

Abstract
Several imprinted genes have been implicated in the regulation of placental function and embryonic growth. On distal mouse chromosome 7, two clusters of imprinted genes, each regulated by its own imprinting center (IC), are separated by a poorly characterized region of 280 kb (the IC1–IC2 interval). We previously generated a mouse line in which this IC1–IC2 interval has been deleted (Del7AI allele) and found that maternal inheritance of this allele results in low birth weights in newborns. Here we report that Del7AI causes a partial loss of Ascl2, a maternally expressed gene in the IC2 cluster, which when knocked out leads to embryonic lethality at midgestation due to a lack of spongiotrophoblast formation. The hypomorphic Ascl2 allele causes embryonic growth restriction and an associated placental phenotype characterized by a reduction in placental weight, reduced spongiotrophoblast population, absence of glycogen cells, and an expanded trophoblast giant cell layer. We also uncovered severe defects in the labyrinth layer of maternal mutants including increased production of the trilaminar labyrinth trophoblast cell types and a disorganized labyrinthine vasculature. Our results have important implications for our understanding of the role played by the spongiotrophoblast layer during placentation and show that regulation of the dosage of the imprinted gene Ascl2 can affect all three layers of the chorioallantoic placenta.
Keywords: Ascl2, Spongiotrophoblast, Placental development, Intrauterine growth restriction, Genomic imprinting
Introduction
In regions of the mammalian genome subject to genomic imprinting, genes tend to be epigenetically regulated in clusters, suggesting that long-range mechanisms coordinate monoallelic expression during development (Jones and Lefebvre, 2009; Reik and Walter, 2001). Distal mouse chromosome 7 (Chr 7), which shares homology with the Beckwith–Wiedemann Syndrome (BWS) region on human chromosome 11p15.5, is approximately 1 Mb in size and contains two imprinted clusters, each regulated by its own cis-acting differentially methylated region (DMR), commonly referred to as imprinting center 1 and imprinting center 2 (IC1 and IC2).
The distal end of the IC1 subdomain and the proximal end of the IC2 subdomain are separated by a distance of about 300 kb. IC1 has been shown to control the maternally expressed H19 gene and paternally expressed Igf2 gene via an epigenetically controlled CTCF-binding insulator element (Bell and Felsenfeld, 2000; Hark et al., 2000). The mechanism of IC2-mediated epigenetic silencing of several genes both centromeric and distal to its position over a relatively broad area (over 800 kb) is not as clear as is the case for IC1 and likely involves multiple components. Specifically, in the IC2 cluster of genes, at least eight protein-coding genes (Ascl2, Tssc4, Cd81, Kcnq1, Cdkn1c, Slc22a18, Phlda2, and Osbpl5) are known to be paternally silenced by the production and elongation of the large ncRNA Kcnq1ot1, transcribed from the unmethylated paternal IC2 (Fitzpatrick et al., 2002; Mancini-Dinardo et al., 2006; Shin et al., 2008). The current paradigm of imprinted regulation in this region proposes that Kcnq1ot1 transcription or the ncRNA itself is responsible for initiating and recruiting repressive H3K9me2, H3K9me3, and H3K27me3 marks specifically in the placenta to the paternal allele along the IC2 domain (Lewis et al., 2004; Pandey et al., 2008; Umlauf et al., 2004; Wagschal et al., 2008).
Even less is known about the large region between the IC1 and IC2 subdomains and information on this intervening region has only recently become available (Lefebvre et al., 2009; Shirohzu et al., 2004). It has been problematic to study because the entire region is repeat-rich and consists mostly of retroelements and tandem repeats, with one known gene, Tyrosine hydroxylase (Th) (Shirohzu et al., 2004). Th-null mice die in utero but can be rescued by supplementing pregnant females with L-DOPA, the next metabolite in catecholamine biosynthesis pathway (Zhou et al., 1995). To study whether the IC1–IC2 interval plays a role in regulating imprinting in this region, we previously generated a ~280 kb deletion of the IC1–IC2 interval called the Del7AI allele, with breakpoints 5′ of Ins2 in the IC1 sub-domain and 3′ of Ascl2 at the proximal end of the IC2 sub-domain (Lefebvre et al., 2009). We reported that the Del7AI deletion was viable upon both paternal and maternal inheritance, where it is recovered in Mendelian ratios, although Del7AI/+ pups were growth retarded at birth (Lefebvre et al., 2009). Homozygous Del7AI/Del7AI mice died in utero due to a lack of Th, consistent with the observed Th-null phenotype. This was demonstrated by showing that homozygous deletion mutants can be rescued to term by supplementing pregnant Del7AI heterozygous females with L-DOPA during gestation (Lefebvre et al., 2009; Zhou et al., 1995). Otherwise, our analysis of the Del7AI allele indicated that the deletion does not perturb acquisition or maintenance of imprinting marks at the flanking imprinting centers, regardless of parental inheritance (Lefebvre et al., 2009).
Several genes in the distal Chr 7 imprinted domain have been implicated in placental development and the regulation of embryonic growth. These include Igf2 (Baker et al., 1993), Ascl2 (Guillemot et al., 1995), Cdkn1c (Andrews et al., 2007; Takahashi et al., 2000), and Phlda2 (Frank et al., 2002; Salas et al., 2004; Tunster et al., 2010). The achaete-scute complex homolog 2 gene Ascl2 codes for a basic helix–loop–helix (bHLH) transcription factor implicated in lineage specification in extra-embryonic tissues (Guillemot et al., 1994) as well as in the adult intestinal epithelium (van der Flier et al., 2009). Although Ascl2 expression in extra-embryonic tissue is imprinted with exclusive transcription from the maternal allele (Guillemot et al., 1995), it is biallelically expressed in adult LGR5-positive stem cells (van der Flier et al., 2009). During development, Ascl2 is first detected during preimplantation stages, with predominant expression in the trophectoderm cells of the blastocyst (Rossant et al., 1998). Following implantation, Ascl2 transcripts are abundant in diploid cells of the ectoplacental cone and chorion (Guillemot et al., 1994; Rossant et al., 1998). In the chorioallantoic placenta, Ascl2 expression becomes restricted to spongiotrophoblast cells, with some patchy expression also detected in the labyrinth layer (Rossant et al., 1998). The role of Ascl2 in the development of the extra-embryonic lineages was previously addressed by generating a null allele of Ascl2 by gene targeting (Guillemot et al., 1994). Ascl2-null conceptuses die of placental failure at E10.0 (Guillemot et al., 1994; Tanaka et al., 1999). Mutant placentae show an absence of the spongiotrophoblast layer, as shown by loss of Tpbpa- and Flt1-positive cells as early as E7.5, as well as an expansion of the trophoblast giant cell layer (Guillemot et al., 1994; Oh et al., 2008).
Here we show that the Del7AI allele, which is associated with low birth weights in maternal heterozygotes, acts as a hypomorphic Ascl2 allele. Since the mutant embryos are viable, although growth retarded, we were able to assess the phenotypic consequences of reduced Ascl2 levels on placental development. Our results show that Ascl2 function is dosage-sensitive and that hypomorphs exhibit developmental abnormalities in the three main layers of the mature placenta.
