

Summary
Cullin4, Ddb1 and Cdt2 are core subunits of the ubiquitin ligase complex CRL4Cdt2, which controls genome stability by targeting Spd1 for degradation during DNA replication and repair in fission yeast. Spd1 has an inhibitory effect on ribonucleotide reductase (RNR), the activity of which is required for deoxynucleotide (dNTP) synthesis. The failure to degrade Spd1 in mutants where CRL4Cdt2 is defective leads to DNA integrity checkpoint activation and dependency. This correlates with a lower dNTP pool. Pools are restored in a spd1-deleted background and this also suppresses checkpoint activation and dependency. We hypothesized that fission yeast with RNR hyperactivity would display a mutator phenotype on their own, but also possibly repress aspects of the phenotype associated with the inability to target Spd1 for degradation. Here, we report that a mutation in the R1 subunit of ribonucleotide reductase cdc22 (cdc22-D57N), which alleviated allosteric feedback inhibition, caused a highly elevated dNTP pool that was further increased by deleting spd1. The Δspd1 cdc22-D57N double mutant had elevated mutation rates and was sensitive to damaging agents that cause DNA strand breaks, demonstrating that Spd1 can protect the genome when dNTP pools are high. In ddb1-deleted cells, cdc22-D57N also potently elevated RNR activity, but failed to allow cell growth independently of the intact checkpoint. Our results provide evidence that excess Spd1 interferes with other functions in addition to its inhibitory effect on ribonucleotide reduction to generate replication stress and genome instability.
Key words: Ribonucleotide reductase, Spd1, Genome instability, Checkpoint kinase, Ddb1
Introduction
Genomic integrity depends on error-free DNA synthesis and repair of damaged DNA, which in turn depends on precise and balanced concentrations of the deoxyribonucleotides. The Cul4-Ddb1 ring ligase (CRL4) is important for genome integrity. The core subunits of CRL4 are Cul4 and Ddb1, and, through a family of adaptor proteins, the complex acts as an E3 ubiquitin ligase that targets substrate proteins for degradation. In mammalian cells, the DDB2 and CSA adaptors direct CRL4 towards substrates important for nucleotide excision repair of UV damaged DNA, whereas the CDT2 adaptor is important for proper S phase as it targets CDT1 for degradation after origin firing (Havens and Walter, 2011). The CRL4 complex is also present in the fission yeast Schizosaccharomyces pombe, but not in the distantly related budding yeast Saccharomyces cerevisiae. In S. pombe, mutations that abrogate the function of CRL4Cdt2 cause increased mutation rates, DNA damage sensitivity, slow progression through S phase, activation of and dependency on the DNA damage and integrity checkpoints, and failure to proceed through premeiotic S phase (Liu et al., 2003; Bondar et al., 2004; Holmberg et al., 2005; Liu et al., 2005; Moss et al., 2010). These parts of the phenotype can be suppressed to varying extents by simultaneous deletion of spd1 (Holmberg et al., 2005), which encodes a small intrinsically disordered protein (Nestoras et al., 2010). Spd1 has inhibitory activity towards the highly conserved and essential enzyme ribonucleotide reductase (RNR) both in vivo and in vitro (Liu et al., 2003; Holmberg et al., 2005; Håkansson et al., 2006). RNR controls the deoxynucleotide (dNTP) pool by reducing ribonucleoside diphosphates to their deoxy forms required for DNA synthesis (Nordlund and Reichard, 2006).
Eukaryotes depend on class 1a RNR, which consists of α and β subunits, R1 and R2. The larger R1 subunit, named Cdc22 in fission yeast, provides the catalytic activity. The smaller R2 subunit, Suc22 in fission yeast, donates reducing power to R1 from a diferric tyrosyl radical (Nordlund and Reichard, 2006). Apart from the active site, R1 also contains two nucleotide-binding allosteric sites: the specificity site S selects the appropriate substrate for reduction to maintain a balanced pool, whereas the allosteric activity site A in the N-terminus controls overall activity by monitoring the dATP:ATP ratio. Recent data suggest that dATP bound to the A-site traps RNR in an inactive oligomeric α6β2 complex, whereas the active and ATP bound form might be a variant α6 complex associated with two or six β-subunits (Hofer et al., 2012). Inactivation of the allosteric activity site by changing the conserved aspartic acid at position 57 for asparagine (D57N) leads to an elevated dNTP pool and a mutator phenotype in a murine T-lymphoma cell line and in budding yeast (Caras and Martin, 1988; Chabes et al., 2003). This demonstrates the importance of keeping the dNTP level below a certain threshold for genomic integrity.
In addition to allosteric feedback control, RNR is regulated at the transcriptional level. In mammalian cells, the R2 protein (known as RRM2) appears to be rate limiting for enzyme activity and the R2 promoter is relieved from repression when cells pass the restriction point in late G1, thus providing sufficient RNR activity for replication (DeGregori et al., 1995; Chabes et al., 2004). In response to DNA damage, a second R2-encoding gene, p53R2 (also known as RRM2B), is induced by the checkpoint protein p53 (Nakano et al., 2000; Tanaka et al., 2000), possibly to provide the cell with sufficient dNTP for repair and thus linking RNR activity to the DNA damage checkpoint. In budding yeast, the RNR1 gene encoding R1 is the major target for cell cycle regulation, but both the R1- and R2-encoding genes are strongly induced upon DNA damage (Elledge and Davis, 1989; Elledge and Davis, 1990; Huang and Elledge, 1997; Huang et al., 1998). Similarly in fission yeast, the R1-encoding cdc22 gene is activated at the G1/S boundary, and both cdc22 and suc22 are induced by replication stress (Lowndes et al., 1992; Fernandez Sarabia et al., 1993; Harris et al., 1996; de Bruin et al., 2006).
The layers of regulation involving small unstructured proteins that directly interfere with RNR activity have so far only been described in the two distantly related yeasts, the budding yeast S. cerevisiae and the fission yeast S. pombe. In budding yeast, the small Dif1 protein associates with R2 and sequesters it in the nucleus when cells are not in S phase, whereas the small inhibitor Sml1 associates directly with R1 to restrict RNR activity. When cells enter S phase, or are subjected to DNA damage, both Dif1 and Sml1 are targeted for degradation by the DNA-structure-dependent checkpoints (Zhao et al., 1998; Chabes et al., 1999; Zhao et al., 2000; Zhao et al., 2001). This allows RNR activation and provides the cell with building blocks for replication and repair. Hence, the DNA structure checkpoint is essential in this organism, even during unperturbed cycles, unless SML1 is deleted (Zhao et al., 1998). In fission yeast Spd1 is a functional ortholog of both Dif1 and Sml1 and shows sequence similarity to the two in short stretches (Lee et al., 2008). Spd1 restricts RNR function by both nuclear sequestration of R2 (Liu et al., 2003) and direct inhibition (Håkansson et al., 2006), possibly by interaction with both RNR subunits (Liu et al., 2003; Håkansson et al., 2006; Nestoras et al., 2010). Spd1 is targeted for degradation through ubiquitylation mediated by the CRL4Cdt2 ubiquitin ligase as cells enter S phase or experience DNA damage (Liu et al., 2003; Bondar et al., 2004; Holmberg et al., 2005). In both cases, Spd1 is recruited to Cul4-Ddb1 through MBF-mediated transcriptional induction of the adaptor Cdt2 (Liu et al., 2005) and association with DNA-bound PCNA (Salguero et al., 2012). Mutants of the CRL4Cdt2 ubiquitin ligase fail to degrade Spd1 in response to S phase entry and DNA damage, with the major consequences of increased mutation rates and damage sensitivity in addition to checkpoint activation and dependency. The CRL mutants also fail to undergo premeiotic S phase (Holmberg et al., 2005). We have previously correlated these effects to a lower dNTP pool and they are, to a large extent, suppressed by deletion of the spd1 gene or overexpression of Suc22, which both restore the dNTP level (Holmberg et al., 2005).
