

Summary
Rgnef (also known as p190RhoGEF or ARHGEF28) is a Rho guanine-nucleotide-exchange factor (GEF) that binds focal adhesion kinase (FAK). FAK is recruited to adhesions and activated by integrin receptors binding to matrix proteins, such as fibronectin (FN). Canonical models place Rgnef downstream of integrin–FAK signaling in regulating Rho GTPase activity and cell movement. Herein, we establish a new, upstream role for Rgnef in enhancing FAK localization to early peripheral adhesions and promoting FAK activation upon FN binding. Rgnef-null mouse embryo fibroblasts (MEFs) exhibit defects in adhesion formation, levels of FAK phosphotyrosine (pY)-397 and FAK localization to peripheral adhesions upon re-plating on FN. Rgnef re-expression rescues these defects, but requires Rgnef–FAK binding. A mutation in the Rgnef pleckstrin homology (PH) domain inhibits adhesion formation, FAK localization, and FAK-Y397 and paxillin-Y118 phosphorylation without disrupting the Rgnef–FAK interaction. A GEF-inactive Rgnef mutant rescues FAK-Y397 phosphorylation and early adhesion localization, but not paxillin-Y118 phosphorylation. This suggests that, downstream of FN binding, paxillin-pY118 requires Rgnef GEF activity through a mechanism distinct from adhesion formation and FAK activation. These results support a scaffolding role for Rgnef in FAK localization and activation at early adhesions in a PH-domain-dependent but GEF-activity-independent manner.
Key words: Rgnef, p190RhoGEF, Arhgef28, FAK, Adhesion, PH domain, GEF
Introduction
The integrin family of transmembrane receptors links the extracellular matrix (ECM) to the intracellular actin cytoskeleton at points of cell–substratum interaction termed focal adhesions (Geiger and Yamada, 2011). In addition to this structural role, integrin clustering can initiate intracellular signaling events that promote cell proliferation, survival and migration in both normal and tumor cells (Desgrosellier and Cheresh, 2010; Schwartz, 2001). One type of signaling event stimulated by integrins is increased tyrosine phosphorylation of cytoskeletal-adaptor as well as signaling proteins (Parsons et al., 2010). As integrins do not possess intrinsic catalytic activity, the signals initiated by ECM–integrin interactions are transduced into cells through the activation of associated proteins (Guo and Giancotti, 2004). Two of the most-studied integrin-activated protein tyrosine kinases (PTKs) are focal adhesion kinase (FAK) and Src (Mitra and Schlaepfer, 2006). Despite two decades of investigation (Parsons, 2003), the molecular mechanisms controlling the recruitment and activation of FAK upon cell binding to ECM proteins such as fibronectin remain incompletely defined.
FAK is composed of an N-terminal FERM (band 4.1, ezrin, radixin, moesin homology) domain, a central PTK region, proline-rich regions and a C-terminal domain that links FAK to integrins through indirect multi-protein binding interactions (Mitra et al., 2005). Models of integrin-mediated FAK activation include receptor clustering and intermolecular Y397 FAK phosphorylation (Toutant et al., 2002) or displacement of regulatory intramolecular FAK FERM binding interactions with the FAK kinase domain, resulting in conformational FAK activation and Y397 FAK autophosphorylation (Lietha et al., 2007). Y397 FAK phosphorylation creates an Src homology (SH)2-binding site for Src leading to the mutual activation of a FAK–Src PTK complex (Wu et al., 2008; Zhao and Guan, 2009). Interestingly, mutational analyses of FAK reveal that intrinsic activity and Y397 phosphorylation are not essential for adhesion localization (Corsi et al., 2009; Lim et al., 2010). Canonical models for the sequence of events associated with focal adhesion formation postulate that ECM binding by integrins triggers the rapid recruitment of cytoskeletal proteins, such as talin and paxillin, to multi-protein complexes formed with the integrin cytoplasmic domains (Geiger and Yamada, 2011). Although FAK is one of the first proteins recruited to integrins upon ECM binding (Miyamoto et al., 1995), mutational analyses of the paxillin- or talin-binding sites on FAK reveal that these interactions contribute to, but are not essential for, adhesion localization of FAK (Lawson et al., 2012; Lawson and Schlaepfer, 2012; Scheswohl et al., 2008). Thus, the molecular mechanisms contributing to FAK recruitment and activation at adhesion sites remain unresolved.
Another important molecule that regulates integrin signaling events is phosphatidylinositol (4,5)-bisphosphate [PtdIns(4,5)P2] generated upon cell adhesion to ECM (McNamee et al., 1993). Proteins such as talin and FAK contain FERM domains that bind PtdIns(4,5)P2 (Cai et al., 2008). Talin interaction with PtdIns(4,5)P2 has been linked to the stabilization of integrin clustering (Saltel et al., 2009) and talin–integrin-mediated force coupling (Legate et al., 2011). Binding of the FAK FERM domain to PtdIns(4,5)P2 can enhance FAK activation in vitro (Cai et al., 2008). When activated, FAK binds to a variety of signaling (Src), adaptor [p130Cas (also known as BCAR1), Grb2], and cytoskeletal proteins (paxillin, cortactin), some of which can also bind to phosphatidylinositol lipids (Schaller, 2010; Schlaepfer and Mitra, 2004). Although multiple and overlapping protein binding interactions might stabilize integrin signaling complexes (FAK binds p130Cas, Src binds p130Cas, and FAK binds Src), a subset of FAK protein binding interactions are unique (Schaller, 2010). One of these is with Rgnef (also known as p190RhoGEF or Arhgef28), a ubiquitous Rho guanine-nucleotide-exchange factor (GEF) that contains central Dbl and pleckstrin homology domains (DH and PH, respectively) linked to Rho GTPase activation and lipid binding, respectively (van Horck et al., 2001). FAK binds to Rgnef residues 1292–1301 and this direct interaction is not shared with other GEFs (Zhai et al., 2003).
Rgnef activates RhoA and RhoC GTPases (Bravo-Cordero et al., 2011; van Horck et al., 2001), which function as bi-molecular switches alternating between inactive GDP- and active GTP-bound states (Hall, 1998). It is the coordination of GEF and GTPase-activating protein (GAP) activity that control cycles of Rho GTPase activation. Cell adhesion to ECM through integrins promotes rapid FAK–Src activation, p190RhoGAP (also known as ARHGAP35) tyrosine phosphorylation, and GAP activation leading to the transient RhoA inhibition during cell spreading (Arthur et al., 2000; Huveneers and Danen, 2009; Ren et al., 2000; Tomar et al., 2009). The formation and maturation of adhesions are required for optimal cell migration and this depends upon RhoA reactivation occurring ∼1 hour after cell adhesion to ECM (Ren et al., 1999). Mutation of the Rgnef DH domain (Y1003A) eliminates Rgnef exchange activity in vitro (van Horck et al., 2001) and prevents Rgnef-mediated activation of RhoA in cells (Lim et al., 2008b). Rgnef knockout and studies with Rgnef-null mouse embryonic fibroblasts (MEFs) confirm that Rgnef is a key regulator of RhoA reactivation and adhesion establishment downstream of integrins (Miller et al., 2012). Studies with MEFs or dominant-negative inhibition of Rgnef–FAK binding in colon cancer cells show that this signaling axis is important for normal and tumor cell motility (Lim et al., 2008b; Miller et al., 2012; Yu et al., 2011).
Herein, we show that Rgnef expression facilitates FAK localization to early adhesion sites and FAK Y397 phosphorylation upon MEF binding to fibronectin (FN). Analyses of Rgnef re-expression in Rgnef-null MEFs show that FAK activation is dependent upon Rgnef–FAK binding and Rgnef-PH domain function. However, Rgnef-mediated FAK Y397 phosphorylation and localization is independent of intrinsic Rgnef GEF activity. Our results support a new, non-canonical, GEF-independent and PH-domain-associated scaffolding role for Rgnef in promoting FAK localization and activation at early adhesions.
Results
Rgnef promotes FAK localization to early adhesions and FAK Y397 phosphorylation
Integrin-stimulated protein tyrosine phosphorylation occurs rapidly upon MEF adhesion to FN and continues during the processes of cell spreading. To elucidate biochemical signaling changes occurring upon cell adhesion to FN, lysates of wild-type (Rgnef+/+) and Rgnef-null (Rgnef−/−) MEFs were made from cells held in suspension or suspended and then re-plated on FN-coated plates for 5, 15 and 30 minutes (Fig. 1A). FAK activation, as measured by increased FAK Y397 phosphorylation, is observed within 5 minutes upon MEF adhesion and reaches a maximum level within 30 minutes during Rgnef+/+ MEF spreading on FN (Fig. 1A; data pooled from three independent experiments in Fig. 1B). Interestingly, FAK Y397 phosphorylation is significantly attenuated in lysates of Rgnef−/− compared to Rgnef+/+ MEFs at 30 minutes on FN (Fig. 1A,B). A similar reduction in FAK Y397 phosphorylation occurs upon Rgnef knockdown in HEY human carcinoma cells when plated on FN (Fig. 1C). These results implicate Rgnef in the early signaling events following integrin binding to FN. Notably, MEF adhesion and spreading between 5 and 30 minutes on FN is associated with low levels of Rgnef and RhoA GTPase activity (Miller et al., 2012; Ren et al., 2000). Rgnef becomes activated by 60 minutes on FN and this corresponds to increased RhoA GTPase activity (Miller et al., 2012). Taken together, decreased FAK Y397 phosphorylation in FN-re-plated Rgnef−/− MEFs at 30 minutes supports a potential GEF-independent role of Rgnef in facilitating FAK activation.
