

Abstract
Purpose
Retinitis pigmentosa (RP) is an inherited retinal dystrophy characterized by extreme genetic and clinical heterogeneity. Thus, the diagnosis is not always easily performed due to phenotypic and genetic overlap. Current clinical practices have focused on the systematic evaluation of a set of known genes for each phenotype, but this approach may fail in patients with inaccurate diagnosis or infrequent genetic cause. In the present study, we investigated the genetic cause of autosomal recessive RP (arRP) in a Spanish family in which the causal mutation has not yet been identified with primer extension technology and resequencing.
Methods
We designed a whole-exome sequencing (WES)-based approach using NimbleGen SeqCap EZ Exome V3 sample preparation kit and the SOLiD 5500×l next-generation sequencing platform. We sequenced the exomes of both unaffected parents and two affected siblings. Exome analysis resulted in the identification of 43,204 variants in the index patient. All variants passing filter criteria were validated with Sanger sequencing to confirm familial segregation and absence in the control population. In silico prediction tools were used to determine mutational impact on protein function and the structure of the identified variants.
Results
Novel Usher syndrome type 2A (USH2A) compound heterozygous mutations, c.4325T>C (p.F1442S) and c.15188T>G (p.L5063R), located in exons 20 and 70, respectively, were identified as probable causative mutations for RP in this family. Family segregation of the variants showed the presence of both mutations in all affected members and in two siblings who were apparently asymptomatic at the time of family ascertainment. Clinical reassessment confirmed the diagnosis of RP in these patients.
Conclusions
Using WES, we identified two heterozygous novel mutations in USH2A as the most likely disease-causing variants in a Spanish family diagnosed with arRP in which the cause of the disease had not yet been identified with commonly used techniques. Our data reinforce the clinical role of WES in the molecular diagnosis of highly heterogeneous genetic diseases where conventional genetic approaches have previously failed in achieving a proper diagnosis.
Introduction
Inherited retinal dystrophies (IRDs) are a group of disorders characterized by progressive dysfunction and death of retinal photoreceptors. Retinitis pigmentosa (RP, MIM# 26800) is the most common form of retinal dystrophy and is characterized by significant clinical and genetic heterogeneity (reviewed in [1]). Patients with RP initially display night blindness followed by tunnel vision due to rod defects, which often progresses to complete blindness when the cones are also affected [2]. Most patients with RP have no associated systemic disease and are considered to have non-syndromic RP. Prevalence of non-syndromic RP is approximately 1 in 4,000 [3]. The condition may segregate as an autosomal dominant RP (24%), autosomal recessive (41%), or X-linked recessive trait (22%). The remaining 12% of cases are presumed to result from non-genetic factors, non-Mendelian inheritance (for example, mitochondrial or de novo mutations) or complex inheritance (digenic or polygenic inheritance) [4]. A few cases, however, have associated non-ocular symptoms and are classified as syndromic RP. The most frequent forms of syndromic RP are Usher and Bardet-Biedl syndromes. Usher syndrome can be divided into type 1 (2% to 6% of RP cases), which has profound congenital deafness and vestibular ataxia, and type 2 (15% of all cases of RP), which has an associated moderate non-progressive hearing loss [5].
To date, 36 genes have been implicated in non-syndromic arRP and three additional loci have been mapped (RetNet). In spite of the intense mapping efforts over the past two decades, mutations detectable in known RP genes have been able to explain only a relatively small percentage of cases. The two most prevalent genes known to cause arRP are eyes shut homolog (EYS; MIM# 602772) [6], which has been reported to account in different population-based studies for up to 18% of arRP [7], and Usher syndrome type 2A (USH2A) (MIM# 608400), found in around 5% of arRP cases [8]. Therefore, many genes have yet to be identified, as well as mutations in regions of known RP genes not routinely analyzed.
