

Abstract
Alzheimer's disease is a progressive, neurodegenerative disease that represents a growing global health crisis. Two major forms of the disease exist: early onset (familial) and late onset (sporadic). Early onset Alzheimer's is rare, accounting for less than 5% of disease burden. It is inherited in Mendelian dominant fashion and is caused by mutations in three genes (APP, PSEN1, and PSEN2). Late onset Alzheimer's is common among individuals over 65 years of age. Heritability of this form of the disease is high (79%), but the etiology is driven by a combination of genetic and environmental factors. A large number of genes have been implicated in the development of late onset Alzheimer's. Examples that have been confirmed by multiple studies include ABCA7, APOE, BIN1, CD2AP, CD33, CLU, CR1, EPHA1, MS4A4A/MS4A4E/MS4A6E, PICALM, and SORL1. Despite tremendous progress over the past three decades, roughly half of the heritability for the late onset of the disease remains unidentified. Finding the remaining genetic factors that contribute to the development of late onset Alzheimer's disease holds the potential to provide novel targets for treatment and prevention, leading to the development of effective strategies to combat this devastating disease.
1. Introduction
The disease that would, upon the suggestion of his colleague Dr. Emil Kraepelin, eventually bear his name was first described by Aloysius “Alois” Alzheimer in 1907, based upon his observations and treatment of a 51-year-old patient, August “D” [1]. In addition to short-term memory loss, symptoms included unusual behavior and the neuropathological characteristics which have become hallmarks of the disease [1]. These features include extracellular plaques formed from cleaved amyloid precursor protein (APP) and intracellular tangles of hyperphosphorylated microtubule associated protein tau (MAPT) [2–4]. APP is an integral membrane protein with wide expression throughout the body, which is concentrated in neuronal synapses [5]. Although the primary function of APP is not fully understood, it has been implicated in neurite extension and synaptic plasticity. The tau protein is expressed primarily in neurons, where it stabilizes microtubules that are responsible for axonal transport [6].
Alzheimer's disease is a progressive, neurodegenerative disease that is ultimately and invariably fatal, unless another cause of death intervenes. Although the clinical course is heterogeneous, there are a number of common features in addition to the characteristic extracellular plaques and neurofibulary tangles. Early symptoms are often mistaken as part of normal aging, or manifestations of stress [7]. The most common symptom that is first noticed is a loss of short-term memory. As the disease progresses, symptoms may include aggression, irritability, confusion, and language problems, as well as loss of long-term memory. In later stages of the disease, patients become withdrawn and ultimately are completely unable to care for themselves [7]. Neuropathological changes include loss of neurons and synapses in the cerebral cortex and certain subcortical regions. This loss results in gross atrophy of the affected regions, including degeneration in the temporal and parietal lobes, as well as parts of the frontal cortex and cingulate gyrus [8]. MRI and PET analyses have documented atrophy of specific brain regions, particularly the hippocampus, as individuals progress from mild cognitive impairment to Alzheimer's disease [9, 10].
Alzheimer's disease is the most common form of age-related neurodegenerative dementia and one of the most serious health problems in the industrialized world. More than 35 million individuals suffer from dementia globally, and the majority of these are diagnosed with Alzheimer's disease. Alzheimer's disease is also a major public health problem in the United States. The Alzheimer's Association reports that in 2011, 5.4 million individuals were diagnosed with Alzheimer's in the US, with an estimated prevalence ranging from 6%–12% of individuals aged 65 years and older [11]. This figure is expected to increase to over 10 million over the next 25 years, as individuals born between 1946 and 1964 begin reaching the age where they are at risk of developing the disease. Alzheimer's is the 6th leading cause of death in America and the 5th leading cause of death for those over 65 [11]. Currently available therapies provide short-term symptomatic relief but do not slow disease progression. In order for novel therapeutic approaches to be created, greater knowledge regarding the underlying etiology of the disease is needed. The financial costs of Alzheimer's currently exceed $183 billion annually in the US, and an additional $210 billion worth of unpaid care is provided by friends and family members of patients [11].
There are two types of Alzheimer's disease: familial (also known as early onset) and sporadic (also known as late onset). Familial Alzheimer's disease is inherited in a Mendelian fashion, with little influence from the environment. In contrast, while genetics play a large role in the development and expression of late onset Alzheimer's disease, nongenetic factors are also very important. To date, there is a large literature documenting that obesity, diabetes, cardiovascular disease, and related factors, as well as cerebral and systemic inflammation increase the risk for late onset Alzheimer's disease, as well as lower the age at which symptoms first appear.
2. Familial Alzheimer's Disease
Familial Alzheimer's disease (FAD) is expressed as a Mendelian trait with dominant inheritance. FAD is relatively rare, accounting for less than 5% of the total Alzheimer's disease burden [11, 14]. The Alzheimer's Association estimates that in 2011, approximately 200,000 individuals were afflicted with FAD in the US. A diagnosis is typically made when patients are in their fifties or sixties. However, there are instances of FAD onset at a much younger age. In one famous and particularly tragic example within an extended Columbian family, disease onset occurs in the mid-forties and as early as the early thirties [14]. The mutation responsible in this Columbian family (Presenilin 1 E280A) is known in other FAD families with a more typical age at onset.
To date, more than 160 highly penetrant but rare mutations have been described in three genes (amyloid precursor protein, presenilin 1, and presenilin 2) that cause familial Alzheimer's disease (Table 1). Each of these genes will be discussed later.
Table 1.
Genes responsible for early onset Alzheimer's disease, including chromosomal location and function.
| Gene name (symbol) | Chromosomal location | Function of encoded protein |
|---|---|---|
| Amyloid precursor protein (APP) | 21q21.3 | Implicated in neuronal development and synaptic formation and repair |
| Presenilin one (PSEN1) | 14q24.2 | Cleavage of the amyloid precursor protein and NOTCH receptor proteins via overlapping, but distinct mechanisms [12, 13] |
| Presenilin two (PSEN2) | 1q42.13 |
2.1. Amyloid Precursor Protein (APP)
The function of APP is not completely understood. However, it has been implicated in neuronal development and synaptic formation and repair and has been shown to be upregulated after neuronal injury. The gene encoding APP is located on chromosome 21q21.3. The gene consists of 19 exons and covers approximately 240 kilobases of DNA. The full-length APP matures in the Golgi and endoplasmic reticulum, prior to being inserted into the plasma membrane. The resulting protein is between 365 and 770 amino acids in length. It is expressed in numerous tissues, and at least eight isoforms are created by alternate splicing of exons 1–13, 13a, and 14–18. The most abundant isoforms are APP695 (exons 1–6, 9–18), APP751 (exons 1–7, 9–18), and APP770 (exons 1–18). All of the transcripts produce a multidomain protein with a single membrane-spanning region. APP751 and APP770 differ from APP695 in that they contain exon seven, which encodes a serine protease inhibitor domain. APP695 is the predominant isoform in neuronal tissues, whereas APP751 is the predominant isoform elsewhere. Full-length APP is processed into a number of fragments through a series of proteolytic cleavages by alpha-, beta- and gamma-secretase. Cleavage by alpha-secretase creates a fragment that is not associated with plaques or Alzheimer's. However, cleavage by beta-, and gamma-secretases creates beta-amyloid, which is encoded by exons 16 and 17 and is 39 to 42 amino acids in length. It is the beta-amyloid fragment that forms the extracellular plaques that are the hallmarks of Alzheimer's disease.
APP was initially implicated in the etiology of Alzheimer's disease by a number of facts. First, the primary component of the extracellular plaques that characterize the disease is the amyloid protein. Tanzi and colleagues and Robakis et al. demonstrated in 1987 that the form of APP that is localized in plaques was derived from a larger protein that is encoded by a gene located on chromosome 21 [15, 16]. Second, chromosome 21 is duplicated in Down's syndrome patients, who typically develop Alzheimer's in the fifth decade of life. An extra copy of the APP gene is duplicated along with the other chromosome 21 genes in Down's syndrome, which further implicated the APP gene in Alzheimer's pathology.
The beta-amyloid that forms the core of amyloid plaques consists of aggregates of a 4.2 kDa polypeptide and is the same in Alzheimer's and older Down's syndrome patients. The existence of this fragment, along with the details of how it is produced from the full-length protein, was resolved in a series of experiments in the mid-eighties by Tanzi, Masters, Robakis, Kang, Glenner, and Wong (Figure 1). These findings laid the groundwork for the amyloid cascade hypothesis of Alzheimer's disease, which postulates that production of beta-amyloid 42 fragments accumulates in the brain with age [17]. Either the resulting plaques, or perhaps the soluble oligomers, are neurotoxic and precipitate hyperphosphorylation of the tau protein, as well as other events that occur downstream in the disease process, including pruning of the dendritic tree and synaptic dysfunction. These changes result in neuronal loss, dementia, and ultimately death.
Figure 1.
