

Abstract
Purpose
X-linked juvenile retinoschisis (XLRS) is a vitreoretinal dystrophy characterized by schisis (splitting) of the inner layers of the neuroretina. Mutations within the retinoschisis (RS1) gene are responsible for this disease. The mutation spectrum consists of amino acid substitutions, splice site variations, small indels, and larger genomic deletions. Clinically, genomic deletions are rarely reported. Here, we characterize two novel full exonic deletions: one encompassing exon 1 and the other spanning exons 4–5 of the RS1 gene. We also report the clinical findings in these patients with XLRS with two different exonic deletions.
Methods
Unrelated XLRS men and boys and their mothers (if available) were enrolled for molecular genetics evaluation. The patients also underwent ophthalmologic examination and in some cases electroretinogram (ERG) recording. All the exons and the flanking intronic regions of the RS1 gene were analyzed with direct sequencing. Two patients with exonic deletions were further evaluated with array comparative genomic hybridization to define the scope of the genomic aberrations. After the deleted genomic region was identified, primer walking followed by direct sequencing was used to determine the exact breakpoints.
Results
Two novel exonic deletions of the RS1 gene were identified: one including exon 1 and the other spanning exons 4 and 5. The exon 1 deletion extends from the 5′ region of the RS1 gene (including the promoter) through intron 1 (c.(−35)-1723_c.51+2664del4472). The exon 4–5 deletion spans introns 3 to intron 5 (c.185–1020_c.522+1844del5764).
Conclusions
Here we report two novel exonic deletions within the RS1 gene locus. We have also described the clinical presentations and hypothesized the genomic mechanisms underlying these schisis phenotypes.
Introduction
X-linked juvenile retinoschisis (XLRS; OMIM 312700) is the most common inherited retinal dystrophy occurring in men. XLRS is the leading cause of juvenile macular degeneration, with an estimated prevalence at 1:5,000 to 1:25,000. XLRS is characterized by the presence of foveomacular cavities in the inner retina [1]. Reduced visual acuity is the most common symptom of patients affected with XLRS. Examination with ophthalmoscopy or optical coherence tomography (OCT) often reveals peripheral retinoschisis, which may lead to retinal detachment in some cases [2,3]. Full field electroretinograms (ERGs) typically show a normal a-wave with reduced amplitude of the b-wave, although a-wave changes have been described in some patients [4]. XLRS exhibits variable expressivity, even among affected individuals in the same family [5]. Female carriers are usually asymptomatic, with normal visual acuity and normal ERG findings. Women who are homozygous for an RS1 mutation are phenotypically similar to affected men [6].
The RS1 gene contains six exons and is located at Xp22.1 [7]. RS1 encodes retinoschisin, a 24 kDa discoidin domain-containing protein present in the photoreceptors and neurons of the inner layers of the retina. Retinoschisin is a cell adhesion protein that maintains the cellular organization and synaptic structure of the retina [8]. More than 189 distinct mutations have been described in the RS1 sequence variation databases (the Human Gene Mutation Database and the Leiden Open Variation Database). Single base substitutions dominate the mutation spectrum followed by larger deletions or inversions. Most of the single base substitutions are missense mutations followed by nonsense and splice site variations. About 10% of the deletions encompass whole exons, and most of these occur over exon 2 of this gene [9].
Many reports have described genotype–phenotype correlations in patients with XLRS, but only a few describe exonic deletions [10-18]. In this study, we describe two distinct deletions and the clinical phenotype associated with these defined genotypes.
Methods
Subjects
Patients with XLRS and their mothers (if available) were examined and enrolled at the National Eye Institute, NIH. Subjects were examined with fundus biomicroscopy and indirect ophthalmoscopy. Best-corrected Snellen visual acuity, Goldmann kinetic perimetry (Haag-Streit, Bern, Switzerland), and optical coherence tomography (Stratus OCT 3 or Cirrus, Carl Zeiss Meditec, Inc., Dublin, CA) were performed. Diagnosis of XLRS was based upon history, the presence of foveal schisis, clinical examination, and electroretinogram findings wherever available. All study protocols were approved by the National Institutes of Health Institutional Review Board, consonant with the tenants of the Declaration of Helsinki, and all the subjects gave informed consent.
