

Abstract
Interactions between proteins and soluble carbohydrates and/or surface displayed glycans are central to countless recognition, attachment and signaling events in biology. The physical chemical features associated with these binding events vary considerably, depending on the biological system of interest. For example, carbohydrate-protein interactions can be stoichiometric or multivalent, the protein receptors can be monomeric or oligomeric, and the specificity of recognition can be highly stringent or rather promiscuous. Equilibrium dissociation constants for carbohydrate binding are known to vary from micromolar to millimolar, with weak interactions being far more prevalent; and individual carbohydrate binding sites can be truly symmetrical or merely homologous, and hence, the affinities of individual sites within a single protein can vary, as can the order of binding. Several factors, including the weak affinities with which glycans bind their protein receptors, the dynamic nature of the glycans themselves, and the non-equivalent interactions among oligomeric carbohydrate receptors, have made NMR an especially powerful tool for studying and defining carbohydrate-protein interactions. Here we describe those NMR approaches that have proven to be the most robust in characterizing these systems, and explain what type of information can (or cannot) be obtained from each. Our goal is to provide to the reader the information necessary for selecting the correct experiment or sets of experiments to characterize their carbohydrate-protein interaction of interest.
INTRODUCTION
Complex glycan or carbohydrate structures are displayed from the surfaces of all cells, including pathogens and viruses. Interactions between glycans and their protein receptors guide many biological processes and recognition events. Illustrating the diversity of biology that carbohydrate interactions control, examples include leukocyte homing that is governed by interactions between sialic acid terminated glycans and L-selectins;1 influenza infections that require binding of the hemagglutinin to Siaα2-6Gal linkages present on human target cells;2,3 tumor cell adhesion and metastasis that can be facilitated by interactions between beta-lactosamine (Gal-GlcNAc) containing glycans and galectins,4,5 some of which are soluble rather than membrane bound; and recruitment of nitrogen fixing bacterial symbionts in the roots of legumes through interactions between chitooligosaccharides and rhizome receptors.6,7 The ranges of physical properties that characterize these carbohydrate-protein interactions are as diverse as the processes they facilitate. In leukocyte homing, dynamic binding and dissociation is necessary to allow the leukocytes to ‘roll’ over the vasculature; hence very high affinity or avidity would be incompatible. At the other end of the spectrum, carbohydrate-protein interactions necessary for cell-cell or virus-cell adhesion and infection may require higher avidity and longer lived interactions.
Many glycan-mediated interactions embody therapeutic targets.8–11 Their detailed chemical and structural descriptions are necessary for the design and synthesis of small molecule and carbohydrate-based inhibitors, and production or development of protein-based inhibitors. While many of the approaches used to characterize carbohydrate-protein interactions are analogous to those used for other systems, such as protein-protein or protein-nucleic acids, there are often times limitations for carbohydrate-based systems that trace to the low abundance and/or relative difficulty of obtaining complex glycans in quantities sufficient for comprehensive studies. In addition, the range of affinities with which carbohydrates bind their protein receptors imposes limitations on the methods that can be used. To this end, solution NMR methods have proven to be extremely useful in defining carbohydrate-protein interactions, and the number of NMR approaches that can be employed enables the study of almost any type of interaction, albeit to varying degrees of ‘resolution’. Several outstanding books or reviews have been published that cover the theory of these NMR-based approaches.12–15 Our goal here is to describe various types of interactions made between carbohydrate-protein interactions (Figure 1), and to provide to the non-specialist experimental approaches that can be taken to study each type, illustrated by recent examples.
Figure 1.

Schematic representation of different types of carbohydrate-protein interactions.
Types of NMR Approaches: Protein-based versus Ligand-based
When setting out to characterize a carbohydrate-protein interaction, several factors must first be considered before deciding on which NMR approach to use. These include the molecular weight of the protein and the carbohydrate, the solubility and behavior of each in solution, and the amount and/or source of each that is available to the researcher. In addition, the level of information that is being sought also is a determining factor. To obtain high-resolution structures of a carbohydrate-protein complex, it will be necessary to uniformly label the protein of interest with 13C and 15N stable isotopes and to obtain full chemical shift assignments of both protein and carbohydrate. Because of the limitations on the size of proteins that can be routinely assigned by NMR using standard labeling techniques, these complexes will ideally fall into the size range of 10–25 kDa. In addition, the protein must be soluble and non-aggregating in aqueous buffers, and milligram quantities of both molecules must be attainable. With ligand-based methods on the other hand, 1H-detected NMR spectra of only the carbohydrate are analyzed, thus obviating the need for larger quantities of labeled protein and protein chemical shift assignments. Situated between these two approaches in terms of the amount of information obtained, and the types of chemical shift assignments and spectrometer time required are methods such as chemical shift mapping of the carbohydrate binding to the protein, or determination of equilibrium dissociation constants (Kds) of very weak interactions by transferred NMR techniques. These are described in more detail below.
