DOI] [PubMed] [Google Scholar]
- 25.Briaud SA, Ding ZM, Michael LH, Entman ML, Daniel S, Ballantyne CM. Leukocyte trafficking and myocardial reperfusion injury in ICAM-1/P-selectin-knockout mice. Am J Physiol Heart Circ Physiol. 2001;280:H60–7. doi: 10.1152/ajpheart.2001.280.1.H60. [DOI] [PubMed] [Google Scholar]
- 26.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–87. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 27.Baskin KK, Taegtmeyer H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ Res. 2011;109:1153–61. doi: 10.1161/CIRCRESAHA.111.252742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 29.Yano T, Miura T, Ikeda Y, Matsuda E, Saito K, Miki T, et al. Intracardiac fibroblasts, but not bone marrow derived cells, are the origin of myofibroblasts in myocardial infarct repair. Cardiovasc Pathol. 2005;14:241–6. doi: 10.1016/j.carpath.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Martinelli R, Gegg M, Longbottom R, Adamson P, Turowski P, Greenwood J. ICAM-1-mediated endothelial nitric oxide synthase activation via calcium and AMP-activated protein kinase is required for transendothelial lymphocyte migration. Mol Biol Cell. 2009;20:995–1005. doi: 10.1091/mbc.E08-06-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Qiu J, Wang X, Xia M. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler Thromb Vasc Biol. 2011;31:2897–908. doi: 10.1161/ATVBAHA.111.237453. [DOI] [PubMed] [Google Scholar]
- 32.van Wijk B, Gunst QD, Moorman AF, van den Hoff MJ. Cardiac regeneration from activated epicardium. PLoS ONE. 2012;7:e44692. doi: 10.1371/journal.pone.0044692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol. 2008;322:208–18. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rich L, Whitteker P. Collagen and picrosirius red staining: a polarized light assessment of fibrillar hue and spatial distribution. Braz J Morphol Sci. 2005;22:97–104. [Google Scholar]
- 35.MacKenna DA, Omens JH, McCulloch AD, Covell JW. Contribution of collagen matrix to passive left ventricular mechanics in isolated rat hearts. Am J Physiol. 1994;266:H1007–18. doi: 10.1152/ajpheart.1994.266.3.H1007. [DOI] [PubMed] [Google Scholar]
- 36.Jelani A, Jugdutt BI. STEMI and heart failure in the elderly: role of adverse remodeling. Heart Fail Rev. 2010;15:513–21. doi: 10.1007/s10741-010-9177-3. [DOI] [PubMed] [Google Scholar]
- 37.Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009;106:18351–6. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800–14. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–9. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 40.Paiva MA, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, Mocanu MM. Transitory activation of AMPK at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc Drugs Ther. 2010;24:25–32. doi: 10.1007/s10557-010-6222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kingma JG, Jr, Simard D, Rouleau JR. Timely administration of AICA riboside reduces reperfusion injury in rabbits. Cardiovasc Res. 1994;28:1003–7. doi: 10.1093/cvr/28.7.1003. [DOI] [PubMed] [Google Scholar]
- 42.Kristiansen SB, Solskov L, Jessen N, Lofgren B, Schmitz O, Nielsen-Kudsk JE, et al. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside increases myocardial glucose uptake during reperfusion and induces late pre-conditioning: potential role of AMP-activated protein kinase. Basic Clin Pharmacol Toxicol. 2009;105:10–6. doi: 10.1111/j.1742-7843.2009.00402.x. [DOI] [PubMed] [Google Scholar]
- 43.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–77. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–3. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 46.Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273–83. doi: 10.1006/excr.1999.4543. [DOI] [PubMed] [Google Scholar]
- 47.Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–41. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 49.Shanley CJ, Gharaee-Kermani M, Sarkar R, Welling TH, Kriegel A, Ford JW, et al. Transforming growth factor-beta 1 increases lysyl oxidase enzyme activity and mRNA in rat aortic smooth muscle cells. J Vasc Surg. 1997;25:446–52. doi: 10.1016/s0741-5214(97)70254-4. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, 3rd, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97:872–9. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 51.Stone JD, Narine A, Shaver PR, Fox JC, Vuncannon JR, Tulis DA. AMP-activated protein kinase inhibits vascular smooth muscle cell proliferation and migration and vascular remodeling following injury. Am J Physiol Heart Circ Physiol. 2013;304:H369–81. doi: 10.1152/ajpheart.00446.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moopanar TR, Xiao XH, Jiang L, Chen ZP, Kemp BE, Allen DG. AICAR inhibits the Na+/H+ exchanger in rat hearts--possible contribution to cardioprotection. Pflugers Arch. 2006;453:147–56. doi: 10.1007/s00424-006-0124-z. [DOI] [PubMed] [Google Scholar]
- 53.Jin J, Mullen TD, Hou Q, Bielawski J, Bielawska A, Zhang X, et al. AMPK inhibitor Compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF7 breast carcinoma cells. J Lipid Res. 2009;50:2389–97. doi: 10.1194/jlr.M900119-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Z, Wang X, He Y, Qi L, Yu L, Xue B, et al. The full capacity of AICAR to reduce obesity-induced inflammation and insulin resistance requires myeloid SIRT1. PLoS ONE. 2012;7:e49935. doi: 10.1371/journal.pone.0049935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaidhu MP, Fediuc S, Anthony NM, So M, Mirpourian M, Perry RL, et al. Prolonged AICAR-induced AMP-kinase activation promotes energy dissipation in white adipocytes: novel mechanisms integrating HSL and ATGL. J Lipid Res. 2009;50:704–15. doi: 10.1194/jlr.M800480-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol. 2009;297:C1041–52. doi: 10.1152/ajpcell.00051.2009. [DOI] [PubMed] [Google Scholar]
- 57.Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem. 2011;286:1–11. doi: 10.1074/jbc.M110.121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaskin FS, Kamada K, Yusof M, Korthuis RJ. 5′-AMP-activated protein kinase activation prevents postischemic leukocyte-endothelial cell adhesive interactions. Am J Physiol Heart Circ Physiol. 2007;292:H326–32. doi: 10.1152/ajpheart.00744.2006. [DOI] [PubMed] [Google Scholar]
- 59.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–82. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 60.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, et al. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282–92. doi: 10.1161/CIRCULATIONAHA.110.953208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Newman MF, Ferguson TB, White JA, Ambrosio G, Koglin J, Nussmeier NA, et al. Effect of adenosine-regulating agent acadesine on morbidity and mortality associated with coronary artery bypass grafting: the RED-CABG randomized controlled trial. Jama. 2012;308:157–64. doi: 10.1001/jama.2012.7633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.