Fig. 1.

Rgnef facilitates FN-stimulated FAK Y397 phosphorylation and FAK localization to early adhesions. (A) Time course of FAK Y397 phosphorylation upon Rgnef+/+ and Rgnef−/− MEF adhesion to FN. Lysates made from suspended (susp) or FN-re-plated (5, 15 and 30 minutes) MEFs were analyzed by phosphospecific immunoblotting to FAK Y397 (pY397) and re-probed for total FAK. Levels of Rgnef and actin are shown as controls. (B) Quantification of FAK pY397 phosphorylation in suspended and FN-re-plated Rgnef+/+ and Rgnef−/− lysates, as shown in panel A, by densitometry and normalized to total FAK levels. Values are means±s.d. of three independent experiments. ***P<0.001, by two-way ANOVA and Bonferroni post-test for multiple comparisons. (C) Rgnef knockdown in HEY ovarian carcinoma cells reduced FN-stimulated FAK activation. Lysates from scrambled (Scr) or shRgnef HEY cells held in suspension or plated on FN for 15 or 30 minutes were immunoblotted for Rgnef, FAK pY397, total FAK and actin. (D) Rgnef+/+ (top) and Rgnef−/− (middle and bottom) MEFs were plated onto glass FN-coated (10 µg/ml) coverslips for 30 minutes. Cells were co-stained with antibodies to paxillin (green) and FAK (red). The merged image shows colocalization (yellow). Inset, enlarged area of peripheral adhesion staining (white box). Scale bar: 10 µm. Middle and bottom panels show Rgnef−/− MEFs containing high and low numbers of peripheral adhesions. (E) MEFs were analyzed for paxillin and FAK colocalization at 30 minutes within peripheral adhesions in 10–12 cells per experimental group on a pixel-by-pixel basis. The Pearson's correlation coefficient, calculated using Cell Profiler, is shown as a box-and-whisker plot (black square, mean; bottom line, 25th percentile; middle line, median; top line, 75th percentile; whiskers, fifth and 95 percentiles). A value of 1 corresponds to 100% colocalization. *P<0.05, Students t-test.
As MEFs bind and spread on FN, small punctate structures termed early adhesions containing clustered integrin receptors form in the peripheral region of cells (Parsons et al., 2010). Early adhesions can be visualized by staining for paxillin, an adaptor protein that binds FAK (Deakin and Turner, 2008). FN-stimulated FAK activation is associated with FAK colocalization with paxillin at early peripheral adhesions (Lawson et al., 2012) and this was visualized by immunofluorescent staining and confocal microscopy of Rgnef+/+ MEFs plated on FN for 30 minutes (Fig. 1D). Surprisingly, in Rgnef−/− MEFs, despite paxillin staining at early adhesions, FAK was not recruited to peripheral Rgnef−/− MEF adhesion sites (Fig. 1D). This loss of FAK recruitment occurred in Rgnef−/− MEFs with both high and low numbers of adhesions (Fig. 1D), and analyses of multiple FN re-plated cells revealed significantly greater paxillin–FAK colocalization at early adhesions in Rgnef+/+ compared to Rgnef−/− MEFs (Fig. 1E). The above data support a role for Rgnef in targeting FAK to early adhesions and in promoting integrin-stimulated FAK activation.
Rgnef−/− MEFs have decreased early adhesion formation
Primary and immortalized Rgnef−/− MEFs form decreased numbers of mature adhesions at 90 to 120 minutes on FN than do wild-type cells (Miller et al., 2012). To determine whether Rgnef loss alters early cell adhesion or spreading, comparisons of Rgnef+/+ and Rgnef−/− MEFs were performed (Fig. 2). At 30 minutes on FN, both Rgnef+/+ and Rgnef−/− MEFs contained equivalent patterns of cortical F-actin staining around the cell periphery but Rgnef−/− MEFs had formed significantly fewer peripheral adhesions (Fig. 2A). No differences in total cell area were observed between Rgnef+/+ and Rgnef−/− MEFs at 30 minutes on FN (Fig. 2B) and equal numbers of cells adhered to plates coated with either 2 or 10 µg/ml FN at 15 or 30 minutes (Fig. 2C). These data show that despite having fewer early adhesions, Rgnef−/− MEFs spread and adhere normally on FN.
Fig. 2.

Rgnef−/− MEFs form fewer peripheral adhesions but exhibit equivalent β1 integrin expression and activity. (A) Left, Rgnef+/+ and Rgnef−/− MEFs were plated onto glass coverslips coated with FN (10 µg/ml) for 30 minutes. Fixed cells were analyzed for FA formation by paxillin staining (green), F-actin staining (phalloidin, red) and nuclei were stained with Hoechst (blue), as shown in the merged image. Scale bar: 10 µm. Right, significantly reduced number of peripheral adhesions as enumerated following thresholding images with Cell Profiler [shown as a box-and-whisker plot; black square, mean; bottom line, 25th percentile; middle line, median; top line, 75th percentile; whiskers, fifth and 95 percentiles]. *P<0.05, Students t-test. (B) Cell area was analyzed in MEFs analyzed in A using ImageJ. No statistical differences were observed. (C) Cellular adhesion was measured in Rgnef+/+ and Rgnef−/− MEFs plated on either 2 or 10 µg/ml FN for either 15 or 30 minutes. Quantification of the number of cells adherent to FN was performed with ImageJ software. No statistical differences were observed. (D) Histogram showing β1 integrin surface expression in Rgnef+/+ (open peak) and Rgnef−/− (gray peak) MEFs as determined by flow cytometry. Secondary-antibody-only staining (black peak) was used as a control. (E) Analysis of active β1 integrin (9EG7 antibody) surface expression in Rgnef+/+ (left) and Rgnef−/− (right) MEFs. A histogram from a representative experiment is shown. Cells were kept in suspension for 45 minutes (dark gray) or plated on 10 µg/ml FN for 30 minutes (open peaks). Secondary-antibody-only staining (solid black) was used as a negative control. MFI, mean fluorescence intensity (MFI) values are shown below histograms. (F) Analysis of active β1 integrin (9EG7, red) and paxillin (green) staining of Rgnef+/+ and Rgnef−/− MEFs plated on 10 µg/ml FN for 30 minutes. Arrows indicate sites of colocalization. Scale bar: 2 µm.
Early adhesion formation can be modulated by alterations in the cell surface expression and activation state of integrins (Truong and Danen, 2009). Although multiple different pairs of integrins can bind FN, the β1 integrin subunit is a main mediator of MEF binding to FN (Wickström et al., 2011). Flow cytometry analyses revealed that Rgnef+/+ and Rgnef−/− MEFs exhibit equivalent β1 integrin surface expression (Fig. 2D). Furthermore, flow cytometry analyses showed increased levels of α5 and αv integrins in Rgnef−/− compared to Rgnef+/+ MEFs (data not shown). This difference is unlikely to account for the lower numbers of adhesions formed in Rgnef−/− MEFs at 30 minutes on FN.
To investigate whether Rgnef loss alters β1 integrin activation, cells were analyzed using a monoclonal antibody (9EG7) that recognizes an activation-specific epitope on the extracellular region of β1 integrin (Lenter et al., 1993). Rgnef+/+ and Rgnef−/− MEFs were either held in suspension or allowed to adhere to FN for 30 minutes before flow cytometry analysis for 9EG7 binding. Compared to the low level of 9EG7 binding to suspended cells (Fig. 2E, gray histogram), FN re-plating equivalently increased 9EG7 binding (Fig. 2E, open histogram) to Rgnef+/+ and Rgnef−/− MEFs as quantified by the mean fluorescence index (MFI). Limited trypsin treatment used to remove cells from matrix prior to flow cytometry could potentially alter integrin detection. Thus, co-staining of FN re-plated cells with 9EG7 and paxillin antibodies were used to determine the potential activation state of β1 integrin at early adhesions. Although this assay is not quantitative, both Rgnef+/+ and Rgnef−/− MEFs exhibit points of 9EG7 co-staining with paxillin at peripheral sites at 30 minutes on FN (Fig. 2F). Taken together, although Rgnef−/− MEFs form fewer early adhesions when plated on FN, this is not associated with detectable changes in cell adhesion, spreading, β1 integrin expression or β1 integrin activation.
