

Abstract
DNA organization and dynamics profoundly affect many biological processes such as gene regulation and DNA repair. In this review, we present the latest studies on DNA mobility in the context of DNA damage. Recent studies demonstrate that DNA mobility is dramatically increased in the presence of double-strand breaks (DSBs) in the yeast Saccharomyces cerevisiae. As a consequence, chromosomes explore a larger nuclear volume, facilitating homologous pairing but also increasing the rate of ectopic recombination. Increased DNA dynamics is dependent on several homologous recombination (HR) proteins and we are just beginning to understand how chromosome dynamics is regulated after DNA damage.
Keywords: DNA mobility, DNA repair, homologous recombination, double-strand break repair
Exploring DNA mobility in living cells
The genetic information contained in DNA molecules is packaged into chromatin and is highly condensed in the cell nucleus. In the early 1900s, the cell biologist Theodor Bovery proposed the idea that chromosomes are not randomly organized in the nucleus but occupy distinct areas named ‘chromosome territories’ [1]. The first experimental evidence for such an organization came in the 1980s when Cremer et al. observed that a laser beam directed on a particular area of mammalian nuclei always reaches the same chromosomes [2]. Over the past 30 years, the organization of chromosomes inside the nucleus has been extensively studied using model systems such as budding yeast [3–5], Drosophila [3,6], mice [7,8], and humans [9,10]. Those studies revealed that genome organization plays an important role in many biological processes such as gene regulation [11], genome integrity [10,12], and cell differentiation [13–15]. For example, there is a correlation between the distance two genes are apart from one another and their probability of translocation [16]. As a consequence, the spatial organization of genes influences which chromosomes recombine to form cancer-promoting translocations [17,18]. In addition, gene positions differ in tumor cells compared with cells from normal tissue, suggesting that the analysis of gene positioning may become a powerful tool for early-stage detection of cancers [19].
Advances in microscopy and image analysis have made it possible to visualize not only the static organization of chromosomes but also their mobility in real time [20]. Despite a higher-order spatial organization, chromosomes can be highly mobile under certain conditions and the movement of genomic loci is associated with a wide range of DNA processes, such as transcription [21], replication [22], and repair [23–25]. Recently, two studies revealed that the mobility of damaged loci is dramatically increased in the presence of DNA damage in budding yeast [24,25]. One consequence of increased chromosome mobility (ICM) after damage is the facilitation of homology search during inter-homolog recombination [24]. Interestingly, undamaged loci are also more mobile, albeit at lower levels, indicating that there is a global increase in DNA mobility following DSB formation [24]. Increased DNA mobility must be controlled because it might promote unwanted translocations between chromosomes and endanger genome stability [26]. In mammalian cells, it is unclear whether chromosomes are more mobile following DSB induction (Table 1). Several studies report increased chromatin mobility after damage [6,8,23,27,28], whereas others do not [7,29,30]. Additionally, when mobility is observed along with local chromatin perturbations, chromatin is rapidly decondensed at the site of the DSB, whereas the DSB site remains relatively immobile over time [31,32].
Table 1.
Different methods used to induce DSB in vivo and study DNA mobility
| Type of DSB | Properties | Organism |
|---|---|---|
| Endonuclease-induced DSB | ||
| I-SceI | The I-SceI cut-site can be
inserted anywhere into the genome and allows the induction of a single DSB in the genome. In budding yeast, the efficiency of the cutting varies between 10% and 75% depending on the locus where the cut-site is inserted. |
Budding yeast [24,25]; NIH
3T3 cells and MEFs [7] |
| HO | Used to create a DSB at the endogenous HO
cut-site on budding yeast chromosome III, or its recognition site can be inserted into the genome at any locus. The efficiency of cutting in budding yeast is 60–80%. |
Budding yeast [24] |
| Chemically induced DSBs | ||
| Zeocin | Creates random single-strand breaks and DSBs in DNA | Budding yeast [25] |
| Radiation-induced DSBs | ||
| UV laser microirradiation | Cells need to be treated with 10 μM
bromodeoxyuridine for 24 h before irradiation to sensitize them to DSB generation by UV-A laser (λ = 337 nm). |
MEFs [31] |
| X-rays | Create localized DSBs. | Drosophila [6] |
| γ-rays | Create random single-strand breaks and DSBs.
The density of lesions can be scaled according to the time of irradiation. |
Budding yeast [24]; U2OS [28];
human MCF7 mammary carcinoma cells; human lung fibroblasts [32]; MEFs [31] |
| α-particles | Create localized DSBs. The density of DSBs can
be scaled according to the energy and charge of the particle used. |
HeLa cells, human primary fibroblasts
and Chinese hamster ovary [23]; U2OS [30] |
In this review, we describe recent studies of chromosome mobility focusing on the context of DNA repair. First, we review results that describe the nature of chromosome motion and discuss its dependence on genome localization as well as effects of the cell cycle. Next, we present recent evidence that DNA damage dramatically modifies genome organization by causing transient ICM. Finally, we discuss the role that different genes play in DNA mobility after DNA damage.
Evidence for constrained diffusion of chromosomes
Chromosome movement can be followed inside the nucleus of living cells using genomic integration of tetO and lacO arrays at specific loci, which are bound by fluorescently tagged repressors [20,33]. From measurements of its position over time, the nuclear space explored by a locus can be calculated using mean-square displacement (MSD) analysis, a standard tool to analyze the motion of particles [34] (Box 1). When a single locus is tracked relative to a fixed point in the nucleus, it is referred to as a MSD. When two moving loci are tracked simultaneously, it is referred to as mean-square change in distance (MSCD) [3]. In the case of confined motion, the locus cannot escape a certain nuclear subvolume, implying that, for long time intervals, the MSD/MSCD covered by the locus is independent of the elapsed time interval. Consequently, the plateau reached by the MSD/MSCD curve is proportional to the confinement volume explored by the locus. In addition, the initial slope of the MSD/MSCD curves is proportional to the diffusion coefficient.
