

Abstract
C. difficile is the most common cause of nosocomial diarrhea in North America and Europe. Genomes of individual strains of C. difficile are highly divergent. To determine how divergent strains respond to environmental changes, the transcriptomes of two historic and two recently isolated hypervirulent strains were analyzed following nutrient shift and osmotic shock. Illumina based RNA-seq was used to sequence these transcriptomes. Our results reveal that although C. difficile strains contain a large number of shared and strain specific genes, the majority of the differentially expressed genes were core genes. We also detected a number of transcriptionally active regions that were not part of the primary genome annotation. Some of these are likely to be small regulatory RNAs.
Introduction
Clostridium difficile is a toxin producing anaerobic bacillus and is the leading cause of hospital associated diarrhea in North America and Europe[1,2]. Clinical presentation of C. difficile infection (CDI) ranges from asymptomatic colonization, mild diarrhea, severe pseudomembranous colitis, paralytic ileus, to sepsis and death[3]. Advanced age, prolonged hospitalization, antibiotic use and acid suppression therapy are some of the risk factors for CDI[3,4].The mortality rate is more than 80% in fulminant cases requiring colectomy[5]. C. difficile produces two toxins, toxin A and toxin B, which are responsible for most of the damage caused to the host[6,7].
During the last decade, there has been a significant increase in the rate of CDI across the United States, Canada, and Europe[8-10].Emergence of more virulent strains and changes in antibiotic treatment regimens are some of the established causes of the increase in CDI [11,12]. Some of these strains, which have the capacity to produce more severe colitis and mortality, have been termed as hypervirulent [13]. These strains belong to PCR ribotype 027[13]. Ribotype 027 strains are also characterized as toxinotype III, North American pulsed field gel electrophoresis type1 (NAP1) and restriction endonuclease analysis group BI and in contrast to historical control strains, are fluoroquinolone resistant[10]. In the recent years, C. difficile infection in the community setting has increased [14]. It is also causing a significant number of infections in food animals[15]. Recent studies have shown the possibility of foodborne transmission of C. difficile, which might explain the spread of C. difficile infections in the general community [16].
Comparative genomic studies have revealed that the genome conservation in C. difficile is very low and this is a major contributing factor in the outcome of infection by a given strain [17-19]. The number of core genes in C. difficile is estimated to be less than 20% of the pangenome[20]. This variation in the genome content could enable C. difficile strains to respond differently to environmental changes. A number of functional genomics studies have been conducted to indentify the differential gene network that controls C. difficile response to environmental changes[21-24]. However, all these studies are based C. difficile 630, a strain isolated in 1985 from Zurich, Switzerland. Genome of C. difficile 630 is the most commonly used reference for functional genomics studies as it was the first strain to be sequenced [17]. Another reason for using C. difficile 630 as reference is that microarray based studies require a high quality complete genome for probe design.
Recent advances in new generation sequencing platforms have revolutionized microbial genomics. Genome sequencing using new generation sequencing platforms has resulted in sequencing of several C. difficile strains from different continents that are old as well as newer hypervirulent isolates[20,25-27]. These comparative genomics studies have revealed that when compared to historic strains (isolated in the 1980s), new hypervirulent strains (isolated after 2000) have undergone several genome changes that result in the hypervirulent phenotype[12,25]. The development of transcriptional profiling using RNA sequencing (RNA-seq) also offers vast improvements over microarray based transcriptional profiling. In order to determine how historic and recently emerged C. difficile strains respond to environmental changes we used RNA-seq to compare the transcriptome of four C. difficile strains after subjecting them to physiologically relevant in vitro stress conditions. In the first condition, cells were shifted from a rich medium [Brain heart infusion broth (BHI)] to a poor medium [Basal defined medium (BDM)] [28] with supplementation of 0.5% sucrose. It has been reported that the presence of glucose and other easily metabolizable carbon sources in the growth medium suppress production of toxin A and toxin B[29,30]. The shift from BHI to BDM was designed to induce multiple nutritional changes and to analyze the impact of those changes in C. difficile pathways. Osmotic shock (shift from BHI to BHI supplemented with 1.5% NaCl) was chosen as the second test condition because C. difficile has been shown to have enhanced host cell adherence following osmotic shock[31]. Since adhesion and colonization of animal tissue by bacteria is important in establishing infection, it is probable that without attachment, C. difficile cannot colonize and will be quickly removed by non-specific host defense mechanisms which include intestinal peristalsis, mucosal cell exfoliation and intestinal mucins[32,33].
Two strains in this comparison were isolated in the 1980s (CD630 and CD196), and two were hypervirulent strains isolated after 2000 (R20291 and QCD_32g58). Although s C. difficile 630 is not a ribotype 027 strain, we included this strain in the comparisons as it is the only strain that was sequenced using Sanger method and to enable cross comparison with the previously reported functional genomics studies. Consistent with the large genome diversity in C. difficile, our transcriptome sequencing results show that differentially expressed core genes in various strains are not identical. We also report expression of several novel transcripts that are not part of the primary genome annotation.
Materials and Methods
C. difficile strains and culturing
We selected four C. difficile strains for transcriptomic comparisons. The characteristics of these strains are given in Table 1. Bacterial culturing was performed inside a Bactron IV anaerobic chamber (Shel Lab, Cornelius, OR). The chamber was filled and purged with an anaerobic gas mixture (10% CO2, 85% N2, 5% H2). A palladium catalyst was used in the chamber to remove any trace of oxygen. All materials used in the anaerobic chamber were pre-reduced. Spores of C. difficile strains CD630, CD196, QCD_32g58 and R20291 were streaked on brain-heart infusion (BHI) agar plates containing 0.1% L-cysteine and taurocholate. The plates were incubated overnight at 37°C. Single colonies from these plates were then used inoculate pre-reduced BHI broth and were incubated at 37°C overnight. Fresh BHI broth was then inoculated by transferring 1% overnight culture. Cultures were incubated at 37°C until the OD600 reached between 0.4 - 0.5. Bacteria were then collected by centrifugation at 2,000 x g for 5 minutes. These cells were then shifted to two physiologically relevant in vitro conditions. In the first condition, cells were subjected to nutrient change by shifting to an equal volume of Basal defined medium (BDM)[28] with supplementation of 0.5% sucrose. This shift was designed to induce multiple nutritional changes and to analyze the impact of those changes in C. difficile pathways. In the second condition, cells were subjected to osmotic shock by shifting to an equal volume of BHI supplemented with 1.5% NaCl. The same number of cells was transferred to fresh BHI as the control group. After incubating for 1 hour at 37°C, twice the volume of RNAprotect bacteria reagent (Qiagen, Valencia, CA) was added to the cultures to halt transcription and RNA degradation, and cells were collected by centrifugation at 2,000 x g for 10 minutes.All experiments were performed as two biological replicates.