Rgnef PH domain binds phosphoinositide lipids
Re-expression of Rgnef rescues the adhesion formation defects of Rgnef−/− MEFs through an undefined mechanism (Miller et al., 2012). The majority of GEF family proteins contain a DH domain (∼200 amino acids) followed by a PH domain (∼100 amino acids) that binds phospholipids and other proteins (Lemmon, 2008; Rossman et al., 2005). Whereas point mutation of the Rgnef DH domain (Y1003A) eliminates Rgnef exchange activity in vitro (van Horck et al., 2001) and prevents Rgnef-mediated activation of RhoA in cells (Lim et al., 2008b), the role of the Rgnef PH domain remains unknown.
Although different PH domains have limited primary amino acid conservation, three-dimensional (3D) structures of PH domains reveal a common β-sheet-folding pattern (Lemmon, 2008). Using the β-sheet structure of the Akt1 PH domain as a template, sequence alignment of the Rgnef PH domain reveals only limited amino acid identities (Fig. 3A). It has been shown that a small basic R/KxR/K motif within PH domain β2 strands can make hydrogen-bonding interactions with phosphate in the phosphatidylinositol headgroup (Thomas et al., 2002). This R/KxR/K motif is conserved in Rgnef at R1098 and K1100, so we expressed and purified recombinant glutathione-S-transferase (GST) fusion proteins of the wild-type (WT) and point-mutated (R1098A, K1100A; A/A) Rgnef DH–PH region (residues 846–1184). GST–Rgnef-DH/PH was used to probe lipid membrane strips in a dot-blot format and bound strongest to PtdIns(4)P, phosphatidic acid and PtdIns(4,5)P2 (Fig. 3B). GST–Rgnef-DH/PH-(A/A) did not show detectable lipid binding and GST–PLCδ1-PH binding to PtdIns(4,5)P2 was used as the positive control (Fig. 3B). Results from Rgnef DH–PH domain 3D homology modeling (Kelley and Sternberg, 2009) illustrate the Rgnef PH domain R1098 and K1100 basic residues are putatively located in a surface-exposed pocket that could accommodate a negatively charged phospholipid head group at membranes (Fig. 3C). In summary, these results support an important role for the Rgnef PH domain and R1098A and K1100A in mediating phosphatidylinositol lipid binding.
Fig. 3.

Rgnef PH domain binds phosphoinositide lipids. (A) Alignment of PH domain residues to the Akt1 crystal structure (Thomas et al., 2002). Secondary structure β-strands (β1 to β7) are shown above the corresponding sequences of all compared PH domains (Akt1, DAPP1, PDK1, PLCδ1, human Rgnef and murine Rgnef) and are highlighted in yellow, and conserved basic residues are highlighted in blue. Phosphatidylinositol-binding residues within the β1–β2 regions are indicated (red asterisks). (B) GST–Rgnef-DH/PH-WT and GST–Rgnef-DH/PH (WT and R1098A K1100A, A/A) were expressed in bacteria and affinity purified as shown below by Coomassie Blue staining. Direct binding of purified GST–PH-domain-containing proteins to Membrane Lipid Strips™ (Eschelon Biosciences) was evaluated by anti-GST immunoblotting. Lipids listed on the left correspond to lipid spots on the left of each strip and lipid names on right correspond to spots on the right side of each strip. GST–PLCδ1-PH binding to PtdIns(4,5)P2 is the positive control. GST–Rgnef-DH/PH-WT reacted strongly to PtdIns(4)P and more weakly to phosphatidic acid and PtdIns(4,5)P2. GST–Rgnef-DH/PH-A/A did not show detectable binding to lipids. (C) Theoretical structure of the Rgnef DH-PH domain based on homology modeling. The DH domain (green) and PH domain (blue) are shown at the plasma membrane. Mutation of the conserved DH domain residue Y1003 (yellow) to an alanine abrogates nucleotide exchange activity. Residues R1098/K1100 (red) in the PH domain are critical for interaction with phosphatidylinositol lipids at the membrane (red). Structural model created in Phyre2 (Kelley and Sternberg, 2009).
Rgnef PH domain function is required for FN-stimulated FAK activation
We then sought to determine whether Rgnef PH domain function contributes to FN-stimulated FAK activation. Rgnef−/− MEFs were transiently transfected with GFP alone, GFP–Rgnef-WT, GFP–Rgnef-Δ1292 (FAK-binding domain deletion), GFP–Rgnef-A/A (full-length protein with R1098A/K1100A point mutations), GFP–Rgnef-PH (residues 1079–1165), GFP–Rgnef-PH-A/A (PH domain containing point mutations only) or GFP–Rgnef-ΔPH (deletion of residues 1079–1165) (Fig. 4A). The GFP constructs were first visualized within the cells by confocal microscopy following re-plating on FN for 30 minutes. GFP–Rgnef-WT, GFP–Rgnef-PH and GFP–Rgnef-Δ1292 localized to distinct areas at the cell periphery during early spreading as in the positive control GFP–PLCδ-PH (supplementary material Fig. S1). In contrast, GFP–Rgnef-ΔPH and GFP–Rgnef-A/A are distributed to the perinuclear region (supplementary material Fig. S1). Co-immunoprecipitation experiments demonstrated that GFP–Rgnef-WT, GFP–Rgnef-A/A and GFP–Rgnef-ΔPH associate with FAK whereas GFP–Rgnef-Δ1292 does not bind FAK (Fig. 4B). These results show that Rgnef–FAK binding is independent of the Rgnef PH domain.
Fig. 4.

Rgnef PH domain mutation prevents FN-stimulated FAK Y397 and paxillin Y118 phosphorylation but not FAK binding to Rgnef. (A) Schematic of GFP–Rgnef constructs. Depicted are the N-terminal leucine-rich (L-rich), zinc finger-like (Zn), Dbl and pleckstrin homology (DH-PH) domains, the FAK-binding region (1292–1301), C-terminal coiled-coil (C-coil) domain and PDZ-binding motif. Schematics of Rgnef-WT, Rgnef-Δ1292 (FAK binding domain 1292–1301 deletion), Rgnef-R1098A/K1100A (A/A) and Rgnef-ΔPH (1079–1165 deletion) are shown. (B) Co-immunoprecipitation (IP) of endogenous FAK with GFP–Rgnef in transfected Rgnef−/− MEFs. GFP IPs from lysates of cells expressing GFP, GFP–Rgnef-WT, GFP–Rgnef-Δ1292, GFP–Rgnef-A/A, GFP–Rgnef-PH, GFP–Rgnef-PH-A/A and GFP–Rgnef-ΔPH were immunoblotted for FAK and re-probed for GFP. GFP, GFP–Rgnef and GFP–Rgnef-PH expression is indicated (arrows). (C) Lysates from Rgnef−/− MEFs transfected with GFP control, GFP–Rgnef-WT, GFP–Rgnef-A/A, GFP–Rgnef-ΔPH and GFP–Rgnef-Δ1292 held in suspension or FN re-plated (30 minutes) were analyzed for FAK Y397 and paxillin Y118 phosphorylation followed by total FAK and paxillin by immunoblotting. Actin levels are shown as a control. (D,E) Quantification of FAK pY397 (D) and paxillin pY118 (E) by densitometry in the re-plating experiments shown in panel C. Values are normalized to total FAK and are means±s.d. of three independent experiments. *P<0.05, ** P<0.01, *** P<0.001 by one-way ANOVA and Tukey-Kramer HSD.
Next, lysates of GFP-Rgnef transfected Rgnef−/− MEFs were analyzed by immunoblotting (Fig. 4C). As expected, GFP–Rgnef-WT significantly increased FAK Y397 phosphorylation after FN re-plating but not in suspended cell conditions (Fig. 4C, data pooled from three independent experiments in Fig. 4D). In addition to being recruited to early adhesions upon ECM binding and integrin activation, paxillin is phosphorylated at Y31 and Y118 by FAK in response to cell adhesion (Deakin and Turner, 2008). Concordantly, increased paxillin Y118 phosphorylation occurred upon FN re-plating of Rgnef−/− MEFs transfected with GFP–Rgnef-WT MEFs but not those transfected with GFP alone (Fig. 4C,E). Rgnef R1098A/K1100A point mutations or deletion of the Rgnef PH domain also prevent enhanced FAK Y397 and paxillin Y118 phosphorylation upon FN re-plating at 30 minutes (Fig. 4C,D). These Rgnef PH domain alterations function equivalently to the loss of Rgnef–FAK binding as analyzed by expression of GFP–Rgnef-Δ1292 in Rgnef−/− MEFs (Fig. 4D,E). Taken together, these results suggest that Rgnef lipid binding is part of the mechanism promoting FAK activation. Importantly, selective disruption of the Rgnef PH domain function prevents Rgnef localization to the cell periphery, FAK activation and paxillin phosphorylation independently from effects on Rgnef–FAK binding.