Here, we present that in fission yeast, alleviation of RNR allosteric feedback by knock-in of a cdc22-D57N mutation resulted in a substantial 6–12-fold increase in the concentrations of the four deoxyribonucleotides. This contrasts with the more modest 2-fold dNTP increase obtained through deletion of the spd1 gene. This demonstrates a very stringent allosteric feedback inhibition of S. pombe RNR. In the double mutant Δspd1 cdc22-D57N we observed a 9–21-fold increase in dNTP levels over wild-type. The elevated dNTP pools in cdc22-D57N and Δspd1 cdc22-D57N cells correlated with increased spontaneous mutation rates and increased sensitivity to DNA damage.
In ddb1-deleted cells that have constitutive levels of Spd1, introduction of cdc22-D57N resulted in a more modest 6-fold increase in dATP levels, which still reflects increased RNR activity. This increase in dATP level is more than double that obtained by deleting spd1 in the Δddb1 background. However, whereas deletion of spd1 in the Δddb1 background suppresses checkpoint dependency and the failure to undergo premeiotic S phase (Holmberg et al., 2005), the presence of cdc22-D57N neither suppressed checkpoint activation and dependency, nor did it fully suppress the meiotic defect. We draw two important conclusions from our data: (1) Spd1 supports genome stability in cdc22-D57N backgrounds, possibly by limiting mutagenic DNA translesion synthesis, and (2) genome instability associated with the failure to ubiquitylate and degrade Spd1 in mutants abrogating CRL4Cdt2 function cannot merely be explained by the inhibition of RNR activity. We propose that the elevated Spd1 levels in Δddb1 cells affect genome stability by interfering with other so far unidentified processes in addition to the inhibition of dNTP synthesis.
Results
Elevated dNTP conferred by cdc22-D57N and Δspd1
We generated a fission yeast strain with inactivated allosteric feedback inhibition of RNR by introducing a G-to-A transition in codon 57 of the wild-type cdc22 genomic locus to encode Cdc22-D57N. This particular change in the large RNR subunit has previously been shown to alleviate allosteric feedback inhibition of RNR in a murine cell line (Caras and Martin, 1988) and in budding yeast (Chabes et al., 2003). We readily obtained several independent clones that encode mutant Cdc22-D57N from the endogenous locus. The cdc22-D57N mutant displayed no obvious phenotype in terms of growth rate and morphology.
To monitor the effect of the cdc22-D57N mutation and the spd1 deletion on RNR activity, we harvested small-molecule extracts from asynchronous exponentially growing cultures and determined the concentration of the dNTPs (summarized in Table 1). On its own, the cdc22-D57N mutation caused a substantial 5.8–12.1-fold elevation in dNTP pools, which is much more pronounced than the effect we observe in a strain deleted for the RNR inhibitor spd1 (1.7–2.6 fold, Table 1). In the double mutant Δspd1 cdc22-D57N we observed an 8.8–21.2-fold increase in the four dNTPs (Table 1). The increase was highest for dATP and lowest for dCTP. Thus, simultaneous inactivation of allosteric feedback inhibition and loss of inhibition mediated by Spd1 had near multiplicative effect on the dATP pool.
Table 1. Deoxynucleotide measurements.
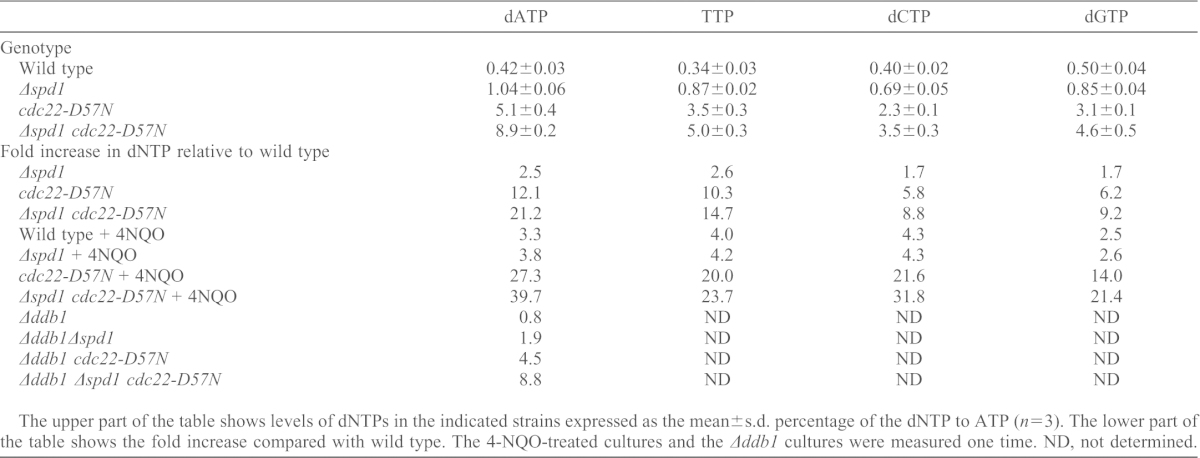
The upper part of the table shows levels of dNTPs in the indicated strains expressed as the mean±s.d. percentage of the dNTP to ATP (n = 3). The lower part of the table shows the fold increase compared with wild type. The 4-NQO-treated cultures and the Δddb1 cultures were measured one time. ND, not determined.
To determine the effects of DNA damage on RNR activity, we compared the dNTP pools in cells either untreated or treated with the DNA damaging agent 4-nitroquinoline 1-oxide (4-NQO) (Table 1). The effect of 4-NQO treatment in wild-type cells led to a 2.5–4.3-fold increase. We observed a slightly smaller increase in 4-NQO-treated Δspd1 cells (1.9–3.2 fold; compare Δspd1 to Δspd1+4-NQO in Table 1), probably because these cells already lack Spd1-mediated RNR inhibition.
The 2-fold increase of dNTPs caused by spd1 deletion contrasts with the more profound induction of the pools in 4-NQO-treated wild-type cells. Thus, we wanted to establish whether the protein levels of Suc22 and Cdc22 in wild-type cells rise to support the increased RNR activity upon γ-irradiation, a treatment previously shown to increase cdc22 and suc22 transcription (Watson et al., 2004). As evident from Fig. 1A, the level of Suc22 remained fairly constant after γ-irradiation, whereas the Cdc22 level increased substantially. Thus, we attribute the damage-induced dNTP increase to Spd1 degradation mediated by CRL4Cdt2 and the increased Cdc22 level, with an overall restriction of the dNTP level imposed by allosteric feedback inhibition.
Fig. 1.