Material and methods
Mice and genotyping
Generation and genotyping of the Del7AI allele were previously described (Lefebvre et al., 2009). The Del7AI allele (Del(7Ascl2-Ins2)1Lef; MGI:3662901) is a ~280 kb deletion of the interval between Ins2 and Ascl2 (from NCBI m37 coordinates 149,868,122 to 150,148,458) obtained by Cre-loxP recombination in germ cells (Lefebvre et al., 2009). The Ascl2-lacZ allele (Ascl2tm1.1Nagy; MGI:2155757) has been described previously (Tanaka et al., 1999). Although the Ascl2 open-reading frame is undisturbed in this allele, the introduction of an IRES-lacZ-pA cassette in the 3′UTR interferes with production of ASCL2, such that Ascl2-lacZ behaves as a null allele (Tanaka et al., 1999). Note that for heterozygous genotypes, the maternal allele is always given first. All animals in this study were maintained on the outbred CD-1 mouse background. All animal experiments were performed under certificate A07-0160 from the UBC Animal Care Committee and complied with the Canadian Council on Animal Care guidelines on the ethical care and use of experimental animals.
In situ hybridization (ISH)
E9.5, E13.5, and E15.5 placentae were dissected in PBS and fixed in fresh 4% paraformaldehyde/1×PBS (RNase free) overnight at 4 °C. For E9.5 placentae, entire conceptuses were fixed to ensure integrity of cryostat sections during the ISH procedure. Antisense and sense strand probes for Ascl2, Tpbpa (4311), Pcdh12, Gcm1 and Cdkn1c were DIG-labeled and used for ISH on 10 μm cryostat sections for E13.5 and E15.5 placentae and 12 μm sections for E9.5 placentae as previously described (Oh-McGinnis et al., 2010). Nuclear fast red was used as a counterstain.
Immunohistochemistry
E7.5 conceptuses as well as E13.5 and E15.5 placentae were dissected in cold PBS and fixed in fresh 4% paraformaldehyde overnight at 4 °C, paraffin-embedded, sectioned (12 μm) and subjected to immunohistochemistry as previously described, using the Vectastain Elite ABC kit and DAB substrate (Vector Labs) (Oh-McGinnis et al., 2010). To detect the PHLDA2 protein in the labyrinth layer, a 1:1000 dilution of the anti-PHLDA2 polyclonal antibody was used (Frank et al., 1999). ASCL2 IHC was performed with the Millipore clone 8F1 monoclonal antibody (Cat. # MAB4417) at a 1:50 dilution (van der Flier et al., 2009). To identify fetal capillaries of the labyrinth layer (Harbers et al., 1996), a 1:25 dilution of rabbit polyclonal anti-laminin antibody (Sigma L9393) was used. Hematoxylin was used as a counterstain.
Periodic acid Schiff stain
E15.5 paraffin sections were deparaffinized, treated with 1% periodic acid for 5 min, washed in tap water, followed in distilled water. The sections were then covered in Schiff’s solution for about 15 min and rinsed in running tap water for about 15 min. Hematoxylin was used as a counterstain and Scott’s Tap water substitute was used to help sharpen the contrast. Sections were then dehydrated and mounted with Entellan mounting medium (EM Science) and cover-slipped with glass.
DAPI stain
E15.5 paraffin sections were deparaffinized and treated with 4′,6-diamidino-2-phenylindole (DAPI) (1:5000 in PBS) for 5 min. Sections were then washed with distilled water, mounted with Vectashield (Vector Labs) and coverslipped with glass.
Expression analysis by quantitative real-time RT-PCR (qRT-PCR)
For total expression level analysis of IC1- and IC2-regulated genes, random-primed cDNA was generated from E9.5 or E15.5 placentae as previously described and cDNA was subject to qRT-PCR (Oh-McGinnis et al., 2010). qRT-PCRs were performed on three to four individual Del7AI/+ placentae, three +/Del7AI placentae and three to six wild type placentae with three technical replicates per individual placentae. All primer sequences for qRT-PCR analyses are available in Supplementary Table 1.
Placental fetal vasculature casts
Fetal vascular corrosion casts of E14.5 placentae were generated by following the procedure previously described (Whiteley et al., 2006). Vascular casts were analyzed by scanning electron microscopy at the UBC Bioimaging Facility.
Results
Reduced placental and embryonic weights in maternal Del7AI/+ heterozygotes
We previously reported that the Del7AI allele is associated with a new imprinted phenotype characterized by a growth restriction of approximately 20% at birth and seen only in maternal Del7AI/+ heterozygous pups (Lefebvre et al., 2009). To assess whether this postnatal phenotype reflects an in utero growth restriction, we measured placental and embryonic weights at E15.5 from reciprocal crosses between wild type (CD-1 outbred strain) and Del7AI heterozygous mice. Consistent with the absence of a growth phenotype at birth, no difference was found between the embryonic or placental weights of the paternal +/Del7AI heterozygotes and their wild type littermates (Fig. 1A). However upon maternal transmission of the deletion, we observed a significant (~28%) reduction in Del7AI/+ placental weights at E15.5 compared to their wild type littermates (Fig. 1B). Weights of Del7AI/+ embryos were concurrently smaller than wild type littermates at this stage but to a lesser degree (~7%). Our results show that maternal inheritance of Del7AI is associated with growth defects during development and suggest that embryonic growth restriction occurs later than E15.5 as a consequence of a primary placental phenotype, as described previously for other imprinted mouse models of intrauterine growth restriction such as the Esx1 knock-out (Li and Behringer, 1998) and the placental-specific loss of Igf2 (Constância et al., 2002).
Fig. 1.

Reduced placental and embryonic weights in maternal Del7AI heterozygotes at E15.5. Scatterplots of individual placental and embryonic weights at E15.5 for cohorts of wild type and Del7AI heterozygous conceptuses obtained from reciprocal crosses. Bars show the average weight ± standard deviation for the given numbers of measurements (n). Results are shown following paternal (A) and maternal (B) transmission of the Del7AI deletion. Differences in wild type and mutant means were evaluated using the two-tailed unpaired Student t test (p values). Asterisks mark mutant results significantly different from those of their wild type littermates (p<0.05).
Both Ascl2 and Phlda2 are perturbed in maternal Del7AI/+ placentae
The imprinted phenotype associated with Del7AI suggests that the deletion interferes with the expression of a maternally expressed placental growth regulator during development. Of the currently annotated protein-coding genes deleted in the Del7AI allele (Ins2 Ensembl isoform Ins-006, Gm6471, and Th) only Th is known to be expressed in the placenta, where it has been reported to be preferentially expressed from the maternal allele (Schulz et al., 2006). However, whereas the embryonic lethality of Del7AI/Del7AI homozygotes can be pharmacologically rescued by provision of exogenous L-DOPA during pregnancy (the product of the reaction catalyzed by TH), such treatment has no effect on the growth restriction phenotype of Del7AI/+ heterozygotes (Lefebvre et al., 2009). These observations suggest that the deletion might interfere with the expression of flanking imprinted genes, several of which have been implicated in placental biology such as Igf2 in the proximal IC1 subdomain(Baker et al., 1993), and Ascl2 (Guillemot et al., 1994), Cdkn1c (Zhang et al., 1998), and Phlda2 (Frank et al., 2002) in the distal IC2 sub-domain. To address this possibility, we analyzed by quantitative reverse transcriptase PCR (qRT-PCR) the expression levels in E9.5 placentae of imprinted genes located proximally (IC1 subdomain) or distally (IC2 subdomain) to the breakpoints of the Del7AI deletion (Figs. 2A–B). In Del7AI/+ placentae, the gene immediately distal of the deletion breakpoint, Ascl2, was significantly lower in the maternal Del7AI/+ placentae compared to wild type littermates, with an overall reduction in expression levels of approximately two-fold (Fig. 2C). In addition, we observed increased Phlda2 expression in maternal Del7AI/+ placentae compared to wild type littermates, also by approximately two-fold (Fig. 2C). The same expression changes at Ascl2 and Phlda2 were also observed in E15.5 placentae (Fig. S1A). The rest of the genes analyzed in the IC2 or IC1 clusters, including Igf2, were unaffected in maternal Del7AI/+ placentae. Unlike what is observed for conceptuses inheriting the deletion maternally, we found no significant difference between +/Del7AI placentae and wild type littermates for the IC1 and IC2 subdomain genes analyzed at this stage (Fig. 2C).