FAK localization to early adhesions requires Rgnef PH and FAK-binding domains
Rgnef loss results in decreased FAK Y397 phosphorylation as well as reduced FAK localization to early adhesions upon FN re-plating. To assess whether the Rgnef PH and FAK-binding domains facilitate FAK localization to early adhesions, GFP–Rgnef fusion proteins were re-expressed in Rgnef−/− MEFs, followed by FAK and paxillin immunofluorescent analyses in cells re-plated on FN for 30 minutes (Fig. 5A). In Rgnef−/− cells expressing GFP–Rgnef-WT, paxillin and FAK strongly colocalize within adhesions at the cell periphery (Fig. 5A, >20 GFP-positive cells analyzed for each condition shown in Fig. 5B). However, in Rgnef−/− MEFs expressing GFP alone, GFP–Rgnef-A/A or GFP–Rgnef-Δ1292, FAK did not significantly colocalize in peripheral adhesions with paxillin at 30 minutes on FN (Fig. 5A,B). Additionally, quantification of peripheral adhesions revealed that Rgnef-WT re-expression significantly increased the number of adhesions formed compared to GFP-transfected Rgnef−/− MEFs at 30 minutes on FN (Fig. 5A, >20 GFP-positive cells analyzed for each condition shown in Fig. 5C). Importantly, no difference in adhesion number was observed upon Rgnef-A/A or Rgnef-Δ1292 re-expression compared to GFP-transfected Rgnef−/− MEFs (Fig. 5A,C). These data indicate that, in addition to binding to lipids and stimulating FAK activation, Rgnef–FAK binding and the integrity of the Rgnef PH domain are important in promoting FAK localization to early adhesions. Rgnef lipid binding and FAK interaction are also important in promoting early peripheral adhesion formation.
Fig. 5.

Rgnef PH domain function facilitates FAK recruitment to peripheral adhesions upon FN re-plating. (A) Rgnef−/− MEFs were transiently transfected with GFP, GFP–Rgnef-WT, GFP–Rgnef-R1098A/K1100A or GFP–Rgnef-Δ1292 and plated onto FN-coated (10 µg/ml) glass coverslips for 30 minutes. GFP-positive cells were co-stained with antibodies to paxillin (pseudocolored green) and FAK (pseudocolored red). Merged images show colocalization (yellow). Inset, enlarged area of peripheral adhesion staining (white box). Scale bar: 10 µm. (B) GFP-positive cells were analyzed for paxillin and FAK colocalization at 30 minute within all peripheral adhesions in at least 20 cells per experimental group (GFP, GFP–Rgnef-WT, GFP–Rgnef-A/A or GFP–Rgnef-Δ1292) on a pixel-by-pixel basis and calculated as a Pearson's correlation coefficient using Cell Profiler (v2.0). A value of 1 corresponds to 100% colocalization. Box and whisker plots show the distribution of the data: black square, mean; bottom line, 25th percentile; middle line, median; top line, 75th percentile; whiskers, 5th and 95th percentiles. ***P<0.001 and *P<0.05 by one-way ANOVA and Tukey-Kramer HSD. (C) Peripheral adhesion number in Rgnef−/− cells expressing GFP, GFP–Rgnef-WT, GFP–Rgnef-A/A or GFP–Rgnef-Δ1292 were enumerated following thresholding of images with Cell Profiler software. *P<0.05, **P<0.01 by one-way ANOVA and Tukey-Kramer HSD.
Rgnef GEF activity regulates FN-stimulated paxillin Y118 but not FAK Y397 phosphorylation
Cell adhesion to FN triggers rapid changes in actin dynamics and myosin-mediated tension generation in the maturation of focal adhesions (Gardel et al., 2010; Parsons et al., 2010). Tension generation in cells is mediated in part by RhoA GTPase activation (Hall, 2005). Rgnef re-expression in Rgnef−/− MEFs showed that Rgnef activates RhoA at 60 to 90 minutes after FN re-plating, but both Rgnef GEF activity and RhoA activity are lowest after 30 minutes on FN (Miller et al., 2012). To test whether Rgnef GEF activity is required for FN-stimulated FAK activation, the effects of Rgnef-WT was compared to the Rgnef GEF-inactive (Y1003A) Dbl domain mutant (Fig. 6A). Equivalent levels of Rgnef-WT and Rgnef-Y1003A were expressed in Rgnef−/− MEFs (Fig. 6B) and lysates were analyzed from suspended and 30 minutes FN re-plated cells by immunoblotting (Fig. 6C). As expected, Rgnef-WT significantly increased FAK Y397 and paxillin Y118 phosphorylation after FN re-plating but not in suspended cells (Fig. 6C, data pooled from three independent experiments in Fig. 6D,E). Interestingly, Rgnef-Y1003A re-expression enhanced FN-stimulated FAK pY397 to the equivalent level to that of Rgnef-WT (Fig. 6C,D). However, Rgnef-Y1003A re-expression did not promote FN-stimulated paxillin Y118 phosphorylation (Fig. 6C,E). To our knowledge, this is the first separation of the FAK and paxillin tyrosine phosphorylation linkage downstream of integrins after FN re-plating. Moreover, these results support a GEF-independent scaffolding role for Rgnef in promoting integrin-stimulated FAK activation. However, Rgnef-mediated events promoting FAK activation are distinct from an Rgnef GEF-dependent role in promoting paxillin Y118 phosphorylation upon FN re-plating.
Fig. 6.

Rgnef GEF activity is required for FN-stimulated paxillin Y118 but not FAK Y397 phosphorylation. (A) Schematic showing the GFP–Rgnef-WT and GFP–Rgnef-Y1003A mutation within the DH domain that prevents GEF activity (van Horck et al., 2001). (B) Rgnef−/− MEFs were transiently transfected with plasmids for GFP, GFP–Rgnef-WT and GFP–Rgnef-Y1003A and analyzed by Rgnef and actin immunoblotting. (C) The transfected Rgnef−/− MEFs in B were held in suspension or FN re-plated (30 minutes), lysed and analyzed for FAK pY397 and paxillin pY118 followed by total FAK and paxillin by immunoblotting. Actin levels are shown as a control. (D,E) Quantification of FAK pY397 (D) and paxillin pY118 (E) by densitometry from the re-plating experiments shown in C. Values are normalized to total FAK and are means±s.d. of three independent experiments. *P<0.05 by one-way ANOVA and Tukey-Kramer HSD.
Rgnef GEF-independent localization of FAK to early adhesions
Given that expression of Rgnef-Y1003A promoted FAK Y397 but not paxillin Y118 phosphorylation upon FN re-plating of Rgnef−/− MEFs (Fig. 6), analyses were performed to determine whether Rgnef-Y1003A also regulates FAK localization to peripheral adhesions (Fig. 7). GFP–Rgnef fusion proteins were re-expressed in Rgnef−/− MEFs, followed by FAK and paxillin immunofluorescent analyses in cells re-plated on FN for 30 minutes (Fig. 7A). As expected, paxillin and FAK strongly colocalize within adhesions at the cell periphery of GFP–Rgnef-WT transfected MEFs (Fig. 7A, >20 GFP-positive cells analyzed for each condition shown in Fig. 7B). Interestingly, Rgnef-Y1003A expression significantly increased FAK colocalization with paxillin at peripheral adhesions compared to GFP-transfected Rgnef−/− MEFs (Fig. 7A,B). Additionally, quantification of peripheral adhesions revealed that Rgnef-WT or Rgnef-Y1003A re-expression significantly increased the number of adhesions formed compared to GFP-transfected Rgnef−/− MEFs at 30 minutes on FN (Fig. 7A, >20 GFP-positive cells analyzed for each condition shown in Fig. 7C). Taken together, these results indicate that Rgnef promotes early adhesion formation, FAK localization to early adhesions and FAK Y397 phosphorylation, in a GEF-independent manner, upon FN re-plating. Notably, the Rgnef GEF-independent role in promoting early adhesion formation is distinct from the Rgnef GEF-dependent enhanced paxillin Y118 phosphorylation.
Fig. 7.