cdc22-D57N does not suppress checkpoint activation and meiosis in Δddb1. (A) Cdc22 levels increase upon γ-irradiation. Western blots showing the levels of Suc22, Cdc22–GFP and Cdc2 in exponentially growing cells exposed to ionizing radiation (500 Gy) at time point zero. Samples were collected at the indicated time points (minutes). (B) Cds1 checkpoint kinase activation by excess Spd1 is not suppressed by cdc22-D57N. As marker for DNA-structure-dependent checkpoint activation, Cds1 kinase activity was monitored after immunoprecipitation from the indicated protein extracts by determining its ability to phosphorylate myelin basic protein (32P-MBP), as shown in the upper panel. The lower panel is a western blot for precipitated Cds1. (C) cdc22-D57N does not suppress checkpoint dependency of Δddb1cells. Serial dilutions of the indicated strains were spotted onto solid rich medium and incubated for 3 days at the indicated temperatures before photography. (D) dNTPs are limiting for repair upon rad3ts inactivation at 36°C. Experiments were performed as in C, but included treatment with MMS as indicated. (E) Formation of Ssb1–GFP foci in Δddb1 cells was suppressed by Δspd1 but not by cdc22-D57N. The Ssb1–GFP fusion protein was expressed from the nmt41 promoter in exponentially growing cells of indicated genotypes and detected by fluorescent microscopy. Yellow arrows point to distinct foci. The percentage of cells with at least one nuclear focus is indicated below the micrographs. (F) Western blot showing Cdt1 and tubulin content in the indicated strains.
Next, we tested whether cdc22-D57N could also elevate RNR activity in ddb1-deleted cells (Table 1). Indeed, the dATP level in Δddb1 cdc22-D57N was elevated 5.6-fold compared with that in the Δddb1 single mutant cells (4.5-fold the level of wild-type cells). This effect was higher than the effect of deleting spd1 (1.9-fold the level when compared with wild type) and again we observed the strongest increase when combining cdc22-D57N and Δspd1.
Elevated dNTP and the DNA damage checkpoint
Because we have previously demonstrated that deletion mutants of the CRL4Cdt2 ubiquitin ligase cause activation of the DNA-structure-dependent checkpoint, presumably owing to reduced dNTP concentrations and slow S phase progression (Holmberg et al., 2005), we tested whether strains with elevated dNTP pools – both in the wild-type and the Δddb1 background – displayed elevated Cds1 checkpoint kinase activity. As evident from Fig. 1B, Cds1 activity was low in wild-type, Δspd1, cdc22-D57N and Δspd1 cdc22-D57N strains. In the Δddb1 background, Cds1 was strongly activated as expected. This activation was fully reversed by co-deletion of spd1 but not by the cdc22-D57N mutation, demonstrating that in terms of Cds1-activation and S phase delay, an inability to degrade Spd1 is epistatic over the cdc22-D57N mutation. This is in contrast to the effects of cdc22-D57N on the dNTP pools (Table 1). Consistently, the synthetic co-lethality between ddb1 loss and inactivation of the Rad3-mediated checkpoint is completely suppressed by additional loss of spd1, but not by the cdc22-D57N mutation (Fig. 1C). Thus, the mere inability to deplete Spd1 in Δddb1 causes a checkpoint dependency that is not bypassed by elevating the dNTP pools with the cdc22-D57N mutation.
The spd1 deletion has been shown to reduce the extreme damage sensitivity of the checkpoint mutant Δrad3, suggesting that dNTP is limiting for repair in the absence of a functional DNA damage checkpoint (Moss et al., 2010). Here, we tested whether cdc22-D57N was also able to have this effect. Indeed, cdc22-D57N was an even better suppressor of the damage sensitivity of rad3ts than Δspd1 at the restrictive temperature (Fig. 1D). Hence, part of the rad3 damage sensitivity can be explained by limiting dNTP pools, but the checkpoint dependency of Δddb1 cannot be explained likewise.
The data above suggest an additional function for Spd1, which is independent of direct regulation of dNTP levels via RNR. We speculated that this was a cause of the dependency of Δddb1 on the rad3 checkpoint, which might be due to spontaneous DNA damage and the presence of single-stranded DNA. To test this hypothesis, we expressed a GFP-tagged version of the DNA replication factor A subunit Ssb1 (Ssb1–GFP) to screen for nuclear GFP-foci built on single-stranded DNA. Ssb1–GFP formed at least one nuclear focus in more than 50% of unperturbed Δddb1 cells, whereas this was reduced to 10% in the Δddb1 Δspd1 background (Fig. 1E). However, cdc22-D57N did not reduce the frequency of Ssb1–GFP foci in Δddb1 cells (60% of the cells presented at least one focus). This is consistent with the failure of cdc22-D57N to rescue the synthetic lethality of Δddb1 rad3ts. The relatively rare foci observed in Δddb1 Δspd1 were smaller than in the two other backgrounds (data not shown). The single mutant cdc22-D57N strain did not present with elevated number of Ssb1–GFP foci (data not shown) consistent with normal Cds1 activity (Fig. 1B). Thus, the elevated pools caused by cdc22-D57N did not prevent the appearance of Ssb1-coated single stranded DNA in Δddb1 cells, whereas deletion of spd1 did.
The DNA replication factor Cdt1 protein is a target of CRL4Cdt2 and it is stabilized in Δddb1 (Ralph et al., 2006) and could be involved in the observed checkpoint activation. Spd1 and Cdt1 in turn could be competing for alternative degradation pathways in Δddb1 cells, so we tested whether deletion of spd1 leads to reduced Cdt1 level in Δddb1, as this could explain the absence of checkpoint activation in Δddb1 Δspd1. However, the dramatic increase in Cdt1 observed in Δddb1 was not reduced by also deleting spd1 (Fig. 1F). We conclude that excess Cdt1 was not the cause of checkpoint activation in Δddb1.
A striking phenotype of deletion mutants of the CRL4Cdt2 complex is their inability to undergo meiosis after mating, due to a failure to initiate premeiotic S phase. Deletion of spd1 in the Δddb1 background completely restores proper meiosis (Holmberg et al., 2005). If the meiotic failure of Δddb1 cells were caused by limiting dNTP levels, it should be suppressed by the cdc22-D57N mutation. However, the cdc22-D57N mutation only partially restored meiosis in Δddb1 cells (11% successful meiosis, see Table 2) indicating that whereas dNTP pools are indeed limiting during Δddb1 meiosis, the inability to degrade Spd1 conferred additional obstacles for a full recovery of meiosis. Again, this is consistent with Spd1 having other targets in addition to RNR activity.
Table 2. Meiosis in Δddb1 is only partially restored by cdc22-D57N.
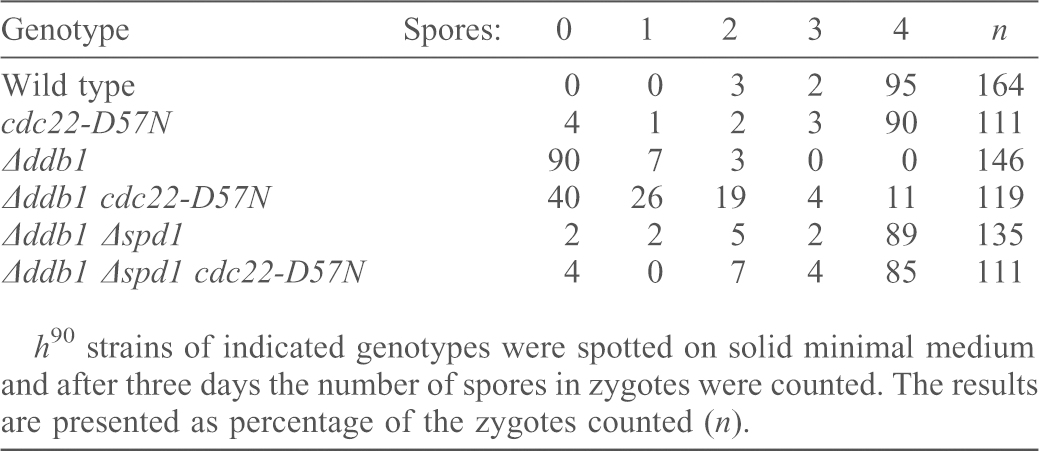
h90 strains of indicated genotypes were spotted on solid minimal medium and after three days the number of spores in zygotes were counted. The results are presented as percentage of the zygotes counted (n).