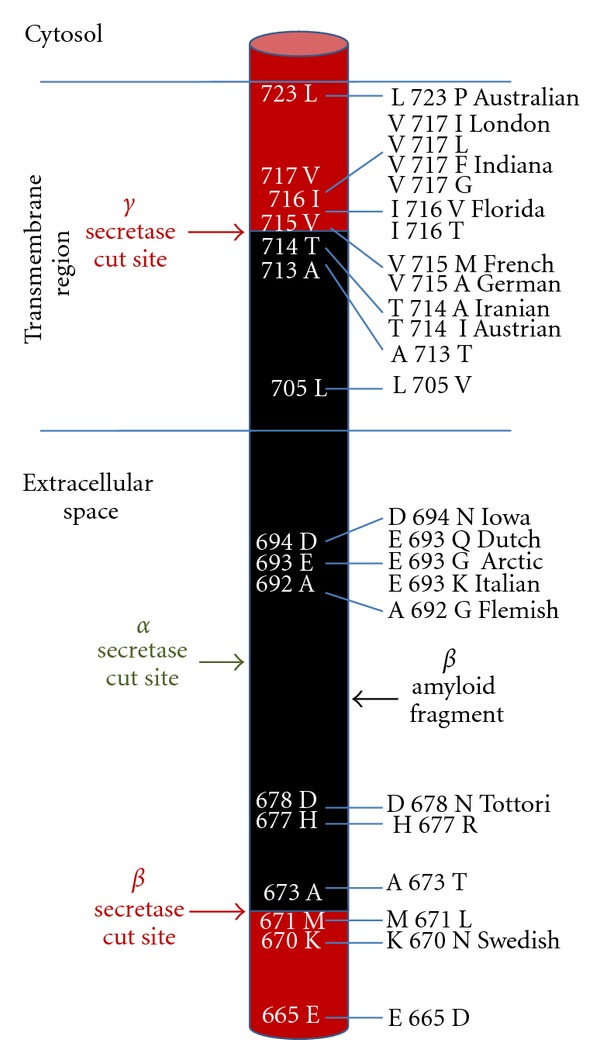
Processing of the amyloid precursor protein (APP). The precursor protein is acted upon by either alpha- or beta-secretase, followed by cleavage by gamma-secretase. Cleavage by beta-secretase allows formation of beta-amyloid (shown in black) by gamma-secretase, while alpha-secretase prevents beta-amyloid formation. Locations of the alpha-, beta-, and gamma-secretase cut sites are shown, along with APP mutations and the names that are associated with certain mutations.
Lustbader et al. demonstrated that beta-amyloid interacted with amyloid beta-binding alcohol dehydrogenase (ABAD) to induce mitochondrial toxicity in Alzheimer's disease patients and in transgenic mice [18]. Crystallography showed that binding of beta-amyloid by ABAD deformed the nicotinamide adenine dinucleotide (NAD) binding site and prevented activation. Furthermore, inhibiting interactions between beta-amyloid and ABAD suppressed the generation of free radicals and beta-amyloid induced apoptosis within neurons.
Linkage was detected between FAD and markers on chromosome 21 near the APP gene in 1987 [19], but subsequent studies failed to detect this association in families with late onset Alzheimer's disease [20]. An association between a missense mutation in the APP gene and FAD in a large kindred was detected in 2006 by Goate [21]. However, nonallelic heterogeneity was detected in this study as well as many others, implicating additional genes in the pathogenesis of familial Alzheimer's disease [20–22].
APP mutations account for a minority of FAD cases but have also been shown to cause cerebral amyloid angiopathy. In a multicenter study of familial and sporadic Alzheimer's disease, Tanzi and colleagues concluded that mutations in the APP gene account for a small portion of FAD cases [23]. In a similar study, Kamino et al. also found that APP mutations account for a small fraction of FAD [24]. In affected members of 2 families with early onset Alzheimer's disease-1, Goate et al. identified a heterozygous mutation in the APP gene (V717I) [25]. Raux and others identified mutations in the APP gene among affected members in five of 31 families with FAD [26]. Combined with the results of earlier studies, the authors estimated that 16% of FAD is attributable to mutations in the APP gene [26].
There have been extensive studies of the effects of APP mutations, particularly those occurring at amino acid 717. Suzuki et al. reported that 3 such mutations (V717I, V717F, and V717G) were consistently associated with a 1.5- to 1.9-fold increase in the generation of longer beta-amyloid fragments, which formed insoluble amyloid fibrils more rapidly than shorter fragments [27]. Yamatsuji and colleagues demonstrated that expression of the cytoplasmic domain of any of the mutations at amino acid 717 (V717I, V717F, and V717G) induced G protein-mediated nucleosomal DNA fragmentation in cultured neuronal cells [28].
In a notable recent discovery, Jonsson and colleagues discovered a rare mutation (A673T) in the APP gene that was protective against Alzheimer's disease [29]. The mutation was discovered in a search for rare coding variants within whole-genome sequence data from 1,795 Icelanders. This A673T mutation was also found to be protective against cognitive decline among elderly without Alzheimer's disease. This substitution was adjacent to the beta-secretase site in APP, and resulted in a 40% reduction in the formation of amyloidogenic peptides.
2.2. Presenilins 1 and 2 (PSEN1 and PSEN2)
The presenilins are important determinants of gamma-secretase activity and are responsible for proteolytic cleavage of the amyloid precursor protein (APP) and NOTCH receptor proteins [12]. Gamma-secretase is a multimeric protein complex consisting of PSEN1 or PSEN2, nicastrin, and APH1 [12]. All mutations in PSEN1 increase cleavage of the amyloid precursor protein by gamma-secretase, thereby increasing the production of the beta-amyloid 42 fragment [13, 30–33]. Presenilin 1 is an evolutionarily conserved membrane protein that is present in Drosophila [34] and C. elegans [35]. The PSEN1 gene, which is located on chromosome 14q24.2, was identified as an Alzheimer's disease locus in 1995 by positional cloning [36]. The PSEN2 gene is located on chromosome 1q42.13 and has significant sequence homology and very similar structural organization to PSEN1 (approximately 60% overall identity). In fact, it was the high degree of sequence identity between the two genes that led to the identification of PSEN2 during the cloning of PSEN1 [37]. However, it seems that the presenilin genes encode proteins with overlapping, but distinct functions. For example, mutations in both presenilin genes result in increased beta-amyloid [30, 32], yet functional PSEN2 does not rescue APP or NOTCH signaling defects observed in PSEN1 null animals [38].
Evidence that presenilin genes were involved in gamma-secretase activity and hence in amyloid precursor protein processing was first obtained in 2000 when Li and colleagues suggested that PSEN1 and PSEN2 comprised the active site of gamma-secretase [39]. In their experiments, gamma-secretase activity coeluted with PSEN1 on gel exclusion chromatography. In addition, they observed that an anti-PSEN1 antibody immunoprecipitated active gamma-secretase. In the same year, Kopan and Goate determined that PSEN1 and PSEN2 both appear to be active gamma-secretase sites that reside in different complexes [40]. The authors proposed that regulation of cleavage may depend on the specific profile of proteins present in the multimeric gamma-secretase complex [40]. Lee et al. reported that nicastrin and presenilin heterodimers associate with APH1A and APH1B to form the gamma-secretase complex that is responsible for the proteolytic cleavage of many membrane proteins, including APP and NOTCH [12].
Cai and colleagues reported that PSEN1 binds and recruits phospholipase D1 (PLD1) to the trans-Golgi network (TGN). Furthermore, expression of PLD1 in mouse neuroblastoma (N2a) cells was found to be inversely correlated with gamma-secretase-mediated beta-amyloid generation [41]. Additional studies by the same lab showed that overexpression of catalytically active PLD1 promoted generation of beta-amyloid-containing vesicles from the TGN [42]. Taken together, these observations showed that PLD1 regulates intracellular trafficking of beta-amyloid, distinct from its effect on gamma-secretase activity.
Activity of PSEN1 and PSEN2 is essential for the formation of beta-amyloid 42. Transgenic mice that overexpressed mutant presenilin-1 showed an increase in beta-amyloid 42. However, mice overexpressing a wildtype version of presenilin-1 did not show a similar increase [31]. These results suggested that mutations in presenilin cause Alzheimer's disease through a deleterious gain of function that increases the amount of beta-amyloid 42 that is deposited in the brain. Support for this model of Alzheimer's etiology was provided by Davis et al. who showed that amyloid deposition was equivalent in the brains of wildtype mice and those with a loss of functional Psen1 allele [43]. Finally, Qian and colleagues showed that the brains of mice carrying the human PSEN1 A246E mutation showed increased levels of beta-amyloid 42 [44]. Citron and colleagues noted that several lines of evidence strongly support the conclusion that progressive cerebral deposition of beta-amyloid protein is a seminal event in familial Alzheimer's disease pathogenesis [32]. Both in vitro and in vivo data demonstrate that FAD-linked presenilin mutations directly or indirectly alter the level of gamma- but not alpha- or beta-secretase, resulting in increased production of beta-amyloid 42 which may lead to cerebral beta-amyloidosis and AD [32].
Nearly 200 variant PSEN1 alleles have been detected (http://www.molgen.ua.ac.be/ADMutations). The vast majority of these are missense mutations, which alter the protein sequence by a single amino acid. Although a few splice-site mutations and insertion-deletion variants have also been discovered, no nonsense mutations, which result in a truncated and nonfunctional protein, have been observed to cause Alzheimer's disease [45, 46].
Relative to PSEN1, a much smaller number of mutations (approximately two dozens) have been observed to date in PSEN2 in families segregating FAD (http://www.molgen.ua.ac.be/ADMutations). Similar to PSEN1, these are missense mutations with a deleterious gain of function effect. The range in age at onset is much more variable within families segregating PSEN2 mutations, relative to those segregating disease alleles in APP or PSEN1 [47–50]. In families that develop FAD due to PSEN2 mutations, the age at onset ranges from 40 to 85 years of age. In contrast, individuals belonging to families that develop Alzheimer's due to mutations in APP or PSEN1 experience an age at disease onset between 35 to 55 (PSEN1) or 45 to 65 years of age (APP).
3. Late Onset Alzheimer's Disease
In contrast to FAD, which is inherited in a Mendelian dominant fashion as described earlier, sporadic or late onset Alzheimer's disease (LOAD) is etiologically heterogeneous and results from a combination of many genetic and environmental factors. Although there is evidence of a strong genetic component to disease risk, a large percentage of the genetic risk remains unidentified. The heritability of LOAD was estimated to be 79% in a large population-based study of Swedish twins [51]. However, until quite recently, only a single gene had been reliably associated with increased risk for Alzheimer's disease. The epsilon allele in the apolipoprotein E gene (APOE4) has been associated with substantially increased risk for LOAD as well as an earlier age of disease onset [52, 53]. A large number of additional polymorphisms have been associated with risk for LOAD, but replication of these associations has been inconsistent.