Table 1. Characteristics of C. difficile strains used in this study.
| Strain name | Ribotype | Isolation year | Isolation country | Hypervirulence |
|---|---|---|---|---|
| CD630 | 13 | 1982 | Switzerland | no |
| CD196 | 27 | 1985 | France | no |
| QCD-32g58 | 27 | 2004 | Canada | yes |
| R20291 | 27 | 2006 | UK | yes |
Isolation of total RNA
Total RNA extraction was performed with TRIzol/RNeasy hybrid RNA extraction protocol. Briefly, the bacterial pellets were re-suspended with 1 ml of TRIzol reagent and were transferred to 2 ml sterile screw-cap microcentrifuge tube. Then 0.5ml of sterile RNase-free 0.1 mm zirconia beads was added to each tube. Cells were homogenized and lysed by bead beating four times in a Mini Bead-Beater (BioSpecProducts, Inc., Bartlesville, OK) for 30 seconds with a gap of 30 seconds. After the chloroform extraction, the aqueous phase was transferred to a 1.5 ml sterile RNase-free micro-centrifuge tube and mixed with an equal volume of 100% ethanol (Sigma). This mix was loaded into an RNeasy column (Qiagen kit) and centrifuged for 30 seconds at 8,000 x g. Washing of the column, DNA digestion and elution steps were performed following the standard Qiagen protocol. Integrity of isolated RNA was estimated using Agilent Bioanalyzer 2100. Only those samples with an RNA Integrity Number (RIN) >9 were used for RNA sequencing.
cDNA library synthesis and sequencing
Ribo-Zero™ rRNA Removal Kit ( Epicentre® Biotechnologies, Madison, WI, USA) was used to purify mRNA from 10µg total RNA. First strand cDNA was synthesized using SuperScript® III (Invitrogen). The second-strand cDNA was synthesized using RNase H (Invitrogen) and DNA polymerase I (New England BioLabs). Then the cDNA libraries were prepared using the Illumina Paired-end Genomic DNA Sample Prep kit (Illumina) following the manufacturer's protocol. For each sample, two libraries were prepared from biological replicates. Each library was then loaded onto flow cell channels of the Illumina High-seq 2000 platform for paired-end 90 bp × 2 sequencing. The average insert size for the paired-end libraries was 200 bp (from 180 to 220 bp). For each strain, six paired-end cDNA libraries were constructed. Therefore twenty four libraries were sequenced across all strains.
RNA-Seq data analysis
For data processing, we used a customized RNA-Seq data analysis pipeline developed at Virginia Bioinformatics Institute (VBI) by combining open source programs. Briefly, the quality of the raw sequence reads was checked using the FastQC program (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). The processed reads were then aligned using Bowtie version 0.12.7 [34] to the corresponding C. difficile reference genome. Read alignments with mapping quality score (MAPQ) < 10 were removed. Cufflinks software package version 1.3.1 [35] was used to assemble transcripts and estimate the relative abundances of the transcripts. Transcript expression levels are estimated as Fragments Per Kilobase per Million mapped reads (FPKM). Cuffdiff [36], a component of Cufflinks was used to calculate transcript expression levels. When compared to control condition, genes with log2 ratio ≥ 1.5 and FDR- adjusted p value less than or equal to 0.05 were considered as differentially expressed. The processed data and raw files from this experiment have been submitted to NCBI Gene expression Omnibus (GEO) under the accession # GSE50497 and NCBI short read archive (SRA) under the accession # SRP029366.
Novel gene discovery
Cufflinks program provides reference annotation based assembly and seeks to build upon available information about the transcriptome of an organism to find novel genes and isoforms [35]. Cufflinks output includes all annotated reference transcripts and any novel genes and isoforms that are assembled. The novel gene transcripts identified by the pipeline can be novel small RNA genes or unannotated CDS. We performed the following steps to categorize these novel transcripts. First, these transcripts were used to search the Rfam database for sRNA matches. Rfam is a comprehensive database containing families of structural RNAs, including non-coding RNA genes as well as cis-regulatory RNA elements[37]. It incorporates literature curated known sRNAs and uses them as seeds to predict sRNAs for sequenced genomes using covariance model[37]. In the second step, the new transcripts were searched against the non-redundant (nr) database using BLASTX to check for any protein coding gene hits. Finally, we used ORF Finder program to verify whether any transcripts are potentially protein coding genes. Assembled transcripts with no BLASTX hits and no ORF assignment were considered as sRNAs.
Functional analysis of differentially expressed genes
Since it is well established that C. difficile strains are known to be highly divergent [18-20], we classified genes in each strain into core, shared and unique categories using OrthoMCL program [38]. We then combined this gene classification with gene expression level data to obtain a comparison of these genes across all strains. Pathologic program in Pathwaytools v 16.0[39] was used to reconstruct the metabolic pathways of strains QCD_32g58, CD196 and R20291. A previously curated high quality pathway annotation for strain CD630 by our group [23] was used to remove false positive pathway predictions in these strains. Omics viewer [40] was used to map the differentially expressed genes onto cellular pathways and to compare differentially expressed pathways across strains. For identifying how gene interaction networks adjust to the stress conditions tested, the differentially expressed genes were mapped to the systems level gene interaction network of C. difficile. The base interaction data for this analysis was obtained from STRING database[41]. Complete interaction data from STRING v 9.0 was downloaded and C. difficile specific interaction data was then extracted using custom Linux shell scripts. The interaction data in STRING database includes both experimental as well as predicted interactions and each interaction is assigned a confidence score. We then selected interactions with confidence score of 400 or above. These would represent medium and high confidence interactions in C. difficile. This interaction network was then imported into Cytoscape [42] for visualization and overlaying of transcriptomic expression data.
qRT-PCR
We used qRT-PCR to verify the expression levels of selected genes. Following the manufacturer’s instructions, 3.0 µg of total RNA isolated from each stress condition was converted to cDNA by using SuperScript III reverse transcriptase (Invitrogen) with random hexamers. The real-time reaction mixture included 12.5 ng cDNA, 200 nM of each of both forward and reverse primers, and 1X SYBR GreenER qPCR SuperMix (Invitrogen). Primers used in this study are listed in the File S1. qPCR was performed in 96-well optical plates using the AB 7500 Real-Time PCR System instrument and software (Invitrogen) and was analyzed by the method previously reported by our group[43].
Results
Genome coverage
C. difficile is an unusual species because the number of conserved genes in a given strain is very low and many of the strains contain a very high number of genes that are unique to that strain[19,20]. It has also undergone very rapid evolution in the last two decades by acquiring several new genes [25]. To understand how historic and recently emerged C. difficile respond to physiological stress, two historical (CD630 and CD196) and two recently emerged (QCD-32g58 and R20291) strains were subjected to nutrient shift and osmotic shock. Gene expression under these conditions was determined using RNA-seq and these results were compared to gene expression levels during growth in BHI (control condition). A total of 24 samples from these conditions were used for paired-end bi-directional Illumina sequencing. Illumina sequence files were converted to Sanger fastq format and rRNAs were filtered. The quality of the sequence data was checked using the FastQC program. The sequence reads were 90 nucleotides in length and the total number of reads per sample was ~ 26.6 million on average. The filtered RNA-Seq sequence reads from the 24 samples were aligned to their corresponding C. difficile reference genome using Bowtie. For all samples analyzed, 88-95% of reads were mapped with MAPQ greater than or equal to 10. The transcript expression levels were estimated as FPKM using Cufflinks[44]. The number of genes expressed (FPKM>0) was calculated for each sample. There was very high correlation between samples when the number of expressed genes was compared between biological replicates (Table 2). We used PATRIC [45] annotations of the C. difficile genomes as references. In this analysis, expression of more than 90% CDS was detected under the three conditions combined (Table 3).