Increased spontaneous mutation rates in Δspd1 cdc22-D57N strains
To test the effects of elevated dNTP pools on genome stability, we measured spontaneous mutation rates. We used the ade6-485 amber mutation, which can revert to ade+ via various base substitutions within the affected codon (Fleck et al., 1999; Kunz and Fleck, 2001; Holmberg et al., 2005). Compared with wild type, mutation rates were not altered in Δspd1 strains (Table 3). They were slightly elevated in cdc22-D57N cells (P = 0.035), and were ∼6-fold higher in the Δspd1 cdc22-D57N double mutant (P<0.0001) (Table 3). In the Δddb1 mutant, which has a slightly reduced dNTP pool owing to the inability to target Spd1 for degradation, we observed a 25-fold increased mutation rate, which was partially suppressed by concomitant inactivation of spd1 (P = 0.005). This is consistent with our previous results (Holmberg et al., 2005). The mutation rate of the Δddb1 cdc22-D57N double mutant was not significantly different from that of Δddb1 cells (P = 0.081), whereas the Δddb1 Δspd1 cdc22-D57N triple mutant showed a further increase (Table 3). Thus, the absence of Spd1 in cells with high dNTP pools (Δspd1 cdc22-D57N and Δddb1 Δspd1 cdc22-D57N) appeared to increase mutagenicity. Notably, Δddb1 cells with high dNTP pools revealed higher mutation rates than Δddb1 cells with low dNTP pools (Δddb1 Δspd1 cdc22-D57N versus Δddb1, P = 0.01). Nevertheless, ddb1-deleted cells are mutation prone in response to fluctuations of the dNTP concentrations either above or below the optimum.
Table 3. Highly elevated dNTP causes increased reversion rates of ade6-485.
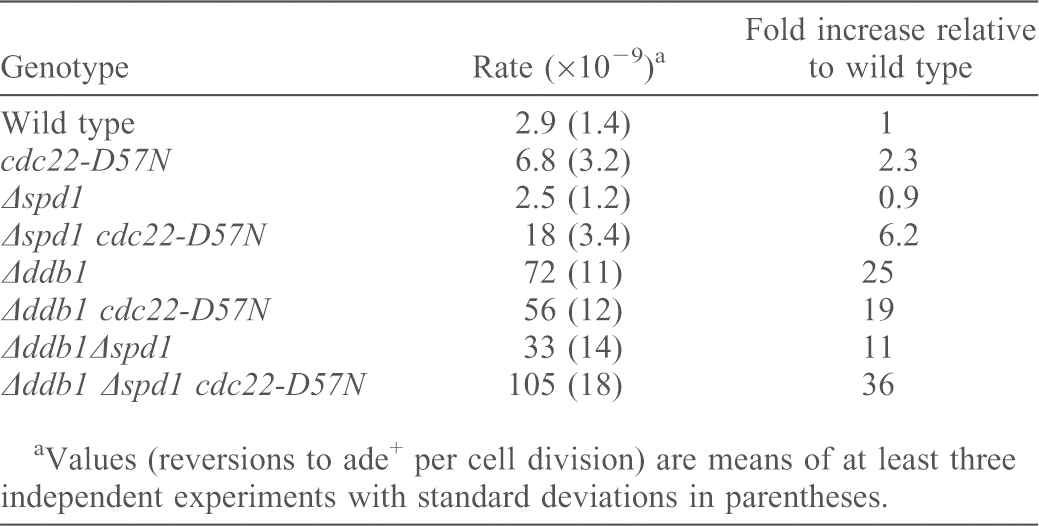
Values (reversions to ade+ per cell division) are means of at least three independent experiments with standard deviations in parentheses.
High dNTP leads to mutagenic translesion synthesis
To establish what caused the increased mutation rate in Δspd1 cdc22-D57N cells, we tested a number of mutants with defects in translesion (TLS) polymerases: deleting Δrev3 (encoding the catalytic subunit of Pol ζ) (Lawrence and Maher, 2001; Waters et al., 2009) in Δspd1 cdc22-D57N background reduced the mutation rate to about half (Table 4; P = 0.0013). A similar effect, although less pronounced, was seen in the Δddb1 background, i.e. the high mutation rate of 1.05×10−7 (reversions to ade+ per cell division) measured in Δddb1 Δspd1 cdc22-D57N strains was reduced to 7.3×10−8 in the quadruple mutant Δddb1 Δrev3 Δspd1 cdc22-D57N (Table 4; P = 0.031). In strains with elevated dNTP pools and active Spd1 (cdc22-D57N and Δddb1 cdc22-D57N), Δrev3 had little effect. The increased mutation rates of spd1-deleted cells with elevated dNTP pools were also suppressed when kpa1 (encoding Pol κ) was deleted (P = 0.021) (Table 4). In contrast, Δrev1, defective for the deoxycytidyl transferase Rev1, did not reduce the mutation rates (Table 4). We conclude that the TLS polymerases Pol ζ and Pol κ contribute to spontaneous mutagenesis in Δspd1 cdc22-D57N cells.
Table 4. Inactivation of rev3 or kpa1 suppresses the mutation rate of ade6-485 in Δspd1 cdc22-D57N strains.
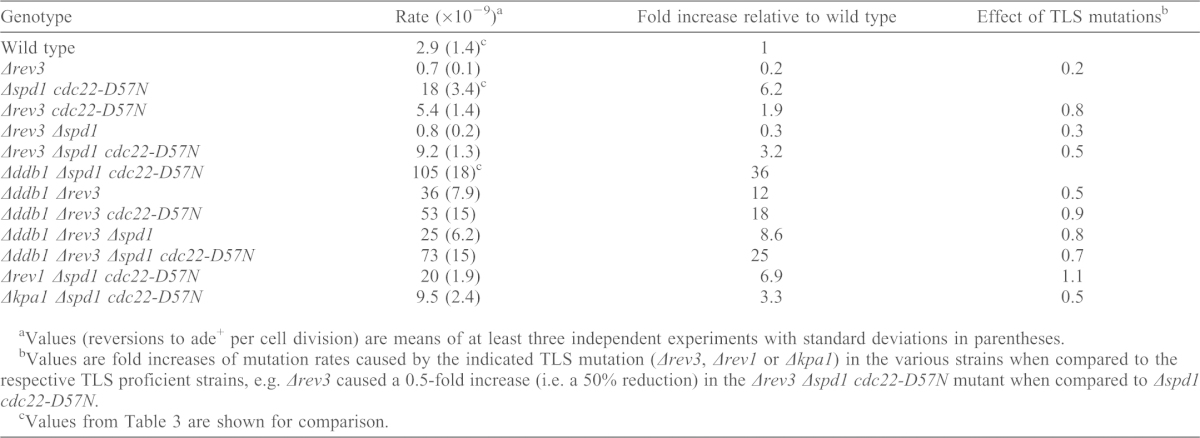
Values (reversions to ade+ per cell division) are means of at least three independent experiments with standard deviations in parentheses.