Box 1. Different methods to analyze chromosome motion in vivo.
MSD. Chromosome mobility can be measured by calculating
MSD = <Δx2(t)> from the position of a tagged locus over time.
In simple liquids, the diffusion of a particle is purely thermal and its MSD depends linearly on time, <Δx2(8)> ~ Dt, where D is the diffusion coefficient of the particle (Brownian motion). In viscoelastic materials, such as polymers, the particle is moving in a more complex environment, which slows it down. In this case, its motion is subdiffusive (or constrained) and <Δx2(8)> ~ tα with α < 1. It is thought that a chromosomal locus exploring the nucleus is confined because of DNA entanglement [70]. A highly confined locus moves in a low elastic environment with very entangled chromatin, whereas a less-confined locus moves in a more elastic chromatin environment. By contrast, a particle transported along a substrate shows directive motion and <Δx2(8)> ~ tα with α > 1. In some cases, the motion of a particle can be a combination of directive motion and thermal diffusion; for example, when a particle moves freely before attaching to a substrate and sliding on it.
To compensate for possible drift during the acquisition of images, the position of a locus has to be corrected for each frame using the 3D position of a fixed point in the nucleus. Many studies use the center of the nucleus, measured by fitting the tagged nuclear membrane to a circle [20]. Other studies use the spindle pole body to serve as a marker of relative nuclear position [24]. Because the center of mass of individual dots can be determined with greater accuracy than the center of mass of an entire nucleus, the second method offers high spatial resolution while minimizing the effects of nuclear translation or rotation.
MSCD. Chromosome mobility can also be quantitated by measuring the distance between two moving loci as a function of time [3,24,38]. This method is named MSCD or distance MSD. An important assumption of this method is that the two moving spots have the same behavior, with similar confinement radii and diffusion coefficients. The confinement radius can be reconstructed from the MSCD plateau if the confining regions are sufficiently far from each other [20].
Consistent with the higher-order structure of the genome, chromosomes undergo confined Brownian motion within a small nuclear subvolume and this confinement is evolutionarily conserved from budding yeast and Drosophila to mammalian cells [4,35–38]. However, the properties of chromatin mobility differ quantitatively between organisms. Human loci have smaller diffusion coefficients than budding yeast , indicating that they are more resistant to motion (Table 2) (4.8 × 10−5 to 1.8 × 10−4 μm2/s for human cells [39–41], 5 × 10–4 to 10−3 μm2/s for budding yeast [3,24,25]). Because human nuclei are around 80 times larger than yeast nuclei, the percentage of the nuclear volume that chromosomes can explore in human cells is much smaller than in yeast. As a consequence, DNA organization is more constrained in mammalian nuclei than in yeast.
Table 2.
Characteristics of DNA mobility under different conditions and in different organisms
| Organism | Cell cycle | Locus | Genotype | Treatment | Confinement radius μm, % of nuclear volume explored |
Nuclear radius (μm) |
Diffusion coefficient (μm2/s) |
Refs |
|---|---|---|---|---|---|---|---|---|
| Yeast, diploid | G1 | LEU2 | WT | None | 0.3, 0.8 | 1.5 | 5 × 10−4 | [3] |
| Yeast, diploid | G1 | LEU2 | WT | Nocodazole | 0.7, 10 | 1.5 | 3 × 10−4 | [3] |
| Yeast, diploid | G1 | CEN plasmid | WT | None | 0.25, 0.5 | 1.5 | 3 × 10−4 | [3] |
| Yeast, haploid | S | ARS4-908 | WT | None | 0.30, 0.4 | 0.9 | – | [4] |
| Yeast, haploid | G1 | ARS4-908 | WT | None | 0.70, 47 | 0.9 | – | [4] |
| Yeast, haploid | S | ARS1413 | WT | None | 0.30, 0.4 | 0.9 | – | [4] |
| Yeast, haploid | G1 | ARS1413 | WT | None | 0.70, 47 | 0.9 | – | [4] |
| Yeast, haploid | S | CEN4 | WT | None | 0.30, 0.4 | 0.9 | – | [4] |
| Yeast, haploid | G1 | CEN4 | WT | None | 0.70, 47 | 0.9 | – | [4] |
| Yeast, haploid | S | ZWF1 | WT | None | 0.46, 13 | 0.9 | 12 × 10−4 | [25] |
| Yeast, haploid | S | ZWF1 | WT | DSB (I-Scel) | 0.70, 47 | 0.9 | 17 × 10−4 | [25] |
| Yeast, haploid | S | ZWF1 | rad51Δ | DSB (I-Scel) | 0.50, 18 | 0.9 | 11 × 10−4 | [25] |
| Yeast, haploid | S | ZWF1 | rad54Δ | DSB (I-Scel) | 0.47, 14 | 0.9 | 13 × 10−4 | [25] |
| Yeast, haploid | S | ZWF1 | rad54 K341A | DSB (I-Scel) | 0.40, 9% | 0.9 | 7 × 10−4 | [25] |
| Yeast, haploid | S | ZWF1 | mec1Δ/sml1Δ | DSB (I-SceI) | 0.36, 6 | 0.9 | 6 × 10−4 | [25] |
| Yeast, haploid | S | ZWF1 | rad53/sml1Δ | DSB (I-SceI) | 0.55, 23 | 0.9 | 9 × 10−4 | [25] |
| Yeast, diploid | S | URA3 | WT | No treatment | 0.45, 2.7 | 1.5 | 14 × 10−4 | [24,25] |
| Yeast, diploid | S | URA3, broken locus | WT | DSB (I-SceI) | 0.99, 29 | 1.5 | 11 × 10−4 | [24,25] |
| Yeast, diploid | S | URA3, intact homolog | WT | DSB (I-SceI) | 0.60, 6.4 | 1.5 | 10 × 10−4 | [24,25] |
| Yeast, diploid | S | URA3 | WT | ~ Four random DSBs |
0.60, 6.4 | 1.5 | 13 × 10−4 | [24,25] |
| Yeast, diploid | S | URA3, damaged locus | rad51Δ | DSB (I-SceI) | 0.455,2.8 | 1.5 | 14 × 10−4 | [24,25] |
| Drosophila | – | X chromosome | WT | None | 0.9, – | – | 20 × 10−4 | [3] |
| Human U2OS cells | – | Telomeres | – | None | 0.23, – | – | 1.8 × 10−4 | [40,41] |
| Human HeLa cells | – | Dense chromatin | – | None | 0.18, – | 4.5 | 0.48 × 10−4 | [39,71] |
| Human HT-1080 | – | Nucleoplasmic loci (5p14 and 3q26.2) | – | None | 0.48, – | – | 1.25 × 10−4 | [38] |
WT, wild type.