Table 2. Spearman correlation between biological replicates of RNA-seq datasets.
| Strain | Control A vs B | BDM A vs B | salt A vs B |
|---|---|---|---|
| CD630 | 0.968 | 0.960 | 0.976 |
| CD196 | 0.970 | 0.963 | 0.977 |
| QCD-32g58 | 0.956 | 0.953 | 0.959 |
| R20291 | 0.961 | 0.947 | 0.959 |
Table 3. Genes expressed in each strain and condition.
| Strain Name | Total number of genes |
Number of genes not expressed
|
% of expressed genes | ||
|---|---|---|---|---|---|
| Control | Nutrient shift | Osmotic shock | |||
| CD630 | 3858 | 274 | 229 | 255 | 94.06 |
| CD196 | 3669 | 164 | 173 | 158 | 95.69 |
| QCD-32g58 | 4224 | 444 | 456 | 421 | 90.03 |
| R20291 | 3754 | 209 | 224 | 191 | 94.91 |
During nutrient shift, C. difficile strains were changed from Brain heart infusion broth (BHI) to Basal defined medium (BDM) for one hour. During osmotic shock, C. difficile was shifted from BHI to BHI supplemented with 1.5% NaCl.
Differential gene expression
We used Cuffdiff [36] to calculate differential expression of transcripts. Genes with log2 ratio ≥ 1.5 and FDR- adjusted p value ≤ 0.05 were considered as differentially expressed. For comparing the differentially expressed genes, we classified genes as core(present in all strains with limited sequence variation), shared (present in some strains) and unique (specific to each strain). Based on orthoMCL clustering of 15 genomes, we had previously defined these gene categories in C. difficile [20]. To update these definitions, we added 7 more publicly available C. difficile genomes and applied orthoMCL across these 22 genomes. OrthoMCL was run with a BLAST E-value cut-off of 1e-5, 50% identity cut-off, 70% length alignment cut-off and an inflation parameter of 1.5. A total of 7650 clusters were identified in all 22 strains combined. Of these, 2563 were core, 2489 were shared and 2598 were unique. We then combined differentially expressed gene list and orthoMCL gene classification. The number of differentially expressed genes across strains and in each condition is listed in Table 4, and the complete list of differentially expressed genes is given in Files S2 and S3. Nutrient shift caused a greater number of differentially expressed genes as compared to osmotic shock The differentially expressed genes were distributed across COG functional categories and across the C. difficile genome (Figure 1). The largest number of differentially expressed genes under nutrient shift belonged to the following COG categories; carbohydrate transport and metabolism (G), amino acid transport and metabolism (E), translation, ribosomal structure and biogenesis (J), energy production and conversion (C), transcription (K), and cell wall/membrane/envelope biogenesis (M). As expected, the highest number of differentially expressed genes during osmotic shock was related to transport functions. These included the following COG categories; amino acid transport and metabolism (E), inorganic ion transport and metabolism (P), carbohydrate transport and metabolism (G), energy production and conversion (C), and cell wall/membrane/envelope biogenesis (M). However, as clearly shown in Table 4, the majority of the differentially expressed CDS were core genes. Surprisingly, only 97 and 6 core genes were differentially expressed across all strains in nutrient shift and osmotic shock respectively (Figure 2). While a subset of differentially expressed core genes overlapped between two or more strains, a very large number of differentially expressed core genes did not show any overlap with other strains. Estimation of expression levels of selected genes using qRT-PCR was in agreement with the trend of fold changes detected using RNA-seq (File S1). The highest number of such differentially expressed non-overlapping core genes was found in strain R20291. Among the core genes differentially expressed across all strains were genes belonging to the phosphotransferase system (PTS). The expression pattern of genes belonging to the PTS system was in agreement with the general properties of PTS. For example, PTS genes associated with utilization of secondary carbon sources such as cellobiose, N-acetylglucosamine, mannose, glucitol, and sorbitol were up-regulated several fold while genes associated with utilization of primary carbon sources such glucose and fructose were highly down-regulated. Ethanolamine genes were also down-regulated. C. difficile toxin genes were not among the differentially expressed genes in either condition tested.
Table 4. Differential expressed CDS genes and core CDS genes.
| Strain |
CD630
|
CD196
|
QCD_32g58
|
R20291 |
||||
|---|---|---|---|---|---|---|---|---|
| Condition | Nutrient shift | Osmotic shock | Nutrient shift | Osmotic shock | Nutrient shift | Osmotic shock | Nutrient shift | Osmotic shock |
| Core | 851 | 234 | 512 | 162 | 765 | 386 | 621 | 301 |
| Shared | 10 | 4 | 27 | 16 | 30 | 41 | 38 | 25 |
| Unique | 27 | 14 | 1 | 0 | 32 | 21 | 1 | 0 |
| Total | 888 | 252 | 540 | 178 | 827 | 449 | 660 | 326 |
Figure 1. Projection of differentially expressed genes from nutrient shift and osmotic shock on C. difficile 630 genome.

RNA-seq data was converted into FPKM values using cufflinks[44]. When compared to control condition, genes with log ratio ≥ 1.5 and FDR- adjusted p value less than or equal to 0.05 were considered as differentially expressed. Circles are numbered from outside to inside. Circle 1 - Molecular clock indicating genome size. Circle 2 - COG gene categories on the forward strand, Circle 3 - COG gene categories on the forward strand. Details of COG color codes in circles 2 & 3 are shown on the right side of the projection. Circle 4 - log2 fold change values of differentially expressed genes under nutrient shift. Circle 5 - log2 fold change values of differentially expressed genes under osmotic shock.
Figure 2. Venn diagram comparing differentially expressed core genes across C. difficile strains.

Core genes in each strain were determined by applying orthoMCL clustering across publicly available C. difficile genomes. When compared to control, genes with log ratio ≥ 1.5 and FDR- adjusted p value less than or equal to 0.05 were considered as differentially expressed. Panel A – differentially expressed core genes under nutrient shift (Shift from Brain heart infusion broth to Basal defined medium). Panel B - differentially expressed core genes under osmotic shock(shift from Brain heart infusion broth to Brain heart infusion broth supplemented with 1.5% NaCl).