Values are fold increases of mutation rates caused by the indicated TLS mutation (Δrev3, Δrev1 or Δkpa1) in the various strains when compared to the respective TLS proficient strains, e.g. Δrev3 caused a 0.5-fold increase (i.e. a 50% reduction) in the Δrev3 Δspd1 cdc22-D57N mutant when compared to Δspd1 cdc22-D57N.
Values from Table 3 are shown for comparison.
Δspd1 cdc22-D57N cells are hypersensitive to DNA damage
There is clearly a complex relationship between Spd1, dNTP levels and the response to DNA damage. To gain further insight into these relationships, we tested cellular sensitivity to the DNA damaging agent methyl methanesulphonate (MMS), which alkylates DNA, and as a consequence of attempts to repair causes stalled replication forks and DNA breaks. Although MMS-treated cdc22-D57N cells could have a slightly reduced survival rate when compared to wild type, the Δspd1 cdc22-D57N double mutant was clearly more sensitive (Fig. 2A). Similarly, the Δddb1 Δspd1 cdc22-D57N triple mutant was more sensitive than the Δddb1 single mutant and Δddb1 strains carrying either the cdc22-D57N mutation or deletion of spd1 (Fig. 2D). We also examined survival of cells treated with camptothecin (CPT), which traps topoisomerase I covalently linked to nicked DNA, preventing re-ligation and thereby causing DNA breaks and stalled replication forks. Although cdc22-D57N cells were only slightly more sensitive than wild-type cells, the Δspd1 cdc22-D57N double mutant was substantially more sensitive (Fig. 2B). We conclude that Δspd1 cells with elevated dNTP pool had an increased sensitivity to MMS and CPT, which led to DNA strand breaks during replication and damaged replication forks, which are primarily processed by homologous recombinational repair. In contrast, cdc22-D57N and Δspd1 cdc22-D57N cells were more resistant to 4-NQO, which causes DNA adducts that are primarily repaired by nucleotide excision repair (Fig. 2C).
Fig. 2.

Cell survival of strains with high dNTP pools in response to DNA-damaging agents. DNA damage sensitivity was evaluated as the ability of the indicated strains to grow on plates containing DNA-damaging drugs. Serial dilutions were spotted onto YEA containing the indicated amounts of drugs and were incubated for 3 days at 30°C. (A) The Δspd1 cdc22-D57N was hypersensitive to MMS, and (B) to CPT. (C) Both cdc22-D57N and Δspd1 cdc22-D57N were more resistant to 4-NQO than wild type. (D) The Δddb1 Δspd1 cdc22-D57N strain was hypersensitive to MMS.
We further tested the various TLS-defective mutations in Δspd1 cdc22-D57N background for sensitivity to MMS (Fig. 3). All TLS single mutants were sensitive. When combined with Δspd1 cdc22-D57N, the triple mutants additionally deleted for Δrev3, Δrev7, Δkpa1 or Δrev1, became more sensitive to MMS than either single mutants or the Δspd1 cdc22-D57N double mutant (Fig. 3). Hence, in contrast to the spontaneous mutations, TLS polymerases appear not to be responsible for DNA damage sensitivity of Δspd1 cdc22-D57N.
Fig. 3.

The DNA damage sensitivity of Δspd1 cdc22-D57N strains was not suppressed by inactivation of factors involved in translesion DNA synthesis. Serial dilutions were spotted onto YEA containing the indicated amounts of MMS and were incubated for 3 days at 30°C. (A) Inactivation of rev3 or rev7 and (B) inactivation of kpa1 or rev1 in Δspd1 cdc22-D57N background rendered cells more sensitive to MMS.
Discussion
Spd1 targets functions other than ribonucleotide reduction
Fission yeast RNR is regulated by transcriptional control, allosteric feedback inhibition and by Spd1. We have shown here that alleviation of the allosteric feedback inhibition of RNR led to a 6–12-fold increase in the dNTP pools. In contrast, deletion of Spd1 only resulted in a 2-fold increase (Table 1). Upon DNA damage, the pools increased by 2.5–4.3-fold in wild-type cells. This demonstrates that the major constraints on RNR activity are conferred by feedback inhibition and limiting protein levels. However, the inability to degrade Spd1 in a timely fashion, i.e. coordinated with progression to S phase, leads to checkpoint activation and dependency, spontaneous mutations, damage sensitivity and failure to progress through premeiotic S phase. All these malfunctions are either completely (as is the case for checkpoint activation and dependency and meiosis) or partially (damage sensitivity and spontaneous mutation rates) suppressed by deletion of spd1 (Liu et al., 2003; Holmberg et al., 2005; Liu et al., 2005).
Importantly, despite the fact that in the Δddb1 background, the cdc22-D57N mutation led to the elevation of the dNTP pool well above the effect of spd1 loss (Table 1), cdc22-D57N completely failed to suppress the checkpoint activation (Cds1 activity in Fig. 1B) and dependency (survival in the rad3ts background in Fig. 1C) in Δddb1 cells, whereas these effects were completely suppressed by spd1 loss. The same pattern was observed when we assayed for the presence of nuclear Ssb1–GFP foci indicating that loss of ddb1 caused replication problems involving areas of single stranded DNA. The high frequency of Ssb1 foci (50%) suggests that this is not limited to S phase but extends into the G2 population, which is consistent with the Δddb1 cells being dependent on both Cds1 and Chk1 for survival (Holmberg et al., 2005). Again, Ssb1 foci formation was suppressed by deleting spd1 but not by elevating the dNTP pools with cdc22-D57N. Thus, our data demonstrates that the inappropriate presence of Spd1 during S phase causes reduced dNTP pools and checkpoint activation, but that, whereas Spd1-mediated dNTP deficiency is repressed by introducing cdc22-D57N, this does not relieve the cells from checkpoint activation and dependency. In concordance with this, the meiotic failure of Δddb1 is completely suppressed by spd1 loss (Holmberg et al., 2005), but is only partly suppressed by cdc22-D57N. On the basis of these results we conclude that, in Δddb1 cells, the toxic presence of Spd1 interferes with functions other than RNR enzyme activity to confer checkpoint activation and dependency.
It is possible that Spd1 targets an as yet undescribed function of RNR, but we rather speculate that Spd1 targets a currently unidentified cellular pathway to perturb DNA replication and genome stability. In line with such a hypothesis, it has recently been published that it is necessary that Spd1 interacts with the DNA polymerase processivity factor PCNA in order to become degraded by CRL4Cdt2 (Salguero et al., 2012). Thus, lack of CRL4Cdt2 might lead to replication stress by Spd1 interfering with PCNA-controlled fork fidelity in addition to its effect on RNR. However, we have so far not been able to detect an interaction between Spd1 and PCNA and it is certainly possible that excess Spd1 with its disordered nature promiscuously exerts multiple low-affinity interactions to cause replication stress. In line with a model of other Spd1 targets than PCNA, the dramatic stabilization in Δddb1 of Cdt1, which is known to interact with PCNA, was not the cause of replication stress.