Fig. 2.
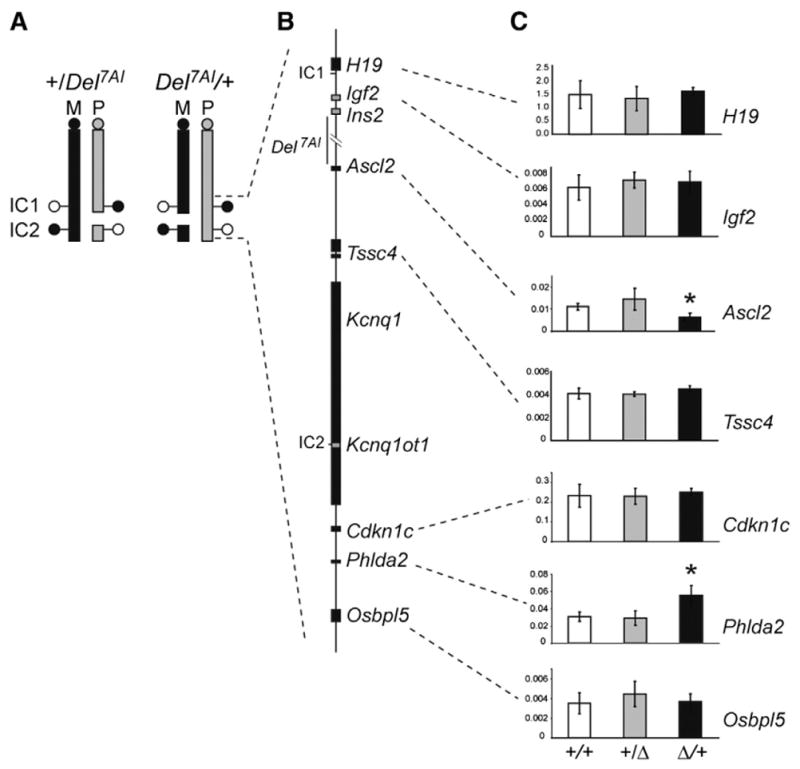
Total expression levels of IC1- and IC2-regulated imprinted genes in reciprocal heterozygous Del7AI placentae at E9.5. (A) Schematic representation of mouse chromosome 7 showing the breakpoints of the Del7AI allele relative to IC1 and IC2. DNA methylation status at the imprinting centers is shown by lollipops (white, unmethylated; black, methylated) on the maternal (M, black) and paternal (P, gray) homologues. (B) Map of the IC1 and IC2 clusters showing selective maternally (black) and paternally (gray) expressed genes. (C) qRT-PCR was performed on E9.5 placentae for the following genes: H19, Igf2, Ascl2, Tssc4, Cdkn1c, Phlda2 and Osbpl5. qRT-PCR was carried out as technical triplicates on three individual placentae (biological replicates) for each genotype and the error bars show the standard deviation for biological triplicates. Expression is relative to the reference housekeeping gene, Peptidylprolyl isomerase A (Ppia). Asterisks indicate mutant values significantly different from wild type values, using two-tailed unpaired Student t test (Ascl2, p = 0.006; Phlda2 p = 0.005).
To confirm the status of Ascl2 and Phlda2 expression in maternal Del7AI/+ placentae using an independent method, we carried out in situ hybridization (ISH) and immunohistochemistry (IHC) on E9.5 and E13.5 Del7AI placentae, respectively. At E9.5, Ascl2 expression is highest in the spongiotrophoblast layer of the developing placenta, with weak expression also detected in the labyrinthine layer (Rossant et al., 1998). We found a general decrease in Ascl2 levels within these placental layers in Del7AI/+ placentae. (Fig. 3A). To ask whether the difference in Ascl2 expression was due to a general decrease in the number of spongiotrophoblast cells where Ascl2 is predominantly expressed, we performed ISH for Tpbpa (4311), a marker of the spongiotrophoblast layer at E9.5 (Lescisin et al., 1988). We found no decrease in the number of Tpbpa-positive cells in mutant placentae at this early stage (Fig. 3B). We noted that Tpbpa levels appeared higher in some (but not all) mutant sections analyzed, but this expression difference was not found to be statistically significant by qRT-PCR analysis (Fig. S1C). As a control, we also performed ISH on another imprinted gene in the IC2 cluster, Cdkn1c, which is critical to placentation and expressed throughout the placenta at this stage, and did not observe any changes in levels or expression patterns (Fig. 3C), consistent with our qRT-PCR results.
Fig. 3.

Abnormal expression of the maternally expressed genes Ascl2 and Phlda2 in Del7AI/+ placentae. The expression patterns of Ascl2 (A), Tpbpa (B) and Cdkn1c (C) were analyzed by ISH on frozen sections of wild type (+/+) and maternal deletion (Del7AI/+) placentae collected at E9.5. Multiple sections from two placentae of each genotype were assessed and representative sections are shown. Sense probes are not shown. The blue stain shows gene expression. Nuclear fast red was used as the counterstain. (D) PHLDA2 IHC at E13.5. The brown stain shows protein expression in the labyrinth. Scale bar: 0.5 mm. sp: spongiotrophoblast layer, lab: labyrinthine layer, d: decidua.
Although Phlda2 (also known as Ipl) is broadly expressed in the ectoplacental cone and chorion at E8.0 (Dunwoodie and Beddington, 2002), it eventually becomes restricted to trophoblast cells of the labyrinthine layer such that by E10.5 it is predominantly expressed in syncytiotrophoblast layer I (SynT-I) with some lower levels in layer II (SynT-II) (Frank et al., 1999, 2002; Simmons et al., 2008a). At E13.5, we observed an overall increase in PHLDA2 protein expression by IHC in Del7AI/+ heterozygotes with a broader distribution of Phlda2-expressing foci throughout the labyrinth (Fig. 3D). Our results show that Del7AI does not have a trans effect on the levels of Igf2 transcribed from the wild type paternal homologue, but rather affects the maternally expressed genes Ascl2 and Phlda2 of the IC2 sub-domain in cis.
Phlda2 upregulation is placental-specific and is also observed in Ascl2-null conceptuses
In light of our results showing misregulation of both Ascl2 and Phlda2 when the deletion is maternally inherited, we sought to distinguish between two possible mechanisms consistent with our observations. In the first model, the deletion could have direct although opposite effects on the expression levels of Ascl2 and Phlda2. According to the second model, Del7AI has a direct effect on Ascl2 levels, reducing them by 50%, and one of the consequences of this hypomorphic Ascl2 allele is an increase in Phlda2 levels in the labyrinth.
Unlike Ascl2 expression which is restricted to the extra-embryonic tissues, Phlda2 is expressed in the developing embryo and was isolated as a mesoderm-enriched cDNA clone from gastrulation stage embryos (Dunwoodie and Beddington, 2002). To determine whether the deletion also affects this embryonic expression of Phlda2, we carried out a Phlda2 qRT-PCR analysis on E13.5 Del7AI/+ embryos. We found no difference in Phlda2 expression levels between Del7AI/+ and wild type embryos (Fig. 4A), suggesting that the Del7AI allele has no impact on embryonic Phlda2 levels.