A search for “Alzheimer's” in the Gene Card database yields 1890 hits. These polymorphisms are located in genes regulating inflammation, oxidative stress, vascular biology, and protease function, among others (a complete list is available at http://www.alzgene.org/). It is likely that the failure to universally reproduce these associations is due at least in part to incomplete phenotypic characterization of study participants, inadequate samples sizes, and incomplete penetrance of risk alleles [54, 55].
4. Genome-Wide Association Studies
Although nearly 2000 genes have been implicated in the etiology of late onset Alzheimer's disease over the past two decades, it was only in 2009 that two large-scale genome-wide association studies replicated associations between LOAD and genes other than APOE (Table 2). In these studies, which were published back to back in Nature Genetics [56, 57], Lambert et al. and Harold and colleagues described associations between late onset Alzheimer's disease and genetic markers in three genes in addition to APOE: clusterin (CLU), complement receptor 1 (CR1), and phosphatidylinositol binding clathrin assembly protein (PICALM). In a study supported by the Welcome Trust and based in Cardiff, UK, Harold et al. employed a two-stage design with 3,941 Alzheimer's cases and 7,948 controls in the first stage, followed by genotyping of the most interesting variants in 2,023 cases and 2,340 controls. Lambert and colleagues conducted a similarly large study based at the Institute Pasteur in Lille, France of 2,032 cases and 5,328 controls, which replicated a European sample of 3,978 Alzheimer's cases and 3,297 controls. These studies were followed in 2010 by a meta-analysis of over 35,000 participants, including 8,371 individuals diagnosed with Alzheimer's disease [58]. In this meta-analysis, Seshadri and colleagues confirmed associations with APOE, CLU, PICALM, and CR1 and identified two new Alzheimer's risk loci: Myc box-dependent-interacting protein 1 (BIN1) and a marker (rs597668) near the EXOC3L2/BLOC1S3/MARK4 gene cluster.
Table 2.
Genes reliably implicated in risk for late-onset Alzheimer's disease, including chromosomal location and function.
| Gene name (symbol) | Chromosomal location | Function of encoded protein |
|---|---|---|
| Apolipoprotein E (APOE) | 19q13.2 | Transportation of lipoproteins, fat-soluble vitamins, and cholesterol |
| Clusterin (CLU) | 8p21 | Chaperone protein |
| Complement receptor 1 (CR1) | 1q32 | Receptor for C3b and C4b complement cleavage fragments, the main system for clearance of complement opsonized immune complexes |
| Phosphatidylinositol binding clathrin assembly protein (PICALM) | 11q14.2 | Membrane retrieval of the synaptic vesicle, intracellular movement of lipids and proteins, and possibly internalization of full length APP from the cell surface |
| Myc box-dependent-interacting protein 1 (BIN1) | 2q14.3 | Tumor suppressor |
| ATP binding cassette transporter 7 (ABCA7) | 19p13.3 | The expression pattern suggests a role for lipid homeostasis and differentiation of immune cells. |
| Membrane-spanning 4-domains, subfamily A (MS4A) | 11q12.2 | Possibly involved in signal transduction or immunological functions |
| Ephrin type-A receptor 1 (EPHA1) | 7q34 | Member of the EPH receptor-tyrosine kinase family, implicated in mediating developmental events of the nervous system |
| CD33 antigen (CD33) | 19q13.3 | Adhesion molecule of myelomonocytic-derived cells |
| CD2 associated protein (CD2AP) | 6p12.3 | A scaffolding molecule that regulates the actin cytoskeleton and vesicle formation |
| Sortilin-related receptor 1 (SORL1) | 11q24.1 | Receptor for ApoE, assists with intracellular trafficking and processing of APP |
It is interesting that BIN1 showed a nonsignificant trend for association in the previous studies by Harold et al. and Lambert et al. The significant signal for BIN1 in the meta-analysis after a nonsignificant trend for association in the two initial GWAS studies was a clear indication of the tremendous sample sizes that are required to detect Alzheimer's risk genes outside of APOE. Current studies underway through the International Genomics of Alzheimer's Project (IGAP) will ultimately evaluate over 60,000 participants including more than 30,000 Alzheimer's cases, roughly twice the size of the previous largest effort by Seshadri et al. [58]. The IGAP consortium was launched in February 2011 as a collaborative effort between the European Alzheimer's Disease Initiative (EADI) from France, the Alzheimer's Disease Genetics Consortium (ADGC) and the neurology subgroup of the Cohorts for Heart and Aging in Genomic Epidemiology (CHARGE) from the United States, and the Genetic and Environmental Risk in Alzheimer's Disease (GERAD) from the United Kingdom. Data and DNA will be contributed by more than 35 individual institutions from Europe and North America.
5. Copy Number Variation (CNV)
With the advent of whole-genome scanning methods, a new perspective on human genetic variation was observed, the widespread variation in the copy number of submicroscopic DNA segments. CNVs offer an alternate genetic marker map for disease association studies. CNVs are major contributors to genetic variance; thus, it is conceivable that they may confer susceptibility to or directly cause disease [59]. CNVs influence gene expression, phenotypic variation and adaptation by altering gene dosage [59]. A recent study of gene expression variation as a model of complex phenotypes found that 18% of gene expression traits were associated with CNVs [60]. With regard to Alzheimer's disease, Brouwers et al. reported a significant association between CNVs in the CR1 gene and increased risk for Alzheimer's [61]. In this study, a Flanders-Belgian cohort of 1,887 individuals was used as a discovery set combined with a French validation cohort of 2,003 individuals. CNV within the gene was observed to produce two CR1 isoforms. One of these was associated with increased Alzheimer's risk (P = 0.0025 for the combined cohorts). The authors hypothesized that the association with the high copy number variant was caused by additional C3b/C4b and cofactor sites [61]. In addition, Szigeti and colleagues detected significant association between a high copy number segment on chromosome 14 (14q11.2) in the region of the olfactory receptor and increased risk for Alzheimer's disease. This finding is of interest due to the early involvement of the olfactory lobes in Alzheimer's neuropathology [62, 63].
6. LOAD Genes
6.1. Apolipoprotein E (APOE)
The APOE protein is 299 amino acids long and is synthesized principally in the liver. In the nervous system, nonneuronal cells (astroglia and microglia) are the primary producers of APOE. It transports lipoproteins, fat-soluble vitamins, and cholesterol into the lymphatic system and then into the blood. APOE is the ligand for the LDL receptor, LRP (LDL-related protein), and VLDL-receptor, all of which are preferentially expressed by neurons. Given its involvement in lipid transport, it is perhaps not surprising that the role for APOE as a disease gene was first recognized with respect to cardiovascular disease [64–66].
The APOE gene is located on chromosome 19q13.2 in a cluster with apolipoprotein C1 and apolipoprotein C2. The gene is on the small side, containing 3,597 nucleotides and four exons [67]. Three primary isoforms exist for apolipoprotein E (E2, E3, and E4), which are encoded by 3 alleles (epsilon 2, 3, and 4). These isoforms differ in the amino acid sequence at 2 sites: residues 112 and 158. At these sites, the E2, E3, and E4 isoforms contain cysteine/cysteine, cysteine/arginine, and arginine/arginine residues, respectively [67–69]. The absolute allele frequencies vary widely by race and ethnicity [70–72], with the E4 allele showing a curvilinear relationship to latitude, with lowest frequency in the mid-latitudes [73]. Eisenberg and colleagues, who reported this relationship, have suggested that it is due to a reduced metabolic rate and hence lowers cholesterol requirements in more moderate climates [73].
The effect size for the APOE4 allele is among the largest for any multifactorial, oligogenic (complex) disease. Individuals homozygous for APOE4 are 10 to 20 times as likely to develop LOAD as APOE4 negative individuals [74, 75], and there is a gene-dosage effect observed between increasing copies of the E4 allele and earlier age of disease onset [76]. In contrast, individuals who carry the APOE2 allele have a lower risk for Alzheimer's disease relative to other genotypes [77]. However, LOAD is clearly a heterogeneous disease, as one third of Alzheimer's patients are APOE4 negative. Interestingly, health risks associated with carriage of the APOE4 allele are not specific to Alzheimer's disease. A worse outcome has been reported for APOE4 carriers following traumatic brain injury [78], selective cardiac bypass surgery [79], and spontaneous intracerebral hemorrhage [80].
Even though the APOE4 allele has one of the greatest effect sizes of any gene associated with an oligogenic disease, the polymorphism accounts for less than half the genetic variance in Alzheimer's disease risk, strongly suggesting that additional Alzheimer's disease genes remain to be identified. Data from several recent large GWAS studies support this contention [56, 57, 81, 82].
6.2. Clusterin (CLU)
Clusterin is a 75-kDa apolipoprotein that is widely expressed throughout the body, particularly in the brain [83]. Structurally, CLU is heterodimeric, comprised of two subunits that are linked by disulfide bonds [84]. These subunits are formed by proteolytic cleavage of the clusterin precursor protein into alpha- and beta-peptide fragments [83]. Clusterin contains two coiled-coil α-helices, which are typical of small heat shock proteins, and it has been suggested that clusterin is in fact a heat shock protein [85]. The CLU gene is located on chromosome 8 (8p21) and has high sequence homology (70%–80% identity) across mammalian taxa. The gene is composed of 9 exons, covering 16Kb of DNA [86]. The name clusterin is derived from an ability to cluster together cells of various types [87]. The former name of this glycoprotein is APOJ, and like its cousin APOE, clusterin has chaperone properties and is able to interact with many different molecules [88]. The similarity to APOE extends to an elevated abundance in brain regions (including the hippocampus and entorhinal cortex) that are affected by the pathology of Alzheimer's disease and cerebrospinal fluid (CSF), as well as being present in amyloid plaques and binding to beta-amyloid [89–94]. Furthermore, both APOE and clusterin have been implicated in clearance of beta-amyloid from neural tissue and CSF [95, 96]. Other identified functions for clusterin include the regulation of complement formation and apoptosis, as well as lipid transport [97]. However, many of these findings have received inconsistent support. The one function that is consistently supported for CLU is to act as a chaperone protein.