Replication stress and cancer
Replication stress and genome instability are hallmarks of cancer and occur from early stages of cancer progression. Early oncogenic mutations often leads to elevated cyclin-dependent kinase activity to drive proliferation at the price of replicative stress, DNA damage and checkpoint activation (Bartkova et al., 2005; Gorgoulis et al., 2005). Recently, reports have demonstrated that activation of certain oncogenes can lead to DNA replication that is not properly supported by nucleotide pools (Bester et al., 2011; Beck et al., 2012). We are intrigued by the very similar outcome in fission yeast cells defective in Spd1 degradation. Functional orthologs of Spd1 have not been identified in mammalian cells yet, probably because their identification is obscured by limited sequence conservation. We speculate that such proteins exist, presumably to regulate RNR activity, but that it is likely that aberrant activity of such proteins can additionally promote oncogenic replicative stress and checkpoint activation independently of the nucleotide pools, as we have demonstrated here is a phenotype of excess Spd1 in fission yeast. Using fission yeast genetics, we are now searching for mammalian proteins that can mimic the effects of Spd1.
Spd1 supports genome stability when dNTP pools are high
Deleting spd1 in the cdc22-D57N background conferred two distinct maladies: spontaneous mutation rates were elevated during unperturbed cycles, and sensitivity towards the DNA damaging agents MMS and CPT was enhanced. We conclude that the 9–21-fold elevated dNTP pools in Δspd1 cdc22-D57N was mutagenic and also led to impaired DNA damage repair. The effect of spd1-deletion on the dNTP pool in the cdc22-D57N background was approximately a 1.5-fold further increase. We cannot rule out that this additional rise is the cause of the mutator phenotype and damage sensitivity of Δspd1 cdc22-D57N cells, but given that the corresponding dNTP pool increase conferred in the Δddb1-background is less dramatic (Table 1), but leads to a comparable increase in mutation rates and sensitivity to drugs that induce DNA strand breaks, we propose that Spd1 protects the genome in the presence of hyperactive RNR. Hence, Spd1 can either support or destabilize genome integrity depending on the genetic background.
Spd1 and the dNTP pool control translesion DNA synthesis
How does the elevated dNTP pool cause mutations in fission yeast? We have demonstrated that the mutation rates are partly suppressed by deleting translesion polymerases. We propose that the highly elevated dNTP pools directly favour the error-prone translesion synthesis, possibly due to the higher Km translesion polymerases exhibit for their nucleotide substrates (Gibbs et al., 1995; Haracska et al., 2001; Johnson et al., 2001; Shimizu et al., 2002), rather than through a direct signalling mechanism that actively recruits translesion polymerases to sites of replication. Spd1 appeared to protect against translesion synthesis in this setting. In agreement with such a model, unperturbed Δspd1 cdc22-D57N cells did not activate the DNA-structure-dependent checkpoint, which has been proposed to regulate translesion synthesis (Kai and Wang, 2003; Frampton et al., 2006). However, the increased damage sensitivity of Δspd1 cdc22-D57N cells did not involve defective translesion synthesis, and in this setting Spd1 supported repair through unknown mechanisms.
Comparison with budding yeast
In budding yeast, RNR regulation is also controlled by small inhibitory proteins, allosteric feedback inhibition and transcriptional regulation, but with important differences. Upon DNA damage in budding yeast, which leads to Sml1 degradation and elevated transcription of the RNR genes, the dNTP pool is induced 6–8 fold (Chabes et al., 2003). This stronger increase demonstrates that allosteric feedback sets in at higher dNTP concentrations than is the case in fission yeast. Consistently, RNR allosteric feedback inhibition has been dubbed ‘relaxed’ in budding yeast (Håkansson et al., 2006). Here, an rnr1-D57N mutation also leads to increased mutations rates, but the 11–17-fold increase of DNA-damage-induced dNTP pools confers hyper resistance to MMS, UV and most notably 4-NQO. Thus, in budding yeast, dNTPs are more limiting for repair, which might be directly related to the observation that allosteric feedback is relaxed in this organism. The picture is different in fission yeast. Here, Spd1 degradation and the transcriptional induction of RNR led to a more modest 2.5–4.3-fold dNTP increase (Table 1), which appeared important for repair. However, as we have demonstrated with the Δspd1 cdc22-D57N mutant, a 9–21-fold increase in the dNTP pools correlated with hypersensitivity to damage by MMS and CPT. Spuriously, repair of damage induced by 4-NQO was stimulated by elevated dNTP and this is also the case in budding yeast. We speculate that continuous exposure to 4-NQO leads to massive adduct formation throughout the cell cycle. This in turn could make the dNTP pool limiting for nucleotide excision repair. We note that strong ectopic expression of rnr1-D57N in budding yeast, which leads to a constitutive 35-fold increase in dNTP pools also confers DNA damage sensitivity, possibly by preventing activation of the DNA damage checkpoint (Chabes and Stillman, 2007). Consistent with important differences in RNR regulation between the two yeasts, we did not observe reduced DNA structure checkpoint activity in Δspd1 cdc22-D57N cells upon damage (supplementary material Fig. S1).
The CRL4Cdt2 E3 ubiquitin ligase is responsible for degradation of the RNR inhibitor Spd1. Interestingly, this ubiquitin ligase becomes important for damage tolerance (Fig. 2D) and mutation avoidance (Table 3) in Δspd1 cdc22-D57N cells with uncontrolled dNTP pools, suggesting that deregulation of other key Ddb1 substrates compromises genome stability particularly when pools are high. The entire CRL4Cdt2 complex is absent in budding yeast, which relies on an essential function of the DNA damage checkpoint for degradation of its RNR inhibitors (Zhao et al., 1998; Zhao et al., 2001). Collectively, these results suggest that budding yeast has evolved its own specific response to DNA damage, where increased dNTP pools are tolerated to facilitate repair reactions.
Materials and Methods
Fission yeast strains
To generate cdc22-D57N, we amplified the cdc22 open-reading frame (ORF) by PCR from the plasmid pcdc22-1 (Fernandez Sarabia et al., 1993) and cloned the amplicon into pGEM4. The D57N mutation was introduced along with a silent mutation that created a nearby DdeI restriction site by site-directed mutagenesis. This mutant ORF fragment was used to cure the temperature-sensitive phenotype of a cdc22-M45 strain. Correct integration of the mutant allele was confirmed by PCR and sequencing. The DdeI site was used to track the D57N allele in subsequent genetic crosses. The rev3 deletion strain was generated by replacing the entire reading frame with hphMX. cds1-2HA6HIS::ura4+ was a kind gift from Paul Russell, The Scripps Research Institute, California, USA (Moser et al., 2000). All other mutant strains are described elsewhere. Crosses between strains to combine mutations were made by using random spore analysis.
dNTP determination
Extracts for nucleoside triphosphate determination were made from 50 ml exponential cultures harvested at 4×106 cells/ml under vacuum onto a 0.22 µm filter, washed once in 50 ml ice-cold water and resuspended in 500 µl 10% trichloroacetic acid (TCA), 15 mM MgCl2. After three freeze–thaw cycles, the supernatants were cleared by centrifugation and extracted seven times with equal volumes of ether. The TCA-cleared extracts were used as source of nucleotides in four different primer extension assays, each specific for a unique deoxyribonucleoside triphosphate essentially as described previously (Roy et al., 1999). The dNTP concentrations were normalized to ATP measured with the luciferase-based ATP determination kit from Biaffin.
Mutation rates
Reversion rates of the ade6 allele 485 were determined by fluctuation tests as described (Kunz and Fleck, 2001). Allele 485 is a C-to-G transversion that reverts to ade+ via base substitutions (Fleck et al., 1999). Each fluctuation test included nine cultures grown in 1.5 ml of yeast extract liquid (YEL). Mutation rates were determined by the method of the median or from the proportion of cultures without ade+ (Luria and Delbrück, 1943; Lea and Coulson, 1949). Fluctuation tests were carried out at least three times.