Rgnef GEF activity is dispensable for FAK localization to and formation of early adhesions. (A) Rgnef−/− MEFs were transiently transfected with GFP (top), GFP–Rgnef-WT (middle) or GFP–Rgnef-Y1003A (bottom) and plated onto FN-coated (10 µg/ml) glass coverslips for 30 minutes. GFP-positive cells were co-stained with antibodies to paxillin (pseudocolored green) and FAK (pseudocolored red). Merged images show colocalization (yellow). Inset, enlarged area of peripheral adhesion staining (white box). Scale bar: 10 µm. (B) Staining was analyzed for paxillin and FAK colocalization at 30 minutes within all peripheral adhesions in at least 20 cells per experimental group on a pixel-by-pixel basis and calculated as a Pearson's correlation coefficient using Cell Profiler. A value of 1 corresponds to 100% colocalization. Box-and-whisker plots show distribution of the data (black square, mean; bottom line, 25th percentile; middle line, median; top line, 75th percentile; whiskers, 5th and 95th percentiles). ***P<0.001 and *P<0.05 by one-way ANOVA and Tukey-Kramer HSD. (C) Paxillin-stained peripheral adhesion numbers in GFP, GFP-Rgnef-WT or GFP-Rgnef-Y1003A expressing cells were enumerated following thresholding images with Cell Profiler software. **P<0.01, ***P<0.001 by one-way ANOVA and Tukey-Kramer HSD.
Discussion
FAK activation is central to the regulation of focal adhesion formation and turnover (Tomar and Schlaepfer, 2009). Following ECM binding and integrin activation, FAK is one of the initial cytoplasmic proteins observed at nascent adhesions (Choi et al., 2011). The FAK C-terminal focal adhesion targeting (FAT) domain binds integrin-associated proteins, such as paxillin and talin, and canonical models suggest that these interactions mediate FAK localization to adhesions (Parsons, 2003). However, FAK mutagenesis experiments show that paxillin or talin binding are not essential for FAK adhesion recruitment (Lawson et al., 2012; Lawson and Schlaepfer, 2012; Scheswohl et al., 2008), and it remains unclear whether other FAK FAT-binding proteins also contribute to FAK adhesion localization. Interestingly, Rgnef binds to the FAK FAT domain (Zhai et al., 2003) and herein we provide evidence for a non-canonical role for Rgnef scaffolding in promoting FAK localization and activation at early adhesions.
This role for Rgnef in promoting FAK activation is independent of Rgnef catalytic GEF activity. However, it is dependent on direct FAK binding and the integrity of the PH domain. Using re-expression studies in cells lacking Rgnef (Miller et al., 2012), we show that Rgnef facilitates integrin-stimulated FAK Y397 phosphorylation and localization of FAK to early sites of cell adhesion formed on FN re-plating. The Rgnef PH domain binds phosphatidylinositol lipids in vitro and is required for FAK localization to early peripheral adhesions. Notably, PH domain mutations disrupting Rgnef lipid binding do not alter the Rgnef–FAK interaction, but do prevent FAK activation downstream of integrins. Moreover, rescue experiments with catalytically inactive Rgnef demonstrate that early FAK activation, localization to adhesions, and peripheral adhesion number is independent of intrinsic Rgnef GEF activity. Unexpectedly, paxillin Y118 phosphorylation required Rgnef catalytic function. These results support the notion of a separate and unconventional function for Rgnef in the regulation of FAK function.
As summarized in a simplistic model (Fig. 8), cell binding to ECM leads to integrin receptors clustering and activation. Signals are generated to increase phosphatidylinositol lipids and we hypothesize that this facilitates PH-domain-mediated Rgnef localization to the peripheral cell membrane near integrin clustering sites during early spreading. This PH-domain-mediated event brings Rgnef-bound FAK into close proximity with adhesion-associated proteins, such as paxillin. This model is consistent with previous studies showing that a complex between FAK and paxillin forms preferentially within adhesions rather than the cytoplasm of cells (Digman et al., 2009). Additionally, FAK facilitates talin recruitment to early peripheral adhesion sites when cells are plated on FN (Lawson et al., 2012). Early FAK activation, as measured by increased Y397 phosphorylation upon FN re-plating, occurs in a tension-independent manner (Lawson et al., 2012) through undefined processes that involve integrin clustering and intermolecular FAK transphosphorylation at Y397 (Toutant et al., 2002). Importantly, our results are in accordance with previous findings that FAK−/− MEFs display increased Rgnef expression and RhoA activation (Lim et al., 2008b) and that MEFs expressing kinase-inactive FAK exhibit abnormalities in adhesion formation (Lim et al., 2010).
Fig. 8.
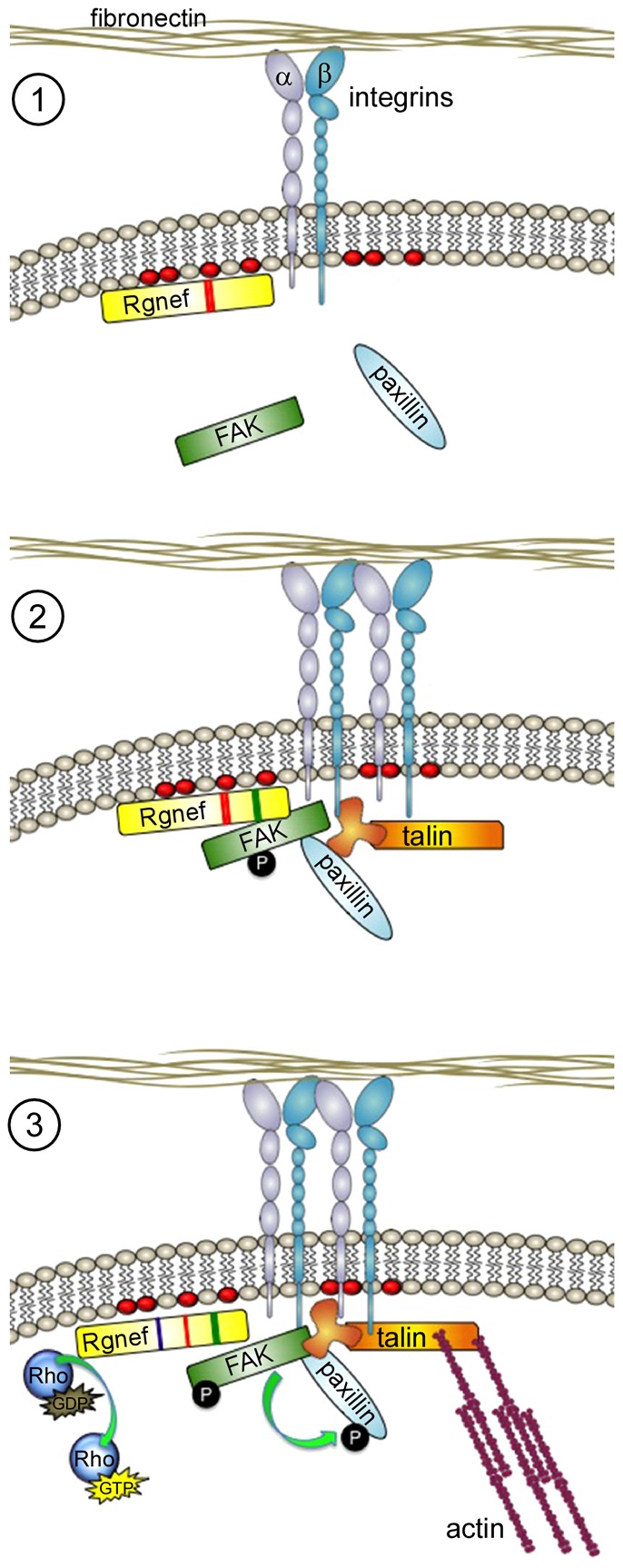
Model of Rgnef GEF-independent and -dependent roles in facilitating integrin-stimulated FAK recruitment and activation at sites of integrin clustering. (1) Following ECM binding and integrin activation, Rgnef might be recruited to the leading edge of cells by increased plasma membrane phosphatidylinositol lipids (red ovals) through the Rgnef PH domain (red region within Rgnef). (2) Binding between Rgnef and FAK through Rgnef residues 1292–1301 (green region within Rgnef) facilitates FAK localization, FAK Y397 phosphorylation and FAK activation at early adhesions containing additional proteins such as paxillin and talin. (3) At later time points, Rgnef GEF activation of RhoA GTPases through its DH domain (blue region within Rgnef) promotes actomyosin tension that facilitates FAK-mediated phosphorylation of paxillin and focal contact maturation.
Despite the finding that FAK−/− MEFs exhibit enhanced adhesion formation, our studies implicate both Rgnef and FAK activation as important in the processes of early adhesion formation needed for the initiation of cell motility. Of note, the number of adhesions in Rgnef−/− MEFs increases proportionally over time, but Rgnef−/− MEFs do not catch up to Rgnef+/+ MEFs by 120 minutes (Miller et al., 2012). Thus, Rgnef−/− adhesion formation is delayed, resulting in fewer and smaller adhesions. The molecular mechanisms responsible for this phenotype remain under investigation, as Rgnef functions as both a scaffold (early) and catalytic GEF (late) in promoting focal adhesion formation. The 60-minute time point on FN seems to be a transition point where Rgnef becomes activate as a GEF (Miller et al., 2012). This is linked to the activation of RhoA, generation of actomyosin contractility, enhancement of paxillin tyrosine phosphorylation and the promotion of focal adhesion maturation.