Chromosome mobility is highly dependent on the locus observed and the stage of the cell cycle. In budding yeast, centromeres and telomeres explore 3–5% of the nuclear volume in the S and G1 phases of haploid or diploid cells [3,4]. Several lines of evidence show that telomeres are confined due to their tethering to the nuclear envelope [5,42]. For example, yeast telomeres interact with the nuclear envelope through the Ku heterodimer and chromosome mobility is greater in Kumutant strains [43]. Similarly, centromeres are highly confined , probably due to their tethering to the spindle pole body (SPB) through microtubules [5]. Unlike centromeres and telomeres, loci in the middle of a chromosome arm explore 4% of the nuclear volume in S-phase cells but up to 43% in G1 haploid cells [4] (Table 1). It is hypothesized that middle-arm regions are more confined during replication due to the presence of multiple replication forks [4,42].
To interpret chromosome movement inside the nucleus, the timescale of observation must be taken into account. In most studies, chromosome motion is analyzed for 1–10 min at time intervals ranging from 1.5 s [4] to 20 s [3] and the results are consistent. However, two interesting studies extended the analysis of interphase chromatin motion to largertimescales and found contradictory results [36,44]. In the first study, chromosome mobility was measured in Drosophila spermatocytes using 10-s time intervals for several minutes or 15–30-min time intervals for several hours and chromatin motion is best described by two different constrained diffusion processes [36]. In both timescales, chromosomes show constrained motion, but the confinement radius increased during the longer measurement time interval (from 0.5 μm for the 30-min interval to 3 μm for the several-hour interval). By contrast, fluorescence recovery after photobleaching (FRAP) experiments in HeLa cells showed that chromatin explores onlysubmicron regionseven after 1 h and the larger confinement radius observed in the Drosophila study [36] was not detected [44]. Reducing acquisition time by using high-speed microscopy to track a locus revealed that its motion is more complicated than simple confined diffusion. In budding yeast [4] and Chinese hamster ovary cells [45], chromatin undergoes apparently confined random motion alternating with so-called fast jumps; that is, a large increase in distance moved in a single time interval compared with the average distance moved during the acquisition. These fast jumps are ATP-dependent and reflect rare events of active diffusion versus passive diffusion similar to Lévy flights [45,46]. Thus, it is clear that more sophisticated and refined mathematical tools need to be developed to more precisely describe chromosome motion.
Evidence for ICM after DNA damage
During the last decade, several studies suggested that damaged chromosomes are more mobile. In budding yeast, Rad52, a central HR protein that binds single-stranded DNA (ssDNA) at processed DSB ends is commonly used as a marker for the presence of a DSB. Haploid cells containing two non-repairable DSBs make a single Rad52 focus most of the time, implying that the broken chromosomes move to the ‘repair center’ [47]. In HeLa cells, after α-particle-induced DSBs, γH2AX foci are more mobile, again supporting the notion that distant DSBs can be juxtaposed [23,28]. Similarly, ICM is associated with uncapped telomeres in mouse cells and this movement is dependent on the 53BP1 repair protein [8]. A role for 53BP1 in long-range V(D)J recombination reactions has also been reported, underlining the importance of properly regulating chromosome mobility [48]. Finally, movement of heterochromatic DSBs toward euchromatin was observed in mouse embryo fibroblasts (MEFs), HeLa cells [27], and Drosophila cells [6]. It was proposed that relocalization of heterochromatic DSBs close to euchromatin regions prevents rearrangements between repetitive DNA sequences present in heterochromatin. Taken together, these studies suggest that chromosome mobility increases significantly in the presence of DSBs. By contrast, in other studies using MEFs [31], HeLa, or U2OS cells [30], DSBs generated by UV laser or γ-irradiation did not significantly alter chromosome mobility. Only energy-dependent local expansion of chromatin was observed around the initial damaged zone immediately after DNA damage [31]. Similarly, in G1-phase NIH3T3 and H2AX−/− MEFs, no mobility increase was observed in the presence of an I-SceI-induced DSB [7]. These contradictory observations in mammalian cells may result from variation between cell lines, the regions of chromatin damaged, or the type of damage induced. In addition, cancer cell lines may not provide a good model system for studying DNA mobility after damage, because genome organization differs between healthy cells and cancer cells [16].