6.3. Phosphatidylinositol Binding Clathrin Assembly Protein (PICALM)
PICALM is a ubiquitously expressed 70 kDa protein that has been implicated in the membrane retrieval of the synaptic vesicle [98]. PICALM is distributed in pre- and postsynaptic structures [99] and functions in clathrin-mediated endocytosis (CME) [100]. The involvement of PICALM in CME is important, as this process is a critical step in the intracellular movement of lipids and proteins [99] as well as internalization of full-length APP from the cell surface in cell culture studies [101]. However, mice that possess nonsense point mutations in the PICALM gene have abnormal hematopoiesis and iron metabolism but do not exhibit abnormal neurological function [102]. The PICALM gene was first identified in studies of myelogenous leukemia as the fusion partner of AF10 in a chromosomal translocation that is observed in acute myeloid leukemia, acute lymphoblastic leukemia, and malignant lymphoma (10;11)(p13;q14) [103]. Meyerholz and colleagues observed that CALM associates with the alpha-appendage domain of the AP2 adaptor via the three peptide motifs 420DPF, 375DIF, and 489FESVF and to a lesser extent with the amino-terminal domain of the clathrin heavy chain [104]. The PICALM gene is located on chromosome 11 (11q14.2), and the first 289 amino acids of the protein have a high degree of homology (81%) to the clathrin assembly protein AP3 [105]. There are three isoforms; the canonical sequence is 652 amino acids in length. Two additional isoforms are generated by deletions of short sequences of amino acids near the 3 ends of the transcript.
6.4. Complement Receptor 1 (CR1)
Complement receptor 1 (also known as CD35, C3b/C4b receptor) is a member of the receptor of complement activation (RCA) family. The CR1 protein is a monomeric type I membrane glycoprotein that is expressed on erythrocytes, leukocytes, and splenic follicular dendritic cells [106]. CR1 is the human receptor for C3b and C4b complement cleavage fragments [107]. As such, CR1 serves as the main system for processing and clearance of complement opsonized immune complexes and mediates cellular binding to particles that are labeled with activated complement [106]. It has been shown that CR1 can act as a negative regulator of the complement cascade, mediate immune adherence and phagocytosis, and inhibit both the classic and alternative complement pathways [106]. The number of CR1 molecules decreases with aging of erythrocytes in normal individuals and is also decreased in pathological conditions such as systemic lupus erythematosus (SLE), HIV infection, some hemolytic anemias, and other conditions featuring immune complexes [108, 109]. The CR1 gene is located within a complex of immunoregulatory genes on chromosome 1 (1q32), known as the regulator of complement activation (RCA) superfamily [110, 111]. The CR1 gene appears to have undergone multiple duplication events. The CR1 glycoprotein exists as four allotypic variants of variable sizes [112]. The most common CR1 isoforms are the “F” and “S” allotypes, which are 250 and 290 kDa, respectively. The size difference is due to the inclusion of a long homologous repeat of 40–50 kDa [112]. Certain alleles of this gene have been statistically associated with an increased risk of developing late onset Alzheimer's disease [56].
6.5. Bridging Integrator 1 (BIN1)
BIN1, which was originally known as Myc box-dependent-interacting protein 1, is a widely expressed protein that interacts with Myc-box regions of the MYC oncoprotein [113]. Structurally, the terminal portion of BIN1 is related to amphiphysin, a cancer-associated autoantigen, and to RVS167, a regulator of the cell cycle in yeast [113]. Based on these observations, Sakamuro and colleagues concluded that BIN1 acts as a tumor suppressor through negative control of the cell cycle. BIN1 is a 70 kDa nuclear protein, as determined by immunoprecipitation and immunofluorescence experiments [113]. The BIN1 gene is located on chromosome 2 (2q14.3). The gene spans 59.3 Kb and contains up to 20 exons [114]. As many as 10 isoforms of BIN1 exist, and these are produced by variable splicing of the mRNA [114]. The largest of these is isoform one, which is expressed exclusively in the brain and concentrated in nerve terminals (NCBI GeneID 274; http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=full_report&list_uids=274). The other smaller isoforms are generated by deletion of downstream exons, particularly 7, 11, 13, and 14 (NCBI GeneID 274).
6.6. ATP Binding Cassette Transporter 7 (ABCA7)
ABCA7 is a 2,146-amino acid protein containing two highly conserved ATP binding cassettes [115]. The gene was first identified in macrophages and is expressed abundantly in myeloid cells, particularly monocytes and granulocytes [115]. Expression is induced by differentiation of monocytes into macrophages [115]. Like other members of the ABC family, ABCA7 expression shows a response to changes in lipid concentration. In macrophages, both mRNA and protein are upregulated in the presence of modified low-density lipoprotein and downregulated by HDL [115]. The ABCA7 gene is located on chromosome 19p13.3 and contains 46 exons and spans about 32 kb [115, 116]. The mRNA is 6.8 kb in length and encodes a polypeptide of 2146 amino acids with a calculated molecular weight of 220 kDa [115].
6.7. Membrane-Spanning 4-Domains, Subfamily A (MS4A)
Three members of this family (MS4A4A, MS4A4E, and MS4A6E) were associated with AD by GWAS analysis [81, 117]. All three of these genes are located on chromosome 11q12.2 [118, 119]. The linkage disequilibrium and genomic structure in the region precludes assignment of the GWAS signal to a precise gene. The MS4A gene family is comprised of 16 genes clustered in a 600 kb region of chromosome 11q12 [119]. Most members of the MS4A gene family encode proteins with four or more transmembrane domains and have cytoplasmic domains at the amino and carboxyl termini, which are typically encoded by distinct exons. The structure and presumed function of the MS4A genes are similar to CD20, the high-affinity IgE receptor beta chain [120]. Ishibashi and colleagues described an MS4A4A mRNA that was predicted to encode a 205-amino acid protein with a conserved phosphorylation site at the intracellular loop [118]. In contrast, Liang and Tedder identified an MS4A4A mRNA that encoded a predicted peptide with 220 amino acids [120]. The same group also predicted that MS4A4E was 220 amino acids in length and was 76% identical to MS4A4A, sharing a high degree of homology with the transmembrane as well as both intracellular domains [119]. Liang and colleagues determined that the MS4A4E gene contained seven exons and spanned more than 23 kb. In contrast to MS4A4A and MS4A4E, as well as most other MS4A family members, MS4A6E encodes a protein with only two transmembrane domains and no carboxyl terminal cytoplasmic domain. As expected, since it contains fewer domains, the MS4A6E protein is smaller at 147 amino acids in length. Liang et al. predicted that MS4A6E contained four exons and spanned only 5 kb.
6.8. Ephrin Type-A Receptor 1 (EPHA1)
The EPH family is one of the largest of the receptor tyrosine kinase families, and members play crucial roles during development, particularly of the nervous system [121, 122]. Several EPH family members have also been implicated in oncogenesis [121]. The family is split into two groups, based upon the nature of the ligand. EPHA receptors bind to GPI-anchored ephrin-A ligands, while EPHB receptors bind to ephrin-B proteins that have a transmembrane and cytoplasmic domain [123]. EPH and ephrin signaling are important for the formation of segmented structures [124] and for the control of axon guidance during development [125]. The EPHA1 protein contains 976 amino acids and is approximately 108 kDa [126]. The EPHA1 gene is located on chromosome 7q34 and contains 18 exons that span a little over 18 kb [127].
6.9. CD 33 Antigen (CD33)
CD33 belongs to a family of cell-surface receptors that are expressed in the peripheral circulation on monocytes and myeloid progenitor cells [128–130]. CD33 is a member of the immunoglobulin superfamily and functions as an adhesion molecule that mediates sialic acid-dependent binding to cells [131, 132]. The protein contains two immunoglobulin-like domains, a transmembrane region and a cytoplasmic tail that has two potential ITIM sequences [131]. CD33 may function as an inhibitory receptor by colligation with CD64 on myeloid cells [133]. CD33 is located on chromosome 19q13.3 [127]. The gene contains seven exons that span 14.2 kb [134]. Alternate splicing of the transcript has been shown to result in two mRNA species of 1.4-1.5 kb and 1.6–1.8 kb [135]. The CD33 protein is 364 amino acids in length and has a mass of approximately 40 kDa [136].
6.10. CD2 Associated Protein (CD2AP)
CD2AP is a docking protein that becomes tyrosine phosphorylated in response to extracellular stimuli such as growth factors or cell-cell interaction and subsequently induces vesicle formation [137]. Ligand binding of CD2AP triggers protein segregation, CD2 clustering, and cytoskeletal polarization [138]. Kim and colleagues identified a CD2AP mutation in the splice acceptor region of exon 7 that was associated with primary focal segmental glomerulosclerosis [139]. There was no stable protein transcribed from the variant allele, suggesting that haploinsufficiency of CD2AP caused the disorder. The protein comprises 639 amino acids and has a deduced molecular mass of approximately 70 kDa [137]. The gene is ubiquitously expressed in adult and fetal human tissues as an approximately 5.4 kb transcript [137].