Fig. 4.
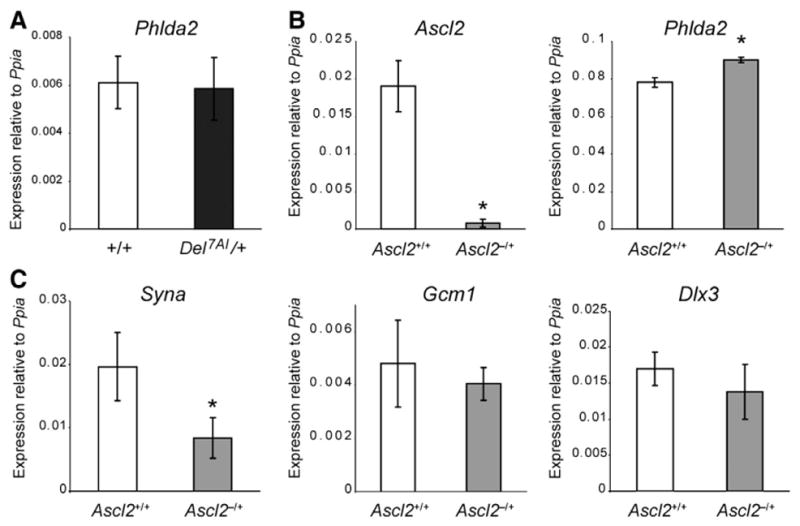
Dysregulation of Phlda2 in Ascl2-null placentae. (A) Phlda2 qRT-PCR on E13.5 embryo was carried out on three technical replicates each from three individual Del7AI/+ embryos and three wild type littermate individual embryos (biological replicates) and the error bars shown are the standard deviation for biological replicates. Expression is relative to Ppia. No significant difference was found between the two genotypes (p = 0.8). Gene expression analysis by qRT-PCR on total E9.5 placental RNA from Ascl2lacZ/+ (Ascl2−/+) mutants and wild type littermates (Ascl2+/+). Analysis is presented for (B) Ascl2 and Phlda2, as well as the labyrinth trophoblast markers Syna, Gcm1, and Dlx3 (C). The results show a significant increase in Phlda2 expression in Ascl2-deficient placentae but not for the syncytiotrophoblast markers Gcm-1 and Dlx3. Syna was also downregulated in mutant placentae. Expression levels are relative to Ppia levels. Asterisks mark mutant levels statistically different from wild type levels (for Ascl2 p = 0.0008; for Phlda2 p = 0.002, and p = 0.012 for Syna).
A prediction from the second model is that one would expect to observe an upregulation of Phlda2 in placentae deficient for Ascl2, before the manifestation of the lethality observed at E10.0 (Guillemot et al., 1994). To address this, we took advantage of an Ascl2-lacZ knock-in mouse line (Ascl2-lacZ allele, Ascl2tm1.1Nagy; MGI: 2155757). Maternal inheritance of the Ascl2-lacZ allele (Ascl2lacZ/+ heterozygotes) results in midgestational lethality due to a lack of spongiotrophoblast formation (Tanaka et al., 1999), a phenotype similar to the one described for the original knock-out allele (Guillemot et al., 1994). Although it was unclear why the insertion of an IRES-lacZ reporter cassette in the 3′UTR of Ascl2 should generate a null allele (Tanaka et al., 1999), we found that Ascl2 mRNA levels are very low in Ascl2lacZ/+ placentae at E9.5 (Fig. 4B). We performed qRT-PCR for Phlda2 on these mutant placentae and found a slight but significant increase (15%) in Phlda2 expression in the mutant placentae compared to wild type littermates (Fig. 4B), establishing that loss of Ascl2 by itself can lead to altered Phlda2 levels in the placenta.
We next investigated whether this increase in Phlda2 expression in Ascl2-deficient placentae was paralleled by a general increase in syncytiotrophoblast markers at E9.5. We performed qRT-PCR for three syncytiotrophoblast markers: syncytin-A (Syna), a marker of syncytiotrophoblast layer I (Simmons et al., 2008a) required for syncytiotrophoblast formation by cell fusion (Dupressoir et al., 2009); Glial cells missing homolog 1 (Gcm1), a marker of syncytiotrophoblast layer II of the labyrinth (Anson-Cartwright et al., 2000; Simmons et al., 2008a), and required for branching morphogenesis in the labyrinth following chorioallantoic fusion (Anson-Cartwright et al., 2000); and Distal-less homeobox 3 (Dlx3), a pan-specific labyrinth trophoblast marker (Simmons et al., 2008a), required for normal morphogenesis of the labyrinthine layer(Morasso et al., 1999). Although we observed a general decrease in these three markers in Ascl2-null placentae at E9.5, significant differences with wild type littermates were only detected for the syncytiotrophoblast layer I gene Syna (Fig. 4C), a marker of the cell population where Phlda2 is predominantly expressed (Frank et al., 2002). Note that although the increase in Phlda2 levels at E9.5 is less pronounced in Ascl2-null (Fig. 4B) then in hypomorphic Del7AI/+ placentae (Fig. 2C), the concomitant decrease in Syna seen in Ascl2-null placentae (Fig. 4B) is not observed in Del7AI/+ samples (Fig. S1B), suggesting a decrease in Phlda2-expressing cells in the null placentae.
Taken together, our analyses of Del7AI/+ embryos and Ascl2-null placentae suggest that Ascl2 operates upstream of Phlda2 in the placenta and that Del7AI acts primarily as an Ascl2 hypomorphic allele. Unlike what has been described for the phenotypes of the Ascl2 KO (Guillemot et al., 1994) and Ascl2-lacZ (Tanaka et al., 1999) alleles, maternal inheritance of Del7AI does not cause an embryonic lethality phenotype and therefore offered us the opportunity to study the consequences of reduced Ascl2 levels on the morphogenesis of the mature placenta.
Increased trophoblast giant cell numbers in Del7AI/+ heterozygotes
One of the phenotypic consequences of loss of Ascl2 is an increase in the number of parietal trophoblast giant cells making up the giant cell layer of the mutant placentae before manifestation of the lethality at E10.0 (Guillemot et al., 1994). We found that reduced levels of Ascl2 in maternal Del7AI heterozygotes are also accompanied by a thicker giant cell layer, readily seen in hematoxylin and eosin (H&E) stained sections (Fig. 5A). At higher magnification, we observed a drastic thickening of the layer, which is usually a single cell layer in wild type placentae, but appears as a thick stack of 4 to 6 giant cells in the mutant placentae at E15.5 (Fig. 5B). DAPI staining confirmed that these cells have large polyploid nuclei, a characteristic of endoreduplicating giant cells (Fig. 5C).
Fig. 5.

Thickening of the giant cell layer in maternal Del7AI heterozygotes. Overall morphology of wild type and Del7AI/+ placentae at E15.5 as revealed by hematoxylin and eosin (H&E) staining (A–B) and DAPI staining (C). Whereas the giant cell layer is hard to detect at lower magnification in wild type placentae (A), a thick layer is clearly visible in the mutants (arrowheads). At higher magnification, the single cell layer seen in wild type placentae (dotted line) is replaced by a 4 to 6 cell layer in the mutants (B). Also noticeable is the absence of clear spongiotrophoblast and trophoblast glycogen cells in proximity of the giant cell layer in the mutants. (C) DAPI staining readily identifies the giant polyploid nuclei of the cells stacked at the giant cell layer in the mutants. Scale bar: 500 μm (A); 100 μm (B–C). d: decidua; TGC: parietal trophoblast giant cells; SpT: spongiotrophoblast; GlyT: trophoblast glycogen cells; lab: labyrinth layer.