It has been shown that myosin II activity and the generation of cell tension can promote FAK-mediated paxillin tyrosine phosphorylation leading to adhesion maturation and cytoskeletion–ECM linkage reinforcement (Pasapera et al., 2010). Interestingly, we find that re-expression of a GEF-inactive Rgnef mutant in Rgnef−/− MEFs rescues FAK Y397 but not paxillin Y118 phosphorylation. This finding is consistent with tension-mediated changes in paxillin tyrosine phosphorylation. However, the ability of GEF-inactive Rgnef to promote FAK activation but not paxillin tyrosine phosphorylation reveals new information on the potential divergent or sequential nature of these phosphorylation events. In addition, and unexpectedly, catalytically inactive Rgnef rescued the ability of Rgnef−/− MEFs to form peripheral adhesions at 30 minutes on FN. This implies that increased early adhesion formation can occur independently of paxillin Y118 phosphorylation. It remains unclear whether these are separate or sequential events, and future analyses addressing the role of GTPase activation downstream of Rgnef will hopefully provide insights into the GEF-dependent and GEF-independent roles of Rgnef in integrin signaling.
Although our studies were performed in MEFs, FAK activation downstream of integrins might be conserved in both normal and tumor cells (Desgrosellier and Cheresh, 2010). The molecular mechanisms controlling FAK activation are of potential clinical relevance owing to the fact that FAK controls various aspects of tumor progression (Zhao and Guan, 2009). Small molecules that act as ATP-competitive inhibitors of FAK activity are in various stages of development and are being tested in human clinical trials (Halder et al., 2007; Roberts et al., 2008; Tanjoni et al., 2010; Walsh et al., 2010; Ward et al., 2013). As Rgnef–FAK signaling promotes colon cancer tumor spread (Yu et al., 2011), and we find that Rgnef knockdown reduces FN-stimulated FAK activation in ovarian carcinoma cells, we hypothesize that Rgnef might be an important contributing mechanism promoting FAK activation in tumor cells.
Additionally, because the Rgnef effectors RhoA and RhoC GTPases have been linked to an invasive cell phenotype (Narumiya et al., 2009), the role of Rgnef might be twofold, regulating both FAK and RhoGTPase activity. In fact, recent studies point to the importance of a RhoA–FAK signaling axis in KRAS-driven non-small cell lung cancer (NSCLC) (Konstantinidou et al., 2013). This study concluded that because RhoA silencing and FAK pharmaceutical inhibition yielded similar effects, that FAK is a main effector of RhoA in those cancers. However, an alternative possibility is that a common target, such as Rgnef, might activate FAK and RhoA in dual independent roles. Future studies will aim to understand the molecular mechanisms behind Rgnef–FAK signaling in tumor progression to better understand how these pathways can be targeted in the future for more effective treatments.
Materials and Methods
Reagents and antibodies
Affinity-purified polyclonal anti-Rgnef antibodies were created as described previously (Miller et al., 2012; Yu et al., 2011). Antibodies to β-actin (AC-17) and α-tubulin (DM1A) were from Sigma (St Louis, MO). Anti-FAK (4.47) was from Millipore (Billerica, MA). Anti-paxillin (clone M107) was from BD Biosciences (San Jose, CA). Anti-GFP for immunoblotting (clone B34) and anti-HA-tag (12CA5) were from Covance (Princeton, NJ). Rabbit polyclonal anti-GFP antibodies against recombinant 6-His-tagged GFP (produced in baculovirus), were affinity-purified and used for immunoprecipitation as described previously (Lim et al., 2008a). Phospho-specific antibodies to FAK Y397 (clone 141-9), paxillin Y118 (44-722G) were from Life Technologies (Carlsbad, CA). Anti-glutathione-S-transferase (GST) antibody (clone 8-326) was from Thermo Scientific (Rockford, IL). Antibodies to integrin β1 (CD29, clone KMI6 and 9EG7), α5 (CD49e, clone MFR5) and αV (CD51, clone RMV7) were from BD Pharmingen (San Jose, CA). Anti-mouse and anti-rabbit horseradish-peroxidase-conjugated secondary antibodies were from Pierce (Rockford, IL). Purified bovine fibronectin (FN) was from Sigma (St Louis, MO).
Cells and plasmids
Large T-antigen immortalized Rgnef−/− and Rgnef+/+ mouse embryo fibroblasts (MEFs) were isolated and maintained on dishes pre-coated with 0.1% gelatin as previously described (Miller et al., 2012). HEY ovarian carcinoma cells were from J. Chien (Mayo Clinic, Rochester, MN) and were maintained in DMEM containing 10% fetal bovine serum, 1 mM nonessential amino acids, 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. Lentiviral short hairpin RNA (shRNA) to either scrambled (Scr) or human Rgnef was created and used as described previously (Yu et al., 2011). Transient transfection of plasmid DNA was performed using JetPrime (Polyplus Transfection, Inc., New York, NY). pcDNA-HA-Rgnef-WT and pcDNA-HA-Rgnef-Δ1292 were created as described previously (Zhai et al., 2003). GST–Rgnef-DH/PH (residues S809 to E1189) was created by PCR using the primers: forward, 5′- ATTAGCGGCCGCATCTTCTCTGTGGATCGACCTCAG-3′ and reverse, 5′-TATAGCGGCCGCTCACTCTGGACAACTTTCTACTGCC-3′ and subcloning into the NotI site of pGEX-4T2. GST–Rgnef-DH/PH-A/A (R1098A/K1100A) was made by site-directed mutagenesis of pGEX-4T2 Rgnef-DH/PH (Stratagene, La Jolla, CA) using the primers: forward: 5′-CTGGAAAACCGCTACGGGTGCCTTCGCAGACATCCTGGCTCTGC-3′ and reverse: 5′-GCAGAGCCAGGATGTCTGCGAAGGCACCCGTAGCGGTTTTCCAG-3′ (bold letters represent the mutated residues). The full-length N-terminal fusion of GFP to Rgnef was created by cloning into pEGFPC2 (Clontech, Mountain View, CA) for Rgnef-WT, Rgnef-Y1003A, and Rgnef-Δ1292-1301 as described previously (Lim et al., 2008b). GFP–Rgnef-ΔPH (deletion of residues 1079–1165) was generated by site-directed mutagenesis using the primers: forward: 5′-AAAGCAGGCGCTGCTGCACACCAATTCCAAGGAGGAGCGGAATA-3′ and reverse: 5′-CCTTGGAATTGGTGTGCAGCAGCGCCTGCTTTCTAAACACGTGG-3′. GFP–Rgnef-R1098A/K1100A (A/A) was made by XhoI/NotI digestion of pGEX4T2-Rgnef-DH/PH (R1098A/K1100A) and subcloning this fragment into pEGFPC2 Rgnef. GFP-Rgnef-PH (residues 1079–1165) was made by digesting pGEX4T2-Rgnef-DH/PH with KpnI and ApaI and subcloning a 300-bp fragment into pEGFPC2. GFP–PLCδ-PH was created as described previously (Field et al., 2005). The mouse Rgnef cDNA sequence corresponds to Genbank number NM_012026. Constructs were verified by DNA sequencing.
Flow cytometry
Cells were lightly trypsinized, enumerated and incubated with primary antibodies to integrins (106 cells/µg antibody) for 20 minutes on ice in 100 µl of PBS followed by pelleting and washing using cold PBS. Allophycocyanin (APC)-conjugated goat anti-rat IgG (eBioscience, San Diego, CA) was used as a secondary antibody, and analyses were performed using a FACS Calibur machine (BD Biosciences, San Jose, CA) with FlowJo software (v. 9.5.2, Ashland, OR). Mouse IgG was used as a negative control.
Cell adhesion
48-well plates (Costar #3548, Sigma, St. Loius, MO) were coated for 2 hours at 37°C with 0, 2 or 10 µg/ml FN in PBS (200 µl), blocked with 5% BSA in PBS for 1 hour, washed with PBS, then warmed to 37°C. MEFs were trypsinized, enumerated, and held in suspension in DMEM with 0.5% BSA at 37°C for 30 minutes. 25,000 cells, suspended in 500 µl medium, were added to each well, incubated for 15 or 30 minutes at 37°C, then plunged in PBS to remove non-adherent cells. Adherent cells were fixed and stained with 0.1% Crystal Violet in 20% methanol-PBS for 30 minutes, washed in tap water, and imaged in phase-contrast using an Olympus (Center Valley, PA) CKX31 microscope and an Infinity 1 camera (Lumenera, Ottowa, ON, Canada) controlled by Infinity Analyze (v. 6.2) software (Ottowa, ON, Canada). One representative image was obtained per well, four replicates were performed per experimental condition, and images were thresholded and cells counted using Image J (v. 1.4, Bethesda, MD). Data are representative of three independent experiments.