Electroretinogram recording
ERGs were recorded according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards 15 with 2.4 cds/m2 flashes to elicit the dark-adapted combined response and the photopic 30-Hz flicker against a 34 cd/m2 background using a visual diagnostic system (UTAS 2000 or Sunburst Ganzfeld Visual Testing System; LKC Technologies, Gaithersburg, MD). Pupils were dilated with topical phenylephrine and tropicamide. Subjects were dark adapted for 30 min before ERG recording began. Burian-Allen electrodes (Hansen Ophthalmic Laboratories, Iowa City, IA) were inserted with the help of artificial tears (Refresh Celluvisc; Allergan, Irvine, CA) for conductivity and subject comfort. Dark-adapted ERG responses were recorded first, followed by 10 min of light adaptation at 34 cd/m2 before photopic testing. The lower limit of normal for all ERG parameters was 2 standard deviation (SD) below the mean response calculated from 96 subjects with normal vision recorded on our ERG systems.
Mutation detection by sequencing
Standard genetic testing for detecting the coding region mutation was performed in the NEI DNA Diagnostic Laboratory as a clinical test using the following protocol. Genomic DNA was extracted from peripheral blood collected through venipuncture into a Lavender top (EDTA) tube and stored in a refrigerator for less than 72 h using the Gentra Puregene kit following the manufacturer’s protocol (Qiagen, Valencia, CA). Coding sequences (exons 1–6) and 25 bp of flanking intronic sequences were amplified with PCR primers located in the introns (the sequences of the primer sets are shown in Appendix 1). The following PCR conditions were applied: 94 °C for 12 min (preliminary denaturation); 32 cycles of denaturation at 94 °C for 40 s, annealing at 63 °C for 30 s, elongation at 72 °C for 30 s, and final synthesis at 72 °C for 10 min. PCR products were purified with ExoSAP-IT following the manufacturer’s protocol (USB, Cleveland, OH) and bidirectionally sequenced on an ABI PRISM 3130×l Genetic Analyzer, using the BigDye Terminator Cycle Sequencing kit following the manufacturer’s protocol (version 3.1; Applied Biosystem, Foster City, CA). Sequence data were analyzed using Sequencher software version 4.8 (Gene Codes Corporation, Ann Arbor, MI).
Array comparative genomic hybridization
Array comparative genomic hybridization (CGH) was performed according to the manufacturer’s protocol with minor modifications on a 720 k oligonucleotide chip (Roche/NimbleGen System, Madison, WI). A commercially available pooled normal control DNA was used (Promega Corporation, Madison, WI) for reference. The patient and the reference DNA were labeled with either Cyanine 3 (Cy-3) or Cyanine 5 (Cy-5) by random priming (Trilink Biotechnologies, San Diego, CA) and then hybridized to the chip via incubation in the MAUI hybridization system (BioMicroSystems, Salt Lake City, UT). After 40 h hybridization at 42 °C, the slides were washed and scanned using a MS200 scanner (Roche/NimbleGen System). NimbleScan version 2.4 and the SignalMap version 1.9 were applied for data analysis (NimbleGen System, Madison, WI) for profile smoothing and breakpoint detection. The genomic locations were retrieved from National Center for Biotechnology Information (NCBI) build 37 (hg 19). Frequently affected regions recently detected as copy number polymorphisms (CNPs) were excluded from data analysis according to the CNP database generated in the University of Oklahoma Health Sciences Center (OUHSC) genetics laboratory and the Database of Genomic Variants.
Deletion mapping
Long-range PCR primers corresponding to the genomic regions before and after the positions of deleted/non-deleted aCGH probes were designed to localize the approximate breakpoints using the GeneRunner 3.01 software. (The sequences of the primer sets are shown in Appendix 2.) Long-range PCRs were performed using the Takara long-range PCR kit (Clontech Laboratories, Mountain View, CA) as previously described [19]. Junction fragments found by PCR were purified with ExoSAP-IT (USB) and sequenced using BigDye Terminator Cycle Sequencing according to the methods for coding region sequencing. Mutations were annotated following the nomenclature recommended by the Human Genome Variation Society (HGVS). Genomic reference sequence was according to the GRCh37.p13 (hg19), and the RS1 gene reference was obtained (GenBank NM_000330.3) from the NCBI database.