ICM favors homologous pairing but also ectopic recombination in budding yeast
Recent studies using budding yeast to investigate the chromosome dynamics of single loci in the presence and absence of DNA damage revealed ‘local’ ICM near a DSB[24,25]. Specific lacO- or tetO-tagged chromosomal loci were tracked in living cells and MSD analysis of their trajectories in vivo was performed. Using haploid yeast cells in which a non-repairable single DSB is induced by the endonuclease I-SceI, two different loci, ZWF1 and ARS425 (middle left arm of chromosome XIV and chromosome IV, respectively), were shown to explore a region four times larger after induction of a single DSB [25] (Table 1). Inducing a single-strand break by Flp-nick in haploid cells did not alter chromosome mobility, indicating that increased mobility near the locus is specific to induction by a DSB [25]. Using diploid yeast cells, the confinement radius of the damaged locus after induction of a single I-SceI DSB is more than double that seen in its absence, allowing the locus to explore a 10 times larger nuclear volume [24]. Although the haploid and diploid studies observed a local increase in mobility, the differences seen in the percentage of nuclear volume explored might be due to ploidy. For diploid cells, the damaged locus is actively searching for a homologous partner, whereas for a non-repairable DSB in haploid cells it is unknown whether the cut chromosome is searching for a target. Interestingly, following a DSB, several studies in yeast have shown that the two broken ends remain together [49–51]. Thus, it is likely that, during homology search, the two ends explore the nucleus as a single unit rather than separately.
To monitor their respective motion during homology search, two differentially marked homologous URA3 loci in diploid yeast cells were tracked after induction of an I-SceI DSB near one of them [24]. After inducing a single DSB, the homologs are paired ten times more often. Analysis of the pairing kinetics showed that, in only 30 min, the percentage of S-phase cells with paired homologs increases from 3% to 30% and that pairing lasts 21 ± 5 min [24].
Interestingly, the undamaged homologous locus is also more mobile after DSB induction, allowing it to explore a 2.4 times larger nuclear volume than in the absence of a DSB (6.4% of the nuclear volume) (Table 1). We refer to this ICM as ‘global’. It is important to keep in mind that the cell must carefully regulate both local and global ICM after DSB induction. For example, ICM correlates with increased rates of ectopic HR in haploid yeast cells [26]. It will be interesting to address how quickly chromosomes return to their initial organization and mobility and determine the mechanisms responsible for this reorganization. Perhaps the failure to slow chromosome mobility and properly reorganize chromosomes promotes cancer-prone rearrangements of the genome.
Genes controlling DNA mobility after DNA damage
The control of DNA mobility has been studied by investigating many of the proteins that associate with DSBs. To this end, several groups have observed the motion of chromosomes in the presence of damage in different deletion backgrounds. Here, we discuss studies where protein modifications and/or genetic mutations affect DNA mobility.
Chromatin remodelers
Chromatin remodelers are multiprotein complexes that use the energy from ATP hydrolysis to change the local structure of chromatin. Defects in chromatin-remodeling activity have been linked to severe diseases such as leukemia and epithelial cancers [52]. Two classes of chromatin remodelers function in DSB repair:INO80 and RSC [53,54]. Several studies have shown that the INO80 complex is recruited to DSBs via the phosphorylation of histone H2A near the DSB [55,56]. The link between local changes in chromatin structure and higher global mobility of the locus is controversial. In MEFs, despite decondensation of the local chromatin surrounding a damaged locus, its mobility did not change significantly, indicating the absence of large-scale motion [31]. By contrast, in haploid budding yeast, constant binding of INO80 to a locus enhances its mobility [26]. The diffusion coefficient, the confinement radius, and the number of large steps taken by the locus all show a significant increase when INO80 is bound. Importantly, ICM requires the ATPase activity of INO80, as shown by the absence of increased DNA mobility in an ATPase-defective mutant, INO80K737A [26]. In addition, increased mobility at a DSB is dependent in part on the INO80 complex, because the mobility of the damaged locus does not increase as much in arp8Δ cells, a nonessential subunit of the complex [26]. Further studies will help elucidate the role of the different chromatin remodelers in DNA mobility.
Chromatin proteins
Highly repetitive sequences are enriched in heterochromatin domains. In the presence of DSBs, the risk of rearrangements between repetitive sequences compromises genome integrity. When a DSB forms in the heterochromatin, several steps occur: DSB detection, checkpoint activation, and resection. In MEFs, HeLa cells [27], and Drosophila cells, these steps are followed by a dramatic increase in the movement of DSBs in heterochromatic regions toward euchromatic regions [6]. This relocalization is thought to prevent rearrangements between repetitive DNA sequences present in heterochromatin. In Drosophila, Rad51 foci form only after the heterochromatic DSBs move to the periphery of the heterochromatic region, a relocalization that is dependent on the Smc5/6 complex, which is recruited by heterochromatin protein 1a. Similarly, in budding yeast, the Smc5/6 complex prevents the formation of DSB-induced Rad52 foci in the nucleolus and suppresses spontaneous recombination of rDNA repeats [57].
Genes essential for regulating chromosome mobility after DNA damage
RAD51
Rad51, a central HR protein, is a homolog of RecA that binds the single-strand tail of a processed DNA end to form a nucleoprotein filament. The Rad51–ssDNA complex, with the help of Rad54, performs the homology search to bind a homologous template and helps restore any lost information caused by the DSB. RAD51 is essential for increasing the mobility of the damaged locus seen in the presence of an I-SceI-induced DSB in both haploid and diploid yeast cells [24,25] (Table 1). In addition, RAD51 deletion inhibits homologous pairing in diploid yeast, probably due to the absence of efficient homology search [24]. In the case of unrepairable DSBs in haploid yeast cells, their movement to the nuclear periphery is dependent on Rad51 [58,59], the histone variant H2A.Z, its SUMO modification, and the DNA-damage checkpoint controlled by Rad9 and Rad24 [59]. The mechanisms promoting homologous pairing in diploid cells and relocalization of unrepairable damaged DNA to the periphery are both Rad51 dependent; however it is unknown whether both processes are controlled similarly. In Drosophila, RAD51 is also important for the increased mobility of heterochromatic DSBs, indicating that the essential role of RAD51 in DNA mobility is conserved in different organisms [6].