6.11. Sortilin-Related Receptor 1 (SORL1)
SORL1 encodes a protein that acts as a cell-surface receptor that binds ApoE and assists in intracellular trafficking of APP [140]. There is also evidence that SORL1 is important for the processing of APP by presenilins and the production of beta-amyloid [141]. The gene is located on chromosome 11q24.1 and encodes a 2,186-amino acid polypeptide that has homology to the RAP binding receptor gp95/sortilin [142]. Support for SORL1 as an Alzheimer's disease risk gene has been mixed, but a recent meta-analysis of previous studies detected a significant association between clusters of polymorphisms in SORL1 and Alzheimer's disease in both Caucasians and Asians [143]. These results, in combination with significant associations between SORL1 polymorphisms and hippocampal atrophy [144], as well as CSF levels of beta amyloid 42 [145], have increased confidence in the gene as an AD candidate.
7. Pathway Analysis
Evidence of the need for additional genetic research into Alzheimer's disease is provided by the fact that despite intensive searching over the past two decades, roughly half of the predicted genetic variation in Alzheimer's disease risk has been identified. Previous efforts have involved family-based linkage studies, population-based genome-wide association studies, and a host of candidate gene association tests. Recently, GWAS methods have been extended to very large numbers of participants. While it is true that one way to identify Alzheimer's genes with vanishingly small effect sizes is to employ ever-increasing numbers of participants in collaborative studies and meta-analyses, this approach relies upon the detection of marginal effects of SNPs within a single haplotype block. Even though sample sizes for recent GWAS studies are in the tens of thousands of participants, which supply an impressive amount of statistical power, this approach remains largely unable to resolve gene-gene interactions, which critically underlie the common gene-common disease hypothesis. Detection of multiple interacting loci requires a more sophisticated analytical approach.
One method for detecting interacting alleles within biological pathways is network enrichment analysis, which has also been termed pathway analysis [146]. This technique was originally developed for the analysis of microarrayed gene expression data [146]. The basis of this analytical method is to identify biological pathways, rather than individual markers or genes that are associated with the outcome of interest [147]. First, standard GWAS data are generated. Next, a gene assignment is made for as many markers as possible, and an adjustment is made for the number of markers per gene. Then, genes are assigned to predefined biological pathways, using databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) or the Gene Otology (GO) network. Finally, pathways or networks are evaluated for a significant overrepresentation of markers associated with the outcome of interest, relative to what would be expected at random.
One of the first applications of pathway analysis to Alzheimer's disease was published by Lambert et al. in 2010 [148]. In this study, Alligator and GenGen/KEGG software packages were used to analyze GWAS data derived from 2,032 Alzheimer's cases and 5,328 controls of French Caucasian ancestry. Both enrichment approaches identified a role for immunological dysfunction in the development of Alzheimer's disease [148]. While this association was far from novel, confirmation of a role for inflammation via pathway analysis supports the utility and value of pathway analysis methods in the study of Alzheimer's disease. In a second study from the same group in the same year, Hong and colleagues reported the involvement of intracellular transmembrane protein transport in Alzheimer's [149]. Of interest in the Hong et al. study was evidence of a functional role for TOMM40 in the development of AD. TOMM40 is the channel-forming subunit of the mitochondrial outer membrane translocase complex. Variation in the length of a poly-T homopolymer in this gene has been implicated in Alzheimer's etiology [150–154]; however, the validity of this association has been controversial due to strong linkage disequilibrium between TOMM40 and APOE, which complicates interpretation of the signal [150, 155, 156]. In a more recent pathway analysis of GWAS data from 742 participants enrolled in the Alzheimer's Disease Neuroimaging Initiative (ADNI) project, Ramanan et al. identified a number of genes, pathways and networks [157]. In this study, the outcome variable was not disease status, but rather a composite memory score that was constructed from the ADNI neuropsychological battery. Using the GSA-SNP software tool, 27 canonical pathways were identified that were over enriched relative to the composite memory score [157]. Among the set of enriched pathways were biological processes known to be related to memory consolidation such as receptor-mediated calcium signaling and long-term potentiation. Additional processes that were enriched against the composite memory score included cell adhesion and neuronal differentiation, as well as glucose signaling and inflammation. Furthermore, a large gene set was identified with MetaCore software that was centered on SP1 transcriptional regulation [157].
Pathway enrichment analysis has only recently been applied to Alzheimer's disease and most of these studies have been conducted in what are now relatively small cohorts. It is likely that as GWAS data are developed for large international cohorts, greater insight will be gained from interrogation of these data by pathway enrichment techniques. The combination of large sample sizes and sophisticated analytical methods such as pathway enrichment analysis is likely to produce many novel targets for the treatment and therapy of Alzheimer's disease.
8. Pharmacogenetics
Antidementia drugs are metabolized by the cytochrome p450 (CYP) gene family [158, 159]. Several of the CYP genes are highly polymorphic, particularly CYP2D6, CYP2C19, CYP2C9, and CYP3A4/5, and allelic frequencies at these loci vary greatly by ethnicity [158]. This is of concern since these allelic variants have substantial functional consequences, which places individuals who are slow metabolizers at risk of severe adverse events. Only 25% of western populations are rapid metabolizers, which places the remaining 75% of individuals who are prescribed antidementia drugs at risk due to overdosing [158]. The proper administration of antidementia drugs that are metabolized by CYP genes is to initially prescribe a low dose and titrate it upwards to the maximum tolerable dose. This presents another concern, in that if the starting dose is not escalated, the maximum effective dose may not be reached. Several excellent reviews of this literature have been provided by Cacabelos and colleagues [158, 160–162].
9. Summary
Alzheimer's disease is a major health problem globally, with massive human and economic costs. Alzheimer's has been one of the most difficult diseases to defeat, and there are currently no proven effective means of cure or prevention. The genetic causes run the entire range from a Mendelian dominant transmission in FAD to risk factors for a complex multifactorial and etiologically heterogeneous disease in LOAD. In addition, a number of genetic polymorphisms are known to impact response to ant-dementia medication through pharmacogenetic effects. While many (perhaps most) causal alleles have been identified for FAD, only roughly half of the genetic variation for LOAD has been reliably identified. Future work toward discovering this missing heritability will likely involve studies of epigenetic phenomena, such as methylation and acetylation, as well as the control of gene expression by micro-RNA species. The advent of genome-wide methods has led to the identification of several risk loci for LOAD, in addition to the well-documented association with the APOE4 allele. However, there is a great need for further study. The advent of genome-wide scanning methods including affordable whole genome sequencing, the assessment of epigenetic mechanisms, and the development of more sophisticated statistical analysis methods will facilitate the identification of additional risk loci for LOAD and lead to the development of effective treatment and prevention strategies for this devastating disease.
References
- 1.Strassnig M, Ganguli M. About a peculiar disease of the cerebral cortex: Alzheimer's original case revisited. Psychiatry. 2005;2:30–33. [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Biochemistry and molecular biology of amyloid β-protein and the mechanism of Alzheimer’s disease. Handbook of Clinical Neurology. 2008;89:245–260. doi: 10.1016/S0072-9752(07)01223-7. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer’s disease: a central role for amyloid. Journal of Neuropathology and Experimental Neurology. 1994;53(5):438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Evolution of neuronal changes in the course of alzheimer’s disease. Journal of Neural Transmission. 1998;(53):127–140. doi: 10.1007/978-3-7091-6467-9_11. [DOI] [PubMed] [Google Scholar]
- 5.Beyreuther K, Dyrks T, Hilbich C, et al. Amyloid precursor protein (APP) and beta A4 amyloid in Alzheimer’s disease and Down syndrome. Progress in clinical and biological research. 1992;379:159–182. [PubMed] [Google Scholar]
- 6.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proceedings of the National Academy of Sciences of the United States of America. 1975;72(5):1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waldemar G, Dubois B, Emre M, et al. Recommendations for the diagnosis and management of Alzheimer’s disease and other disorders associated with dementia: EFNS guideline. European Journal of Neurology. 2007;14(1):e1–e26. doi: 10.1111/j.1468-1331.2006.01605.x. [DOI] [PubMed] [Google Scholar]
- 8.Wenk GL. Neuropathologic changes in Alzheimer’s disease. Journal of Clinical Psychiatry. 2003;64(Supplement 9):7–10. [PubMed] [Google Scholar]