Drug sensitivity assays
Strains were grown to stationary phase in YEL adjusted to 107 cells/ml and then diluted in three 1∶10 consecutive steps. 10 µl of each dilution were spotted on yeast extract agar (YEA) plates without drugs and on plates containing MMS, CPT or 4-NQO. Plates were incubated for 3 days at 30°C and photographed.
Cds1 kinase assay
Strains carrying HA-tagged Cds1 were grown to 5×106 cells/ml and whole-cell extracts were made as described previously (Lindsay et al., 1998). 500 µg protein was used for immunoprecipitation of Cds1 with anti-HA 12CA5 antibody. Precipitated material was assayed for kinase activity towards MBP as described previously (Lindsay et al., 1998).
Rad3 genetic interactions
We tested for dependency on the intact Rad3 DNA damage checkpoint by use of a rad3 temperature sensitive allele rad3ts, which has a functional checkpoint at 25°C but an inactive checkpoint at 35°C. Cells were adjusted to 106 cells/ml and then diluted in three 1∶10 consecutive steps. 5 µl of each dilution were spotted on two YEA plates, which were incubated for three days at 25°C or 35C° before photography.
Cdt1 western blot
Cells were grown 5×106 cells/ml and whole-cell extracts were made as described previously (Lindsay et al., 1998). The proteins were size-separated by SDS-PAGE, blotted onto nitrocellulose and incubated with anti-Cdt1 and anti-tubulin antibodies. Secondary antibodies were coupled to peroxidase and the signal developed with ECL+ according to the manufacturer's instructions (Amersham).
Fluorescence microscopy
Ssb1–GFP foci were visualized on an AxioImager Z1 (Carl Zeiss MicroImaging) microscope using filter 41017 from Chroma. The light source was an HXP 120 metal halide lamp (Carl Zeiss MicroImaging). Pictures were processed with Volocity software (PerkinElmer).
Supplementary Material
Acknowledgments
We thank Karin Holm for excellent technical assistance, Chris McInerney and Paul Russell for plasmids and strains, Jacqueline Hayles for the anti-Cdt1 antibody, and Michael Lisby for instructions in the use of his Zeiss fluorescence microscope.
Footnotes
Author contributions
O.N. initiated the work by generating the cdc22-D57N strain. O.F., C.H., R.V-H. and A.W. performed the experiments. O.F., O.N., A.M.C. and C.H. contributed conceptual directions and interpretation of the results. O.F. and C.H. wrote the manuscript.
Funding
This work was supported by the Danish Cancer Society (to O.N. and C.H.); the UK Medical Research Council (to A.M.C. and A.W.); and the National Institute for Social Care and Health Research-Cancer Genetics Biomedical Research Unit and North West Cancer Research (to O.F.). Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.132837/-/DC1
References
- Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J. M., Lukas C. et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 10.1038/nature03482 [DOI] [PubMed] [Google Scholar]
- Beck H., Nähse-Kumpf V., Larsen M. S., O'Hanlon K. A., Patzke S., Holmberg C., Mejlvang J., Groth A., Nielsen O., Syljuåsen R. G. et al. (2012). Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 32, 4226–4236 10.1128/MCB.00412-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester A. C., Roniger M., Oren Y. S., Im M. M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D. S., Kerem B. (2011). Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446 10.1016/j.cell.2011.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar T., Ponomarev A., Raychaudhuri P. (2004). Ddb1 is required for the proteolysis of the Schizosaccharomyces pombe replication inhibitor Spd1 during S phase and after DNA damage. J. Biol. Chem. 279, 9937–9943 10.1074/jbc.M312570200 [DOI] [PubMed] [Google Scholar]
- Caras I. W., Martin D. W., Jr (1988). Molecular cloning of the cDNA for a mutant mouse ribonucleotide reductase M1 that produces a dominant mutator phenotype in mammalian cells. Mol. Cell. Biol. 8, 2698–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A., Stillman B. (2007). Constitutively high dNTP concentration inhibits cell cycle progression and the DNA damage checkpoint in yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 104, 1183–1188 10.1073/pnas.0610585104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A., Domkin V., Thelander L. (1999). Yeast Sml1, a protein inhibitor of ribonucleotide reductase. J. Biol. Chem. 274, 36679–36683 10.1074/jbc.274.51.36679 [DOI] [PubMed] [Google Scholar]
- Chabes A., Georgieva B., Domkin V., Zhao X., Rothstein R., Thelander L. (2003). Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112, 391–401 10.1016/S0092-8674(03)00075-8 [DOI] [PubMed] [Google Scholar]
- Chabes A. L., Björklund S., Thelander L. (2004). S Phase-specific transcription of the mouse ribonucleotide reductase R2 gene requires both a proximal repressive E2F-binding site and an upstream promoter activating region. J. Biol. Chem. 279, 10796–10807 10.1074/jbc.M312482200 [DOI] [PubMed] [Google Scholar]
- de Bruin R. A., Kalashnikova T. I., Chahwan C., McDonald W. H., Wohlschlegel J., Yates J., 3rd, Russell P., Wittenberg C. (2006). Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol. Cell 23, 483–496 10.1016/j.molcel.2006.06.025 [DOI] [PubMed] [Google Scholar]
- DeGregori J., Kowalik T., Nevins J. R. (1995). Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 15, 4215–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge S. J., Davis R. W. (1989). DNA damage induction of ribonucleotide reductase. Mol. Cell. Biol. 9, 4932–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge S. J., Davis R. W. (1990). Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes Dev. 4, 740–751 10.1101/gad.4.5.740 [DOI] [PubMed] [Google Scholar]
- Fernandez Sarabia M. J., McInerny C., Harris P., Gordon C., Fantes P. (1993). The cell cycle genes cdc22+ and suc22+ of the fission yeast Schizosaccharomyces pombe encode the large and small subunits of ribonucleotide reductase. Mol. Gen. Genet. 238, 241–251 [DOI] [PubMed] [Google Scholar]
- Fleck O., Lehmann E., Schär P., Kohli J. (1999). Involvement of nucleotide-excision repair in msh2 pms1-independent mismatch repair. Nat. Genet. 21, 314–317 10.1038/6838 [DOI] [PubMed] [Google Scholar]
- Frampton J., Irmisch A., Green C. M., Neiss A., Trickey M., Ulrich H. D., Furuya K., Watts F. Z., Carr A. M., Lehmann A. R. (2006). Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol. Biol. Cell 17, 2976–2985 10.1091/mbc.E05-11-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs P. E., Borden A., Lawrence C. W. (1995). The T-T pyrimidine (6-4) pyrimidinone UV photoproduct is much less mutagenic in yeast than in Escherichia coli. Nucleic Acids Res. 23, 1919–1922 10.1093/nar/23.11.1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V. G., Vassiliou L. V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R. A., Jr, Kastrinakis N. G., Levy B. et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 10.1038/nature03485 [DOI] [PubMed] [Google Scholar]
- Håkansson P., Dahl L., Chilkova O., Domkin V., Thelander L. (2006). The Schizosaccharomyces pombe replication inhibitor Spd1 regulates ribonucleotide reductase activity and dNTPs by binding to the large Cdc22 subunit. J. Biol. Chem. 281, 1778–1783 10.1074/jbc.M511716200 [DOI] [PubMed] [Google Scholar]