USH2A, located on chromosome 1q41, is the most commonly mutated gene in Usher syndrome type 2 (USH2) [9]. Genetic overlap between arRP and Usher syndrome exists as mutations in the USH2A and clarin 1(CLRN1) genes are found in both disorders [10,11]. USH2A presents two alternatively spliced isoforms: a short isoform named “a,” consisting of 21 exons, and a long isoform named “b,” consisting of 51 additional exons at the 3′ end of the short isoform [12-14]. The isoform a is predicted to be secreted to the extracellular matrix, whereas the isoform b is anchored on the cell membrane. The long variant is the predominant form in the retina [15]. The proteins encoded, also known as usherins, are multidomain proteins. The isoform b is composed of three regions: a large extracellular region consisting of an N-terminal signal peptide, laminin G-like domain (LamGL), laminin domain N-terminal (LamNT), laminin-type EGF-like modules (EGF-Lam), fibronectin type III (FN3) repeats, and laminin G domains (LamG), a transmembrane region (TM), and a cytoplasmic C-terminal domain containing a PDZ-binding motif. In mammalian photoreceptors, usherin is localized to the connecting cilia [16,17], where it is likely to be involved in cargo delivery from the inner segment to the outer segment of the photoreceptor cell [14-16,18]. Together, these data suggest that usherin is crucial for the long-term maintenance of photoreceptors. Mutations in USH2A are spread throughout the 72 exons and their flanking intronic regions, and consist of nonsense and missense mutations, deletions, duplications, large rearrangements, and splicing variants (UMDUSHbases) [19,20]. In spite of the significant number of publications pointing to USH2A as one of the most common mutated genes in arRP, phenotype–genotype correlations have not yet been established.
Current diagnostic strategies for RP include the use of diverse techniques. Commercially available genotyping microarrays are based on arrayed primer extension technology (APEX, Asper Ophthalmics, Tartu, Estonia), which enable the simultaneous screening of multiple genes, but they can detect only a fixed number of known mutations [21]. Custom-designed resequencing microarrays are a valuable alternative that allow the detection of novel mutations but are limited to a known set of genes [22]. Moreover, since these technologies have been designed specifically to approach a particular inheritance pattern, achieving a molecular diagnosis in simplex cases and/or overlapping phenotypes may be hampered. Sanger sequencing of known genes is still the most reliable method in determining the genetic cause, but this approach is not affordable due to the large genetic heterogeneity of RP. Most of these limitations can now be overcome with the implementation of massive sequencing technologies that have facilitated the discovery of many causative genes and gene variants of complex traits.
Here, we have conducted whole-exome sequencing to identify the disease-causing gene in a Spanish family diagnosed with arRP in which the cause of the disease had not yet been identified with primer extension technology and resequencing. We have found two novel USH2A compound heterozygous mutations consistent with being the cause of disease in this family. These mutations expand the mutant spectrum of USH2A in patients with arRP and further support the use of exome sequencing in the genetic diagnosis of genomic disorders with extreme phenotypic and genetic overlap.
Methods
Subjects and clinical assessment
This study involved nine individuals, five males and four females, aged 41 to 59, from a Spanish family with an inheritance pattern and phenotypic features consistent with arRP derived from the Department of Ophthalmology at our hospital. The study was performed in accordance with the tenets of the Declaration of Helsinki [23] and the ethical guidelines of our institution. A group of 200 matching control individuals was also recruited. Written informed consent was obtained from all participants. Clinical diagnosis of retinitis pigmentosa was based on visual acuity, fundus photography, and computerized testing of central and peripheral visual fields. Typical initial symptoms include night blindness, restriction of visual field, bone spicule-like pigmentation, attenuation of retinal vessels, and waxy disc pallor. Balance and hearing examinations were normal.
DNA extraction
All subjects underwent peripheral blood extraction for genomic DNA isolation from leukocytes using the MagNA Pure LC system (Roche, Indianapolis, IN) according to the manufacturer’s instructions. DNA samples were stored at −80 °C until used. DNA integrity was evaluated with 1% agarose gel electrophoresis.