PROTEIN DETECTED METHODS
Overview
Protein NMR methods can be used to obtain valuable information about carbohydrate-protein interactions. The most commonly used examples include determining high resolution structures of carbohydrate-binding proteins in complex with their ligands, mapping carbohydrate binding site/s onto 3-dimensional structures of proteins, and identifying carbohydrate ligands and determining fine carbohydrate specificity for a given protein. Because these are protein-detected methods that rely on comparing differences between NMR spectra of proteins in their free and carbohydrate-bound state, it is necessary to assign the backbone amide (1HN) resonances of a uniformly 15N-labeled recombinant protein. Due to the cost associated with mammalian cell culture media, recombinant proteins to be used for NMR studies are almost always expressed in standard E. coli-based systems using T7-inducible plasmids.16 Growth of the bacteria and expression of the protein in minimal media supplemented with 15N-NH4Cl and/or 13C-glucose, followed by an optimized purification protocol usually supplies sufficient quantities of labeled protein to prepare multiple NMR samples. The amount of uniformly labeled protein required will depend on the NMR instrumentation and amount of spectrometer time available. On a 500 or 600 MHz NMR equipped with a room temperature 5 mm probe, approximately 300 μL of a 0.1–1 mM sample is optimal, with the upper concentration being necessary only for determining 3-dimensional structures of a complex. If equipped with a 1.7 mm cryogenically cooled, triple resonance ‘micro’ probe, as little as 30 μL of a low μM solution may be sufficient. Numerous NMR experiments and software programs that assist with assignments and analysis are available, making it possible to complete backbone assignments for many 10–20 kDa proteins within one week. If a 3-dimensional solution structure is to be determined, additional experiments that provide side chain 1H and 13C assignments must be recorded, as will a series of 15N– and 13C–separated NOESY spectra that will provide distance restraints necessary for structure calculations. Acquisition and analysis of these additional data sets can take weeks to months.14,17 Common to any of the above protein-detected approaches is the need for the protein to be soluble at high micromolar concentrations.
Mapping Carbohydrate Binding Site/s onto a Protein
By recording and interpreting a series of NMR experiments that select for couplings between different pairs or combinations of backbone 13Cα, 13CO, 15NH and HN, and 13Cβ resonances (examples include CBCACONH, CBCANH, HNHA, HNCO experiments), one can rapidly obtain full resonance assignments for the backbone amide 1H and 15N resonances of a carbohydrate binding protein. Together with the 3-dimensional structure of the protein, this allows one to identify and then map the carbohydrate binding site/s onto the protein structure by recording 1H-15N correlation spectra in the absence and presence of carbohydrate. In addition to identifying the carbohydrate binding site/s by chemical shift perturbations, the types of amino acids and therefore the chemical features contributing to carbohydrate binding will be revealed. These experiments are best carried out by titrating carbohydrate into the protein sample and recording 1H-15N HSQC spectra after each addition. If sufficient amounts of glycan are available, it is recommended to continue the titration until a stoichiometric excess of ligand has been added to the solution. By monitoring the chemical shift perturbations over the course of a titration, this data set also will reveal whether the ligand is in slow, intermediate or fast exchange with the protein; and the stoichiometry of binding. Examples that illustrate the diversity of the types of interactions that can be revealed by NMR are outlined next.
Bivalent, 2-site binding: cyanovirin-N
One example of a carbohydrate-protein complex that exemplifies several of the phenomena mentioned above is the HIV-inactivating protein cyanovirin-N (CV-N)18 binding to α(1-2)mannobiose-containing glycans. CV-N was one of the first glycan-dependent HIV entry inhibitors identified.19 On account of its remarkable potency and novel protein fold, numerous structural, functional and chemical studies have been undertaken with this protein.20–27 Using NMR, we solved the solution structure of CV-N and thus had in hand full chemical shift assignments.20 After demonstrating that CV-N was a lectin that binds high mannose N-linked oligosaccharides, we performed an NMR based screen by recording 1H-15N HSQC spectra of CV-N each in the presence of 1 eq of the di- and tri-saccharide fragments comprising oligomannose-9 (Figure 2a).18,24 Overlays of each of these NMR spectra onto the spectrum of free CV-N demonstrated that CV-N had strict specificity for α(1-2)mannobiose and did not bind to any of the fragments that corresponded to core regions of Man-9. After identifying the carbohydrate ligand, we performed a titration by adding sub-stoichiometric amounts of Manα1-2Man to 15N-CV-N. 1H-15N HSQC spectra of each of these solutions showed that CV-N contained not one but two carbohydrate binding sites, both of which are specific for Manα1-2Man. In addition, the spectra clearly revealed that the two carbohydrate binding sites differed in their affinity for the disaccharide, and were therefore non-equivalent (Figure 2b). This was determined by the following NMR observations: first, upon titrating sub-stoichiometric amounts of Manα1-2Man to 15N-CV-N, two sets of peaks were present in the 1H-15N HSQC spectra (Figure 2c). As a number of peaks belonging to free CV-N diminished in intensity, a new set of peaks comprising 18 residues appeared, and these mapped to one domain of the pseudo-symmetric protein. After 1 eq of disaccharide had been added, each of the resonances belonging to residues in this site had disappeared and only resonances assigned to the bound protein were present. This proved that CV-N contained one high affinity Manα1-2Man binding site that is in slow exchange on the NMR time scale. With further addition of Manα1-2Man to CV-N, a second set of resonances that mapped to the opposite side of the protein was perturbed (Figure 2d). However, rather than moving to a new set of cross peaks, this second set of resonances either broadened, disappeared, or moved very slightly in chemical shift. No further changes in the NMR spectra were detected after addition of a 10-fold excess of disaccharide. The latter result proved that CV-N contains a second lower affinity Manα1-2Man binding site. NMR was later used to solve a solution structure of the 2:1 Manα1-2Man:CV-N complex, showing that the binding was governed by many hydrogen bonds.28 It is noteworthy that identification of the binding site residues by chemical shift mapping, together with microcalorimetry, correctly defined binding as being dominated by electrostatic interactions.24
Figure 2.