Biochemical analyses
Cells were transfected with plasmids and analyzed after 48 hours. For re-plating or imaging experiments, cells were starved (0.5% serum) for 16 hours at sub-confluent densities, and treated with 0.06% trypsin and 2 mM EDTA in PBS (2.5 minutes at 37°C). Trypsin was inactivated by addition of soybean trypsin inhibitor (0.5 mg/ml) with 0.25% BSA in DMEM, collected by centrifugation, resuspended in DMEM with 0.5% BSA, and held at 37°C (2×105 cells/ml) for 45 minutes prior to re-plating or protein extraction. Acid-washed glass coverslips or plastic culture dishes were coated with FN (10 µg/ml in PBS) overnight, blocked with 1% BSA in PBS for 30 minutes and pre-heated to 37°C prior to use in cell experiments. Total protein lysates were prepared after the indicated times in extraction buffer [50 mM Hepes, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 100 mM NaF, 1.5 mM MgCl2, 1 mM EGTA, 10 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 10% glycerol and protease inhibitors (Roche)]. Immunoprecipitation and immunoblotting were performed as described previously (Miller et al., 2012). Relative changes in FAK Y397 or paxillin Y118 phosphorylation under suspended or FN re-plated cell conditions were calculated from densitometry analyses of immunoblots using Image J (v1.4, Bethesda, MD) and mean values were determined from three independent re-plating experiments.
Immunofluorescence
Cells re-plated for the indicated time on FN-coated (10 µg/ml) acid-washed glass coverslips were fixed in 3.7% paraformaldehyde (10 minutes), permeabilized with 0.1% Triton X-100 in PBS (10 minutes) and incubated in blocking buffer (2% BSA in PBS) for 1 hour. Anti-paxillin (1∶300) and anti-FAK (1∶1000) antibodies were diluted in blocking buffer and incubated overnight at 4°C. Coverslips were washed in PBS, incubated with Alexa-Fluor-488, -594 or -647-conjugated goat anti-mouse or -rabbit secondary antibodies (1∶300), 4′-6-diamidino-2-phenylindole (DAPI) or Hoechst 33342 (Invitrogen, Carlsbad, CA) diluted in blocking buffer (30 minutes), and mounted using Vectashield (Vector Labs, Burlingame, CA). Images were acquired sequentially using a mercury lamp source, multiband dichroic, single-band exciter and single band emitter filter sets (Chroma, Brattleboro, VT) by using dual filter wheels, an Olympus IX81 spinning disc confocal microscope at 60× (PlanApo, N.A. 1.42) and an Orca-AG camera (Hamamatsu, Japan) controlled by Slidebook (v5.0) software (Denver, CO). Files were cropped, pseudocolored and adjusted using Adobe Photoshop CS6 (San Jose, CA). Peripheral adhesions were determined by analyzing adhesions only 5–10 µm from the edge of the cell using Cell Profiler v2.0 (www.cellprofiler.org) by creating a pipeline containing the ‘LoadImages’, ‘RescaleIntensity’, ‘IdentifyPrimaryObjects’, ‘MeasureCorrelation’ and ‘ExportToSpreadsheet’ modules through which all images were analyzed. The degree of association exhibited by patterns of fluorescence between paxillin and FAK was measured on a pixel-by-pixel basis and calculated as a Pearson's correlation coefficient using the ‘measure correlations’ module. A value of −1 indicates inverse correlation and a value of 1 corresponds to 100% colocalization. Adhesion number was calculated during the above analysis with Cell Profiler.
Lipid binding
GST–Rgnef-DH/PH fusion protein expression was induced in BL-21 pLysS bacteria for 5 hours at 30°C, affinity purified using a BioRad Profinia™ Protein Purification System (Hercules, CA), buffer exchanged, concentrated by centrifugation (Amicon, Millipore) and stored at 4°C. Membrane lipid strips™ from Eschelon Biosciences were blocked by incubation with 3% BSA with 0.1% Tween-20 in PBS for 1 hour followed by addition of 5 µg/ml GST–PLC-δ1-PH (Eschelon, Salt Lake City, UT), GST–Rgnef-DH/PH or GST–Rgnef-DH/PH (R1098A/K1100A) for 1 hour at room temperature as per the manufacturer's instructions. Membranes were washed and binding was detected by anti-GST antibody immunoblotting and chemiluminescent detection.
Statistical analyses
Data were analyzed using Student's t-tests, or one- or two-way ANOVA and post hoc Tukey-Kramer honestly significant difference (HSD) where indicated with GraphPad Prism (v. 5.0d, La Jolla, CA). Significance was taken as P<0.05.
Supplementary Material
Acknowledgments
We thank members of the Schlaepfer laboratory for expert technical assistance, helpful discussions, and critical reading. GFP-PLCγ-PH was generously provided by Seth Field (UCSD). The authors declare that they have no conflict of interest.
Footnotes
Author contributions
N.L.G.M. and D.D.S. conceived the project. N.L.G.M. designed experiments. N.L.G.M., C.L., E.G.K., I.T., and S.U. executed experiments. All authors interpreted the data. N.L.G.M. and D.D.S. wrote the manuscript.
Funding
This work was supported by the National Institutes of Health [grant number CA180769 to D.D.S.]. N.L.G.M. was supported by the National Institutes of Health [grant number 1F32CA159558]. C.L. was supported in part by the Canadian Institutes of Health Research [grant number 200810MFE-193594139144]. I.T. was supported by a grant from Susan G. Komen for the Cure [grant number KG111237]. Deposited in PMC for release 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.135509/-/DC1
References
- Arthur W. T., Petch L. A., Burridge K. (2000). Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr. Biol. 10, 719–722 10.1016/S0960-9822(00)00537-6 [DOI] [PubMed] [Google Scholar]
- Bravo-Cordero J. J., Oser M., Chen X., Eddy R., Hodgson L., Condeelis J. (2011). A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr. Biol. 21, 635–644 10.1016/j.cub.2011.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X., Lietha D., Ceccarelli D. F., Karginov A. V., Rajfur Z., Jacobson K., Hahn K. M., Eck M. J., Schaller M. D. (2008). Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol. Cell. Biol. 28, 201–214 10.1128/MCB.01324-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C. K., Zareno J., Digman M. A., Gratton E., Horwitz A. R. (2011). Cross-correlated fluctuation analysis reveals phosphorylation-regulated paxillin-FAK complexes in nascent adhesions. Biophys. J. 100, 583–592 10.1016/j.bpj.2010.12.3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsi J. M., Houbron C., Billuart P., Brunet I., Bouvrée K., Eichmann A., Girault J. A., Enslen H. (2009). Autophosphorylation-independent and -dependent functions of focal adhesion kinase during development. J. Biol. Chem. 284, 34769–34776 10.1074/jbc.M109.067280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin N. O., Turner C. E. (2008). Paxillin comes of age. J. Cell Sci. 121, 2435–2444 10.1242/jcs.018044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrosellier J. S., Cheresh D. A. (2010). Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22 10.1038/nrc2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman M. A., Wiseman P. W., Choi C., Horwitz A. R., Gratton E. (2009). Stoichiometry of molecular complexes at adhesions in living cells. Proc. Natl. Acad. Sci. USA 106, 2170–2175 10.1073/pnas.0806036106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field S. J., Madson N., Kerr M. L., Galbraith K. A., Kennedy C. E., Tahiliani M., Wilkins A., Cantley L. C. (2005). PtdIns(4,5)P2 functions at the cleavage furrow during cytokinesis. Curr. Biol. 15, 1407–1412 10.1016/j.cub.2005.06.059 [DOI] [PubMed] [Google Scholar]
- Gardel M. L., Schneider I. C., Aratyn-Schaus Y., Waterman C. M. (2010). Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 10.1146/annurev.cellbio.011209.122036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B., Yamada K. M. (2011). Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 3, a005033 10.1101/cshperspect.a005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Giancotti F. G. (2004). Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 5, 816–826 10.1038/nrm1490 [DOI] [PubMed] [Google Scholar]
- Halder J., Lin Y. G., Merritt W. M., Spannuth W. A., Nick A. M., Honda T., Kamat A. A., Han L. Y., Kim T. J., Lu C. et al. (2007). Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 67, 10976–10983 10.1158/0008-5472.CAN-07-2667 [DOI] [PubMed] [Google Scholar]
- Hall A. (1998). Rho GTPases and the actin cytoskeleton. Science 279, 509–514 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- Hall A. (2005). Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 33, 891–895 10.1042/BST20050891 [DOI] [PubMed] [Google Scholar]
- Huveneers S., Danen E. H. (2009). Adhesion signaling - crosstalk between integrins, Src and Rho. J. Cell Sci. 122, 1059–1069 10.1242/jcs.039446 [DOI] [PubMed] [Google Scholar]