RAD54
The RAD54 gene is also essential for increasing the mobility of an unrepairable damaged locus in haploid yeast cells [25]. Disrupting the ATPase activity of Rad54 (K341A) blocks ICM in the presence of a DSB. Interestingly, Rad54 is not required for the pairing of the mating type locus (MAT) and the silent donor locus on chromosome III during mating-type switching, a process that is initiated by a site-specific DSB at MAT [60]. It is possible that Rad54 is required only for large-scale homology search and not for intrachromosomal pairing. In mammalian cells, a Rad54 ATPase mutation blocks the redistribution of foci from the nuclear periphery. The role of Rad54 has not yet been tested in diploid yeast cells.
SAE2
Sae2 acts upstream of Rad51 and is involved in the resection of broken DNA ends for both enzymatically generated and ‘dirty’ ends [61]. In sae2Δ cells, the formation of Rad52 foci at the sites of damage is delayed and they last longer [62]. Unlike RAD51, SAE2 is not essential for ICM in the presence of DSBs [24]. In sae2Δ cells, the increase in both pairing and chromosome mobility is simply delayed by approximately 2 h [24].
RAD9, SML1, MEC1, and RAD53
Rad9, a cell-cycle checkpoint protein activated by the Mec1/Tel1 kinase cascade in a phosphorylation-dependent manner, recruits Rad53 to the site of DSBs [63]. Rad9 contains a BRCT domain roughly equivalent to human MDC1, BRCA1, and 53BP1 [64,65]. Increased mobility of an I-SceI-induced DSB is suppressed in haploid rad9Δ cells [25] (Table 1). MEC1 and SML1, but not RAD53, are essential for increasing the mobility of the damaged locus [25]. Thus, some checkpoint proteins that act earlier than Rad51 nucleoprotein filament formation are essential for increasing the mobility of the damaged locus. In Drosophila, heterochromatin expansion and relocalization requires checkpoint and resection proteins, indicating that their essential role in DNA mobility is conserved in different organisms [6].
53BP1
In mouse cells, increased chromosome movements are associated with uncapped telomeres and this movement is dependent on the 53BP1 repair protein [8]. By contrast, U2OS cells with decreased 53BP1 levels still exhibit increased DNA mobility in the presence of ionizing radiation-induced DSBs [28]. These conflicting results may reflect the difference between an uncapped telomere, which is formed from repetitive DNA that binds sequence-specific proteins, and a DSB [66]. Additional studies are needed to clarify the role of 53BP1 in DNA mobility after damage.
Taken together, these results show that many genes involved in homologous recombination are essential for increased DNA mobility in the presence of a DSB (Figure 1). However, to have a comprehensive view of the mechanisms controlling DNA mobility in the context of DNA damage, it will be important to test many other genes. Furthermore, because most yeast studies used Rad52 as a marker for the presence of DSBs, it will be important to use tagged proteins that act upstream to learn more about DNA mobility before the formation of Rad52 foci.
Figure 1.
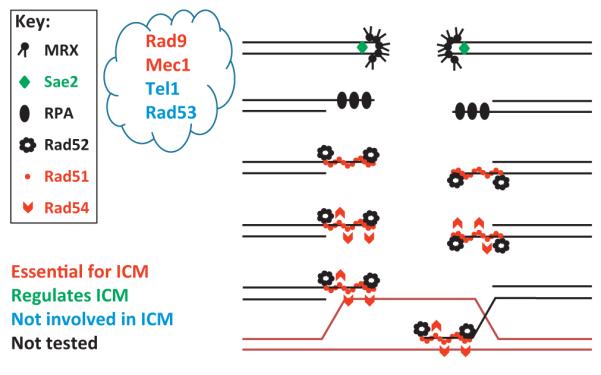
Depiction of the different homologous recombination (HR) proteins and their role in increasing the local mobility of the damaged site. Red: recombination proteins that are essential for increased chromosome mobility (ICM). Green: recombination proteins that regulate ICM by changing its timing. Black: recombination proteins that are not involved in ICM. Grey: recombination proteins that have not yet been tested. It is unknown precisely at which steps the proteins in the ‘cloud’ act.
General increased mobility after DNA damage
ICM after DNA damage is not an intrinsic property of homologous pairing. In diploid yeast, after induction of four random DSBs per nucleus by γ-irradiation, undamaged loci explore a 2.4 times larger nuclear volume than in the absence of irradiation [24]. In addition, after the induction of approximately 20 DSBs, the chromosomes explore almost the entire nucleus (Figure 2). Inducing DSBs on chromosome III using the HO endonuclease also increases the mobility of undamaged loci on chromosome V, indicating that general ICMis not specific to γ-irradiation-induced DSBs [24]. Interestingly, induction of a DSB at the ZWF1 locus in a haploid yeast cell did not increase the mobility of an undamaged locus on another chromosome [25]. Although the results of some of these studies differ, probably due to differences in cell ploidy, it may be that the presence of a single DSB does not significantly induce general ICM, whereas the induction of two or more DSBs alters chromosome mobility across the whole genome [67].
Figure 2.