NMR characterization of Manα1-2Man binding to CV-N. a) Chemical structures of Man9GlcNAc2 (Man-9) and Manα1-2Man; b) location of two symmetrically located binding sites on CV-N, identified by chemical shift mapping; c, d) chemical shift perturbations occurring with addition of 1 and 2 eq Manα1-2Man.
Monovalent binding with intermediate affinity: microvirin-N
Carbohydrate binding proteins generally bind their carbohydrate ligands with weak affinities in the mM range. Often the low affinity is compensated for by oligomerization of the protein, which can in turn participate in multivalent interactions. An interesting example of a monovalent lectin that binds its carbohydrate ligand with micromolar affinity is the cyanobacterial protein microvirin (MVN),29 a homolog of CV-N. MVN was shown by glycan array profiling and NMR titrations to bind very selectively to Manα1-2Man, and therefore Man-8 and Man-9 (Figure 2a).29,30 Prior to determining a solution structure of an MVN-mannobiose complex, NMR titrations were performed using up to a 25-fold excess of Manα1-2Man (Figure 3a). These data revealed that MVN contains only one carbohydrate binding site. This was confirmed by NMR titrations with the branched oligomannosides Man-8 and Man-9, where addition of these complex carbohydrates induced similar chemical shift perturbations as the disaccharides, but did not lead to polymerization as observed for CV-N.30 Last, we used NMR methods to solve the solution structure of MVN in complex with this disaccharide (Figure 3b).30 Crystallization attempts with MVN and carbohydrate-MVN complexes were unsuccessful; for this protein, structures and carbohydrate recognition profiles could only be obtained by NMR.
Figure 3.

Chemical shift mapping and stoichiometry of α1,2-mannobiose binding to microvirin (MVN). a) Chemical shift perturbations as a function of residue number. No perturbations were observed for the second domain; b) location of the carbohydrate binding site on MVN, colored as in panel (a).
Binding in a carbohydrate chain length-dependent manner: the bactericidal lectin RegIII
The bactericidal lectin RegIII is a small, secreted C-type lectin that kills Gram-positive bacteria in the small intestine. Through the use of NMR Lehotzky et al. determined the molecular basis for peptidoglycan recognition.31 In particular, 1H-15N HSQC spectra of RegIII in the presence of various monosaccharides or short oligosaccharides representing peptidoglycan or other polysaccharide fragments demonstrated that this lectin only bound to chitooligosaccharides [(β1-4GlcNAc)n]. NMR titrations employing different chitooligosaccharides further showed that binding affinity for carbohydrate ligands was dependent on carbohydrate chain length. A Kd value of 5 mM was determined for RegIII binding to chitopentaose through NMR titrations and chemical shift perturbations. In this system binding is very weak with ligand binding occurring in fast exchange on the NMR time scale. This gives rise to small but measureable changes in 1H and 15N resonances in the NMR titration spectra. The combined data led this group to propose a binding model wherein the carbohydrate binding domains of RegIII bind and jump along an extended polysaccharide chain of peptidoglycan (Figure 1d), a phenomenon that can reduce dissociation rates and increase apparent binding affinities.32,33 This system provides an elegant example of the value of NMR in characterizing carbohydrate-protein interactions that in turn lead to new biology. In other systems, binding would not have been detected, and the observed discrimination between carbohydrate chain length would have gone unnoticed.
Intermolecular Cross-Linking
Another capability of NMR is to detect aggregation that can occur when carbohydrate-binding proteins participate in intermolecular cross-linking with branched oligosaccharides. This is a form of a multivalent interaction that occurs when the distance between two carbohydrate-binding sites on a multivalent protein is greater than the distance between the two arms of the glycan, and therefore does not support chelation (Figure 1e). This can lead to different outcomes; if a protein that contains more than one carbohydrate binding site is able to engage each branch of a complex carbohydrate through different sites on the protein, then a polymerization event can occur. When performing an NMR titration on this type of system, as larger molecular weight species are formed, the peaks in a 1H-15N correlation spectrum will either broaden, or disappear altogether, due to the much faster relaxation of very large molecular weight species that are undectable by NMR. For CV-N, this was readily apparent after addition of only 0.1–0.2 eq Man9GlcNAc2 to the solution, at which point peaks were barely detectable in the NMR spectra.18
These examples demonstrate that having the ability to detect carbohydrate binding to a protein by analysis of 1H-15N correlation spectra can be a powerful approach to characterize a wide variety of carbohydrate-binding interactions. Although it requires an initial investment in making backbone 1H and 15N chemical shift assignments, the method provides a rich amount of data for the study of these systems.