Reduced spongiotrophoblast cell numbers and absence of trophoblast glycogen cells in mutant placentae
The main reported phenotype caused by Ascl2 deficiency is the absence of a spongiotrophoblast layer (also known as junctional zone) (Guillemot et al., 1994). Although at the stage of the lethality this layer is mostly composed of spongiotrophoblast cells, past mid-gestation a second cell population, the trophoblast glycogen cells starts to populate this layer and eventually migrate into the decidua (Coan et al., 2006). We found that both of these cell types were severely affected in Del7AI/+ placentae.
In histological sections, the labyrinth appears directly juxtaposed to the expanded giant cell layer in the mutants, with no evidence of spongiotrophoblast or glycogen cells at E15.5, within the spongiotrophoblast layer or above the giant cell layer (Fig. 5B). The trophoblast gene Tpbpa is an early marker of spongiotrophoblast cells (Lescisin et al., 1988) but is eventually also expressed in trophoblast glycogen cells starting at E11.5 (Teesalu et al., 1998). We analyzed the expression of Tpbpa by in situ hybridization (ISH) on frozen sections of wild type and Del7AI/+ placentae at E15.5. In the wild type placenta, Tpbpa is strongly expressed in a broad population of spongiotrophoblast cells overlying the labyrinth as well as in trophoblast glycogen cells within the spongiotrophoblast layer and within the decidua (Fig. 6A). In the mutant placentae, only a thin layer of Tpbpa-expressing spongiotrophoblast cells is detected, with no sign of glycogen cells. This conspicuous absence of glycogen cells was also confirmed by periodic acid Schiff (PAS) staining (Fig. 6D), which detects glycogen producing cells (Adamson et al., 2002), as well as by ISH for two other markers of trophoblast glycogen cells, Pcdh12 (Rampon et al., 2005) (Fig. 6B) and Cdkn1c (Georgiades et al., 2002) (Fig. 6C). Cdkn1c is expressed at low levels in the labyrinth in both Del7AI/+ and wild type placentae, but is most highly expressed in glycogen cells in the maternal decidua, where signal is detected in wild type but not in Del7AI/+ placentae (Fig. 6C). We investigated the spongiotrophoblast to labyrinth ratio and we found that although the labyrinth area of Del7AI/+ placentae is similar to wild type, the spongiotrophoblast is reduced by about six-fold in the mutants.
Fig. 6.

Abnormal spongiotrophoblast and glygocen cell phenotype in maternal Del7AI heterozygotes at E15.5. (A) Tpbpa, (B) Pcdh12, and (C) Cdkn1c ISH performed on E15.5 placentae. Multiple sections from two placentae of each genotype were assessed and representative sections are shown. Sense probes not shown. The blue stain shows gene expression. (D) Periodic acid Schiff (PAS) stain revealing glycogen cells. Dark pink/purplish stain shows glycogen staining. Scale bar: 0.5 mm. d: decidua; TGC: parietal trophoblast giant cells; SpT: spongiotrophoblast; GlyT: trophoblast glycogen cells; lab: labyrinth layer.
Our results show that the correct dosage of Ascl2 levels is critical for proper development of the spongiotrophoblast layer and suggest that most of the weight loss observed in mutant placentae is attributable to a total absence of glycogen cells and a drastic decrease in the spongiotrophoblast cell population.
Dysmorphogenesis of the labyrinth layer in mutant placentae
Although Ascl2 is not expressed in the labyrinth layer of the mature placenta, we have shown that the absence or decreased levels of Ascl2 lead to an upregulation of Phlda2, which is expressed at high levels in syncytiotrophoblast layer I (SynT-I) of the trilaminar labyrinthine trophoblast from E10.5 to E12.5, after which stage a sharp decrease in PHLDA2-positive cells is observed (Frank et al., 2002). To assess whether other trophoblast cell types of the labyrinth were also affected in Del7AI/+ placentae at E15.5, we analyzed the expression of Placental lactogen-II (Prl3b1/Pl-II) and Gcm1 by ISH. At this stage, Prl3b1 is expressed in three different trophoblast populations: parietal trophoblast giant cells (the polyploid cells of the giant cell layer), spongiotrophoblast, as well as the mononucleated giant cells lining the maternal blood sinusoids in the labyrinth (sinusoidal trophoblast giant cells) (Simmons et al., 2008b). In the mutant placentae, expression of Prl3b1 is mostly seen in the thickened giant cell layer in the basal region, but also reveals a marked increased in the density of sinusoidal giant cells within the labyrinth (Fig. 7A). A similar result was obtained for Gcm1 (Fig. 7B), a marker of the labyrinth syncytiotrophoblast layer II (Simmons et al., 2008a). In light of the close functional and cellular interactions between the labyrinth trophoblast layers and the fetal vasculature, we also analyzed the architecture of fetal blood vessels in mutant placentae by immunostaining for laminin, a marker of the basement membrane associated with fetal endothelial cells. Our results again highlight important defects in the structure of the labyrinth, characterized by a denser network of fetal blood vessels in the mutant labyrinth (Fig. 7C). Note that despite these abnormalities in labyrinthine structures, the mutant placentae do not show evidence for increased thickness between the fetal and maternal circulations (Fig. S3).
Fig. 7.

Dysmorphogenesis of the labyrinth layer in Del7AI/+ placentae. ISH for (A) Prl3b1 and (B) Gcm1 on frozen sections of wild type and mutant placentae at E15.5. Multiple sections from two placentae of each genotype were assessed and representative sections are shown. Sense probes are not shown. The blue stain shows gene expression. Prl3b1 is expressed in parietal trophoblast giant cells, spongiotrophoblast and mononucleated sinusoidal trophoblast giant cells of the labyrinth. In the mutants, the spongiotrophoblast staining is replaced by a thicker giant cell layer, and the labyrinth signal is denser. Gcm1 is a marker of the syncytiotrophoblast II of the labyrinth. As seen for Phlda2 (expressed in syncytiotrophoblast I) and Prl3b1, we observed a denser network of Gcm1-expressing cells in the mutant placenta. (C) IHC for laminin marks the basement membrane of the fetal blood vessels and revealed a denser network of capillaries in the mutant labyrinth. The brown stain shows protein expression. (D–E) Feto-placental vasculature of the maternal Del7AI heterozygotes. Representative scanning electron micrographs of E14.5 fetal vascular corrosion casts. (D) Lateral view; (E) top view from maternal side. Four fetal vasculature casts were assessed per genotype. Scale bar: 1 mm.
To investigate further the defects in labyrinthine vasculature suggested by the differences in laminin stain as well as labyrinth trophoblast cell types, we performed fetal vasculature corrosion cast experiments (Adamson et al., 2002; Whiteley et al., 2006). Both a lateral view and maternal view of the Del7AI/+ labyrinth revealed a greater degree of disorganization compared to wild type (Figs. 7D–E). In the wild type placenta, the labyrinth is subdivided into functional units of capillary beds arranged in separate villous branches, each served by one arteriole and one venule. In the maternal Del7AI/+ heterozygotes, however, these subdivisions of the capillary branching were not clear and individual functional units could not be dissected from the mutant resin casts (Fig. 7D). Observation of the maternal side of the fetal vasculature reveals this phenotype in an even more dramatic way (Fig. 7E). In the wild type placenta, the discrete functional unit of capillaries are clearly demarcated and separated by spaces filled by finger-like projections of the spongiotrophoblast as well as maternal blood space. In the mutants, this organization is lost and fetal blood vessels fill the entire diameter of the placenta. Our results suggest that sub-optimum levels of Ascl2 not only affect the spongiotropoblast and trophoblast glycogen cells, but also lead to expansion of trophoblast cell types in the giant cell layer and labyrinth.