Previous molecular genetic analysis:
The DNA sample from the index patient was first analyzed and excluded for known mutations by applying commercially available microarray analysis (Asper Biotech, Tartu, Estonia). In addition, pathogenic variants in ceramide kinase-like protein (CERKL), cyclic nucleotide gated channel alpha 1 (CNGA1), crumbs homolog 1 (Drosophila) (CRB1), eyes shut homolog (Drosophila) (EYS), isocitrate dehydrogenase 3 (NAD+) beta (IDH3B), lecithin retinol acyltransferase (phosphatidylcholine--retinol O-acyltransferase) (LRAT), c-mer proto-oncogene tyrosine kinase (MERTK), nuclear receptor subfamily 2, group E, member 3 (NR2E3), phosphodiesterase 6B, cGMP-specific, rod, beta (PDE6B), progressive rod-cone degeneration (PRCD), prominin 1 (PROM1), retinal G protein coupled receptor (RGR), rhodopsin (RHO), retinaldehyde binding protein 1 (RLBP1), retinal pigment epithelium-specific protein 65 kDa (RPE65), and tubby like protein 1 (TULP1) were excluded with a custom genome resequencing microarray experiment [22].
Whole-exome sequencing
Library preparation and exome capture were performed according to a protocol based on the Baylor College of Medicine protocol version 2.1 with several modifications. Briefly, 5 µg of input genomic DNA was sheared, end-repaired, and ligated with specific adaptors. A fragment size distribution ranging from 160 bp to 180 bp after shearing and 200 bp to 250 bp after adaptor ligation was verified with Bioanalyzer (Agilent Technologies, Santa Clara, CA). The library was amplified by precapture linker-mediated PCR (LM-PCR) using FastStart High Fidelity PCR System (Roche). After purification, 2 µg LM-PCR product were hybridized to SeqCap EZ Exome libraries V3 (Roche Nimblegen, Madison, WI). After washing, amplification was performed with post-capture LM-PCR using FastStart High Fidelity PCR System (Roche). Capture enrichment was measured with qPCR according to the NimbleGen protocol. The successfully captured DNA was measured with Quant-iT PicoGreen dsDNA reagent (Invitrogen, Carlsbad, CA) and subjected to standard sample preparation procedures for sequencing with the SOLiD 5500×l platform as recommended by the manufacturer (Applied Biosystems, Foster City, CA). Shortly, emulsion PCR was performed on E80 scale (about 1 billion template beads) using a concentration of 0.616 pM of enriched captured DNA. After breaking and enrichment, about 276 million enriched template beads were sequenced per lane on a six-lane SOLiD 5500×l slide.
Variant analysis
SOLiD 5500×l reads were aligned against the human genome reference (hg19) using the program Blat-like Fast Accurate Search Tool (BFAST). Properly mapped reads were filtered with the SAMtools package, which was also used to sort and index SAM files. For prediction of variants (variant calling), only reads mapping to a unique position in the reference genome were used. Variants were identified with the Genome Analysis Toolkit (GATK) software taking into account the single nucleotide polymorphisms obtained from the Single Nucleotide Polymorphism Database (dbSNP, National Center for Biotechnology Information), and 1,000 Genomes project. Secondary analysis was performed with a custom script that uses ANNOVAR [24], SIFT (Sorting Intolerant From Tolerant), and PolyPhen-2 (Polymorphism Phenotyping v2) for annotating the variants. Annotated, non-synonymous variants found in affected individuals were compared to variants present in the non-affected relatives. Finally, the remaining variants were compared with variants obtained from a group of healthy controls from the same local population as the family under study. Variants present in affected individuals but not in healthy individuals of the family or in the control local population were ranked based on the analysis of the interactome to generate a list of candidate genes. The nomenclature for the variations observed at DNA (cds) level followed the recommendations of the Human Genome Variation Society [25], where nucleotide +1 is the A of the ATG translation initiation codon in the USH2A reference sequence NM_206933.2.