CARBOHYDRATE LIGAND BASED APPROACHES
Overview
In ligand based NMR approaches, the most commonly used experiments employ 1H NMR methods and rely on the transfer of NMR parameters from the macromolecule to the smaller molecular weight ligand.12,13 In these systems, large molecular weight receptors have certain properties, including fast relaxation, slow rotation and translation, and efficient spin diffusion. Small molecular weight ligands in contrast relax slowly and rotate and translate more rapidly than large proteins. Ligand based NMR methods take advantage of these differences in the NMR parameters of protein and ligand. The NMR samples used in ligand-based experiments contain on the order of 1–50 μM protein in the presence of approximately 20- to 100-fold excess of the ligand. (Typical ligand concetrations range from 0.2–2 mM.) As the ligand is freely exchanging between the bound and free state, it retains the NMR properties of its larger protein receptor. These differences can be detected by NMR and provide information about the bound state of the ligand. For the study of carbohydrate-protein interactions, ligand based NMR approaches offer several advantages over receptor-based NMR methods. There is no need to introduce labels to either protein or carbohydrate; these methods do not require chemical shift assignments of the protein thus reducing instrument and data analysis time; relatively small amounts of protein are needed; and there is no size limit imposed on the protein, indeed some methods can even be carried out with cells or viral particles. However, to obtain the greatest amount of information from ligand-based NMR experiments, 1H chemical shift assignments of the ligand must be made using standard NMR approaches. Finally, ligand based approaches are amenable to studying carbohydrate-protein interactions that span a wide range of affinities, typically from high nanomolar to millimolar Kds.
Mapping Receptor Interaction Sites onto Glycans using STD NMR
One of the most commonly used NMR methods for detecting ligand binding is Saturation Transfer Difference (STD) NMR.12,34 This method works on the same premise of spin-diffusion as described above. When performing STD NMR, two spectra are recorded. In the ‘on-resonance’ spectrum, the protein is selectively saturated by applying radio frequency (RF) pulses on a region of the spectrum that contains protein, but not ligand resonances. Saturation of the protein is transferred to the ligand in a distance dependent manner so that protons in closest proximity to the protein will experience the largest effects. In the ‘off-resonance’ or reference spectrum, the saturation RF pulse is applied to a region where signals are absent. Subtraction of the on-resonance spectrum from the off-resonance spectrum gives a difference spectrum wherein the 1H signals present correspond to protons at the interface of the complex (Figure 4). Because the peak intensities in the difference spectrum are proportional to the distance between protons belonging to the ligand and those belonging to the protein, STD NMR allows mapping of the epitope used by the carbohydrate to bind its protein receptor.34 STD NMR has proven to be an invaluable method for the study of carbohydrate-protein interactions. Below we provide several examples that illustrate the multiple applications in which STD NMR can be used.
Figure 4.
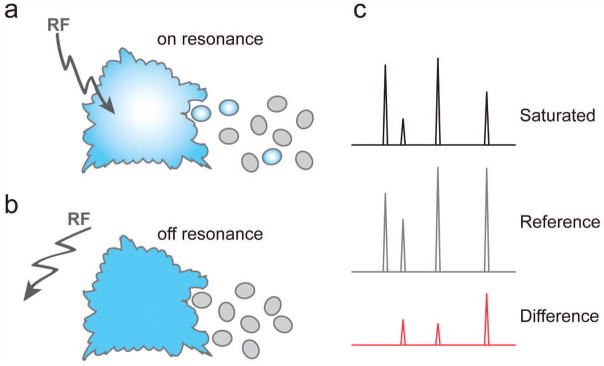
Schematic of and STD NMR experiment. Depiction of a) on-resonance (saturated) and b) off-resonance (control) experiments where saturation diffuses to the ligand in the on-resonance experiment. c) Subtraction of the spectra give a difference spectrum where the peak intensities are proportional to the distance of the 1Hs from the receptor.
Detecting weak interactions: carbohydrate recognition and specificity by the potent HIV neutralizing antibody PG16
Very recently, as part of several large HIV vaccine efforts, two classes of potent, broadly neutralizing monoclonal antibodies (bNAbs) have been discovered.35,36 These bNAbs recognize the HIV envelope glycoprotein gp120. They are highly unusual in that they have a strict requirement for glycan-dependent interactions. The glycans they target are located on or proximal to the variable loops V1/V2 or V3. While it could be shown using standard screening approaches such as glycan array profiling or ELISAs that one group of neutralizing mAbs (including 2G1237 and PGT12838) binds V3 high mannose oligosaccharides with high affinities, the glycan preferences for another set of neutralizing antibodies that bind V1/V2 could not be determined using commonly employed methods (see below).36 In fact, binding of the PG9- and PG16-like antibodies could not even be detected using higher density carbohydrate displays39 and carbohydrate-linked dendrons.40 We turned to STD NMR and used a glycan- and glycan fragment-screening approach to show that the HIV neutralizing mAb PG16 recognizes both complex-type and hybrid-type glycans (Figure 5). The NMR also revealed that the interaction is dependent on the presence of a terminal sialic acid residue, and is specific for an α2-6 sialic acid linkage. This finding was verified by x-ray crystallography and mutagenesis of the glycan binding sites of PG16.41
Figure 5.

STD NMR spectrum of a complex-type glycan binding to the HIV-1 neutralizing antibody PG16.41 The difference spectrum and chemical shift assignments of the glycan demonstrate recognition of complex-type glycan through the Sial-Gal termini.