- Kelley L. A., Sternberg M. J. (2009). Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- Konstantinidou G., Ramadori G., Torti F., Kangasniemi K., Ramirez R. E., Cai Y., Behrens C., Dellinger M. T., Brekken R. A., Wistuba I. I. et al. (2013). RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov 3, 444–457 10.1158/2159-8290.CD-12-0388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson C., Schlaepfer D. D. (2012). Integrin adhesions: who's on first? What's on second? Connections between FAK and talin. Cell Adh. Migr. 6, 302–306 10.4161/cam.20488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson C., Lim S. T., Uryu S., Chen X. L., Calderwood D. A., Schlaepfer D. D. (2012). FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 196, 223–232 10.1083/jcb.201108078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate K. R., Takahashi S., Bonakdar N., Fabry B., Boettiger D., Zent R., Fässler R. (2011). Integrin adhesion and force coupling are independently regulated by localized PtdIns(4,5)2 synthesis. EMBO J. 30, 4539–4553 10.1038/emboj.2011.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M. A. (2008). Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- Lenter M., Uhlig H., Hamann A., Jenö P., Imhof B., Vestweber D. (1993). A monoclonal antibody against an activation epitope on mouse integrin chain beta 1 blocks adhesion of lymphocytes to the endothelial integrin alpha 6 beta 1. Proc. Natl. Acad. Sci. USA 90, 9051–9055 10.1073/pnas.90.19.9051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietha D., Cai X., Ceccarelli D. F., Li Y., Schaller M. D., Eck M. J. (2007). Structural basis for the autoinhibition of focal adhesion kinase. Cell 129, 1177–1187 10.1016/j.cell.2007.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. T., Chen X. L., Lim Y., Hanson D. A., Vo T. T., Howerton K., Larocque N., Fisher S. J., Schlaepfer D. D., Ilic D. (2008a). Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 29, 9–22 10.1016/j.molcel.2007.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim Y., Lim S. T., Tomar A., Gardel M., Bernard-Trifilo J. A., Chen X. L., Uryu S. A., Canete-Soler R., Zhai J., Lin H. et al. (2008b). PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J. Cell Biol. 180, 187–203 10.1083/jcb.200708194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. T., Chen X. L., Tomar A., Miller N. L., Yoo J., Schlaepfer D. D. (2010). Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J. Biol. Chem. 285, 21526–21536 10.1074/jbc.M110.129999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee H. P., Ingber D. E., Schwartz M. A. (1993). Adhesion to fibronectin stimulates inositol lipid synthesis and enhances PDGF-induced inositol lipid breakdown. J. Cell Biol. 121, 673–678 10.1083/jcb.121.3.673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller N. L., Lawson C., Chen X. L., Lim S. T., Schlaepfer D. D. (2012). Rgnef (p190RhoGEF) knockout inhibits RhoA activity, focal adhesion establishment, and cell motility downstream of integrins. PLoS ONE 7, e37830 10.1371/journal.pone.0037830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S. K., Schlaepfer D. D. (2006). Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523 10.1016/j.ceb.2006.08.011 [DOI] [PubMed] [Google Scholar]
- Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005). Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Teramoto H., Coso O. A., Gutkind J. S., Burbelo P. D., Akiyama S. K., Yamada K. M. (1995). Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J. Cell Biol. 131, 791–805 10.1083/jcb.131.3.791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S., Tanji M., Ishizaki T. (2009). Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28, 65–76 10.1007/s10555-008-9170-7 [DOI] [PubMed] [Google Scholar]
- Parsons J. T. (2003). Focal adhesion kinase: the first ten years. J. Cell Sci. 116, 1409–1416 10.1242/jcs.00373 [DOI] [PubMed] [Google Scholar]
- Parsons J. T., Horwitz A. R., Schwartz M. A. (2010). Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 11, 633–643 10.1038/nrm2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., Waterman C. M. (2010). Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 10.1083/jcb.200906012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X. D., Kiosses W. B., Schwartz M. A. (1999). Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18, 578–585 10.1093/emboj/18.3.578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X. D., Kiosses W. B., Sieg D. J., Otey C. A., Schlaepfer D. D., Schwartz M. A. (2000). Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113, 3673–3678 [DOI] [PubMed] [Google Scholar]
- Roberts W. G., Ung E., Whalen P., Cooper B., Hulford C., Autry C., Richter D., Emerson E., Lin J., Kath J. et al. (2008). Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 68, 1935–1944 10.1158/0008-5472.CAN-07-5155 [DOI] [PubMed] [Google Scholar]
- Rossman K. L., Der C. J., Sondek J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- Saltel F., Mortier E., Hytönen V. P., Jacquier M. C., Zimmermann P., Vogel V., Liu W., Wehrle-Haller B. (2009). New PI(4,5)P2- and membrane proximal integrin-binding motifs in the talin head control beta3-integrin clustering. J. Cell Biol. 187, 715–731 10.1083/jcb.200908134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M. D. (2010). Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell Sci. 123, 1007–1013 10.1242/jcs.045112 [DOI] [PubMed] [Google Scholar]
- Scheswohl D. M., Harrell J. R., Rajfur Z., Gao G., Campbell S. L., Schaller M. D. (2008). Multiple paxillin binding sites regulate FAK function. J. Mol. Signal. 3, 1 10.1186/1750-2187-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D. D., Mitra S. K. (2004). Multiple connections link FAK to cell motility and invasion. Curr. Opin. Genet. Dev. 14, 92–101 10.1016/j.gde.2003.12.002 [DOI] [PubMed] [Google Scholar]
- Schwartz M. A. (2001). Integrin signaling revisited. Trends Cell Biol. 11, 466–470 10.1016/S0962-8924(01)02152-3 [DOI] [PubMed] [Google Scholar]
- Tanjoni I., Walsh C., Uryu S., Tomar A., Nam J. O., Mielgo A., Lim S. T., Liang C., Koenig M., Sun C. et al. (2010). PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 9, 764–777 10.4161/cbt.9.10.11434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. C., Deak M., Alessi D. R., van Aalten D. M. (2002). High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr. Biol. 12, 1256–1262 10.1016/S0960-9822(02)00972-7 [DOI] [PubMed] [Google Scholar]
- Tomar A., Schlaepfer D. D. (2009). Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell Biol. 21, 676–683 10.1016/j.ceb.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar A., Lim S. T., Lim Y., Schlaepfer D. D. (2009). A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J. Cell Sci. 122, 1852–1862 10.1242/jcs.046870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toutant M., Costa A., Studler J. M., Kadaré G., Carnaud M., Girault J. A. (2002). Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol. Cell. Biol. 22, 7731–7743 10.1128/MCB.22.22.7731-7743.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong H., Danen E. H. (2009). Integrin switching modulates adhesion dynamics and cell migration. Cell Adh. Migr. 3, 179–181 10.4161/cam.3.2.8036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horck F. P., Ahmadian M. R., Haeusler L. C., Moolenaar W. H., Kranenburg O. (2001). Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J. Biol. Chem. 276, 4948–4956 10.1074/jbc.M003839200 [DOI] [PubMed] [Google Scholar]
- Walsh C., Tanjoni I., Uryu S., Tomar A., Nam J. O., Luo H., Phillips A., Patel N., Kwok C., McMahon G. et al. (2010). Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. Ther. 9, 778–790 10.4161/cbt.9.10.11433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward K. K., Tancioni I., Lawson C., Miller N. L., Jean C., Chen X. L., Uryu S., Kim J., Tarin D., Stupack D. G. et al. (2013). Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin. Exp. Metastasis 30, 579–594 10.1007/s10585-012-9562-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickström S. A., Radovanac K., Fässler R. (2011). Genetic analyses of integrin signaling. Cold Spring Harb. Perspect. Biol. 3, a005116 10.1101/cshperspect.a005116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Bernard-Trifilo J. A., Lim Y., Lim S. T., Mitra S. K., Uryu S., Chen M., Pallen C. J., Cheung N. K., Mikolon D. et al. (2008). Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene 27, 1439–1448 10.1038/sj.onc.1210770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. G., Nam J. O., Miller N. L., Tanjoni I., Walsh C., Shi L., Kim L., Chen X. L., Tomar A., Lim S. T. et al. (2011). p190RhoGEF (Rgnef) promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res. 71, 360–370 10.1158/0008-5472.CAN-10-2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai J., Lin H., Nie Z., Wu J., Cañete-Soler R., Schlaepfer W. W., Schlaepfer D. D. (2003). Direct interaction of focal adhesion kinase with p190RhoGEF. J. Biol. Chem. 278, 24865–24873 10.1074/jbc.M302381200 [DOI] [PubMed] [Google Scholar]
- Zhao J., Guan J. L. (2009). Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 28, 35–49 10.1007/s10555-008-9165-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.