Since majority of core genes in C. difficile are associated with cellular pathways, we analyzed how the tested stress conditions modulate pathways of C. difficile. Omics viewer [40] in Pathway tools was used to map differentially expressed genes to cellular pathways. We used enrichment analysis to determine pathways that changed significantly during nutrient shift. Fishers exact test with a p-value of >0.05 was used as the cutoff threshold. This analysis revealed that 20 pathways were significantly enriched in at least one strain (Table 5). Some of the pathways that were differentially expressed in all strains included pathway of gluconeogensis, folate transformation, plamitate biosynthesis and pyruvate fermentation (Figure 3). Using the same in vitro conditions used in this study, we have previously reported the proteomic profile of strains compared in this study[43]. There was very good overlap between the number of differentially expressed pathways detected in that study and current results. Some of the differentially expressed genes such as folD, gapA and fchA are multifunctional enzymes associated with more than one pathway. In contrast to nutrition shift, osmotic shock had only minimal impact on pathways. Only six genes (CD0022, CD0079, CD0087, CD0177, CD0627A, CD0628) were differentially expressed across all strains during osmotic shock. Among these, CD0079 and CD0087, which code for ribosomal proteins, were down-regulated. The other four genes, which code for membrane proteins, were up-regulated.
Table 5. List of differentially enriched pathways during nutrient shift.
| Pathway | p-value | Genes associated |
|---|---|---|
| Carbohydrates Biosynthesis | 1.04E-07 | CD0118, CD0886, CD2318, cooS, eno, fbp, fchA, fhs, folD, gapA, gapB, glgA, glgC, gpmI, pgi, pgk, pmi, pyc, pykF, rkpK, tpiA |
| Sugars Biosynthesis | 3.21E-07 | CD0118, CD0886, cooS, eno, fbp, fchA, fhs, folD, gapA, gapB, gpmI, pgi, pgk, pmi, pyc, pykF, rkpK, tpiA |
| Gluconeogenesis | 8.20E-07 | CD0118, cooS, eno, fbp, fchA, fhs, folD, gapA, gapB, gpmI, pgi, pgk, pyc, pykF, tpiA |
| Generation of Precursor Metabolites and Energy | 1.01E-05 | abfD, abfH, abfT, adhE, adhE, bcd2, buk, cat1, CD0118, CD0715, CD2379, crt2, ctfB, eno, fbp, gapA, gapB, gpmI, hydA, pfkA, pgi, pgk, plfB, ptb, pykF, rpe, rpiB1, rpiB2, thlA1, tkt, tkt', tpiA |
| Adenosine nucleotides de novo biosynthesis | 4.81E-05 | adk, atpA, atpB, atpC, atpD, atpF, atpG, atpH, atpI, ntpA, ntpB, ntpC, ntpD, ntpE, ntpK, purA |
| Fermentation | 1.82E-04 | abfD, abfH, abfT, adhE, adhE, bcd2, buk, cat1, CD0118, CD0715, CD2379, crt2, ctfB, eno, gapA, gapB, gpmI, pgi, pgk, plfB, ptb, pykF, rpe, thlA1 |
| Superpathway of glycolysis and Entner-Doudoroff | 2.49E-04 | eno, fbp, gapA, gapB, gpmI, pfkA, pgi, pgk, pykF, tpiA |
| Amines and Polyamines Degradation | 9.96E-04 | abfD, abfH, abfT, bcd2, CD1585, eutB, eutC, gabT, gluD, nanA, nanE, sucD |
| Purine Nucleotide Biosynthesis | 0.001257477 | adk, atpA, atpB, atpC, atpD, atpF, atpG, atpH, atpI, CD0489, hpt, iunH, ntpA, ntpB, ntpC, ntpD, ntpE, ntpK, purA, pyrH, pyrR, upp, xpt |
| Aspartate superpathway | 0.002091068 | aspD, CD1339, CD2382, CD2828, metE, nadA, nadB, nadC |
| C1 Compounds Utilization and Assimilation | 0.002151781 | CD0118, cooS, fchA, fhs, folD, plfB |
| 4-Aminobutyrate Degradation | 0.002258078 | abfD, abfH, abfT, bcd2, gabT, gluD, sucD |
| Pyruvate Fermentation | 0.004278705 | adhE, adhE, bcd2, buk, cat1, CD0118, CD0715, CD2379, crt2, ctfB, plfB, ptb, thlA1 |
| Reductive acetyl coenzyme A pathway | 0.005518962 | cooS, fchA, fhs, folD |
| Heterolactic fermentation | 0.005744886 | adhE, adhE, eno, gapA, gapB, gpmI, pgi, pgk, pykF, rpe |
| 4-aminobutyrate degradation | 0.006648169 | abfD, abfH, abfT, bcd2, gabT, gluD |
| Folate transformations | 0.006927081 | CD3456, fchA, fhs, folD, glyA |
| Methionine Biosynthesis | 0.007449931 | aspD, CD1339, CD2382, CD2828, dapG, hom2, lysC, malY, metE |
| Purine Nucleotide de novo Biosynthesis | 0.009883003 | adk, atpA, atpB, atpC, atpD, atpF, atpG, atpH, atpI, CD0489, ntpA, ntpB, ntpC, ntpD, ntpE, ntpK, purA |
| Degradation/Utilization/Assimilation | 0.012817157 | abfD, abfH, abfT, adhE, adhE, aspD, bcd2, cat1, CD0118, CD0723, CD1339, CD1585, CD2318, CD2382, CD2819, CD2828, CD3477, cooS, crt2, ctfB, eutB, eutC, fchA, fhs, folD, fruK, gabT, garR, glgP, glpK1, glsA, gluD, glyA, grdC, grdD, grdE, gutD, mdeA, nanA, nanE, pgmB, phnA, plfB, pmi, prdA, prdF, rluB, sucD, thlA1 |
Genes in bold were also found to be differentially expressed in our previous proteomics study.
Figure 3. Overview of differentially expressed C. difficile pathways.

Pathways of C. difficile were reconstructed using pathway tools. Amino acids, carbohydrates, proteins, purines, pyrimidines, cofactors, tRNAs and other components in the pathways are coded as per the scheme given on the top right hand side of the figure. Pathways on the left side are biosynthetic pathways, middle are central intermediary metabolism pathways, right side are catabolic pathways and the group on the extreme right are reactions associated with the state of the cell. The following color scheme in the pathways represent; blue - present in at least one strain, red - present in all strains, green - differentially expressed in all strains. Numbers in the pathways denote the following differentially expressed pathways; 1- Gluconeogensis, 2 - Folate transformation, 3- Biotin-carboxyl carrier assembly, 4 - Plamitate biosynthesis, 5 - Pyruvate fermentation, 6 - Fermentation of butanote, 7- Lysine fermentation acetate, 8- Aminobutyrate degradation, 9- Degradation/Utilization/Assimilation.