Quantifying carbohydrate-protein interactions by STD NMR
In fragment based drug discovery programs, STD NMR is often used simply to detect binding, that is, to give a positive or negative read out.13,42 STD NMR can also be used to determine Kd values. Two general approaches are employed to obtain quantitative information, namely competition STD NMR experiments, and single ligand titrations. In competition STD NMR, the affinity of a ligand can be determined by competing it with another ligand of the same protein whose Kd value is already known.43, 34 The method is especially useful because a wide range of Kd values can be determined, including nM affinities. Generally, high affinity ligands (nM Kds) cannot be detected directly by STD NMR due to the ligand being in slow exchange between its bound and free states on the NMR time scale.12 However, detection of very high-affinity binding ligand with Kd values less than 5 nM can be measured by competing the high affinity ligand with a low affinity ligand.44 Determining binding affinities by competition STD NMR has several requirements, however. First, a ligand having a known Kd is required for use as a reference competitor, and second, the ligand under study should bind to the same binding site on the macromolecule as that of the reference molecule.
In practice, when studying a carbohydrate-protein interaction, a ligand with a known Kd may not be available for your protein of interest. Meyer et al. recently described a single ligand titration STD NMR approach that can be used to determine Kds for any small molecule ligand. In this method, STD NMR spectra are acquired on samples where the ligand concentration is varied while keeping the concentration of the protein constant. Regression analysis of the effect on the STD amplification factors (ASTD) as a function of ligand concentration gives the Kd.45 More recently, Angulo et al., described an improved single ligand titration STD NMR method that addresses competing factors such as relaxation and ligand rebinding, both of which can affect accurate affinity measurements. In their approach they obtained STD amplification factors at zero saturation time (A0STD) from the initial growth rates of the amplification factors with respect to saturation time. Precise Kd values could in turn be obtained from a regression plot of the effect on A0STD at varying ligand concentrations.46
Single ligand titrations: determining Kd values for the HIV neutralizing antibody PG9 binding to Man5GlcNAc2
The HIV-neutralizing antibody PG9 provides a second example of a bNAb whose epitope contains glycans critical for activity. PG9 recognizes a glycopeptide epitope consisting of two β-strands of the V1/V2 domain of HIV-1 gp120, and two glycans linked to Asn 160 (N160), and either Asn 173 (N173) or Asn 156 (N156). As with PG16, attempts to study glycan interactions between PG9 and carbohydrate ligands were unsuccessful using glycan array profiling, SPR and ITC. We used single ligand NMR titrations to determine the affinity of Man5GlcNAc2 binding to PG9. STD NMR spectra were acquired on samples containing 15 μM FAB PG9 and varying concentrations of Man5GlcNAc2, ranging from 0.6 mM to 2 mM, at 0.2 s saturation time. STD amplification factors (ASTD) were obtained by integrating isolated STD signals of N-acetyl protons using the equation ASTD = (I0-ISAT)I0-1 ([Lt]/[P]) where Lt and P are the concentrations of Man5GlcNAc2 and PG9, respectively; and I0 and ISAT are the signal intensities in the reference and saturated spectra, respectively. A Kd of 1.6±0.91 mM for Man5GlcNAc2 binding to PG9 was determined by fitting the ASTD values as the function of ligand concentration to a single-site binding curve according to the equation y=Bmax/Kd + x, where x is the concentration of ligand, and Bmax represents the plateau of the curve (Figure 6).
Figure 6.

Single ligand titration STD NMR of Man5GlcNAc2 binding to the HIV-1 neutralizing antibody PG9. a) Difference spectrum indicates recognition of oligomannose; b) integration and c) fitting of the N-acetyl methyl signals allows determination of the Kd of binding.56
It is significant that these glycan dependent antibodies are among the most promising group of HIV-1 neutralizing antibodies discovered to date. NMR methods have proven to be essential to detecting and defining their carbohydrate-dependent interactions.
Determining specificities: human blood group antigens binding to norovirus protruding domain (P domain)
Human noroviruses are the leading cause of human gastroenteritis outbreaks. Their outer P domains are known to bind to human blood group antigens (HBGAs). To elucidate the properties of norovirus binding to HBGAs, crystal structures of norovirus P domain in the presence of different HBGAs or HBGA fragments were solved by crystallography.47,48 Surprisingly the structures showed that the H-type 2 disaccharide α-L-fucose(1-2)-β-D-galactose, H-type 2 trisaccharide β-L-fucose(1-2)-β-D-galactose(1-4)-2-N-acetyl-β-D-glucosamine, and citrate, bound to the P-domain at the same site and in nearly superimposable modes. The affinities of these interactions were too weak to be detected by other methods. Quantitative STD NMR measurements were used to determine the binding affinities of these ligands, each of which binds the P domain protein with mM Kd values.48 In addition, because β-fucose does not exist in natural HBGAs but was present in the bound trisaccharide ligand, STD NMR was used to investigate whether specificity occurred for one anomer over another. In a standard STD NMR spectrum of L-fucose in the presence of the P domain protein, strong enhancements were observed only for the unnatural β-anomer (blue labels, Figure 7), and were at least 10 times stronger than those for α-fucose. Although that result must be confirmed in the context of viral particles, it provides a clear example of the selectivity of ligands that can be revealed by STD NMR.