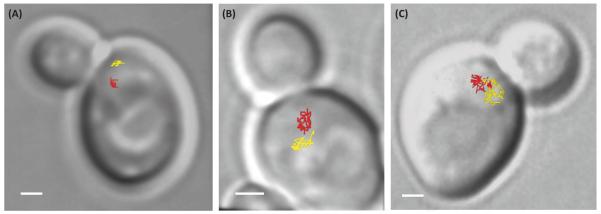
Examples of the dynamics of URA3 loci (chromosome V) in budding yeast as a function of the number of double-strand breaks (DSBs) in the nucleus. The lines indicate a 2D projection of the trajectories of the two URA3 loci taken at between 10- and 30-s time intervals for approximately 15 min. (A) In the absence of DSBs, the two homologous loci are distant and explore only 3% of the nuclear volume. (B) In the presence of one to four DSBs induced on chromosomes III, the mobility of the URA3 loci increases and each locus can explore 11% of the nuclear volume. (C) After about 20 random γ-irradiation-induced DSBs per nucleus (200 Gy), URA3 loci explore almost the entire nuclear volume and their trajectories overlap. The scale is 1 μm. These three examples illustrate that more DSBs in the nucleus induce greater DNA mobility, thereby increasing the probability of collisions between loci. With permission from [72].
To explain how the mobility of undamaged chromosomes increases after DNA damage, we propose two models, which are not mutually exclusive (Figure 3). One we name the ‘collision model’, in which collisions between the damaged locus and undamaged loci are sufficient to provoke genome-wide increased mobility. In this model, the damaged locus ‘hits’ many double-stranded DNA (dsDNA) sequences during the homology search. Two lines of evidence support this model. First, during in vitro homology search, Rad54 facilitates chromatin remodeling [68] and unwinding of the intact dsDNA that is being tested for homology [69]. Second, the recruitment of the chromatin remodeler INO80 to a specific locus increases its mobility in the absence of DSBs [26]. Taking this evidence together, we propose that simple collisions of the nucleoprotein filament, covered by chromatin remodelers, with potential dsDNA targets in nearby chromosomes might be sufficient to provoke general ICM across the entire genome. The presence of several DSBs and thus several nucleoprotein filaments in the nucleus would then increase the number of collisions with undamaged chromatin and further increase general ICM. Importantly, in the collision model, the presence of a nucleoprotein filament is required to increase the mobility of undamaged loci. Thus, in this model, the increased mobility of both the damaged locus and undamaged loci comes from the same pathway.
Figure 3.
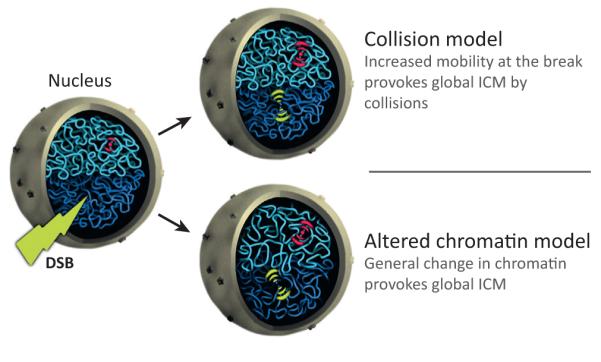
The ‘collision’ and ‘altered chromatin’ models for increased chromosome mobility (ICM). A cut view of the nucleus is depicted. Dark blue represents the general chromatin state of one chromosome and light blue that of another. The mobility of a single intact locus on the light blue chromosome is colored red and the surrounding waves represent its mobility (from one to three, which reflect different confinement radii). Before a double-strand break (DSB), there is limited mobility (one wave on each side of the red focus). After a DSB, the mobility of the broken locus increases locally (three yellow waves on each side). In the collision model, the mobility of the intact red locus increases (shown as two red waves on each side of the locus) due to collisions with the broken locus provoking global ICM. In the altered chromatin model, the state of the chromatin, depicted as less entangled, changes globally as a result of DNA damage. This change in state increases the mobility of the whole genome (represented by the two waves surrounding the red locus) independently of the local ICM at the damaged locus (three yellow waves on each side).
An alternative model, the ‘altered chromatin model’, proposes that general ICM following DNA damage results from changes in the flexibility/topology of chromatin throughout the genome at both damaged and undamaged loci, probably controlled by DNA damage-sensing proteins (Figure 3). Unlike the collision model, in the altered chromatin model general ICM does not depend on nucleoprotein filament formation and thus RAD51. Indeed, changes in chromatin flexibility would occur on both damaged and undamaged DNA before the formation of the nucleoprotein filament. To explain the greater mobility of broken ends near the damaged site (local ICM), we propose that additional local mobility at the damaged locus depends on the presence of the Rad51 nucleoprotein filament. The altered chromatin model is consistent with the fact that local ICM depends on RAD51, RAD54, and SAE2 as well as MEC1 and RAD9, but not on RAD53 [24,25]. Importantly, the model predicts that local ICM at the site of the damage and general ICM across the whole genome are dependent on two separate pathways. Further genetic experiments are necessary to better understand the mechanism of ICM.
Concluding remarks
Recent studies have shown that chromosome movement is important in the context of DNA repair. In budding yeast, the damaged locus is the most mobile following a DSB (local ICM), but undamaged loci are also affected (general ICM). It is unknown whether ICM after DNA damage is due to physical collisions between the damaged locus and the rest of the genome (collision model) or due to a general change in chromatin flexibility (altered chromatin model). To elucidate the mechanisms controlling ICM, it will be important to conduct comprehensive studies to find mutations that alter chromosome mobility. In larger eukaryotes, ICM appears to be dependent on cell type, level of differentiation, and the state of the chromatin that is damaged. After the damage is repaired, it is also important to understand how chromosomes reorganize. Because chromosome mobility increases dramatically after DNA damage, there must be a mechanism to regulate this reorganization so that the normal cell cycle can resume. Perhaps this reorganization is linked to the completion of cellular checkpoint functions. Ultimately, ICM may be indicative of an underlying cellular problem and its detection may lead to new ways of diagnosing diseases.