Verification and assessment of the pathogenicity of variants
The novel identified variants were subsequently verified and screened in 200 healthy matched control subjects with Sanger sequencing. Cosegregation analysis was performed in available family members DNA samples. Specific primers encompassing USH2A exons 20 and 70 were designed (Table 1) using the Primer3 software [26]. To evaluate the pathogenicity of the novel variants, we analyzed the potential impact of a given variant on the function or structure of the encoded protein based on conservation, physical properties of the amino acids, or possible occurrence in regulatory or splicing motifs using bioinformatic tools PolyPhen-2, SIFT, and Splicing site Mutation by Berkeley Drosophila Genome Project (BDGP) website [27]. Evolutionary conservation across species was assessed through the alignment of orthologous USH2A protein sequences (chimpanzee, dog, bovine, mouse, rat, chicken, and zebrafish) with the human USH2A protein sequence, using HomoloGene and Clustal Omega (ClustalO) Tool. The HOPE server was used to analyze and predict the structural variations in mutant USH2A [28].
Table 1. Primer sequences used to amplify USH2A exons 20 and 70 from gDNA samples.
| Primer Name | Sequence (5′-3′) | Tm (°C) | Direction | Amplicon Length (bp) |
|---|---|---|---|---|
| USH2A Ex20-F |
TCCTAATGGTGGTTGGCAAT |
60 |
Sense |
382 |
| USH2A Ex20-R |
AGTGAGGGAGGAGAAGACAAA |
58 |
Antisense |
382 |
| USH2A Ex70-F |
CTACAGCGAGCTGTGGTTCA |
60 |
Sense |
362 |
| USH2A Ex70-R | GCAGCCAAAGTTGAGAAAGC | 60 | Antisense | 362 |
Results
Clinical findings in affected siblings
The age of the patients ranged from 41 to 59 years at the time of diagnosis, indicating late onset RP. Affected members of the family II:1, II:3, and II:5 (Figure 1) exhibited typical clinical features of RP, including bilateral visual loss, initial hemeralopia, and restriction of visual field leaving only the central 10° functional. Fundus examination of patient II:5 showed typical signs of RP with intraretinal bone spicule-like pigment formation and preserved posterior pole. Individuals II:2 and II:7 were clinically reevaluated upon identification of potentially causative genetic variants. On examination, both patients showed initial RP symptoms, including attenuated retinal vessels, waxy pallor of the optic discs, and bone spicule-like pigment around the midperiphery.
Figure 1.
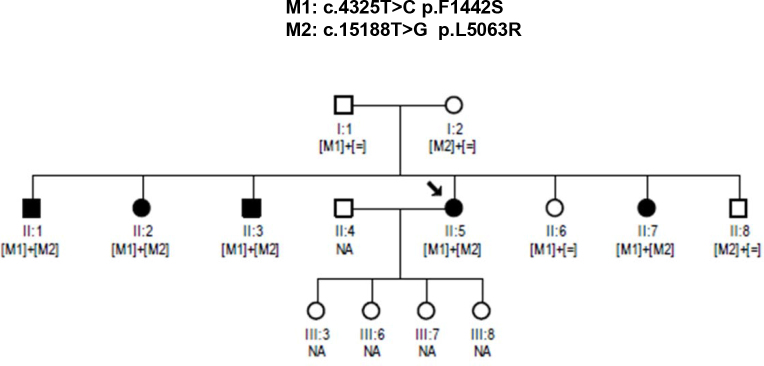
Pedigree with autosomal recessive retinitis pigmentosa showing cosegregation of the compound heterozygous changes, c.4325 T>C and c.15188 T>G within the USH2A gene in all affected members. Black symbols represent clinically affected subjects. Open symbols represent unaffected subjects. Patient II:5 (proband) is indicated with a black arrow. NA, not available.