Results
Ophthalmological evaluation
Clinical examinations were performed in the NEI Ophthalmic Genetics Clinic, and molecular characterization of XLRS1 was performed in the NEI DNA Diagnostic Laboratory. This study was approved by the NIH Combined NeuroScience (CNS) Institute Review Board, and all patients gave informed consent consistent with the Declaration of Helsinki. Patient 645 was evaluated for XLRS at age 7, and patient 22 was examined at age 21. The clinical findings are summarized in Table 1. Patient 645 showed evidence of foveal schisis in both eyes, with the right eye demonstrating more significant and extensive schisis changes (Figure 1). Patient 22 presented with more symmetric foveal schisis. Neither patient had significant bullous schisis in the retinal periphery, although patient 645 had shallow schisis superiorly and inferotemporally in both eyes. Patient 22 did not have significant peripheral schisis. Although patient 645 was young at the time of examination and was not able to cooperate with an ERG, patient 22 had an ERG without the characteristic findings of an electronegative b-wave, defined as b-wave responses having smaller amplitude than a-wave amplitudes.
Table 1. Clinical presentation of XLRS patient #645 and patient #22 with retinal schisis.
| Clinical criteria | Patient #645 | Patient #22 |
|---|---|---|
| Age (yrs) when data recorded |
7 |
21 |
| Age (yrs) when symptoms noticed |
7 |
5 |
| BCVA-OD, OS |
20/800, 20/50 |
20/50+1, 20/40–1 |
| Central visual field defect present (both OD and OS) |
Yes. OD |
Yes. Both OD and OS |
| Foveal/macular cystic changes (both OD and OS) |
Yes. Both OD and OS |
Yes. Both OD and OS |
| Macular atrophy (both OD and OS) |
No |
No |
| Peripheral visual field defect present (both OD and OS) |
Yes. Inferonasally OD and OS |
No |
| Peripheral schisis (both OD and OS) |
Yes, peripheral flat schisis superiorly and inferotemporally OS>OD. |
No |
| Retinal detachment present or treated (both OD and OS) |
No |
No |
| Inheritance Pattern based on family history |
Only the proband is affected |
Only the proband is affected |
| ERG Dark-adapted combined/mixed response wave values- OD |
Not performed |
a wave=237,
b wave=312(μV)* |
| ERG Dark-adapted combined/mixed response wave values- OS | Not performed | a wave=240, b wave=318(μV)* |
*Compared to NEI clinical normal lower bound of 188 microvolt and 375 microvolt (mean, −2 SD, 96 subjects) for a wave and b-wave amplitudes, respectively.
Figure 1.
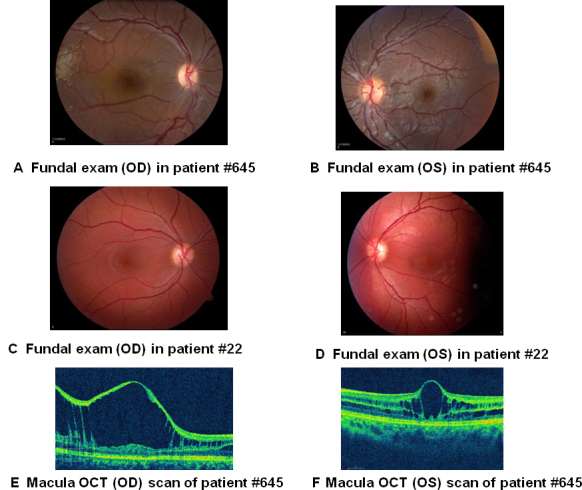
Fundus photographs and optical coherence tomography scans demonstrated foveal schisis. A: Fundal exam (OD) in patient 645. B: Fundal exam (OS) in patient 645. C: Fundal exam (OD) in patient 22. D: Fundal exam (OS) in patient 22. E, F: Optical coherence tomograms through a horizontal section of the left and right eyes of patient 645 showed classic foveal schisis.