Acknowledgments
The authors thank P. Thorpe, M. Lisby, I. Cisse, and Vincent Recamier for helpful comments on this manuscript and Myles Marshall for drawing Figure 3. This work was financially supported by an EMBO Long-Term fellowship (J.M-H.), a Marie Curie International Outgoing Fellowship (J.M-H.), the Bettencourt Foundation (J.M-H.), a postdoctoral award from the Philippe Foundation (J.M-H.), and a grant from the NIH (GM67055 to R.R.).
References
- 1.Strickfaden H, et al. 4D chromatin dynamics in cycling cells: Theodor Boveri’s hypotheses revisited. Nucleus. 2010;1:284–297. doi: 10.4161/nucl.1.3.11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cremer C, et al. Detection of laser–UV microirradiation-induced DNA photolesions by immunofluorescent staining. Hum. Genet. 1980;54:107–110. doi: 10.1007/BF00279058. [DOI] [PubMed] [Google Scholar]
- 3.Marshall WF, et al. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. 1997;7:930–939. doi: 10.1016/s0960-9822(06)00412-x. [DOI] [PubMed] [Google Scholar]
- 4.Heun P, et al. Chromosome dynamics in the yeast interphase nucleus. Science. 2001;294:2181–2186. doi: 10.1126/science.1065366. [DOI] [PubMed] [Google Scholar]
- 5.Berger AB, et al. High-resolution statistical mapping reveals gene territories in live yeast. Nat. Methods. 2008;5:1031–1037. doi: 10.1038/nmeth.1266. [DOI] [PubMed] [Google Scholar]
- 6.Chiolo I, et al. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. 2011;144:732–744. doi: 10.1016/j.cell.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soutoglou E, et al. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimitrova N, et al. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dostie J, Bickmore WA. Chromosome organization in the nucleus - charting new territory across the Hi-Cs. Curr. Opin. Genet. Dev. 2012;22:125–131. doi: 10.1016/j.gde.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Meaburn KJ, Misteli T. Cell biology: chromosome territories. Nature. 2007;445:379–781. doi: 10.1038/445379a. [DOI] [PubMed] [Google Scholar]
- 11.Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 12.Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 13.Meshorer E, et al. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masui O, et al. Live-cell chromosome dynamics and outcome of X chromosome pairing events during ES cell differentiation. Cell. 2011;145:447–458. doi: 10.1016/j.cell.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinde E, et al. Tracking the mechanical dynamics of human embryonic stem cell chromatin. Epigenetics Chromatin. 2012;5:20. doi: 10.1186/1756-8935-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roix JJ, et al. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 2003;34:287–291. doi: 10.1038/ng1177. [DOI] [PubMed] [Google Scholar]
- 17.Meaburn KJ, et al. Spatial genome organization in the formation of chromosomal translocations. Semin. Cancer Biol. 2007;17:80–90. doi: 10.1016/j.semcancer.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misteli T. The inner life of the genome. Sci. Am. 2011;304:66–73. doi: 10.1038/scientificamerican0211-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meister P, et al. Visualizing yeast chromosomes and nuclear architecture. Methods Enzymol. 2010;470:535–567. doi: 10.1016/S0076-6879(10)70021-5. [DOI] [PubMed] [Google Scholar]
- 21.Chuang CH, et al. Long-range directional movement of an interphase chromosome site. Curr. Biol. 2006;16:825–831. doi: 10.1016/j.cub.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 22.Kitamura E, et al. Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell. 2006;125:1297–1308. doi: 10.1016/j.cell.2006.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aten JA, et al. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 2004;303:92–95. doi: 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- 24.Mine-Hattab J, Rothstein R. Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 2012;14:510–517. doi: 10.1038/ncb2472. [DOI] [PubMed] [Google Scholar]
- 25.Dion V, et al. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 2012;14:502–509. doi: 10.1038/ncb2465. [DOI] [PubMed] [Google Scholar]
- 26.Neumann FR, et al. Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev. 2012;26:369–383. doi: 10.1101/gad.176156.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakob B, et al. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011;39:6489–6499. doi: 10.1093/nar/gkr230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krawczyk PM, et al. Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 2012;125:2127–2133. doi: 10.1242/jcs.089847. [DOI] [PubMed] [Google Scholar]
- 29.Nelms BE, et al. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–592. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- 30.Jakob B, et al. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl. Acad. Sci. U.S.A. 2009;106:3172–3177. doi: 10.1073/pnas.0810987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kruhlak MJ, et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falk M, et al. Chromatin dynamics during DSB repair. Biochim. Biophys. Acta. 2007;1773:1534–1545. doi: 10.1016/j.bbamcr.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Strukov YG, Belmont AS. Development of mammalian cell lines with lac operator-tagged chromosomes. CSH Protoc. 2008 doi: 10.1101/pdb.prot4903. pdb.prot 4903. [DOI] [PubMed] [Google Scholar]
- 34.Qian H, et al. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 1991;60:910–921. doi: 10.1016/S0006-3495(91)82125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazsuzawa N, et al. Constrained, random, and independent motion of Texas-red labeled chromatin in living interphase PtK2 cells. Acta Histochem. Cytochem. 2000;33:419–427. [Google Scholar]
- 36.Vazquez J, et al. Multiple regimes of constrained chromosome motion are regulated in the interphase Drosophila nucleus. Curr. Biol. 2001;11:1227–1239. doi: 10.1016/s0960-9822(01)00390-6. [DOI] [PubMed] [Google Scholar]