Discussion
We previously reported that maternal inheritance of a deletion of the ~280 kb intergenic region between the two imprinted domains on distal Chr 7 is associated with reduced birth weights (Lefebvre et al., 2009). Here we show that upon maternal inheritance of Del7AI, the expression levels of the genes in the IC1 and IC2 subdomains are unaffected with the exception of a decrease in Ascl2 expression. This is accompanied by a reduction in placental weight and a severe placental phenotype characterized by a reduced spongiotrophoblast, absence of trophoblast glycogen cells, expanded giant cell layer and a disorganized labyrinth. We also observed an increase in Phlda2 expression in Del7AI/+ placentae, which we propose is a secondary effect of decreased Ascl2 expression (see below).
We have previously demonstrated that inheritance of Del7AI does not affect the transcription of the ncRNA Kcnq1ot1 (Lefebvre et al., 2009). The analysis of IC2-regulated genes presented here also argues against a global effect of the deletion on the entire cluster of imprinted genes. The mechanism whereby Ascl2 is decreased in Del7AI/+ heterozygotes is currently under investigation. One possibility is that the Del7AI allele deletes a placental-specific enhancer of Ascl2 expression on the maternal allele. However, to date no such enhancers have been identified in the IC1–IC2 interval, although an Ascl2 transgene rescuing the lethality of Ascl2 deficiency and encompassing ~60 kb of our deletion has recently been described (Rentsendorj et al., 2010).
The role of the spongiotrophoblast in the mouse placenta is unclear but it is essential for development, since a complete lack of spongiotrophoblast leads to midgestational lethality (Guillemot et al., 1994; Oh et al., 2008). Previous work on Ascl2 has established that it plays an important role in the formation of the spongiotrophoblast, together with a negative role on trophoblast giant cell differentiation from diploid ectoplacental precursors (Guillemot et al., 1995). Maternal inheritance of the Ascl2 null allele results in midgestational lethality due to a complete lack of spongiotrophoblast formation (Guillemot et al., 1994). The Del7AI allele gave us the opportunity to assess the phenotypic consequences in the mature placenta associated with suboptimal Ascl2 levels. We show that the dosage of Ascl2 levels is critical for normal placentation and that reduced Ascl2 results in a decreased spongiotrophoblast population, lack of trophoblast glycogen cells, an expanded giant cell layer, a reduction in placental size, and embryonic growth restriction. Nevertheless, these suboptimal Ascl2 levels can still result in viable pups, although growth retarded.
One of the drastic phenotypes described here is the disorganization of the fetal vasculature. Observed from the maternal side, the vascular bed is seen as a punctate structure with regularly spaced openings organizing the underlying network of fetal capillaries. In the mutants with a stunted spongiotrophoblast layer, this organization is lost and the labyrinth fills the entire placenta. Our results therefore suggest that finger-like projections of the spongiotrophoblast within the labyrinth play an important role, structurally and perhaps also functionally via the maintenance of an organized vascular network for proper feto-maternal exchanges. Although this aspect of the phenotype has not been studied in detail here, it is likely that the observed changes in fetal capillary beds and labyrinth trophoblast densities would affect the expansion of the maternal blood spaces.
The phenotype studied here describes a functional placenta with an apparent total loss of trophoblast glycogen cells. Both the developmental origin and function of this cell lineage is currently unknown (Coan et al., 2006). Based on the observation that these glycogen-rich vacuolated cells are first detected past midgestation within the spongiotrophoblast layer and that they express markers of spongiotrophoblast (such as Igf2 (Redline et al., 1993) and Tpbpa (Lescisin et al., 1988)) it has been proposed that they may represent a specific subtype of spongiotrophoblast cells. However it has been shown that Pcdh12, which is specifically expressed in trophoblast glycogen cells past mid-gestation (Rampon et al., 2005), also labels a specific sub-population of cells within the ectoplacental cone at E7.5 (Bouillot et al., 2006). This led the authors to suggest that trophoblast glycogen cells and spongiotrophoblast might represent distinct trophoblast lineages both emerging from Ascl2-positive ectoplacental cone (EPC) precursors. Our results clearly show that Ascl2 is required for formation of the trophoblast glycogen cell lineage. Whether this is mediated via a direct requirement for Ascl2 in precursors of the lineage or an indirect effect via EPC or spongiotrophoblast factors themselves dependent on Ascl2 will have to be addressed by lineage-specific knock out experiments.
With regards to the function of trophoblast glycogen cells during development, our results suggest that this trophoblast lineage is not required for parturition, as previously suggested (Coan et al., 2006), at least not within the context of mixed litters composed of wild type and mutant conceptuses, as studied here. It might be informative to analyze litters composed entirely of mutant conceptuses, an experiment which would require embryo transfers since the homozygous Del7AI/Del7AI are not viable (Lefebvre et al., 2009). However, it is tempting to suggest that the absence of trophoblast glycogen cells documented in Del7AI/+ heterozygotes might contribute to the embryonic growth retardation phenotype observed. Although our results are suggestive of a correlation between the absence of trophoblast glycogen cells and embryonic growth restriction, we cannot exclude the contributions of the abnormalities described in other placental trophoblast cell types to the embryonic phenotype. Therefore a definitive demonstration of the role of the trophoblast glycogen cells on embryonic growth is still lacking and would require the ablation of this specific lineage during development.
An intriguing aspect of our work is the finding that in addition to Ascl2, a second IC2-regulated gene, the imprinted gene Phlda2, is also affected in deletion heterozygotes. However the transcriptional effects at both genes is opposite such that Phlda2 is upregulated in the mutants. We considered the possibility that the deletion might have a direct effect on Phlda2, for instance via the removal of a silencer, but several of our results are not consistent with such a model. The first argument against this model is the structure of the allele itself. The deletion has a distal breakpoint less than five kilobase pairs away from Ascl2, whereas Phlda2 is located more than half a megabase away from this breakpoint. Furthermore, both Tssc4 and Cdkn1c, located in between Ascl2 and Phlda2 are not affected by Del7AI. Second, the deletion has no effect on the embryonic expression of Phlda2 and up-regulation is only observed in the placentae of Del7AI/+ mutants. Third, we showed that the levels of Phlda2 are also elevated in Ascl2-null placentae, suggesting that Ascl2 acts upstream of Phlda2 and exerts negative effects on Phlda2-expressing cells, the syncytiotrophoblast layer I. Finally, our analysis of markers for the other two trophoblast cell types of the labyrinth layer at E15.5 also suggests an increased cellularity in these lineages in the mature placenta. This would suggest that a decreased dosage in Ascl2 and the ensuing underdevelopment of the spongiotrophoblast have broader impacts on chorionic ectoderm-derived lineages. Our results are consistent with a model proposing that, as for the trophoblast giant cell lineage, Ascl2 plays an inhibitory role on the differentiation of the trilaminar trophoblast cell lineages of the labyrinth and that normal development of the placenta requires an appropriate balance of positive and negative signals governing the ratio of spongiotrophoblast and labyrinth trophoblast precursors. Such a model could also help to explain previously published intriguing results on the effects of varying Phlda2 gene dosage. Phlda2 knockout mice are viable but their placentae are characterized by placentomegaly, an expanded spongiotrophoblast layer and increased number of trophoblast glycogen cells, but no fetal overgrowth (Frank et al., 2002). Although this has not been addressed by the authors, we propose that increased cellularity in the spongiotrophoblast layer of Phlda2-null placentae might be mediated via an increase in Ascl2 levels. Conversely, overexpression of Phlda2 from a BAC transgene leads to a reduced spongiotrophoblast layer and defects in trophoblast glycogen cells, changes which are accompanied by a reduction in Ascl2 levels (Tunster et al., 2010). Together, these studies and our results suggest a previously unrecognized relationship between Ascl2 and Phlda2. Both are expressed in the ectoplacental cone (EPC) and extra-embryonic ectoderm in early development, two layers which are brought in close proximity with the collapse of the ectoplacental cavity at E8.0 (Dunwoodie and Beddington, 2002; Guillemot et al., 1994). We have confirmed that ASCL2 and PHLDA2 are co-expressed in the EPC at E7.5 (Fig. S2), arguing against a direct repression of Phlda2 by ASCL2 at those early stages. Our results and those of others (Frank et al., 2002; Salas et al., 2004; Tunster et al., 2010) could be reconciled by proposing that each of these two genes play positive roles in the maintenance of different precursor populations within these layers, and that reduced levels in one are accompanied by increased levels in the other, the proper balance assuring development of a fully functional placenta.