- 9.Desikan RS, Cabral HJ, Hess CP, et al. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimers disease. Brain. 2009;132(8):2048–2057. doi: 10.1093/brain/awp123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frisoni GB, Fox NC, Jack CR, Jr., Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nature Reviews Neurology. 2010;6(2):67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alzheimer's Association. 2012 Alzheimer's disease facts and figures. Alzheimer's #38; Dementia. 2012;8:131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Lee SF, Shah S, Li H, Yu C, Han W, Yu G. Mammalian APH-1 interacts with presenilin and nicastrin and is required for intramembrane proteolysis of amyloid-β precursor protein and Notch. Journal of Biological Chemistry. 2002;277(47):45013–45019. doi: 10.1074/jbc.M208164200. [DOI] [PubMed] [Google Scholar]
- 13.Martins RN, Turner BA, Carroll RT, et al. High levels of amyloid-β protein from S182 (Glu246) familial Alzheimer’s cells. NeuroReport. 1995;7(1):217–220. [PubMed] [Google Scholar]
- 14.Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. The Lancet Neurology. 2011;10(3):213–220. doi: 10.1016/S1474-4422(10)70323-9. [DOI] [PubMed] [Google Scholar]
- 15.Tanzi RE, Gusella JF, Watkins PC, et al. Amyloid β protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235(4791):880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 16.Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(12):4190–4194. doi: 10.1073/pnas.84.12.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends in Neurosciences. 1997;20(4):154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 18.Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 19.St George-Hyslop PH, Tanzi RE, Polinsky RJ, et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science. 1987;235(4791):885–890. doi: 10.1126/science.2880399. [DOI] [PubMed] [Google Scholar]
- 20.St George-Hyslop PH, Haines JL, Farrer LA, et al. Genetic linkage studies suggest that Alzheimer’s disease is not a single homogeneous disorder. Nature. 1990;347(6289):194–197. doi: 10.1038/347194a0. [DOI] [PubMed] [Google Scholar]
- 21.Goate A. Segregation of a missense mutation in the amyloid β-protein precursor gene with familial Alzheimer’s disease. Journal of Alzheimer’s Disease. 2006;9(3):341–347. doi: 10.3233/jad-2006-9s338. [DOI] [PubMed] [Google Scholar]
- 22.Schellenberg GD, Bird TD, Wijsman EM, et al. Absence of linkage of chromosome 21q21 markers to familial Alzheimer’s disease. Science. 1988;241(4872):1507–1510. doi: 10.1126/science.3420406. [DOI] [PubMed] [Google Scholar]
- 23.Tanzi RE, Vaula G, Romano DM, et al. Assessment of amyloid β-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. American Journal of Human Genetics. 1992;51(2):273–282. [PMC free article] [PubMed] [Google Scholar]
- 24.Kamino K, Orr HT, Payami H, et al. Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. American Journal of Human Genetics. 1992;51(5):998–1014. [PMC free article] [PubMed] [Google Scholar]
- 25.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 26.Raux G, Guyant-Maréchal L, Martin C, et al. Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: an update. Journal of Medical Genetics. 2005;42(10):793–795. doi: 10.1136/jmg.2005.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki N, Cheung TT, Cai XD, et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994;264(5163):1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 28.Yamatsuji T, Matsui T, Okamoto T, et al. G protein-mediated neuronal DNA fragmentation induced by familial Alzheimer’s disease-associated mutants of APP. Science. 1996;272(5266):1349–1352. doi: 10.1126/science.272.5266.1349. [DOI] [PubMed] [Google Scholar]
- 29.Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 30.Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Medicine. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 31.Duff K, Eckman C, Zehr C, et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383(6602):710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 32.Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nature Medicine. 1997;3(1):67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 33.St George-Hyslop PH, Petit A. Molecular biology and genetics of Alzheimer’s disease. Comptes Rendus Biologies. 2005;328(2):119–130. doi: 10.1016/j.crvi.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 34.Boulianne GL, Livne-Bar I, Humphreys JM, et al. Cloning and characterization of the Drosophila presenilin homologue. NeuroReport. 1997;8(4):1025–1029. doi: 10.1097/00001756-199703030-00041. [DOI] [PubMed] [Google Scholar]
- 35.Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans s182 Alzheimer’s disease gene. Nature. 1995;377(6547):351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 36.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 37.Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376(6543):775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 38.Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(21):11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li YM, Xu M, Lai MT, et al. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405(6787):689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 40.Kopan R, Goate A. A common enzyme connects Notch signaling and Alzheimer’s disease. Genes and Development. 2000;14(22):2799–2806. doi: 10.1101/gad.836900. [DOI] [PubMed] [Google Scholar]
- 41.Cai D, Netzer WJ, Zhong M, et al. Presenilin-1 uses phospholipase D1 as a negative regulator of β-amyloid formation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1941–1946. doi: 10.1073/pnas.0510708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai D, Zhong M, Wang R, et al. Phospholipase D1 corrects impaired βAPP trafficking and neurite outgrowth in familial Alzheimer’s disease-linked presenilin-1 mutant neurons. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1936–1940. doi: 10.1073/pnas.0510710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis JA, Naruse S, Chen H, et al. An Alzheimer’s disease-linked PS1 variant rescues the developmental abnormalities of PS1-deficient embryos. Neuron. 1998;20(3):603–609. doi: 10.1016/s0896-6273(00)80998-8. [DOI] [PubMed] [Google Scholar]
- 44.Qian S, Jiang P, Guan XM, et al. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Aβ1-42/43 expression. Neuron. 1998;20(3):611–617. doi: 10.1016/s0896-6273(00)80999-x. [DOI] [PubMed] [Google Scholar]
- 45.Sato S, Kamino K, Miki T, et al. Splicing mutation of presenilin-1 gene for early-onset familial Alzheimer’s disease. Human Mutation. 1998;10(Supplement 1):S91–S94. doi: 10.1002/humu.1380110131. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Tur J, Froelich S, Prihar G, et al. A mutation in Alzheimer’s disease destroying a splice acceptor site in the presenilin-1 gene. NeuroReport. 1995;7(1):297–301. [PubMed] [Google Scholar]
- 47.Sherrington R, Froelich S, Sorbi S, et al. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Human Molecular Genetics. 1996;5(7):985–988. doi: 10.1093/hmg/5.7.985. [DOI] [PubMed] [Google Scholar]
- 48.Bird TD, Levy-Lahad E, Poorkaj P, et al. Wide range in age of onset for chromosome 1-related familial Alzheimer’s disease. Annals of Neurology. 1996;40(6):932–936. doi: 10.1002/ana.410400619. [DOI] [PubMed] [Google Scholar]
- 49.Bird TD, Lampe TH, Nemens EJ, Miner GW, Sumi SM, Schellenberg GD. Familial Alzheimer’s disease in American descendants of the volga germans: probable genetic founder effect. Annals of Neurology. 1988;23(1):25–31. doi: 10.1002/ana.410230106. [DOI] [PubMed] [Google Scholar]
- 50.Bird TD, Sumi SM, Nemens EJ, et al. Phenotypic heterogeneity in familial Alzheimer’s disease: a study of 24 kindreds. Annals of Neurology. 1989;25(1):12–25. doi: 10.1002/ana.410250104. [DOI] [PubMed] [Google Scholar]
- 51.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Archives of General Psychiatry. 2006;63(2):168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 52.Pericak-Vance MA, Bebout JL, Gaskell PC, Jr., et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. American Journal of Human Genetics. 1991;48(6):1034–1050. [PMC free article] [PubMed] [Google Scholar]
- 53.Roses AD, Saunders AM. APOE is a major susceptibility gene for Alzheimer’s disease. Current Opinion in Biotechnology. 1994;5(6):663–667. doi: 10.1016/0958-1669(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 54.Colhoun HM, McKeigue PM, Smith GD. Problems of reporting genetic associations with complex outcomes. The Lancet. 2003;361(9360):865–872. doi: 10.1016/s0140-6736(03)12715-8. [DOI] [PubMed] [Google Scholar]
- 55.Ioannidis JPA, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nature Genetics. 2001;29(3):306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 56.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 57.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Journal of the American Medical Association. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stranger BE, Forrest MS, Dunning M, et al. Relative impact of nucleotide and copy number variation on gene phenotypes. Science. 2007;315(5813):848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brouwers N, Van Cauwenberghe C, Engelborghs S, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Molecular Psychiatry. 2012;17:223–233. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pearson RCA. Cortical connections and the pathology of Alzheimer’s disease. Neurodegeneration. 1996;5(4):429–434. doi: 10.1006/neur.1996.0058. [DOI] [PubMed] [Google Scholar]
- 63.Thomann PA, Dos Santos V, Seidl U, Toro P, Essig M, Schröder J. MRI-derived atrophy of the olfactory bulb and tract in mild cognitive impairment and Alzheimer’s disease. Journal of Alzheimer’s Disease. 2009;17(1):213–221. doi: 10.3233/JAD-2009-1036. [DOI] [PubMed] [Google Scholar]
- 64.Tan MH, Weldon KL, Albers JJ, et al. Serum HDL-cholesterol, apo-A-I and apo-E levels in patients with abnormal coronary arteries. Clinical and Investigative Medicine. 1980;3(3-4):225–232. [PubMed] [Google Scholar]
- 65.Zannis VI, Breslow JL. Apolipoprotein E. Molecular and Cellular Biochemistry. 1982;42(1):3–20. doi: 10.1007/BF00223534. [DOI] [PubMed] [Google Scholar]
- 66.Morrisett JD, Kim HS, Patsch JR. Genetic susceptibility and resistance to diet-induced atherosclerosis and hyperlipoproteinemia. Arteriosclerosis. 1982;2(4):312–324. doi: 10.1161/01.atv.2.4.312. [DOI] [PubMed] [Google Scholar]
- 67.Rall SC, Jr., Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. Journal of Biological Chemistry. 1982;257(8):4171–4178. [PubMed] [Google Scholar]