Genetic testing
Patients 645 and 22 had PCR failures for amplification of exon 1 and exons 4 and 5 PCRs, respectively, despite multiple attempts, and were selected for further genomic analysis. Routine genetic testing in these patients did not reveal any mutation other than the failure of the respective exons to amplify by PCR, and maternal DNA analysis did not identify a mutation or variation (data not shown and primer information available in Appendix 1). Array CGH (aCGH) was performed to investigate the possibility of large genomic deletions and to exclude the possibility of unlikely primer region variations. The aCGH testing identified deletions in the patients and copy number reductions in the maternal DNA’s within the RS1 gene locus. For patient 645, the location described by the array was arr Xp22.13(18,689,722–18,690,832)×0 hg19, which includes the promoter region and exon 1. For patient 22, the location described by the array was arr Xp22.13(18,661,426–18,665,836)×0 hg19, which includes exons 4–5 of the RS1 gene (data not shown). Although these results defined the deleted genomic regions, they could not accurately delineate the sizes of the deletions because of the large distances between the nearest flanking positive probes and the adjacent probe within the deletion. This distance was about 5 kb from the next positive probe on each side for patient 645 (distal BP: 3′ :18,684,315–18,689,722 // 18,690,832–18,694,318: 5′ proximal BP, hg 19), which gives a 10 kb region of uncertainty for the breakpoints. Similarly, the uncertainty for breakpoints was also about 10 kb in patient 22 (distal BP: 3′ :18,658,747–18,661,426 // 18,665,836–18,668,498: 5′ proximal BP, hg 19).
Identification of breakpoints in the patients
To further define the breakpoints, four forward and five reverse primers crossing the exon 1 deletion region and three forward and three reverse primers for the exon 4/5 deletion region were designed to include the undeleted and possible deleted regions (Figure 2A and Figure 3A; primer information in Appendix 2). With long-range PCR using different combinations of designed primers, we identified the junctional fragments in both cases (Figure 2B, lanes 5 and 6 for patient 645; Figure 3B, lane 3 for patient 22). Further sequencing of the junction fragments clearly identified the breakpoints in both cases (Figure 2C and Figure 3C). In patient 645, genomic DNA breaks at genomic position 18,691,946 and joins at 18,687,473 on chromosome X (X:18687474_18691945del; HGVS annotation: c.(−35)-1723_ c.51+2664del4472). In patient 22, genomic DNA breaks at 18,666,471 and joins at the nucleotide position 18,660,706 on chromosome X, deleting intron 3 to intron 5 of the RS1 gene (X:18660707_18666470del; HGVS annotation: c.185–1020_c.522+1844del5764). Maternal carrier status was also confirmed in both cases (Figure 2B, lanes 2 and 3, for maternal heterozygous mutation carrier status of exon 1 deletion; Figure 3B, lane 2, for maternal heterozygous mutation carrier status of exons 4–5 deletion).
Figure 2.
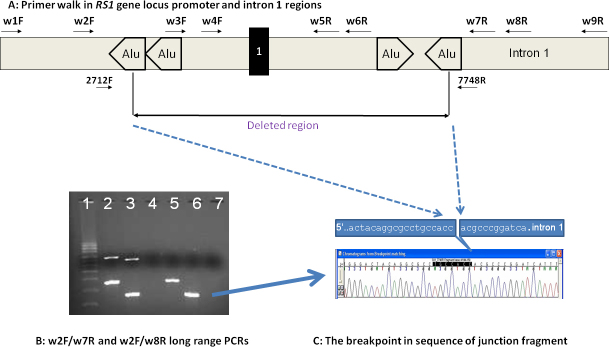
Determination of breakpoints in the RS1 gene gross genomic deletion in patient #645 and confirmation of maternal carrier status. A: Schematic diagram of the RS1 gene promoter region for primer walk in patient #645 (not scaled). The results of long range PCR using different combinations of designed primers (Appendix 2) are not shown. B: PCR products were analyzed by using pre-cast 1% agarose gels stained with ethidium bromide (SeaKem® Gold Agarose, Lonza Rockland Inc, Rockland, ME). This gel shows the amplification of the deletion junction fragment in the patient. Lane 1 was loaded with supercoiled DNA Ladder (0.01 mg/ml, Life technologies, Grand Island, CA). Lanes 2, 4 and 5 used primer pair w2F-w8R. Lanes 3, 6 and 7 used primer pair w2F-W7R. The mother's DNA was loaded in lanes 2 and 3. The Patient #645’s (son) DNA was loaded in lanes 5 and 6. Lanes 4 and 7 were non-DNA water as PCR controls. The top bands in lanes 2 and 3 represent the wild type fragments with an estimated size of about 6-7 kb. The lower bands in lanes 2, 3, 5, and 6 represent the junction fragments with estimated sizes of 3.5 kb and 2.6 kb respectively. The blue arrow indicates the isolated fragment used for sequencing. C: Junction fragments were sequenced to determine the exact breakpoints in exon 1 of patient #645 (reverse direction sequencing by primer 7748R is shown).