Figure 7.

STD NMR spectrum of α/β L-fucose bound to norovirus P domain protein. Binding of the unnatural β-anomer dominates binding to the protein.48
Conformational studies of protein-bound carbohydrates using trNOE
Ligand-based approaches can also be used to determine the conformation of a ligand when bound to its protein partner through the use of transferred nuclear Overhauser effects (NOE). The NOE is a cross relaxation effect that occurs between spatially proximal protons, and is dependent on the correlation time (τc) of the molecule. In macromolecules, which have large correlation times and are rapidly relaxing, cross relaxation is fast and NOEs evolve within a 200–300 ms mixing time giving rise to cross peaks that have the same sign as the diagonal in a 2D NOESY spectrum (negative peaks). Small molecules on the other hand have low cross relaxation rates and require longer mixing times (0.5–2 s), and generally show positive NOEs. In a sample containing an excess of ligand to protein, the observed cross relaxation is an average of the free and bound states. The faster cross relaxation in the bound state can contribute the majority of the NOE, especially with short mixing times. This effect is known as the transferred NOE (trNOE) and allows for transfer of cross relaxation information between two nuclei in their bound to free states.15,49 In a trNOESY spectrum, the sign of the cross peaks of the ligand become negative, and confirm binding to the protein receptor. By integrating the volume of the trNOE cross peaks, inter proton distances can be derived allowing for determination of the overall geometry and 3-dimensional conformation of the ligand in the bound state. For carbohydrates, many of which are inherently flexible and resist crystallization, trNOESY data may be the only way to obtain 3-dimensional information of the bound conformation.50
NMR Diffusion Experiments
Binding of carbohydrates to their protein receptors can also be studied by measuring translational diffusion coefficients (D) using Diffusion Ordered SpectroscopY (DOSY).51 Small molecules diffuse with faster rates than large protein receptors. Therefore, an observed change in D of a carbohydrate in its free state versus D of a carbohydrate in its protein-bound state provides information on binding. This method has an advantage in that it can be used to determine binding without having to assign the chemical shifts for either the carbohydrate or protein. Pulsed field gradient-based DOSY experiments have been extensively used to directly measure the diffusion coefficient of molecules to identify binding and determine equilibrium dissociation constants.52,53 An additional NMR method that has found use in ligand-based screening, but less so for carbohydrate interactions, includes waterLOGSY.54 In this method RF pulses are applied to the water signal, and bulk water magnetization is transferred to the ligand for the detection of ligand binding.
Complimentary methods used in carbohydrate-protein interactions, in brief
There are a number of other approaches that can be used to detect carbohydrate-protein interactions. The utility of each will of course depend on the information being sought, and the abundance and state of the glycan and protein of interest. If each molecule is available in milligram quantities and binding constants are the only information being sought, ITC would be a logical first choice. If the protein is of low abundance and the glycan is plentiful, surface Plasmon resonance (SPR) or bio-layer interferometry (BLI) can be good options. When using SPR, BLI, or any method that requires immobilization of the protein onto a solid support, the choice of immobilization chemistry must be considered carefully. Covalent attachment to a solid surface can be detrimental to the overall structure or activity of, or binding site accessibility in, carbohydrate binding proteins. If the glycan can be modified to contain a chemical linker, preferably at the reducing end, immobilization of the carbohydrate may be another option. In our experience however, it is difficult to eliminate non-specific binding of carbohydrate binding proteins to cellulose surfaces.
Another very useful method for detecting carbohydrate binding and recognition profiles involves screening a labeled protein over a glycan microarray. Like their nucleotide counterparts, glycan microarrays are typically constructed from activated glass slides to which large libraries of chemically modified glycans have been attached.55 This technology has been made freely available to academic researchers through the Consortium for Functional Glycomics (www.functionalglycomics.org). Glycan array screening provides a nice compliment to the other methods discussed here: while glycan profiling provides abundant information about binding to complex glycans or glycan fragments, it does not yield binding constants or kinetics, or structural insight about the interface of the protein involved in the interaction.
SUMMARY
In closing, a number of recent technological developments that allow for rapid screening of protein-carbohydrate interactions, identification of glycan-dependent antibodies, and the use of cell lines that produce aberrantly glycosylated proteins, to name a few, have refueled the research areas of glycobiology, carbohydrate chemistry and chemical biology. Fundamental to controlling or manipulating these important interactions is an understanding of the structural basis for glycan recognition and binding. The flexibility that is inherent to complex glycans often precludes crystallization of both free and protein-bound glycan; and the weak affinities associated with many carbohydrate-protein interactions means that some might go undetected. NMR methods continue to play an important role in characterizing these complex interactions. We have discussed here the applicability of NMR to this field of research, and provided background and examples that should make the method accessible to researchers interested in glycan-dependent interactions.
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institutes of Health (NIDDK and NIAID).