- 68.Weisgraber KH, Rall SC, Jr., Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. Journal of Biological Chemistry. 1981;256(17):9077–9083. [PubMed] [Google Scholar]
- 69.Rall SC, Jr., Weisgraber KH, Innerarity TL, Mahley RW. Structural basis for receptor binding heterogeneity of apolipoprotein E from type III hyperlipoproteinemic subjects. Proceedings of the National Academy of Sciences of the United States of America. 1982;79(15):4696–4700. doi: 10.1073/pnas.79.15.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maestre G, Ottman R, Stern Y, et al. Apolipoprotein E and Alzheimer’s disease: ethnic variation in genotypic risks. Annals of Neurology. 1995;37(2):254–259. doi: 10.1002/ana.410370217. [DOI] [PubMed] [Google Scholar]
- 71.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. Journal of the American Medical Association. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- 72.Haan MN, Mungas DM, Gonzalez HM, Ortiz TA, Acharya A, Jagust WJ. Prevalence of dementia in older Latinos: the influence of type 2 diabetes mellitus, stroke and genetic factors. Journal of the American Geriatrics Society. 2003;51(2):169–177. doi: 10.1046/j.1532-5415.2003.51054.x. [DOI] [PubMed] [Google Scholar]
- 73.Eisenberg DTA, Kuzawa CW, Hayes MG. Worldwide allele frequencies of the human apolipoprotein E gene: climate, local adaptations, and evolutionary history. American Journal of Physical Anthropology. 2010;143(1):100–111. doi: 10.1002/ajpa.21298. [DOI] [PubMed] [Google Scholar]
- 74.Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 75.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annual Review of Medicine. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 76.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 77.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nature Genetics. 1994;7(2):180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 78.Mayeux R, Ottman R, Maestre G, et al. Synergistic effects of traumatic head injury and apolipoprotein-ε4 in patients with Alzheimer’s disease. Neurology. 1995;45(3):555–557. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 79.Newman MF, Croughwell ND, Blumenthal JA, et al. Predictors of cognitive decline after cardiac operation. Annals of Thoracic Surgery. 1995;59(5):1326–1330. doi: 10.1016/0003-4975(95)00076-w. [DOI] [PubMed] [Google Scholar]
- 80.Alberts MJ, Graffagnino C, McClenny C, et al. ApoE genotype and survival from intracerebral haemorrhage. The Lancet. 1995;346(8974, article 575) doi: 10.1016/s0140-6736(95)91411-0. [DOI] [PubMed] [Google Scholar]
- 81.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature Genetics. 2011;43(5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Archives of Neurology. 2010;67(12):1473–1484. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jones SE, Jomary C. Clusterin. International Journal of Biochemistry and Cell Biology. 2002;34(5):427–431. doi: 10.1016/s1357-2725(01)00155-8. [DOI] [PubMed] [Google Scholar]
- 84.De Silva HV, Stuart WD, Duvic CR, et al. A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. Journal of Biological Chemistry. 1990;265(22):13240–13247. [PubMed] [Google Scholar]
- 85.Humphreys DT, Carver JA, Easterbrook-Smith SB, Wilson MR. Clusterin has chaperone-like activity similar to that of small heat shock proteins. Journal of Biological Chemistry. 1999;274(11):6875–6881. doi: 10.1074/jbc.274.11.6875. [DOI] [PubMed] [Google Scholar]
- 86.Wong P, Taillefer D, Lakins J, Pineault J, Chader G, Tenniswood M. Molecular characterization of human TRPM-2/clusterin, a gene associated with sperm maturation, apoptosis and neurodegeneration. European Journal of Biochemistry. 1994;221(3):917–925. doi: 10.1111/j.1432-1033.1994.tb18807.x. [DOI] [PubMed] [Google Scholar]
- 87.Fritz IB, Burdzy K, Setchell B, Blaschuk O. Ram rete testis fluid contains a protein (clusterin) which influences cell-cell interactions in vitro. Biology of Reproduction. 1983;28(5):1173–1188. doi: 10.1095/biolreprod28.5.1173. [DOI] [PubMed] [Google Scholar]
- 88.Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer's disease. Microscopy Research and Technique. 2000;50:305–315. doi: 10.1002/1097-0029(20000815)50:4<305::AID-JEMT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 89.Giannakopoulos P, Kövari E, French LE, Viard I, Hof PR, Bouras C. Possible neuroprotective role of clusterin in Alzheimer’s disease: a quantitative immunocytochemical study. Acta Neuropathologica. 1998;95(4):387–394. doi: 10.1007/s004010050815. [DOI] [PubMed] [Google Scholar]
- 90.Liang WS, Dunckley T, Beach TG, et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiological Genomics. 2008;33(2):240–256. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McGeer PL, Kawamata T, Walker DG. Distribution of clusterin in Alzheimer brain tissue. Brain Research. 1992;579(2):337–341. doi: 10.1016/0006-8993(92)90071-g. [DOI] [PubMed] [Google Scholar]
- 92.Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer's disease. Microscopy Research and Technique. 2000;50:305–315. doi: 10.1002/1097-0029(20000815)50:4<305::AID-JEMT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 93.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Research Reviews. 2009;61(2):89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 94.May PC, Lampert-Etchells M, Johnson SA, Poirier J, Masters JN, Finch CE. Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer’s disease and in response to experimental lesions in rat. Neuron. 1990;5(6):831–839. doi: 10.1016/0896-6273(90)90342-d. [DOI] [PubMed] [Google Scholar]
- 95.Bartl MM, Luckenbach T, Bergner O, Ullrich O, Koch-Brandt C. Multiple receptors mediate apoJ-dependent clearance of cellular debris into nonprofessional phagocytes. Experimental Cell Research. 2001;271(1):130–141. doi: 10.1006/excr.2001.5358. [DOI] [PubMed] [Google Scholar]
- 96.Bell RD, Sagare AP, Friedman AE, et al. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. Journal of Cerebral Blood Flow and Metabolism. 2007;27(5):909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wilson MR, Easterbrook-Smith SB. Clusterin is a secreted mammalian chaperone. Trends in Biochemical Sciences. 2000;25(3):95–98. doi: 10.1016/s0968-0004(99)01534-0. [DOI] [PubMed] [Google Scholar]
- 98.Harel A, Wu F, Mattson MP, Morris CM, Yao PJ. Evidence for CALM in directing VAMP2 trafficking. Traffic. 2008;9(3):417–429. doi: 10.1111/j.1600-0854.2007.00694.x. [DOI] [PubMed] [Google Scholar]
- 99.Tebar F, Bohlander SK, Sorkin A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Molecular Biology of the Cell. 1999;10(8):2687–2702. doi: 10.1091/mbc.10.8.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nature cell biology. 2009;11(12):1399–1410. doi: 10.1038/ncb1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer β/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. Journal of Biological Chemistry. 1993;268(1):608–612. [PubMed] [Google Scholar]
- 102.Klebig ML, Wall MD, Potter MD, Rowe EL, Carpenter DA, Rinchik EM. Mutations in the clathrin-assembly gene Picalm are responsible for the hematopoietic and iron metabolism abnormalities in fit1 mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8360–8365. doi: 10.1073/pnas.1432634100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Archangelo LF, Gläsner J, Krause A, Bohlander SK. The novel CALM interactor CATS influences the subcellular localization of the leukemogenic fusion protein CALM/AF10. Oncogene. 2006;25(29):4099–4109. doi: 10.1038/sj.onc.1209438. [DOI] [PubMed] [Google Scholar]
- 104.Meyerholz A, Hinrichsen L, Groos S, Esk PC, Brandes G, Ungewickell EJ. Effect of clathrin assembly lymphoid myeloid leukemia protein depletion on clathrin coat formation. Traffic. 2005;6(12):1225–1234. doi: 10.1111/j.1600-0854.2005.00355.x. [DOI] [PubMed] [Google Scholar]
- 105.Dreyling MH, Martinez-Climent JA, Zheng M, Mao J, Rowley JD, Bohlander SK. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(10):4804–4809. doi: 10.1073/pnas.93.10.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilson JG, Andriopoulos NA, Fearon DT. CR1 and the cell membrane proteins that bind C3 and C4. A basic and clinical review. Immunologic Research. 1987;6(3):192–209. doi: 10.1007/BF02918091. [DOI] [PubMed] [Google Scholar]
- 107.Fearon DT, Wong WW. Complement ligand-receptor interactions that mediate biological responses. Annual Review of Immunology. 1983;1:243–271. doi: 10.1146/annurev.iy.01.040183.001331. [DOI] [PubMed] [Google Scholar]
- 108.Minota S, Terai C, Nojima Y. Low C3b receptor reactivity on erythrocytes from patients with systemic lupus erythematosus detected by immune adherence hemagglutination and radioimmunoassays with monoclonal antibody. Arthritis and Rheumatism. 1984;27(12):1329–1335. doi: 10.1002/art.1780271202. [DOI] [PubMed] [Google Scholar]
- 109.Iida K, Mornaghi R, Nussenzweig V. Complement receptor (CR1) deficiency in erythrocytes from patients with systemic lupus erythematosus. Journal of Experimental Medicine. 1982;155(5):1427–1438. doi: 10.1084/jem.155.5.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weis JH, Morton CC, Bruns GAP. A complement receptor locus: genes encoding C3b/C4b receptor and C3d/Epstein-Barr virus receptor map to 1q32. Journal of Immunology. 1987;138(1):312–315. [PubMed] [Google Scholar]
- 111.Moulds JM, Nickells MW, Moulds JJ, Brown MC, Atkinson JP. The C3b/C4b receptor is recognized by the Knops, McCoy, Swain-Langley, and York blood group antisera. Journal of Experimental Medicine. 1991;173(5):1159–1163. doi: 10.1084/jem.173.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wong WW, Cahill JM, Rosen MD, et al. Structure of the human CR1 gene. Molecular basis of the structural and quantitative polymorphisms and identification of a new CR1-like allele. Journal of Experimental Medicine. 1989;169(3):847–863. doi: 10.1084/jem.169.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nature Genetics. 1996;14(1):69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 114.Nicot AS, Toussaint A, Tosch V, et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nature Genetics. 2007;39(9):1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 115.Kaminski WE, Orsó E, Diederich W, Klucken J, Drobnik W, Schmitz G. Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7) Biochemical and Biophysical Research Communications. 2000;273(2):532–538. doi: 10.1006/bbrc.2000.2954. [DOI] [PubMed] [Google Scholar]