Figure 3.
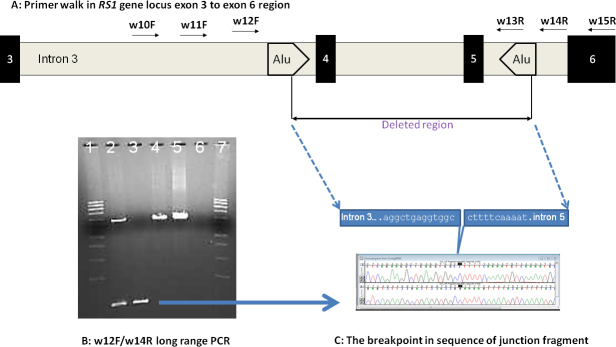
Determination of breakpoints in the RS1 gene gross genomic deletion in patient #22 and confirmation of maternal carrier status. A: Schematic Diagram of RS1 gene exon 3 to exon 6 region for primer walk in patient #22 (not scaled). The results of long range PCR using different combinations of designed primers (Appendix 2) are not shown. B: PCR products using primer pair w12F/w14R were analyzed by using pre-cast 1% agarose gels stained with ethidium bromide (SeaKem® Gold Agarose, Lonza Rockland Inc, Rockland, ME). The gel shows the amplification of the deletion junction fragment in the patient. Lanes 1 & 7 were loaded with supercoiled DNA Ladder (0.01 mg/ml, Life technologies, Grand Island, CA). Lane 2 used the DNA from patient's mother. Lane 3 used DNA from patient #22 (son). Lanes 4 & 5 used DNA from two unrelated samples as wild type PCR product references. Lane 6 used non-DNA water as PCR control. The top band in lanes 2, 4, and 5 represents the wild type fragment with an estimated size of about 7 kb. The lower band represents the junction fragment with an estimated size of about 0.8 kb. The blue arrow indicates the isolated fragment used for sequencing. C: Junction fragments were sequenced to determine the exact breakpoints in intron 3 and intron 5 and results of sequencing by using primers w12F (forward direction, top) and w14R (reverse direction, bottom) are shown.
Discussion
In this report, we identified two gross genomic deletions within the RS1 gene locus in two affected patients with XLRS (overall study strategy is represented in a flowchart Figure 4). The c.(−35)-1723_c.51+2664del4472 deletion in patient 645 removed a portion or the whole promoter region and the entire exon 1 of the RS1 gene. It is thus predicted to result in a null allele with no XLRS protein products produced. Patients with XLRS with gross genomic deletion including exon 1 deletions have previously been reported to have severe phenotypes [17]. Patient 645 has a clinical presentation similar to other patients described with exon 1 deletions. The c.185–1020_c.522+1844del5764 deletion in patient 22 removed a genomic region that includes exons 4 and 5. In patient 22, the absence of exons 4 and 5 would be predicted to result in splicing of exon 3 to exon 6, resulting in the glutamic acid at codon position 62 being replaced by 87 spurious amino acids followed by a stop codon (p.E62Gfs*87). Presumably, the new transcript with this additional length of in-frame coding sequence would not be subjected to nonsense-mediated decay since this transcript terminates in the final exon. A previous study revealed that premature termination in the final exon may not affect the stability of mutated transcripts [20]. In this regard, although patient 22 was evaluated at age 21, he presented with a mild phenotype relative to patient 645 (Table 1).