References
- 1.Varki A. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- 2.Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Nature. 2006;440:435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 3.Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, Raguram S, Tumpey TM, Sasisekharan V, Sasisekharan R. Nat Biotechnol. 2008;26:107–113. doi: 10.1038/nbt1375. [DOI] [PubMed] [Google Scholar]
- 4.Liu FT, Rabinovich GA. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 5.Takenaka Y, Fukumori T, Raz A. Glycoconj J. 2004;19:543–549. doi: 10.1023/B:GLYC.0000014084.01324.15. [DOI] [PubMed] [Google Scholar]
- 6.Mulder L, Lefebvre B, Cullimore J, Imberty A. Glycobiology. 2006;16:801–809. doi: 10.1093/glycob/cwl006. [DOI] [PubMed] [Google Scholar]
- 7.Long SR. Plant Cell. 1996;8:1885–1898. doi: 10.1105/tpc.8.10.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balzarini J. Antiviral Res. 2006;71:237–247. doi: 10.1016/j.antiviral.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Balzarini J, Van Herrewege Y, Vermeire K, Vanham G, Schols D. Mol Pharmacol. 2007;71:3–11. doi: 10.1124/mol.106.030155. [DOI] [PubMed] [Google Scholar]
- 10.Fuster MM, Esko JD. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 11.Rek A, Krenn E, Kungl AJ. Br J Pharmacol. 2009;157:686–694. doi: 10.1111/j.1476-5381.2009.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer B, Peters T. Angew Chem Int Ed Engl. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 13.Lepre CA, Moore JM, Peng JW. Chem Rev. 2004;104:3641–3676. doi: 10.1021/cr030409h. [DOI] [PubMed] [Google Scholar]
- 14.Cavanagh J, Fairbrother WJ, Palmer AG, III, Skelton NJ, Rance M. Protein NMR Spectroscopy, Principles and Practice. Elsevier Academic Press; 2005. [Google Scholar]
- 15.Campbell AP, Sykes BD. Annu Rev Biophys Biomol Struct. 1993;22:99–122. doi: 10.1146/annurev.bb.22.060193.000531. [DOI] [PubMed] [Google Scholar]
- 16.Sastry M, Bewley CA, Kwong PD. Adv Exp Med Biol. 2012;992:197–211. doi: 10.1007/978-94-007-4954-2_11. [DOI] [PubMed] [Google Scholar]
- 17.Nietlispach D, Mott HR, Stott KM, Nielsen PR, Thiru A, Laue ED. Methods Mol Biol. 2004;278:255–288. doi: 10.1385/1-59259-809-9:255. [DOI] [PubMed] [Google Scholar]
- 18.Bewley CA, Otero-Quintero S. J Am Chem Soc. 2001;123:3892–3902. doi: 10.1021/ja004040e. [DOI] [PubMed] [Google Scholar]
- 19.Boyd MR, Gustafson KR, McMahon JB, Shoemaker RH, O’Keefe BR, Mori T, Gulakowski RJ, Wu L, Rivera MI, Laurencot CM, Currens MJ, Cardellina JH, 2nd, Buckheit RW, Jr, Nara PL, Pannell LK, Sowder RC, 2nd, Henderson LE. Antimicrob Agents Chemother. 1997;41:1521–1530. doi: 10.1128/aac.41.7.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bewley CA, Gustafson KR, Boyd MR, Covell DG, Bax A, Clore GM, Gronenborn AM. Nat Struct Biol. 1998;5:571–578. doi: 10.1038/828. [DOI] [PubMed] [Google Scholar]
- 21.Yang F, Bewley CA, Louis JM, Gustafson KR, Boyd MR, Gronenborn AM, Clore GM, Wlodawer A. J Mol Biol. 1999;288:403–412. doi: 10.1006/jmbi.1999.2693. [DOI] [PubMed] [Google Scholar]
- 22.Bewley CA. J Am Chem Soc. 2001;123:1014–1015. doi: 10.1021/ja005714o. [DOI] [PubMed] [Google Scholar]
- 23.Kelley BS, Chang LC, Bewley CA. J Am Chem Soc. 2002;124:3210–3211. doi: 10.1021/ja025537m. [DOI] [PubMed] [Google Scholar]
- 24.Bewley CA, Kiyonaka S, Hamachi I. Journal of Molecular Biology. 2002;322:881–889. doi: 10.1016/s0022-2836(02)00842-2. [DOI] [PubMed] [Google Scholar]
- 25.Alexandre KB, Gray ES, Mufhandu H, McMahon JB, Chakauya E, O’Keefe BR, Chikwamba R, Morris L. Virology. 2012;423:175–186. doi: 10.1016/j.virol.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keeffe JR, Gnanapragasam PN, Gillespie SK, Yong J, Bjorkman PJ, Mayo SL. Proc Natl Acad Sci U S A. 2011;108:14079–14084. doi: 10.1073/pnas.1108777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koharudin LM, Liu L, Gronenborn AM. Proc Natl Acad Sci U S A. 2013;110:7702–7707. doi: 10.1073/pnas.1300327110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bewley CA. Structure. 2001;9:931–940. doi: 10.1016/s0969-2126(01)00653-0. [DOI] [PubMed] [Google Scholar]
- 29.Kehr JC, Zilliges Y, Springer A, Disney MD, Ratner DD, Bouchier C, Seeberger PH, de Marsac NT, Dittmann E. Mol Microbiol. 2006;59:893–906. doi: 10.1111/j.1365-2958.2005.05001.x. [DOI] [PubMed] [Google Scholar]