Identification of two novel mutations in USH2A by whole-exome sequencing
We performed whole-exome sequencing of four members of the family (individuals I:1, I:2, II:1, and II:3) using Roche NimbleGen SeqCap EZ Exome V3 sample preparation kit and SOLiD 5500×l. After duplicated reads were removed, sequences were aligned to the human genome reference sequence (build hg19) using BFAST [29] (0.7.0a,”-a 2” mode) and a maximum of two mismatches allowed. Variants were called and annotated using the GATK [30] (release 1.4, Best Practices V3 filters and “DP<6”) and ANNOVAR [24] (release 2012 March 08) software packages. A mean of 43,463 SNVs per sample were identified (Table 2). Inspection of candidate variants resulted in the identification of two novel compound heterozygous mutations in the coding region (exons 20 and 70) of the long isoform of the USH2A gene at genomic positions 1:216363636 (c.4325T>C) and 1:215807910 (c.15188T>G). No other pathogenic mutations were found.
Table 2. Number of candidate variants filtered against dbSNP and 1000 Genomes public variation databases.
| Single nucleotide variants (SNVs | Individuals |
|||
|---|---|---|---|---|
| II:3 | II:1 | I:1 | I:2 | |
| Total SNVs |
43,204 |
43,201 |
43,476 |
43,974 |
| Non-synonymous SNVs |
5850 |
5833 |
5888 |
5883 |
| Filtered dbSNP |
5752 |
5716 |
5781 |
5769 |
| Filtered dbSNP and 1000 g |
5697 |
5662 |
5735 |
5712 |
| Filtered dbSNP and 1000 g and predicted deleterious | 356 | 346 | 350 | 361 |
Variants validation
Primers that specifically amplify exons 20 and 70 were used for amplification and sequencing of USH2A variants. All available members of the pedigree and 200 matched control individuals were screened for the USH2A c.4325T>C and c.15188T>G mutations. Sanger sequencing confirmed cosegregation of both mutations with RP in this pedigree (Figure 2). In addition, two siblings not clinically diagnosed with RP at the time of enrollment (II:2 and II:7) harbored the same combination of USH2A mutations. Both siblings underwent clinical reevaluation revealing symptoms and signs of RP, such as bone spicule-like pigmentation in the midperiphery of both eyes, attenuation of the retinal vessels and pale waxy discs. Clinically unaffected family members had only one of the two heterozygous mutations, consistent with the expected genotype–phenotype correlation. The variants were not reported in the 1,000 Genomes database or in any other single nucleotide polymorphism database. We also did not find these changes in 200 control individuals.
Figure 2.

The USH2A gene harbors mutations in exons 20 and 70 of subjects with autosomal recessive retinitis pigmentosa. A: The USH2A gene structure is composed of 72 exons. B: Chromatograms of normal USH2A DNA sequence and subjects with autosomal recessive retinitis pigmentosa (arRP) show mutations at the c.4325 and c.15188 nucleotide positions in USH2A exons 20 and 70, respectively.
Assessment of the pathogenicity of variants
The novel mutations resulted in a substitution of a phenylalanine for a serine at protein position 1442 (p.F1442S) and a leucine for an arginine at protein position 5063 (p.L5063R). The comparative amino acid sequence analysis in HomoloGene and ClustalO showed complete conservation at both positions (Figure 3). To predict whether a novel missense variant was deleterious, we used the combined results of two different computer algorithms: SIFT and PolyPhen. Both changes were predicted to be pathogenic (SIFT score=0.01, PolyPhen score=0.9). Together, these observations support that these changes are pathogenic variants.
Figure 3.

Schematic representation of USH2A protein structure encompassing the interspecies conservation of mutated residues. Isoform a is an N-terminal fragment of isoform b. NCBI HomoloGene-generated amino acid sequences were aligned using Clustal Omega software. The predicted orthologs show the conservation of the F1442 and L5063 residues. The F1442S mutation is located within the fourth fibronectin domain type III common for both isoforms, while the L5063R variant locates in the C-terminal transmembrane domain exclusive of isoform b. SP, signal peptide; LamGL, LamG-like jellyroll fold; Lam NT, laminin N-terminal; EGFLam, laminin-type EGF-like (LE); FN3, fibronectin type-III; LamG, laminin G; TM, transmembrane; PDB, PDZ-binding domain.