- 37.Edelmann P, et al. Morphology and dynamics of chromosome territories in living cells. Biochim. Biophys. Acta. 2001;1551:M29–M40. doi: 10.1016/s0304-419x(01)00023-3. [DOI] [PubMed] [Google Scholar]
- 38.Chubb JR, et al. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. 2002;12:439–445. doi: 10.1016/s0960-9822(02)00695-4. [DOI] [PubMed] [Google Scholar]
- 39.Gorisch SM, et al. Mobility of multi-subunit complexes in the nucleus: accessibility and dynamics of chromatin subcompartments. Histochem. Cell Biol. 2005;123:217–228. doi: 10.1007/s00418-005-0752-y. [DOI] [PubMed] [Google Scholar]
- 40.Jegou T, et al. Dynamics of telomeres and promyelocytic leukemia nuclear bodies in a telomerase-negative human cell line. Mol. Biol. Cell. 2009;20:2070–2082. doi: 10.1091/mbc.E08-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molenaar C, et al. Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J. 2003;22:6631–6641. doi: 10.1093/emboj/cdg633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heun P, et al. The positioning and dynamics of origins of replication in the budding yeast nucleus. J. Cell Biol. 2001;152:385–400. doi: 10.1083/jcb.152.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gasser SM. Visualizing chromatin dynamics in interphase nuclei. Science. 2002;296:1412–1416. doi: 10.1126/science.1067703. [DOI] [PubMed] [Google Scholar]
- 44.Abney JR, et al. Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J. Cell Biol. 1997;137:1459–1468. doi: 10.1083/jcb.137.7.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levi V, et al. Chromatin dynamics in interphase cells revealed by tracking in a two-photon excitation microscope. Biophys. J. 2005;89:4275–4285. doi: 10.1529/biophysj.105.066670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weber SC, et al. Nonthermal ATP-dependent fluctuations contribute to the in vivo motion of chromosomal loci. Proc. Natl. Acad. Sci. U.S.A. 2012;109:7338–7343. doi: 10.1073/pnas.1119505109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lisby M, et al. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 48.Difilippantonio S, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lisby M, et al. Cell cycle-regulated centers of DNA double-strand break repair. Cell Cycle. 2003;2:479–483. [PubMed] [Google Scholar]
- 50.Lobachev K, et al. Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr. Biol. 2004;14:2107–2112. doi: 10.1016/j.cub.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 51.Kaye JA, et al. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr. Biol. 2004;14:2096–2106. doi: 10.1016/j.cub.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 52.Guasconi V, Ait-Si-Ali S. Chromatin dynamics and cancer. Cancer Biol. Ther. 2004;3:825–830. doi: 10.4161/cbt.3.9.1102. [DOI] [PubMed] [Google Scholar]
- 53.van Attikum H, Gasser SM. ATP-dependent chromatin remodeling and DNA double-strand break repair. Cell Cycle. 2005;4:1011–1014. doi: 10.4161/cc.4.8.1887. [DOI] [PubMed] [Google Scholar]
- 54.Saha A, et al. Chromatin remodelling: the industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 55.van Attikum H, et al. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 56.Morrison AJ, et al. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 57.Torres-Rosell J, et al. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007;9:923–931. doi: 10.1038/ncb1619. [DOI] [PubMed] [Google Scholar]
- 58.Oza P, et al. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009;23:912–927. doi: 10.1101/gad.1782209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalocsay M, et al. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell. 2009;33:335–343. doi: 10.1016/j.molcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 60.Houston PL, Broach JR. The dynamics of homologous pairing during mating type interconversion in budding yeast. PLoS Genet. 2006;2:e98. doi: 10.1371/journal.pgen.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 62.Lisby M, et al. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 63.Finn K, et al. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 2012;69:1447–1473. doi: 10.1007/s00018-011-0875-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stucki M, Jackson SP. MDC1/NFBD1: a key regulator of the DNA damage response in higher eukaryotes. DNA Repair (Amst.) 2004;3:953–957. doi: 10.1016/j.dnarep.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 65.Mochan TA, et al. 53BP1, an activator of ATM in response to DNA damage. DNA Repair (Amst.) 2004;3:945–952. doi: 10.1016/j.dnarep.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 66.Dewar JM, Lydall D. Similarities and differences between “uncapped” telomeres and DNA double-strand breaks. Chromosoma. 2012;121:117–130. doi: 10.1007/s00412-011-0357-2. [DOI] [PubMed] [Google Scholar]
- 67.Dion V, Gasser SM. Chromatin movement in the maintenance of genome stability. Cell. 2013;152:1355–1364. doi: 10.1016/j.cell.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 68.Alexeev A, et al. Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat. Struct. Biol. 2003;10:182–186. doi: 10.1038/nsb901. [DOI] [PubMed] [Google Scholar]
- 69.Mazin AV, et al. Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell. 2000;6:583–592. doi: 10.1016/s1097-2765(00)00057-5. [DOI] [PubMed] [Google Scholar]
- 70.Kawamura R, et al. Mitotic chromosomes are constrained by topoisomerase II-sensitive DNA entanglements. J. Cell Biol. 2010;188:653–663. doi: 10.1083/jcb.200910085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maul GG, Deaven L. Quantitative determination of nuclear pore complexes in cycling cells with differing DNA content. J. Cell Biol. 1977;73:748–760. doi: 10.1083/jcb.73.3.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mine-Hattab J, Rothstein R. DNA repair: finding the perfect match. Med. Sci. (Paris) 2012;28:714–716. doi: 10.1051/medsci/2012288014. (in French) [DOI] [PubMed] [Google Scholar]