Figure 4.
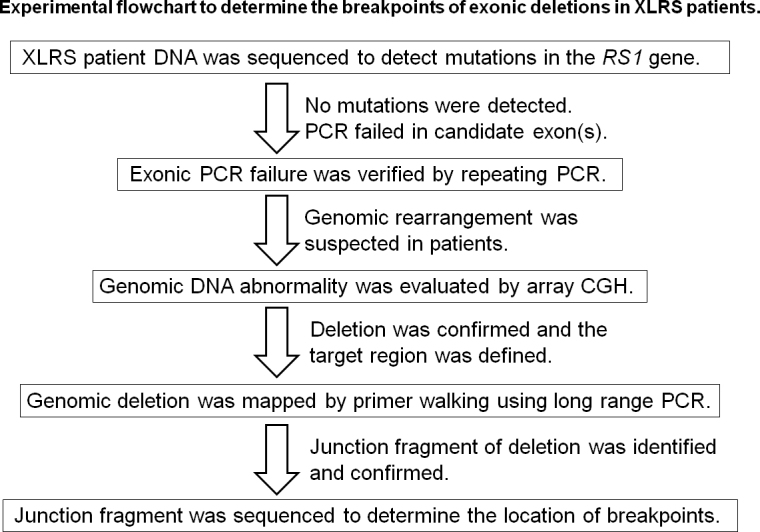
Experimental flowchart to determine the breakpoints of exonic deletions in patients with XLRS.
The NEI DNA Diagnostic Laboratory is a CLIA–certified clinical laboratory, providing service to the NEI Ophthalmic Genetics Clinic and the eye research community. Clinical testing of XLRS has identified mutations in the RS1 gene for most patients. These include missense, nonsense, indels, splicing error mutations and gross genomic deletions. According to the literature, mutations cannot be found in about 9% of patients with XLRS [9]. Of the reported mutations, gross genomic deletions were rare except those including exon 2, which have been more frequently reported compared to other genomic alterations in the RS1 gene (exon 2 deletion was reported in 11 probands out of a total of 198 probands tested at the NEI DNA Diagnostic Laboratory).
In the literature, exon 1 deletions were usually determined based on PCR failures or confirmed with Southern blot analysis [17,18]. Few reports have determined the breakpoints at the genomic region [21]. In our examination of the breakpoints, we found that the breakpoints were within two Alu elements within the promoter region and intron 1 in patient 645. The two breakpoints were exactly 12 bases downstream from the mid-A stretch region sequences within two Alu elements, respectively (Appendix 3). From further analysis of the neighboring intronic sequence, we found that the two involved Alu sequences lie in opposite orientation to the RS1 gene. There is also a secondary Alu element downstream of the promoter Alu element and a secondary Alu element upstream of the intron Alu element. The promoter secondary Alu element was in the same direction, and the other lies in the opposite direction (same orientation as the RS1 gene). We propose that the deletion may involve a hairpin structure from these secondary Alu elements. During replication, the potential hairpin from these secondary Alu elements may induce a mismatch and crossover between the two neighboring Alu elements and lead to a deletion. We have no evidence to support this hypothesis, but a recent study of more than 20 genes found evidence that the high content of transposable elements causes increased frequency of gene disruption by gross deletions in human disease [22]. We have also been screening the RS1 introns in patients with XLRS in whom we did not find a mutation in the coding region. Using multiple overlapped long-range PCR analysis to find gross insertions/deletions for potential homologous recombinations between sister chromosomes, we did not find any evidence of genomic rearrangement in these patients (data not shown). We found that the breakpoints of deletion in patient 22 were also located in two Alu elements, but with no sequence/structure similarity within the Alu components. No mechanism was revealed based on our analysis.