Pathways in a microbial cell are interconnected to form a system wide interaction network. Subsections of the system wide network control bacterial cell response to physiological changes. To determine the gene interaction network that is activated during nutrient shift, we mapped the differentially expressed genes to C. difficile interaction network. The system wide interaction data for C. difficile was downloaded from STRING[41]. We then extracted interaction data that were associated with nutrient shift in all strains. This network contained 2149 nodes (genes) and 14186 edges (interactions). A complete list of these nodes and interaction confidence scores is given in File S4. When differential expression data was overlaid on this network, we found that up-regulated and down-regulated genes were scattered all over the network (Figure 4). However, the mean S. P. betweeness of node couples in strain R20291 specific interaction clusters were higher than other strains.
Figure 4. Differentially expressed genes under nutrient shift mapped to C. difficile interaction network.

The shapes of the nodes denote strains as per the scheme given on top left hand side. The color of the code represents expression fold change. Scale of the fold change is given on top right hand side of the figure.
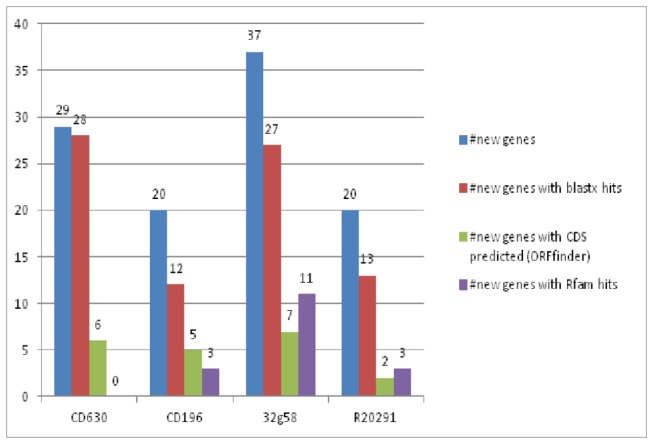
Novel gene discovery
Cufflinks assembles transcripts and maps them to the annotated genes of a genome. If the transcripts do not map to known genes of that genome, then these transcripts could be novel genes. After assembly to known annotated genes, we identified a total of 106 new gene transcripts in the intergenic regions of the four strains compared (File S5). These transcripts could be genes that were missed during genome annotation or could be novel small RNA genes. To determine this, first we searched the Rfam database for sRNA that matched these novel transcripts. Then using BLASTX, we searched non-redundant (nr) database with the new transcripts to check for any protein coding gene hits. We also made ORF predictions using the ORFfinder program to see if any transcripts are potentially protein coding genes. A total 80 transcripts had significant BLASTX hits with evalue cutoff of 0.001 (File S6). For the transcripts that do not have ORFs, they are likely to be sRNAs. The results are summarized in Figure 5. There were a total of 17 genes that matched Rfam small RNAs. Ten of them matched RF01327 (CRISPR-DR14), three matched RF01051 (GEMM_RNA_motif), two matched RF01786 (c-di-GMP-II), one matched RF00230 (T-box), and one to RF00504 (Glycine).
Figure 5. Summary of new genes and potential small RNAs identified in C. difficile strains.
Discussion
In the recent years, C. difficile has emerged as emerged as a serious human pathogen. Very high genome diversity among C. difficile strains is a contributing factor towards this [18,19,25,26]. The core genome content of C. difficile is less than 20% of the pangenome[20]. This core genome content is unusually low because core genome of other highly divergent pathogens such as Streptococcus agalactiae is at least 80% of the pangenome [46,47]. Despite this massive genome diversity, functional genomics studies in C. difficile have mostly been done using strain 630, which is a historic strain isolated from a human patient in the 1980s[21-24]. Recently isolated strains of C. difficile are more infectious and virulent than old strains[25,48]. To determine how these old and recent isolates adjust to physiological stress and to provide a better definition of C. difficile transcriptome across strains, we performed in vitro transcriptome profiling of two old and two recent hypervirulent strains. Nutritional shift from brain heart infusion broth (rich medium) to basal minimal medium was chosen as the first in vitro stress condition. This was chosen because C. difficile toxins are known to be up-regulated several fold when the growth medium contains difficult to metabolize carbon and nitrogen sources[29,49]. In the second stress condition, cells were shifted to BHI supplemented with 1.5% NaCl to induce osmotic shock. This was selected as the second test condition because C. difficile has been shown to have enhanced host cell adherence following osmotic shock [31,32,50]. We used Illumina Hiseq 2000 based RNA-seq to sequence the C. difficile transcriptome from these test conditions. When compared to microarray based transcriptome analysis, RNA-seq offers several advantages such as not being limited to detecting transcripts that correspond to existing annotations, low or no background signal, a very large dynamic range and very high reproducibility[51]. Consistent with this, under both in vitro conditions tested, we detected transcripts for more than 90% CDS(Table 3). This provides much higher resolution than the results we obtained in our previous microarray based transcriptome analysis of C. difficile[22,23].
When differentially expressed genes were classified as core, shared and unique based on orthoMCL ortholog clustering, we find that the majority of the differentially expressed CDS were core genes(Table 4). Since each C. difficile strain contains a large number of shared and strain specific genes, the low level of differential expression we detected in this study is surprising. This could be because many of the strain specific genes are virulence factors or antibiotic resistance genes that provide niche adaptation[18,25]. These genes may not have been expressed in the conditions tested in this study since nutritional switch and osmotic shock are basic metabolic functions. When the differentially expressed core genes were compared across strains (Figure 2), there was not much overlap between the genes. A total of 97 and 6 core genes were differentially expressed across all strains during nutrient shift and osmotic shock respectively. It is widely known that nutritional availability in the medium determines the amount of toxin produced by various C. difficile strains[52-57]. In C. difficile, a cluster of genes known as the pathogenicity locus (PaLoc) contains toxin A and B genes and three accessory genes, including tcdD and tcdC, which are thought to code for the positive and negative regulators of toxin expression, respectively[58]. Little is known about how other genes in individual C. difficile strains interact with the genes in PaLoc. The large number of uniquely expressed core genes we found in this study could be a mechanism by which individual C. difficile strain adjust to nutrient changes and produce variable levels of toxins. The number of differentially expressed genes during osmotic shock was less than that of nutrient shift. Differentially expressed genes following osmotic shock included genes such as GroEL, RecA, CspG, and CspF. This is consistent with previous findings that genes such as GroEL are up-regulated during osmotic shock and increases C. difficile adhesion to host cells[32,50,59].
To understand how nutritional shift modulates cellular pathways, differentially expressed genes were mapped to C. difficile pathways. Enrichment analysis revealed that 20 pathways were significantly changed during nutrient shift (Table 5). Using the same strains used in this study, we had previously reported the proteome profile of C. difficile during nutrient shift and osmotic shock[43]. Pathways found to be differentially expressed at the protoemics level correlated well with RNA-seq results(Table 5). However, the number of differentially expressed genes detected using RNA-seq was larger when compared to the number of pathways detected at the proteome level. This could be due to the differences in the resolution of the technologies and the depth of sequencing. The number of transcripts that can be detected in one single lane RNA-seq run is far more than the number of peptides that can be detected using TMT based nanoLC–MS/MS proteome sequencing. In this study, each cDNA library was sequenced using one full flow cell without multiplexing. In our previous proteomics study, four samples were multiplexed in one sequencing reaction. These factors could account for the differences in the number of differentially expressed genes detected in both studies.