Structural analysis of USH2A mutations
Structural analysis of USH2A performed with the HOPE server [28] suggests that the original wild-type residues and the newly introduced mutant residues differ in size, charge, and hydrophobicity values. No solved three-dimensional structure or modeling template was found for these mutations. The missense mutant p.F1442S residue, buried in a fibronectin type III domain (Figure 3), is predicted to disturb the core structure of this domain and thus affect its binding properties. Similarly, HOPE predictions showed that the mutant p.L5063R residue, located in a region annotated as a transmembrane domain, is smaller than the wild-type residue and causes the replacement of a neutral residue for a positively charged residue. These differences in charge and size are predicted to affect the hydrophobic interactions with the membrane lipids.
Discussion
In this report, a Spanish family with five affected siblings is described. The mode of inheritance and the main clinical features correspond to late onset autosomal recessive RP. Exome analysis of two affected siblings and healthy parents led to the identification of two novel compound heterozygous mutations in USH2A segregating with the disease. Consistent with the phenotype of patients with USH2A mutations, the affected members of the pedigree developed retinal degeneration. Depending on the mutation, Usher genes can cause RP without hearing loss or profound deafness without RP. Hearing tests were normal. Mutations in USH2A are responsible for a wide spectrum of phenotypes, ranging from non-syndromic RP to full-blown USH2 [31,32]. Our result is consistent with previous findings indicating that mutations in USH2A may cause retinitis pigmentosa without hearing loss [8]. Although many studies have dissected USH2A functional properties [33-36], genotype–phenotype correlations are still not well understood. It has been hypothesized that mutations identified in patients with USH2 result in complete inactivation of the corresponding mutant protein whereas patients with non-syndromic RP can also carry severe mutations, but never in both alleles [37].
Among the variants identified in this pedigree, the USH2A p.F1442S mutation is located in the extracellular region within the fourth fibronectin domain type III. The USH2A large extracellular domain is projected into the pericilliary matrix and may interact with the connecting cilium to fulfil important structural or signaling roles [15]. We propose that this mutation might affect specific connections between USH2A and its network of interacting protein(s). The second identified mutation, p.L5063R, represents the most downstream pathogenic mutation described to date (HGMD) and the first located in the transmembrane domain of USH2A. Since a change in the polarity (non-polar to polar) may affect insertion of the protein into the membrane, we speculated with the possibility that the replacement of a leucine (non-polar) to an arginine (polar, basic) may result in defective USH2A membrane anchorage. Furthermore, the p.F1442S change affects long and short isoforms while the p.L5063R variant affects only the long isoform of USH2A. Since the long variant is the predominant form in the retina [15] and both copies of the large protein are defective in affected individuals, we support the hypothesis that non-syndromic RP might be caused by two missense mutations in the long isoform. The residual function of usherin would be enough for proper functioning of the stereocilia of the inner ear but insufficient for photoreceptors’ integrity maintenance. This is in agreement with the low transcript levels of the long isoform in the cochlea [14].
The index patient investigated here had previously undergone selected genotyping (APEX analysis, arRP panel) and custom resequencing microarray [22], but both mutational screening approaches failed to identify the underlying gene defect in this family. Genetic underlying defects may be overlooked if the method used for the analysis is confined to the screening of a set of mutations in particular genes. The resequencing chip allows the identification of novel mutations, but regrettably, USH2A was not tiled onto our array. Since a significant number of mutations in the RP genes affect only a single family or a few, a reliable diagnostic system able to detect novel mutations, even in genes not previously associated with the phenotype, must be implemented. Exome sequencing has proven to be an important diagnostic tool for disorders that are characterized by significant genetic heterogeneity. In the case of RP, apart from the high number of mutations involved, different mutations in one gene can cause different phenotypes, and the same mutation can exhibit intrafamilial and interfamilial phenotypic variability. Thus, exome sequencing makes feasible investigating the role of second-site mutations that may be modulating the expression of the arRP phenotype.
In summary, two novel compound heterozygous mutations in USH2A have been identified with exome sequencing. These results highlight the clinical heterogeneity of RP and demonstrate that exome sequencing is a valuable tool for comprehensive genetic diagnosis particularly in patients in which conventional testing failed to detect mutations.