In DNA diagnostics, homozygous or hemizygous deletions crossing the target genomic region can be difficult to detect, but generally lead to a PCR failure in analysis of X-linked diseases. However, without further analysis, heterozygotes of such a deletion would not be detected, which cannot guide mutation segregation analysis. In addition, genomic variations may occasionally be located within the PCR primer region, which may lead to allele dropout during PCR/sequencing analysis. These scenarios can be detected only with further analysis using methods such as nested primer PCR, copy number studies such as array CGH, multiplex ligation-dependent probe amplification, real-time PCR. These confirmatory tests have their own limitations. However, determining the junction fragments and/or breakpoints is still the ultimate goal and the gold standard. It should be applied as a good practice in clinical DNA diagnosis. In this study, confirmation of the maternal carrier status excluded germline mosaicism, which is important for genetic counseling of affected families.
Acknowledgments
This research was supported by the US National Institutes of Health (NIH), National Eye Institute (NEI) extramural, clinical, and intramural programs. The authors would like to thank the NEI/NIH eyeGENE® Coordinating Center, Kerry Goetz, Melissa Reeves, Vida Ndiforv, Sally Vitez, and Alexandra Garafalo for their assistance and technical support during the course of this study.
Appendix 1. Primer pairs used to screen RS1 gene for mutations.
To access the data, click or select the words “Appendix 1.”
Appendix 2. Primer pairs used for determining the location of breakpoints in patients with partial gene deletions.
To access the data, click or select the words “Appendix 2.”
Appendix 3. The two Alu elements spanning the breakpoints of the exon 1 deletion.
To access the data, click or select the words “Appendix 3.”
References
- 1.George ND, Yates JR, Moore AT. X linked retinoschisis. Br J Ophthalmol. 1995;79:697–702. doi: 10.1136/bjo.79.7.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Condon GP, Brownstein S, Wang N-S, Kearns AF, Ewing CC. Congenital hereditary (juvenile X-linked) retinoschisis: histopathologic and ultrastructural findings in three eyes. Arch Ophthalmol. 1986;104:576–83. doi: 10.1001/archopht.1986.01050160132029. [DOI] [PubMed] [Google Scholar]
- 3.Forsius H, Krause U, Helve J, Vuopala V, Mustonen E, Vainio-Mattila B, Fellman J, Eriksson AW. Visual acuity in 183 cases of X-chromosomal retinoschisis. Can J Ophthalmol. 1973;8:385–93. [PubMed] [Google Scholar]
- 4.Pimenides D, George NDL, Yates JRW, Bradshaw K, Roberts SA, Moore AT, Trump D. X-linked retinoschisis: clinical phenotype and RS1 genotype in 86 UK patients. J Med Genet. 2005;42:e35. doi: 10.1136/jmg.2004.029769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol. 1996;114:274–80. doi: 10.1001/archopht.1996.01100130270007. [DOI] [PubMed] [Google Scholar]
- 6.Ali A, Feroze AH, Rizvi ZH, Rehman TU. Consanguineous marriage resulting in homozygous occurrence of X-linked retinoschisis in girls. Am J Ophthalmol. 2003;136:767–9. doi: 10.1016/s0002-9394(03)00448-3. [DOI] [PubMed] [Google Scholar]
- 7.Sauer CG, Gehrig A. Warneke-Wittstock Ret al. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 1997;17:164–70. doi: 10.1038/ng1097-164. [DOI] [PubMed] [Google Scholar]
- 8.Wu WW, Molday RS. Defective discoidin domain structure, subunit assembly, and endoplasmic reticulum processing of retinoschisin are primary mechanisms responsible for X-linked retinoschisis. J Biol Chem. 2003;278:28139–46. doi: 10.1074/jbc.M302464200. [DOI] [PubMed] [Google Scholar]
- 9.The Retinoschisis Consortium Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis (XLRS). Hum Mol Genet. 1998;7:1185–92. doi: 10.1093/hmg/7.7.1185. [DOI] [PubMed] [Google Scholar]