It has been proposed that elevated levels of PHLDA2 are associated with low birth weights (Apostolidou et al., 2007; Mcminn et al., 2006). Although the ASCL2 gene might not be imprinted in human placenta (Miyamoto et al., 2002), our results show that ASCL2 function is dosage sensitive and suggest that ASCL2 mutations could be involved in the etiology of these cases of intrauterine growth restriction (IUGR) associated with upregulation of PHLDA2. The Del7AI mouse line will provide a valuable model for the study of IUGR in the mouse, including its consequences on maternal physiology during pregnancy and postnatal adult phenotypes in growth retarded pups.
Supplementary Material
Acknowledgments
The authors thank Dr. Jay Cross for the Pcdh12 and Gcm1 cDNA clones; Dr. Rosalind John for the Cdkn1c cDNA clone; Dr. Ben Tycko for the PHLDA2 antibody; Dr. Janet Rossant for the Tpbpa cDNA clone and Dr. Andras Nagy for the Ascl2-lacZ mouse line. This work was funded by a grant from the Canadian Institutes of Health Research (CIHR) to LL (MOP-82863). LL holds a Canada Research Chair. ROM is supported by a Four Year Doctoral Fellowship from UBC.
Footnotes
Author contributions
ROM performed most of the experiments and wrote the manuscript. ABB and ROM performed the fetal vasculature casting experiments. ABB performed the IHC analyses at E7.5. LL designed and directed the study and wrote the manuscript.
References
- Adamson S, Lu Y, Whiteley KJ, Holmyard D, Hemberger M, Pfarrer C, Cross J. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev Biol. 2002;250:358–373. doi: 10.1016/s0012-1606(02)90773-6. [DOI] [PubMed] [Google Scholar]
- Andrews S, Wood M, Tunster S, Barton S, Surani M, John R. Cdkn1c (p57Kip2) is the major regulator of embryonic growth within its imprinted domain on mouse distal chromosome 7. BMC Dev Biol. 2007;7:53. doi: 10.1186/1471-213X-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson-Cartwright L, Dawson K, Holmyard D, Fisher SJ, Lazzarini RA, Cross JC. The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat Genet. 2000;25:311–314. doi: 10.1038/77076. [DOI] [PubMed] [Google Scholar]
- Apostolidou S, Abu-Amero S, O’Donoghue K, Frost J, Olafsdottir O, Chavele KM, Whittaker JC, Loughna P, Stanier P, Moore GE. Elevated placental expression of the imprinted PHLDA2 gene is associated with low birth weight. J Mol Med. 2007;85:379–387. doi: 10.1007/s00109-006-0131-8. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Bouillot S, Rampon C, Tillet E, Huber P. Tracing the glycogen cells with protocadherin 12 during mouse placenta development. Placenta. 2006;27:882–888. doi: 10.1016/j.placenta.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Coan PM, Conroy N, Burton GJ, Ferguson-Smith AC. Origin and characteristics of glycogen cells in the developing murine placenta. Dev Dyn. 2006;235:3280–3294. doi: 10.1002/dvdy.20981. [DOI] [PubMed] [Google Scholar]
- Constância M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417:945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- Dunwoodie SL, Beddington RS. The expression of the imprinted gene Ipl is restricted to extra-embryonic tissues and embryonic lateral mesoderm during early mouse development. Int J Dev Biol. 2002;46:459–466. [PubMed] [Google Scholar]
- Dupressoir A, Vernochet C, Bawa O, Harper F, Pierron G, Opolon P, Heidmann T. Syncytin-A knockout mice demonstrate the critical role in placentation of a fusogenic, endogenous retrovirus-derived, envelope gene. Proc Natl Acad Sci USA. 2009;106:12127–12132. doi: 10.1073/pnas.0902925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick G, Soloway P, Higgins M. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002;32:426–431. doi: 10.1038/ng988. [DOI] [PubMed] [Google Scholar]
- Frank D, Mendelsohn CL, Ciccone E, Svensson K, Ohlsson R, Tycko B. A novel pleckstrin homology-related gene family defined by Ipl/Tssc3, TDAG51, and Tih1: tissue-specific expression, chromosomal location, and parental imprinting. Mamm Genome. 1999;10:1150–1159. doi: 10.1007/s003359901182. [DOI] [PubMed] [Google Scholar]
- Frank D, Fortino W, Clark L, Musalo R, Wang W, Saxena A, Li CM, Reik W, Ludwig T, Tycko B. Placental overgrowth in mice lacking the imprinted gene Ipl. Proc Natl Acad Sci USA. 2002;99:7490–7495. doi: 10.1073/pnas.122039999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta. 2002;23:3–19. doi: 10.1053/plac.2001.0738. [DOI] [PubMed] [Google Scholar]
- Guillemot F, Nagy A, Auerbach A, Rossant J, Joyner AL. Essential role of Mash-2 in extraembryonic development. Nature. 1994;371:333–336. doi: 10.1038/371333a0. [DOI] [PubMed] [Google Scholar]
- Guillemot F, Caspary T, Tilghman S, Copeland NG, Gilbert DJ, Jenkins NA, Anderson DJ, Joyner AL, Rossant J, Nagy A. Genomic imprinting of Mash2, a mouse gene required for trophoblast development. Nat Genet. 1995;9:235–242. doi: 10.1038/ng0395-235. [DOI] [PubMed] [Google Scholar]
- Harbers K, Müller U, Grams A, Li E, Jaenisch R, Franz T. Provirus integration into a gene encoding a ubiquitin-conjugating enzyme results in a placental defect and embryonic lethality. Proc Natl Acad Sci USA. 1996;93:12412–12417. doi: 10.1073/pnas.93.22.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman S. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- Jones MJ, Lefebvre L. An imprinted GFP insertion reveals long-range epigenetic regulation in embryonic lineages. Dev Biol. 2009;336:42–52. doi: 10.1016/j.ydbio.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre L, Mar L, Bogutz A, Oh-McGinnis R, Mandegar MA, Paderova J, Gertsenstein M, Squire JA, Nagy A. The interval between Ins2 and Ascl2 is dispensable for imprinting centre function in the murine Beckwith–Wiedemann region. Hum Mol Genet. 2009;18:4255–4267. doi: 10.1093/hmg/ddp379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescisin KR, Varmuza S, Rossant J. Isolation and characterization of a novel trophoblast-specific cDNA in the mouse. Genes Dev. 1988;2:1639–1646. doi: 10.1101/gad.2.12a.1639. [DOI] [PubMed] [Google Scholar]
- Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, Higgins M, Feil R, Reik W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet. 2004;36:1291–1295. doi: 10.1038/ng1468. [DOI] [PubMed] [Google Scholar]
- Li Y, Behringer R. Esx1 is an X-chromosome-imprinted regulator of placental development and fetal growth. Nat Genet. 1998;20:309–311. doi: 10.1038/3129. [DOI] [PubMed] [Google Scholar]