Despite the differences in the number of genes detected at the transcriptome and proteome levels, a number core functions that control metabolism were common in both data sets. Phosphotransferase system (PTS) is composed of a cluster of genes that regulate the switch between utilization of primary and secondary carbon sources [60-62]. Several genes belonging to PTS were upregulated in both the in RNA-seq as well as proteome sequence data sets. In contrast, ethanolamine utilization genes were found to be down-regulated in both datasets. Members of the genus Clostridium can use ethanolamine as a source of carbon or nitrogen[63]. Host diet and gut epithelial cells are an important source of ethanolamine for bacteria[63,64]. Although the defined medium we used in this study does not contain any ethanolamine, BHI contains large amount of animal cell derived nutrients. This could contribute significant amounts of ethanolamine in BHI and the observed down-regulation of ethanolamine proteins could be a result of this concentration difference.
When differentially expressed genes under nutrient shift were overlaid on the C. difficile gene interaction network, the differentially expressed genes from all four strains were scattered across the network(Figure 4). However, when compared to other strains, the average S. P. betweeness of the node couples differentially expressed in strain R20291 was more than other strains. The S. P. betweeness is a node centrality index. The S. P. betweenness of a node in a biological network is an indicator of the relevance of a protein’s functional capability to hold communicating proteins together[65]. The S. P. betweenness of a protein effectively indicates the capability of a protein to recruit? distant proteins[65]. Strain R20291 is a hypervirulent strain associated with a severe outbreak and several mortalities in Aylesbury, UK[25]. Although it produces very low numbers of spores[66], it is highly infectious[25]. A more efficient gene interaction network indicated by increased S. P. betweeness in node pairs of R20291 might enable this strain to respond to environmental changes more efficiently. This could translate to better survival within a host.
One of the advantages of the RNA-seq technology is the ability to detect transcription of genes that are not part of the primary genome annotation[51]. Consistent with this, we detected a number of transcriptionally active regions in all four C. difficile strains that were not part of the genome annotation of the respective strain (File S5). BLASTX searches showed that the majority of these assembled transcripts were annotated as protein coding genes in other C. difficile genomes or other bacterial species (File S6). This shows that RNA-seq could detect genes that were missed by microbial annotation engines and can be used to improve the genome annotation. When searched against Rfam database, some of the transcriptionally active regions did match small RNAs. A recent study has found that small noncoding RNAs (sRNAs) in C. difficile are involved in the regulation of motility and biofilm formation[67]. Therefore, the small RNAs detected in this study could be involved in the regulation of nutritional switch and osmolarity sensing in C. difficile. Further work is required to ascertain this possibility.
Supporting Information
Details of primers used for qPCR and comparison of qPCR and RNA-seq data for select genes.
(XLSX)
Details of differentially expressed genes during nutrient shift. Data for strains QCD-32g58, R20291, CD196 and CD630 are given in separate worksheets named after these strains.
(XLSX)
Details of differentially expressed genes during osmotic shock. Data for strains QCD-32g58, R20291, CD196 and CD630 are given in separate worksheets named after these strains.
(XLSX)
Protein interaction data for C. difficile genes involved in nutrient shift. : Nodes are represented by NCBI locus tags for the CDS. The combined interaction score represents cumulative interaction score for each edge.
(XLSX)
Details of new gene transcripts identified among the compared strains.
(XLSX)
Details of BLASTX searches for the new gene transcripts.
(XLSX)
Funding Statement
This work was partially supported by a subcontract (project no. 62078/A001) from the Virginia Tech, which has been funded from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272200900040C, a grant (NYCV-478820) from the USDA Animal Health and Disease Research Program and a grant (NYC-478499) from the USDA formula fund. No additional external funding was received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Rupnik M, Wilcox MH, Gerding DN (2009) Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7: 526-536. doi: 10.1038/nrmicro2164. PubMed: 19528959. [DOI] [PubMed] [Google Scholar]
- 2. Gerding DN (2012) Clostridium difficile infection prevention: biotherapeutics, immunologics, and vaccines. Discov Med 13: 75-83. PubMed: 22284786. [PubMed] [Google Scholar]
- 3. Loo VG, Bourgault AM, Poirier L, Lamothe F, Michaud S et al. (2011) Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med 365: 1693-1703. doi: 10.1056/NEJMoa1012413. PubMed: 22047560. [DOI] [PubMed] [Google Scholar]
- 4. Pacheco SM, Johnson S (2013) Important clinical advances in the understanding of Clostridium difficile infection. Curr Opin Gastroenterol 29: 42-48. doi: 10.1097/MOG.0b013e32835a68d4. PubMed: 23207596. [DOI] [PubMed] [Google Scholar]
- 5. Norén T (2010) Clostridium difficile and the disease it causes. Methods Mol Biol 646: 9-35. doi: 10.1007/978-1-60327-365-7_2. PubMed: 20597000. [DOI] [PubMed] [Google Scholar]
- 6. Lyerly DM, Krivan HC, Wilkins TD (1988) Clostridium difficile: its disease and toxins. Clin Microbiol Rev 1: 1-18. PubMed: 3144429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rupnik M, Dupuy B, Fairweather NF, Gerding DN, Johnson S et al. (2005) (2005) Revised nomenclature of Clostridium difficile toxins and associated genes. J Med Microbiol 54: 113-117. doi: 10.1099/jmm.0.45810-0. PubMed: 15673503. [DOI] [PubMed] [Google Scholar]
- 8. Chitnis AS, Holzbauer SM, Belflower RM, Winston LG, Bamberg WM et al. (2013) Epidemiology of Community-Associated Clostridium difficile Infection, 2009 Through 2011. JAMA. Intern Med 173: 1359-1367. [DOI] [PubMed] [Google Scholar]
- 9. Gerding DN (2010) Global epidemiology of Clostridium difficile infection in 2010. Infect Control Hosp Epidemiol 31 Suppl 1: S32-S34. doi: 10.1086/655998. PubMed: 20929364. [DOI] [PubMed] [Google Scholar]
- 10. Jones AM, Kuijper EJ, Wilcox MH (2013) Clostridium difficile: a European perspective. J Infect 66: 115-128. doi: 10.1016/j.jinf.2012.10.019. PubMed: 23103666. [DOI] [PubMed] [Google Scholar]
- 11. McDonald LC, Killgore GE, Thompson A, Owens RC Jr., Kazakova SV et al. (2005) An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353: 2433-2441. doi: 10.1056/NEJMoa051590. PubMed: 16322603. [DOI] [PubMed] [Google Scholar]