- 10.Shinoda K, Mashima Y, Ishida S, Oguchi Y. Severe juvenile retinoschisis associated with a 33-bps deletion in XLRS1 gene. Ophthalmic Genet. 1999;20:57–61. doi: 10.1076/opge.20.1.57.2303. [DOI] [PubMed] [Google Scholar]
- 11.Eksandh LC, Ponjavic V, Ayyagari R, Bingham EL, Hiriyanna KT, Andreasson S, Ehinger B, Sieving PA. Phenotypic expression of juvenile X-linked retinoschisis in Swedish families with different mutations in the XLRS1 gene. Arch Ophthalmol. 2000;118:1098–104. doi: 10.1001/archopht.118.8.1098. [DOI] [PubMed] [Google Scholar]
- 12.Hiraoka M, Trese MT, Shastry BS. X–Linked juvenile retinoschisis associated with a 4-base pair insertion at codon 55 of the XLRS1 gene. Biochem Biophys Res Commun. 2000;268:370–2. doi: 10.1006/bbrc.2000.2133. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura M, Ito S, Terasaki H, Miyake Y. Japanese X-linked juvenile retinoschisis: conflict of phenotype and genotype with novel mutations in the XLRS1 gene. Arch Ophthalmol. 2001;119:1553–4. [PubMed] [Google Scholar]
- 14.Shinoda K, Ishida S, Oguchi Y, Mashima Y. Clinical characteristics of 14 Japanese patients with X-linked juvenile retinoschisis associated with XLRS1 mutation. Ophthalmic Genet. 2000;21:171–80. [PubMed] [Google Scholar]
- 15.Simonelli F, Cennamo G, Ziviello C, Testa F, de Crecchio G, Nesti A, Manitto MP, Ciccodicola A, Banfi S, Brancato R, Rinaldi E. Clinical features of X linked juvenile retinoschisis associated with new mutations in the XLRS1 gene in Italian families. Br J Ophthalmol. 2003;87:1130–4. doi: 10.1136/bjo.87.9.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sieving PA, Bingham EL, Kemp J, Richards J, Hiriyanna K. Juvenile X-linked retinoschisis from XLRS1 Arg213Trp mutation with preservation of the electroretinogram scotopic b-wave process. Am J Ophthalmol. 1999;128:179–84. doi: 10.1016/s0002-9394(99)00144-0. [DOI] [PubMed] [Google Scholar]
- 17.Chan WM, Choy KW, Wang J, Lam DS, Yip WW, Fu W, Pang CP. Two cases of X-linked juvenile retinoschisis with different optical coherence tomography findings and RS1 gene mutations. Clin Experiment Ophthalmol. 2004;32:429–32. doi: 10.1111/j.1442-9071.2004.00820.x. [DOI] [PubMed] [Google Scholar]
- 18.Eksandh L, Andréasson S, Abrahamson M. Juvenile X-linked retinoschisis with normal scotopic b-wave in the electroretinogram at an early stage of the disease. Ophthalmic Genet. 2005;26:111–7. doi: 10.1080/13816810500228688. [DOI] [PubMed] [Google Scholar]
- 19.Song J, Smaoui N, Ayyagari R, Stiles D, Benhamed S, MacDonald IM, Daiger SP, Tumminia SJ, Hejtmancik F, Wang X. High-Throughput Retina-Array for Screening 93 Genes Involved in Inherited Retinal Dystrophy. Invest Ophthalmol Vis Sci. 2011;52:9053–60. doi: 10.1167/iovs.11-7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hentze MW, Kulozik AE. A perfect message: RNA surveillance and nonsense-mediated decay. Cell. 1999;96:307–10. doi: 10.1016/s0092-8674(00)80542-5. [DOI] [PubMed] [Google Scholar]
- 21.Huopaniemi L, Tyynismaa H, Rantala A, Rosenberg T, Alitalo T. Characterization of two unusual RS1 gene deletions segregating in Danish retinoschisis families. Hum Mutat. 2000;16:307–14. doi: 10.1002/1098-1004(200010)16:4<307::AID-HUMU3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 22.van Zelm MC, Geertsema C, Nieuwenhuis N, Ridder DD, Conley ME, Schiff C, Tezcan I, Bernatowska E, Hartwig NG, Sanders EAM, Litzman J, Kondratenko I, Dongen JJMV, Burg MVD. Gross Deletions Involving IGHM, BTK, or Artemis: A Model for Genomic Lesions Mediated by Transposable Elements. Am J Hum Genet. 2008;82:320–32. doi: 10.1016/j.ajhg.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]