- Mancini-Dinardo D, Steele SJ, Levorse J, Ingram RS, Tilghman S. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006;20:1268–1282. doi: 10.1101/gad.1416906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcminn J, Wei M, Schupf N, Cusmai J, Johnson E, Smith A, Weksberg R, Thaker H, Tycko B. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta. 2006;27:540–549. doi: 10.1016/j.placenta.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Hasuike S, Jinno Y, Soejima H, Yun K, Miura K, Ishikawa M, Niikawa N. The human ASCL2 gene escaping genomic imprinting and its expression pattern. J Assist Reprod Genet. 2002;19:240–244. doi: 10.1023/A:1015362903486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morasso MI, Grinberg A, Robinson G, Sargent TD, Mahon KA. Placental failure in mice lacking the homeobox gene Dlx3. Proc Natl Acad Sci USA. 1999;96:162–167. doi: 10.1073/pnas.96.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh R, Ho R, Mar L, Gertsenstein M, Paderova J, Hsien J, Squire JA, Higgins M, Nagy A, Lefebvre L. Epigenetic and phenotypic consequences of a truncation disrupting the imprinted domain on distal mouse chromosome 7. Mol Cell Biol. 2008;28:1092–1103. doi: 10.1128/MCB.01019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-McGinnis R, Bogutz AB, Lee KY, Higgins M, Lefebvre L. Rescue of placental phenotype in a mechanistic model of Beckwith–Wiedemann syndrome. BMC Dev Biol. 2010;10:50. doi: 10.1186/1471-213X-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Rampon C, Prandini MH, Bouillot S, Pointu H, Tillet E, Frank R, Vernet M, Huber P. Protocadherin 12 (VE-cadherin 2) is expressed in endothelial, trophoblast, and mesangial cells. Exp Cell Res. 2005;302:48–60. doi: 10.1016/j.yexcr.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Redline RW, Chernicky CL, Tan HQ, Ilan J. Differential expression of insulin-like growth factor-II in specific regions of the late (post day 9.5) murine placenta. Mol Reprod Dev. 1993;36:121–129. doi: 10.1002/mrd.1080360202. [DOI] [PubMed] [Google Scholar]
- Reik W, Walter J. Evolution of imprinting mechanisms: the battle of the sexes begins in the zygote. Nat Genet. 2001;27:255–256. doi: 10.1038/85804. [DOI] [PubMed] [Google Scholar]
- Rentsendorj A, Mohan S, Szabó P, Mann J. A genomic imprinting defect in mice traced to a single gene. Genetics. 2010;186:917–927. doi: 10.1534/genetics.110.118802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J, Guillemot F, Tanaka M, Latham K, Gertenstein M, Nagy A. Mash2 is expressed in oogenesis and preimplantation development but is not required for blastocyst formation. Mech Dev. 1998;73:183–191. doi: 10.1016/s0925-4773(98)00051-3. [DOI] [PubMed] [Google Scholar]
- Salas M, John R, Saxena A, Barton S, Frank D, Fitzpatrick G, Higgins M, Tycko B. Placental growth retardation due to loss of imprinting of Phlda2. Mech Dev. 2004;121:1199–1210. doi: 10.1016/j.mod.2004.05.017. [DOI] [PubMed] [Google Scholar]
- Schulz R, Menheniott TR, Woodfine K, Wood A, Choi J, Oakey R. Chromosome-wide identification of novel imprinted genes using microarrays and uniparental disomies. Nucleic Acids Res. 2006;34:e88. doi: 10.1093/nar/gkl461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Fitzpatrick G, Higgins M. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J. 2008;27:168–178. doi: 10.1038/sj.emboj.7601960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirohzu H, Yokomine T, Sato C, Kato R, Toyoda A, Purbowasito W, Suda C, Mukai T, Hattori M, Okumura K, Sakaki Y, Sasaki H. A 210-kb segment of tandem repeats and retroelements located between imprinted subdomains of mouse distal chromosome 7. DNA Res. 2004;11:325–334. doi: 10.1093/dnares/11.5.325. [DOI] [PubMed] [Google Scholar]
- Simmons DG, Natale DR, Begay V, Hughes M, Leutz A, Cross J. Early patterning of the chorion leads to the trilaminar trophoblast cell structure in the placental labyrinth. Development. 2008a;135:2083–2091. doi: 10.1242/dev.020099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DG, Rawn S, Davies A, Hughes M, Cross J. Spatial and temporal expression of the 23 murine Prolactin/Placental Lactogen-related genes is not associated with their position in the locus. BMC Genomics. 2008b;9:352. doi: 10.1186/1471-2164-9-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Kobayashi T, Kanayama N. p57(Kip2) regulates the proper development of labyrinthine and spongiotrophoblasts. Mol Hum Reprod. 2000;6:1019–1025. doi: 10.1093/molehr/6.11.1019. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Puchyr M, Gertsenstein M, Harpal K, Jaenisch R, Rossant J, Nagy A. Parental origin-specific expression of Mash2 is established at the time of implantation with its imprinting mechanism highly resistant to genome-wide demethylation. Mech Dev. 1999;87:129–142. doi: 10.1016/s0925-4773(99)00158-6. [DOI] [PubMed] [Google Scholar]
- Teesalu T, Blasi F, Talarico D. Expression and function of the urokinase type plasminogen activator during mouse hemochorial placental development. Dev Dyn. 1998;213:27–38. doi: 10.1002/(SICI)1097-0177(199809)213:1<27::AID-AJA3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Tunster SJ, Tycko B, John RM. The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol Cell Biol. 2010;30:295–306. doi: 10.1128/MCB.00662-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet. 2004;36:1296–1300. doi: 10.1038/ng1467. [DOI] [PubMed] [Google Scholar]
- van der Flier LG, van Gijn ME, Hatzis P, Kujala P, Haegebarth A, Stange DE, Begthel H, van den Born M, Guryev V, Oving I, van Es JH, Barker N, Peters PJ, van de Wetering M, Clevers H. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–912. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- Wagschal A, Sutherland HG, Woodfine K, Henckel A, Chebli K, Schulz R, Oakey R, Bickmore WA, Feil R. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol Cell Biol. 2008;28:1104–1113. doi: 10.1128/MCB.01111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley KJ, Pfarrer CD, Adamson S. Vascular corrosion casting of the uteroplacental and fetoplacental vasculature in mice. Meth Mol Med. 2006;121:371–392. doi: 10.1385/1-59259-983-4:369. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wong C, DePinho RA, Harper JW, Elledge SJ. Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev. 1998;12:3162–3167. doi: 10.1101/gad.12.20.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QY, Quaife CJ, Palmiter RD. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature. 1995;374:640–643. doi: 10.1038/374640a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.