- 30.Shahzad-ul-Hussan S, Gustchina E, Ghirlando R, Clore GM, Bewley CA. J Biol Chem. 2011;286:20788–20796. doi: 10.1074/jbc.M111.232678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehotzky RE, Partch CL, Mukherjee S, Cash HL, Goldman WE, Gardner KH, Hooper LV. Proc Natl Acad Sci U S A. 2010;107:7722–7727. doi: 10.1073/pnas.0909449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dam TK, Gerken TA, Brewer CF. Biochemistry. 2009;48:3822–3827. doi: 10.1021/bi9002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dam TK, Brewer CF. Biochemistry. 2008;47:8470–8476. doi: 10.1021/bi801208b. [DOI] [PubMed] [Google Scholar]
- 34.Mayer M, Meyer B. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 35.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Protocol GPI, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Protocol GPI, Koff WC, Wilson IA, Burton DR, Poignard P. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, Kelly JW, Rudd PM, Dwek RA, Katinger H, Burton DR, Wilson IA. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- 38.Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, Depetris R, Katpally U, Marozsan A, Cupo A, Maloveste S, Liu Y, McBride R, Ito Y, Sanders RW, Ogohara C, Paulson JC, Feizi T, Scanlan CN, Wong CH, Moore JP, Olson WC, Ward AB, Poignard P, Schief WR, Burton DR, Wilson IA. Science. 2011;334:1097–1103. doi: 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tseng SY, Wang CC, Lin CW, Chen CL, Yu WY, Chen CH, Wu CY, Wong CH. Chem Asian J. 2008;3:1395–1405. doi: 10.1002/asia.200800154. [DOI] [PubMed] [Google Scholar]
- 40.Wang SK, Liang PH, Astronomo RD, Hsu TL, Hsieh SL, Burton DR, Wong CH. Proc Natl Acad Sci U S A. 2008;105:3690–3695. doi: 10.1073/pnas.0712326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pancera M, Shahzad-Ul-Hussan S, Doria-Rose NA, McLellan JS, Bailer RT, Dai K, Loesgen S, Louder MK, Staupe RP, Yang Y, Zhang B, Parks R, Eudailey J, Lloyd KE, Blinn J, Alam SM, Haynes BF, Amin MN, Wang LX, Burton DR, Koff WC, Nabel GJ, Mascola JR, Bewley CA, Kwong PD. Nat Struct Mol Biol. 2013 doi: 10.1038/nsmb.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalvit C. Drug Discov Today. 2009;14:1051–1057. doi: 10.1016/j.drudis.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 43.Meinecke R, Meyer B. J Med Chem. 2001;44:3059–3065. doi: 10.1021/jm0109154. [DOI] [PubMed] [Google Scholar]
- 44.Wang YS, Liu D, Wyss DF. Magn Reson Chem. 2004;42:485–489. doi: 10.1002/mrc.1381. [DOI] [PubMed] [Google Scholar]
- 45.Meyer B, Klein J, Mayer M, Meinecke R, Moller H, Neffe A, Schuster O, Wulfken J, Ding Y, Knaie O, Labbe J, Palcic MM, Hindsgaul O, Wagner B, Ernst B. Ernst Schering Res Found Workshop; 2004. pp. 149–167. [DOI] [PubMed] [Google Scholar]
- 46.Angulo J, Enriquez-Navas PM, Nieto PM. Chemistry. 2010;16:7803–7812. doi: 10.1002/chem.200903528. [DOI] [PubMed] [Google Scholar]
- 47.Hansman GS, Biertumpfel C, Georgiev I, McLellan JS, Chen L, Zhou T, Katayama K, Kwong PD. J Virol. 2011;85:6687–6701. doi: 10.1128/JVI.00246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansman GS, Shahzad-Ul-Hussan S, McLellan JS, Chuang GY, Georgiev I, Shimoike T, Katayama K, Bewley CA, Kwong PD. J Virol. 2012;86:284–292. doi: 10.1128/JVI.05909-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clore GM, Gronenborn AM. J Magn Res. 1982;48:16. [Google Scholar]
- 50.Roldos V, Canada FJ, Jimenez-Barbero J. Chembiochem. 2011;12:990–1005. doi: 10.1002/cbic.201000705. [DOI] [PubMed] [Google Scholar]
- 51.Morris KF, Johnson CSJ. J Am Chem Soc. 1992;114:3. [Google Scholar]
- 52.Lucas LH, Larive CK. Concepts in Magnetic Resonance. 2004;20A:24–41. [Google Scholar]
- 53.Fielding L. Curr Top Med Chem. 2003;3:39–53. doi: 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- 54.Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B. Journal of Biomolecular NMR. 2001;21:11. doi: 10.1023/a:1013302231549. [DOI] [PubMed] [Google Scholar]
- 55.Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC. Proc Natl Acad Sci U S A. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, O’Dell S, Patel N, Shahzad-ul-Hussan S, Yang Y, Zhang B, Zhou T, Zhu J, Boyington JC, Chuang GY, Diwanji D, Georgiev I, Kwon YD, Lee D, Louder MK, Moquin S, Schmidt SD, Yang ZY, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Burton DR, Koff WC, Walker LM, Phogat S, Wyatt R, Orwenyo J, Wang LX, Arthos J, Bewley CA, Mascola JR, Nabel GJ, Schief WR, Ward AB, Wilson IA, Kwong PD. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]