- 12. He M, Miyajima F, Roberts P, Ellison L, Pickard DJ et al. (2013) Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 45: 109-113. PubMed: 23222960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Warny M, Pepin J, Fang A, Killgore G, Thompson A et al. (2005) Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366: 1079-1084. doi: 10.1016/S0140-6736(05)67420-X. PubMed: 16182895. [DOI] [PubMed] [Google Scholar]
- 14. Khanna S, Pardi DS, Aronson SL, Kammer PP, Orenstein R et al. (2012) The epidemiology of community-acquired Clostridium difficile infection: a population-based study. Am J Gastroenterol 107: 89-95. doi: 10.1038/ajg.2011.398. PubMed: 22108454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Songer JG, Anderson MA (2006) Clostridium difficile: an important pathogen of food animals. Anaerobe 12: 1-4. doi: 10.1016/j.anaerobe.2005.09.001. PubMed: 16701605. [DOI] [PubMed] [Google Scholar]
- 16. Gould LH, Limbago B (2010) Clostridium difficile in food and domestic animals: a new foodborne pathogen? Clin Infect Dis Off Publ Infect Dis Soc Am 51: 577-582. doi: 10.1086/655692. PubMed: 20642351. [DOI] [PubMed] [Google Scholar]
- 17. Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N et al. (2006) The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38: 779-786. doi: 10.1038/ng1830. PubMed: 16804543. [DOI] [PubMed] [Google Scholar]
- 18. Stabler RA, Gerding DN, Songer JG, Drudy D, Brazier JS et al. (2006) Comparative Phylogenomics of Clostridium difficile Reveals Clade Specificity and Microevolution of Hypervirulent Strains. J Bacteriol 188: 7297-7305. doi: 10.1128/JB.00664-06. PubMed: 17015669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janvilisri T, Scaria J, Thompson AD, Nicholson A, Limbago BM et al. (2009) Microarray identification of Clostridium difficile core components and divergent regions associated with host origin. J Bacteriol 191: 3881-3891. doi: 10.1128/JB.00222-09. PubMed: 19376880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scaria J, Ponnala L, Janvilisri T, Yan W, Mueller LA et al. (2010) Analysis of ultra low genome conservation in Clostridium difficile. PLOS ONE 5: e15147. doi: 10.1371/journal.pone.0015147. PubMed: 21170335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emerson JE, Stabler RA, Wren BW, Fairweather NF (2008) Microarray analysis of the transcriptional responses of Clostridium difficile to environmental and antibiotic stress. J Med Microbiol 57: 757-764. doi: 10.1099/jmm.0.47657-0. PubMed: 18480334. [DOI] [PubMed] [Google Scholar]
- 22. Janvilisri T, Scaria J, Chang YF (2010) Transcriptional profiling of Clostridium difficile and Caco-2 cells during infection. J Infect Dis 202: 282-290. doi: 10.1086/653484. PubMed: 20521945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scaria J, Janvilisri T, Fubini S, Gleed RD, McDonough SP et al. (2011) Clostridium difficile transcriptome analysis using pig ligated loop model reveals modulation of pathways not modulated in vitro. J Infect Dis 203: 1613-1620. doi: 10.1093/infdis/jir112. PubMed: 21592991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ternan NG, Jain S, Srivastava M, McMullan G (2012) Comparative transcriptional analysis of clinically relevant heat stress response in Clostridium difficile strain 630. PLOS ONE 7: e42410. doi: 10.1371/journal.pone.0042410. PubMed: 22860125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stabler RA, He M, Dawson L, Martin M, Valiente E et al. (2009) Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10: R102. doi: 10.1186/gb-2009-10-9-r102. PubMed: 19781061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF et al. (2010) Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A 107: 7527-7532. doi: 10.1073/pnas.0914322107. PubMed: 20368420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forgetta V, Oughton MT, Marquis P, Brukner I, Blanchette R et al. (2011) Fourteen-genome comparison identifies DNA markers for severe-disease-associated strains of Clostridium difficile. J Clin Microbiol 49: 2230-2238. doi: 10.1128/JCM.00391-11. PubMed: 21508155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karasawa T, Ikoma S, Yamakawa K, Nakamura S (1995) A defined growth medium for Clostridium difficile. Microbiology 141 ( 2): 371-375. doi: 10.1099/13500872-141-2-371. [DOI] [PubMed] [Google Scholar]
- 29. Karlsson S, Burman LG, Akerlund T (2008) Induction of toxins in Clostridium difficile is associated with dramatic changes of its metabolism. Microbiology 154: 3430-3436. doi: 10.1099/mic.0.2008/019778-0. PubMed: 18957596. [DOI] [PubMed] [Google Scholar]
- 30. Antunes A, Martin-Verstraete I, Dupuy B (2011) CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79: 882-899. doi: 10.1111/j.1365-2958.2010.07495.x. PubMed: 21299645. [DOI] [PubMed] [Google Scholar]
- 31. Waligora AJ, Barc MC, Bourlioux P, Collignon A, Karjalainen T (1999) Clostridium difficile cell attachment is modified by environmental factors. Appl Environ Microbiol 65: 4234-4238. PubMed: 10473442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hennequin C, Porcheray F, Waligora-Dupriet A, Collignon A, Barc M et al. (2001) GroEL (Hsp60) of Clostridium difficile is involved in cell adherence. Microbiology 147: 87-96. PubMed: 11160803. [DOI] [PubMed] [Google Scholar]
- 33. Corfield AP, Myerscough N, Longman R, Sylvester P, Arul S et al. (2000) Mucins and mucosal protection in the gastrointestinal tract: new prospects for mucins in the pathology of gastrointestinal disease. Gut 47: 589-594. doi: 10.1136/gut.47.4.589. PubMed: 10986224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. doi: 10.1186/gb-2009-10-3-r25. PubMed: 19261174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roberts A, Pimentel H, Trapnell C, Pachter L (2011) Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 27: 2325-2329. doi: 10.1093/bioinformatics/btr355. PubMed: 21697122. [DOI] [PubMed] [Google Scholar]
- 36. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL et al. (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31: 46-53. PubMed: 23222703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burge SW, Daub J, Eberhardt R, Tate J, Barquist L et al. (2013) Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41: D226-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li L, Stoeckert CJ Jr., Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178-2189. doi: 10.1101/gr.1224503. PubMed: 12952885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karp PD, Paley S, Romero P (2002) The Pathway Tools software. Bioinformatics 18 Suppl 1: S225-S232. doi: 10.1093/bioinformatics/18.suppl_1.S225. PubMed: 12169551. [DOI] [PubMed] [Google Scholar]