Acknowledgments
The authors are grateful to the patients that have participated in this study. The project was financially supported by the Instituto de Salud Carlos III (ISCIII), Spanish Ministry of Economy and Competitiveness, Spain (PI11–02923), Regional Ministry of Economy, Innovation, Science and Employment of the Autonomous Government of Andalusia (CTS-03687), Regional Ministry of Health of the Autonomous Government of Andalusia (PI10–0154), and to Fundación Ramón Areces (CIVP16A1856). The CIBER de Enfermedades Raras is an initiative of the ISCIII. The Medical Genome Project is an initiative of the Regional Ministry of Health of the Autonomous Government of Andalusia supported by the “Programa Nacional de Proyectos de investigación Aplicada,” I+D+i 2008, “Subprograma de actuaciones Científicas y Tecnológicas en Parques Científicos y Tecnológicos ACTEPARQ (PCT-30000-2009-12), INNPLANTA (PCT-300000-2010-007) and FEDER.”
BIBLIOGRAPHY
- 1.Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12:238–49. doi: 10.2174/138920211795860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neveling K, den Hollander AI, Cremers FP, Collin RW. Identification and analysis of inherited retinal disease genes. Methods Mol Biol. 2013;935:3–23. doi: 10.1007/978-1-62703-080-9_1. [DOI] [PubMed] [Google Scholar]
- 3.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–84. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 2012;25:42–9. doi: 10.1097/WCO.0b013e32834ef8b2. [DOI] [PubMed] [Google Scholar]
- 6.Abd El-Aziz MM, Barragan I, O'Driscoll CA, Goodstadt L, Prigmore E, Borrego S, Mena M, Pieras JI, El-Ashry MF, Safieh LA, Shah A, Cheetham ME, Carter NP, Chakarova C, Ponting CP, Bhattacharya SS, Antinolo G. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. 2008;40:1285–7. doi: 10.1038/ng.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barragán I, Borrego S, Pieras JI, Gonzalez-del Pozo M, Santoyo J, Ayuso C, Baiget M, Millan JM, Mena M, Abd El-Aziz MM, Audo I, Zeitz C, Littink KW, Dopazo J, Bhattacharya SS, Antinolo G. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum Mutat. 2010;31:E1772–800. doi: 10.1002/humu.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernal S, Ayuso C, Antinolo G, Gimenez A, Borrego S, Trujillo MJ, Marcos I, Calaf M, Del Rio E, Baiget M. Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet. 2003;40:e8. doi: 10.1136/jmg.40.1.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGee TL, Seyedahmadi BJ, Sweeney MO, Dryja TP, Berson EL. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J Med Genet. 2010;47:499–506. doi: 10.1136/jmg.2009.075143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66:1975–8. doi: 10.1086/302926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan MI, Kersten FF, Azam M, Collin RW, Hussain A, Shah ST, Keunen JE, Kremer H, Cremers FP, Qamar R, den Hollander AI. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2011;118:1444–8. doi: 10.1016/j.ophtha.2010.10.047. [DOI] [PubMed] [Google Scholar]
- 12.Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, Cremers C, Davenport S, Moller C, Talmadge CB, Beisel KW, Tamayo M, Morton CC, Swaroop A, Kimberling WJ, Sumegi J. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–7. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- 13.Weston MD, Eudy JD, Fujita S, Yao S, Usami S, Cremers C, Greenberg J, Ramesar R, Martini A, Moller C, Smith RJ, Sumegi J, Kimberling WJ. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000;66:1199–210. doi: 10.1086/302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, Cremers FP, Cremers CW, Kremer H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004;74:738–44. doi: 10.1086/383096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, Li T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci USA. 2007;104:4413–8. doi: 10.1073/pnas.0610950104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006;83:97–119. doi: 10.1016/j.exer.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–93. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 18.Maerker T, van Wijk E, Overlack N, Kersten FF, McGee J, Goldmann T, Sehn E, Roepman R, Walsh EJ, Kremer H, Wolfrum U. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 19.Aller E, Jaijo T, Beneyto M, Najera C, Oltra S, Ayuso C, Baiget M, Carballo M, Antinolo G, Valverde D, Moreno F, Vilela C, Collado D, Perez-Garrigues H, Navea A, Millan JM. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006;43:e55. doi: 10.1136/jmg.2006.041764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baux D, Faugere V, Larrieu L, Le Guedard-Mereuze S, Hamroun D, Beroud C, Malcolm S, Claustres M, Roux AF. UMD-USHbases: a comprehensive set of databases to record and analyse pathogenic mutations and unclassified variants in seven Usher syndrome causing genes. Hum Mutat. 2008;29:E76–87. doi: 10.1002/humu.20780. [DOI] [PubMed] [Google Scholar]
- 21.Jaijo T, Aller E, Garcia-Garcia G, Aparisi MJ, Bernal S, Avila-Fernandez A, Barragan I, Baiget M, Ayuso C, Antinolo G, Diaz-Llopis M, Kulm M, Beneyto M, Najera C, Millan JM. Microarray-based mutation analysis of 183 Spanish families with Usher syndrome. Invest Ophthalmol Vis Sci. 2010;51:1311–7. doi: 10.1167/iovs.09-4085. [DOI] [PubMed] [Google Scholar]
- 22.González-del Pozo M, Borrego S, Barragan I, Pieras JI, Santoyo J, Matamala N, Naranjo B, Dopazo J, Antinolo G. Mutation screening of multiple genes in Spanish patients with autosomal recessive retinitis pigmentosa by targeted resequencing. PLoS ONE. 2011;6:e27894. doi: 10.1371/journal.pone.0027894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.World Medical Association declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA. 1997;277:925–6. [PubMed] [Google Scholar]
- 24.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horaitis O, Cotton RG. The challenge of documenting mutation across the genome: the human genome variation society approach. Hum Mutat. 2004;23:447–52. doi: 10.1002/humu.20038. [DOI] [PubMed] [Google Scholar]
- 26.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 27.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–23. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 28.Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS ONE. 2009;4:e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu XZ, Hope C, Liang CY, Zou JM, Xu LR, Cole T, Mueller RF, Bundey S, Nance W, Steel KP, Brown SD. A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am J Hum Genet. 1999;64:1221–5. doi: 10.1086/302332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernal S, Meda C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, Del Rio E, Baiget M. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype–phenotype correlation. Clin Genet. 2005;68:204–14. doi: 10.1111/j.1399-0004.2005.00481.x. [DOI] [PubMed] [Google Scholar]
- 33.Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–56. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- 34.Reiners J, van Wijk E, Marker T, Zimmermann U, Jurgens K, te Brinke H, Overlack N, Roepman R, Knipper M, Kremer H, Wolfrum U. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum Mol Genet. 2005;14:3933–43. doi: 10.1093/hmg/ddi417. [DOI] [PubMed] [Google Scholar]
- 35.van Wijk E, Kersten FF, Kartono A, Mans DA, Brandwijk K, Letteboer SJ, Peters TA, Marker T, Yan X, Cremers CW, Cremers FP. Wolfrum, U., Roepman, R., and Kremer, H. Usher syndrome and Leber congenital amaurosis are molecularly linked via a novel isoform of the centrosomal ninein-like protein. Hum Mol Genet. 2009;18:51–64. doi: 10.1093/hmg/ddn312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, Schafer E, Roux AF, Dafinger C, Bernd A, Zrenner E, Claustres M, Blanco B, Nurnberg G, Nurnberg P, Ruland R, Westerfield M, Benzing T, Bolz HJ. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest. 2010;120:1812–23. doi: 10.1172/JCI39715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaiserman N, Obolensky A, Banin E, Sharon D. Novel USH2A mutations in Israeli patients with retinitis pigmentosa and Usher syndrome type 2. Arch Ophthalmol. 2007;125:219–24. doi: 10.1001/archopht.125.2.219. [DOI] [PubMed] [Google Scholar]