- 116.Broccardo C, Osorio J, Luciani MF, et al. Comparative analysis of the promoter structure and genomic organization of the human and mouse ABCA7 gene encoding a novel ABCA transporter. Cytogenetics and Cell Genetics. 2001;92(3-4):264–270. doi: 10.1159/000056914. [DOI] [PubMed] [Google Scholar]
- 117.Antúnez C, Boada M, González-Pérez A, et al. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Medicine. 2011;3(5, article 33) doi: 10.1186/gm249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ishibashi K, Suzuki M, Sasaki S, Imai M. Identification of a new multigene four-transmembrane family (MS4A) related to CD20, HTm4 and β subunit of the high-affinity IgE receptor. Gene. 2001;264(1):87–93. doi: 10.1016/s0378-1119(00)00598-9. [DOI] [PubMed] [Google Scholar]
- 119.Liang Y, Buckley TR, Tu L, Langdon SD, Tedder TF. Structural organization of the human MS4A gene cluster on Chromosome 11q12. Immunogenetics. 2001;53(5):357–368. doi: 10.1007/s002510100339. [DOI] [PubMed] [Google Scholar]
- 120.Liang Y, Tedder TF. Identification of a CD20-, FcεRIβ-, and HTm4-related gene family: sixteen new MS4A family members expressed in human and mouse. Genomics. 2001;72(2):119–127. doi: 10.1006/geno.2000.6472. [DOI] [PubMed] [Google Scholar]
- 121.Coulthard MG, Lickliter JD, Subanesan N, et al. Characterization of the EphA1 receptor tyrosine kinase: expression in epithelial tissues. Growth Factors. 2001;18(4):303–317. doi: 10.3109/08977190109029118. [DOI] [PubMed] [Google Scholar]
- 122.Zhou R. The Eph family receptors and ligands. Pharmacology and Therapeutics. 1998;77(3):151–181. doi: 10.1016/s0163-7258(97)00112-5. [DOI] [PubMed] [Google Scholar]
- 123.Wilkinson DG. Eph receptors and ephrins: regulators of guidance and assembly. International Review of Cytology. 2000;196:177–244. doi: 10.1016/s0074-7696(00)96005-4. [DOI] [PubMed] [Google Scholar]
- 124.Xu Q, Mellitzer G, Wilkinson DG. Roles of Eph receptors and ephrins in segmental patterning. Philosophical transactions of the Royal Society of London B. 2000;355(1399):993–1002. doi: 10.1098/rstb.2000.0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Holder N, Klein R. Eph receptors and ephrins: effectors of morphogenesis. Development. 1999;126(10):2033–2044. doi: 10.1242/dev.126.10.2033. [DOI] [PubMed] [Google Scholar]
- 126.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nature Reviews Molecular Cell Biology. 2002;3(7):475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 127.Owshalimpur D, Kelley MJ. Genomic structure of the EPHA1 receptor tyrosine kinase gene. Molecular and Cellular Probes. 1999;13(3):169–173. doi: 10.1006/mcpr.1999.0228. [DOI] [PubMed] [Google Scholar]
- 128.Cao H, Crocker PR. Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology. 2011;132(1):18–26. doi: 10.1111/j.1365-2567.2010.03368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nature Reviews Immunology. 2007;7(4):255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 130.Crocker PR, Varki A. Siglecs, sialic acids and innate immunity. Trends in Immunology. 2001;22(6):337–342. doi: 10.1016/s1471-4906(01)01930-5. [DOI] [PubMed] [Google Scholar]
- 131.Ulyanova T, Blasioli J, Woodford-Thomas TA, Thomas ML. The sialoadhesin CD33 is a myeloid-specific inhibitory receptor. European Journal of Immunology. 1999;29:3440–3449. doi: 10.1002/(SICI)1521-4141(199911)29:11<3440::AID-IMMU3440>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 132.Freeman SD, Kelm S, Barber EK, Crocker PR. Characterization of CD33 as a new member of the Sialoadhesin family of cellular interaction molecules. Blood. 1995;85(8):2005–2012. [PubMed] [Google Scholar]
- 133.Taylor VC, Buckley CD, Douglas M, Cody AJ, Simmons DL, Freeman SD. The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. Journal of Biological Chemistry. 1999;274(17):11505–11512. doi: 10.1074/jbc.274.17.11505. [DOI] [PubMed] [Google Scholar]
- 134.Yousef GM, Ordon MH, Foussias G, Diamandis EP. Genomic organization of the siglec gene locus on chromosome 19q13.4 and cloning of two new siglec pseudogenes. Gene. 2002;286(2):259–270. doi: 10.1016/s0378-1119(02)00432-8. [DOI] [PubMed] [Google Scholar]
- 135.Hernández-Caselles T, Martínez-Esparza M, Pérez-Oliva AB, et al. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: two isoforms of CD33 are generated by alternative splicing. Journal of Leukocyte Biology. 2006;79(1):46–58. doi: 10.1189/jlb.0205096. [DOI] [PubMed] [Google Scholar]
- 136.Simmons D, Seed B. Isolation of a cDNA encoding CD33, a differentiation antigen of myeloid progenitor cells. Journal of Immunology. 1988;141(8):2797–2800. [PubMed] [Google Scholar]
- 137.Kirsch KH, Georgescu MM, Ishimaru S, Hanafusa H. CMS: an adapter molecule involved in cytoskeletal rearrangements. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(11):6211–6216. doi: 10.1073/pnas.96.11.6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dustin ML, Olszowy MW, Holdorf AD, et al. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell. 1998;94(5):667–677. doi: 10.1016/s0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- 139.Kim JH, Wu H, Green G, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300(5623):1298–1300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 140.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nature Genetics. 2007;39(2):168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Andersen OM, Reiche J, Schmidt V, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(38):13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jacobsen L, Madsen P, Moestrup SK, et al. Molecular characterization of a novel human hybrid-type receptor that binds the α2-macroglobulin receptor-associated protein. Journal of Biological Chemistry. 1996;271(49):31379–31383. doi: 10.1074/jbc.271.49.31379. [DOI] [PubMed] [Google Scholar]
- 143.Reitz C, Cheng R, Rogaeva E, et al. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Archives of Neurology. 2011;68(1):99–106. doi: 10.1001/archneurol.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cuenco KT, Lunetta KL, Baldwin CT, et al. Association of distinct variants in SORL1 with cerebrovascular and neurodegenerative changes related to Alzheimer disease. Archives of Neurology. 2008;65(12):1640–1648. doi: 10.1001/archneur.65.12.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kölsch H, Jessen F, Wiltfang J, et al. Influence of SORL1 gene variants: association with CSF amyloid-β products in probable Alzheimer’s disease. Neuroscience Letters. 2008;440(1):68–71. doi: 10.1016/j.neulet.2008.05.049. [DOI] [PubMed] [Google Scholar]
- 146.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Torkamani A, Topol EJ, Schork NJ. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92(5):265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lambert JC, Grenier-Boley B, Chouraki V, et al. Implication of the immune system in Alzheimer’s disease: evidence from genome-wide pathway analysis. Journal of Alzheimer’s Disease. 2010;20(4):1107–1118. doi: 10.3233/JAD-2010-100018. [DOI] [PubMed] [Google Scholar]
- 149.Hong MG, Alexeyenko A, Lambert JC, Amouyel P, Prince JA. Genome-wide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease. Journal of Human Genetics. 2010;55(10):707–709. doi: 10.1038/jhg.2010.92. [DOI] [PubMed] [Google Scholar]
- 150.Deelen J, Beekman M, Uh HW, et al. Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell. 2011;10(4):686–698. doi: 10.1111/j.1474-9726.2011.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bekris LM, Millard SP, Galloway NM, et al. Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. Journal of Alzheimer’s Disease. 2008;13(3):255–266. doi: 10.3233/jad-2008-13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bekris LM, Galloway NM, Montine TJ, Schellenberg GD, Yu CE. APOE mRNA and protein expression in postmortem brain are modulated by an extended haplotype structure. American Journal of Medical Genetics B. 2010;153(2):409–417. doi: 10.1002/ajmg.b.30993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Roses AD. An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Archives of Neurology. 2010;67(5):536–541. doi: 10.1001/archneurol.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Roses AD, Lutz MW, Amrine-Madsen H, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics Journal. 2010;10(5):375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jun G, Vardarajan BN, Buros J, et al. Comprehensive search for Alzheimer disease susceptibility loci in the APOE region. Archives of Neurology. 2012;69(10):1270–1279. doi: 10.1001/archneurol.2012.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yu CE, Seltman H, Peskind ER, et al. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89(6):655–665. doi: 10.1016/j.ygeno.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ramanan VK, Kim S, Holohan K, et al. Genome-wide pathway analysis of memory impairment in the Alzheimer's disease neuroimaging initiative (ADNI) cohort implicates gene candidatescanonical pathways, and networks. doi: 10.1007/s11682-012-9196-x. Brain Imaging and Behavior. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cacabelos R, Martinez R, Fernandez-Novoa L, et al. Genomics of dementia: APOE- and CYP2D6-related pharmacogenetics. International Journal of Alzheimer's Disease. 2012;2012:37 pages. doi: 10.1155/2012/518901.518901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cacabelos R, Llovo R, Fraile C, Fernández-Novoa L. Pharmacogenetic aspects of therapy with cholinesterase inhibitors: the role of CYP2D6 in Alzheimer’s disease pharmacogenetics. Current Alzheimer Research. 2007;4(4):479–500. doi: 10.2174/156720507781788846. [DOI] [PubMed] [Google Scholar]
- 160.Cacabelos R. Pharmacogenomics and therapeutic strategies for dementia. Expert Review of Molecular Diagnostics. 2009;9(6):567–611. doi: 10.1586/erm.09.42. [DOI] [PubMed] [Google Scholar]
- 161.Cacabelos R. Pharmacogenetic basis for therapeutic optimization in Alzheimer’s disease. Molecular Diagnosis and Therapy. 2007;11(6):385–405. doi: 10.1007/BF03256262. [DOI] [PubMed] [Google Scholar]
- 162.Cacabelos R, Martinez-Bouza R, Carril JC, et al. Genomics and pharmacogenomics of brain disorders. Current Pharmaceutical Biotechnology. 2012;13:674–725. doi: 10.2174/138920112799857576. [DOI] [PubMed] [Google Scholar]