- Haracska L., Unk I., Johnson R. E., Johansson E., Burgers P. M., Prakash S., Prakash L. (2001). Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 15, 945–954 10.1101/gad.882301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris P., Kersey P. J., McInerny C. J., Fantes P. A. (1996). Cell cycle, DNA damage and heat shock regulate suc22+ expression in fission yeast. Mol. Gen. Genet. 252, 284–291 [DOI] [PubMed] [Google Scholar]
- Havens C. G., Walter J. C. (2011). Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 25, 1568–1582 10.1101/gad.2068611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer A., Crona M., Logan D. T., Sjöberg B. M. (2012). DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 10.3109/10409238.2011.630372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg C., Fleck O., Hansen H. A., Liu C., Slaaby R., Carr A. M., Nielsen O. (2005). Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev. 19, 853–862 10.1101/gad.329905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Elledge S. J. (1997). Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol. Cell. Biol. 17, 6105–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Zhou Z., Elledge S. J. (1998). The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94, 595–605 10.1016/S0092-8674(00)81601-3 [DOI] [PubMed] [Google Scholar]
- Johnson R. E., Haracska L., Prakash S., Prakash L. (2001). Role of DNA polymerase zeta in the bypass of a (6-4) TT photoproduct. Mol. Cell. Biol. 21, 3558–3563 10.1128/MCB.21.10.3558-3563.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M., Wang T. S. (2003). Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 17, 64–76 10.1101/gad.1043203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz C., Fleck O. (2001). Role of the DNA repair nucleases Rad13, Rad2 and Uve1 of Schizosaccharomyces pombe in mismatch correction. J. Mol. Biol. 313, 241–253 10.1006/jmbi.2001.5054 [DOI] [PubMed] [Google Scholar]
- Lawrence C. W., Maher V. M. (2001). Mutagenesis in eukaryotes dependent on DNA polymerase zeta and Rev1p. Philos. Trans. R. Soc. B 356, 41–46 10.1098/rstb.2000.0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea D. E., Coulson C. A. (1949). The dristribution of the numbers of mutants in bacterial populations. J. Genet. 49, 264–285 10.1007/BF02986080 [DOI] [PubMed] [Google Scholar]
- Lee Y. D., Wang J., Stubbe J., Elledge S. J. (2008). Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol. Cell 32, 70–80 10.1016/j.molcel.2008.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H. D., Griffiths D. J., Edwards R. J., Christensen P. U., Murray J. M., Osman F., Walworth N., Carr A. M. (1998). S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 12, 382–395 10.1101/gad.12.3.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Powell K. A., Mundt K., Wu L., Carr A. M., Caspari T. (2003). Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev. 17, 1130–1140 10.1101/gad.1090803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Poitelea M., Watson A., Yoshida S. H., Shimoda C., Holmberg C., Nielsen O., Carr A. M. (2005). Transactivation of Schizosaccharomyces pombe cdt2+ stimulates a Pcu4-Ddb1-CSN ubiquitin ligase. EMBO J. 24, 3940–3951 10.1038/sj.emboj.7600854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes N. F., McInerny C. J., Johnson A. L., Fantes P. A., Johnston L. H. (1992). Control of DNA synthesis genes in fission yeast by the cell-cycle gene cdc10+. Nature 355, 449–453 10.1038/355449a0 [DOI] [PubMed] [Google Scholar]
- Luria S. E., Delbrück M. (1943). Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 28, 491–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser B. A., Brondello J. M., Baber-Furnari B., Russell P. (2000). Mechanism of caffeine-induced checkpoint override in fission yeast. Mol. Cell. Biol. 20, 4288–4294 10.1128/MCB.20.12.4288-4294.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J., Tinline-Purvis H., Walker C. A., Folkes L. K., Stratford M. R., Hayles J., Hoe K. L., Kim D. U., Park H. O., Kearsey S. E. et al. (2010). Break-induced ATR and Ddb1-Cul4(Cdt)2 ubiquitin ligase-dependent nucleotide synthesis promotes homologous recombination repair in fission yeast. Genes Dev. 24, 2705–2716 10.1101/gad.1970810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K., Bálint E., Ashcroft M., Vousden K. H. (2000). A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene 19, 4283–4289 10.1038/sj.onc.1203774 [DOI] [PubMed] [Google Scholar]
- Nestoras K., Mohammed A. H., Schreurs A. S., Fleck O., Watson A. T., Poitelea M., O'Shea C., Chahwan C., Holmberg C., Kragelund B. B. et al. (2010). Regulation of ribonucleotide reductase by Spd1 involves multiple mechanisms. Genes Dev. 24, 1145–1159 10.1101/gad.561910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund P., Reichard P. (2006). Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 10.1146/annurev.biochem.75.103004.142443 [DOI] [PubMed] [Google Scholar]
- Ralph E., Boye E., Kearsey S. E. (2006). DNA damage induces Cdt1 proteolysis in fission yeast through a pathway dependent on Cdt2 and Ddb1. EMBO Rep. 7, 1134–1139 10.1038/sj.embor.7400827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B., Beuneu C., Roux P., Buc H., Lemaire G., Lepoivre M. (1999). Simultaneous determination of pyrimidine or purine deoxyribonucleoside triphosphates using a polymerase assay. Anal. Biochem. 269, 403–409 10.1006/abio.1999.4051 [DOI] [PubMed] [Google Scholar]
- Salguero I., Guarino E., Shepherd M. E., Deegan T. D., Havens C. G., MacNeill S. A., Walter J. C., Kearsey S. E. (2012). Ribonucleotide reductase activity is coupled to DNA synthesis via proliferating cell nuclear antigen. Curr. Biol. 22, 720–726 10.1016/j.cub.2012.02.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K., Hashimoto K., Kirchner J. M., Nakai W., Nishikawa H., Resnick M. A., Sugino A. (2002). Fidelity of DNA polymerase epsilon holoenzyme from budding yeast Saccharomyces cerevisiae. J. Biol. Chem. 277, 37422–37429 10.1074/jbc.M204476200 [DOI] [PubMed] [Google Scholar]
- Tanaka H., Arakawa H., Yamaguchi T., Shiraishi K., Fukuda S., Matsui K., Takei Y., Nakamura Y. (2000). A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 404, 42–49 10.1038/35003506 [DOI] [PubMed] [Google Scholar]
- Waters L. S., Minesinger B. K., Wiltrout M. E., D'Souza S., Woodruff R. V., Walker G. C. (2009). Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154 10.1128/MMBR.00034-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson A., Mata J., Bähler J., Carr A., Humphrey T. (2004). Global gene expression responses of fission yeast to ionizing radiation. Mol. Biol. Cell 15, 851–860 10.1091/mbc.E03-08-0569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Muller E. G., Rothstein R. (1998). A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 2, 329–340 10.1016/S1097-2765(00)80277-4 [DOI] [PubMed] [Google Scholar]
- Zhao X., Georgieva B., Chabes A., Domkin V., Ippel J. H., Schleucher J., Wijmenga S., Thelander L., Rothstein R. (2000). Mutational and structural analyses of the ribonucleotide reductase inhibitor Sml1 define its Rnr1 interaction domain whose inactivation allows suppression of mec1 and rad53 lethality. Mol. Cell. Biol. 20, 9076–9083 10.1128/MCB.20.23.9076-9083.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Chabes A., Domkin V., Thelander L., Rothstein R. (2001). The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 20, 3544–3553 10.1093/emboj/20.13.3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.