- 40. Paley SM, Karp PD (2006) The Pathway Tools cellular overview diagram and Omics Viewer. Nucleic Acids Res 34: 3771-3778. doi: 10.1093/nar/gkl334. PubMed: 16893960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A et al. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39: D561-D568. doi: 10.1093/nar/gkq973. PubMed: 21045058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27: 431-432. doi: 10.1093/bioinformatics/btq675. PubMed: 21149340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen JW, Scaria J, Mao C, Sobral B, Zhang S et al. (2013) Proteomic Comparison of Historic and Recently Emerged Hypervirulent Clostridium difficile Strains. J Proteome Res 12: 1151-1161. doi: 10.1021/pr3007528. PubMed: 23298230. [DOI] [PubMed] [Google Scholar]
- 44. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511-515. doi: 10.1038/nbt.1621. PubMed: 20436464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP et al. (2011) PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun 79: 4286-4298. doi: 10.1128/IAI.00207-11. PubMed: 21896772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D et al. (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proc Natl Acad Sci U S A 102: 13950-13955. doi: 10.1073/pnas.0506758102. PubMed: 16172379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tettelin H, Riley D, Cattuto C, Medini D (2008) Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol 11: 472-477. doi: 10.1016/j.mib.2008.09.006. PubMed: 19086349. [DOI] [PubMed] [Google Scholar]
- 48. Stabler RA, Dawson LF, Phua LT, Wren BW (2008) Comparative analysis of BI/NAP1/027 hypervirulent strains reveals novel toxin B-encoding gene (tcdB) sequences. J Med Microbiol 57: 771-775. doi: 10.1099/jmm.0.47743-0. PubMed: 18480336. [DOI] [PubMed] [Google Scholar]
- 49. Karlsson S, Lindberg A, Norin E, Burman LG, Akerlund T (2000) Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun 68: 5881-5888. doi: 10.1128/IAI.68.10.5881-5888.2000. PubMed: 10992498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waligora AJ, Hennequin C, Mullany P, Bourlioux P, Collignon A et al. (2001) Characterization of a cell surface protein of Clostridium difficile with adhesive properties. Infect Immun 69: 2144-2153. doi: 10.1128/IAI.69.4.2144-2153.2001. PubMed: 11254569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57-63. doi: 10.1038/nrg2484. PubMed: 19015660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamakawa K, Kamiya S, Meng XQ, Karasawa T, Nakamura S (1994) Toxin production by Clostridium difficile in a defined medium with limited amino acids. J Med Microbiol 41: 319-323. doi: 10.1099/00222615-41-5-319. PubMed: 7966203. [DOI] [PubMed] [Google Scholar]
- 53. Karasawa T, Maegawa T, Nojiri T, Yamakawa K, Nakamura S (1997) Effect of arginine on toxin production by Clostridium difficile in defined medium. Microbiol Immunol 41: 581-585. PubMed: 9310936. [DOI] [PubMed] [Google Scholar]
- 54. Ikeda D, Karasawa T, Yamakawa K, Tanaka R, Namiki M et al. (1998) Effect of isoleucine on toxin production by Clostridium difficile in a defined medium. Zentralbl Bakteriol Int J Med Microbiol 287: 375-386. doi: 10.1016/S0934-8840(98)80174-6. [DOI] [PubMed] [Google Scholar]
- 55. Yamakawa K, Karasawa T, Ohta T, Hayashi H, Nakamura S (1998) Inhibition of enhanced toxin production by Clostridium difficile in biotin-limited conditions. J Med Microbiol 47: 767-771. doi: 10.1099/00222615-47-9-767. PubMed: 9736158. [DOI] [PubMed] [Google Scholar]
- 56. Maegawa T, Karasawa T, Ohta T, Wang X, Kato H et al. (2002) Linkage between toxin production and purine biosynthesis in Clostridium difficile. J Med Microbiol 51: 34-41. PubMed: 11800470. [DOI] [PubMed] [Google Scholar]
- 57. Lei XH, Bochner BR (2013) Using phenotype microarrays to determine culture conditions that induce or repress toxin production by Clostridium difficile and other microorganisms. PLOS ONE 8: e56545. doi: 10.1371/journal.pone.0056545. PubMed: 23437164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Spigaglia P, Mastrantonio P (2002) Molecular Analysis of the Pathogenicity Locus and Polymorphism in the Putative Negative Regulator of Toxin Production (TcdC) among Clostridium difficile Clinical Isolates. J Clin Microbiol 40: 3470-3475. doi: 10.1128/JCM.40.9.3470-3475.2002. PubMed: 12202595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hennequin C, Collignon A, Karjalainen T (2001) Analysis of expression of GroEL (Hsp60) of Clostridium difficile in response to stress. Microb Pathog 31: 255-260. doi: 10.1006/mpat.2001.0468. PubMed: 11710845. [DOI] [PubMed] [Google Scholar]
- 60. Stock JB, Waygood EB, Meadow ND, Postma PW, Roseman S (1982) Sugar transport by the bacterial phosphotransferase system. The glucose receptors of the Salmonella typhimurium phosphotransferase system. J Biol Chem 257: 14543-14552. PubMed: 6292227. [PubMed] [Google Scholar]
- 61. Dupuy B, Sonenshein AL (1998) Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27: 107-120. doi: 10.1046/j.1365-2958.1998.00663.x. PubMed: 9466260. [DOI] [PubMed] [Google Scholar]
- 62. Peterkofsky A, Wang G, Seok YJ (2006) Parallel PTS systems. Arch Biochem Biophys 453: 101-107. doi: 10.1016/j.abb.2006.01.004. PubMed: 17022110. [DOI] [PubMed] [Google Scholar]
- 63. Garsin DA (2010) Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol 8: 290-295. doi: 10.1038/nrmicro2334. PubMed: 20234377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roof DM, Roth JR (1988) Ethanolamine utilization in Salmonella typhimurium. J Bacteriol 170: 3855-3863. PubMed: 3045078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Scardoni G, Petterlini M, Laudanna C (2009) Analyzing biological network parameters with CentiScaPe. Bioinformatics 25: 2857-2859. doi: 10.1093/bioinformatics/btp517. PubMed: 19729372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burns DA, Heap JT, Minton NP (2010) The diverse sporulation characteristics of Clostridium difficile clinical isolates are not associated with type. Anaerobe 16: 618-622. doi: 10.1016/j.anaerobe.2010.10.001. PubMed: 20950700. [DOI] [PubMed] [Google Scholar]
- 67. Soutourina OA, Monot M, Boudry P, Saujet L, Pichon C et al. (2013) Genome-Wide Identification of Regulatory RNAs in the Human Pathogen Clostridium difficile. PLOS Genet 9: e1003493 PubMed: 23675309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of primers used for qPCR and comparison of qPCR and RNA-seq data for select genes.
(XLSX)
Details of differentially expressed genes during nutrient shift. Data for strains QCD-32g58, R20291, CD196 and CD630 are given in separate worksheets named after these strains.
(XLSX)
Details of differentially expressed genes during osmotic shock. Data for strains QCD-32g58, R20291, CD196 and CD630 are given in separate worksheets named after these strains.
(XLSX)
Protein interaction data for C. difficile genes involved in nutrient shift. : Nodes are represented by NCBI locus tags for the CDS. The combined interaction score represents cumulative interaction score for each edge.
(XLSX)
Details of new gene transcripts identified among the compared strains.
(XLSX)
Details of BLASTX searches for the new gene transcripts.
(XLSX)