

Background: The involvement of phosphorylation of endothelin receptor A (ETA) in its desensitization and internalization is unclear.
Results: Phosphorylation-deficient ETA mutants display defective regulation, whereas receptor internalization is not affected.
Conclusion: GRK2-mediated phosphorylation contributes to ETA desensitization, but not to ETA internalization.
Significance: This work contributes to the understanding of the complex GRK2-mediated ETA regulation involving multiple mechanisms.
Keywords: Endothelin, G Protein-coupled Receptor (GPCR), Phosphorylation, Receptor Desensitization, Receptor Endocytosis, Receptor Regulation
Abstract
Endothelin receptor A (ETA), a G protein-coupled receptor, mediates endothelin signaling, which is regulated by GRK2. Three Ser and seven Thr residues recently proven to be phosphoacceptor sites are located in the C-terminal extremity (CTE) of the receptor following its palmitoylation site. We created various phosphorylation-deficient ETA mutants. The phospholipase C activity of mutant receptors in HEK-293 cells was analyzed during continuous endothelin stimulation to investigate the impact of phosphorylation sites on ETA desensitization. Total deletion of phosphoacceptor sites in the CTE affected proper receptor regulation. However, proximal and distal phosphoacceptor sites both turned out to be sufficient to induce WT-like desensitization. Overexpression of the Gαq coupling-deficient mutant GRK2-D110A suppressed ETA-WT signaling but failed to decrease phospholipase C activity mediated by the phosphorylation-deficient mutant ETA-6PD. In contrast, GRK2-WT acted on both receptors, whereas the kinase-inactive mutant GRK2-D110A/K220R failed to inhibit signaling of ETA-WT and ETA-6PD. This demonstrates that ETA desensitization involves at least two autonomous GRK2-mediated components: 1) a phosphorylation-independent signal decrease mediated by blocking of Gαq and 2) a mechanism involving phosphorylation of Ser and Thr residues in the CTE of the receptor in a redundant fashion, able to incorporate either proximal or distal phosphoacceptor sites. High level transfection of GRK2 variants influenced signaling of ETA-WT and ETA-6PD and hints at an additional phosphorylation-independent regulatory mechanism. Furthermore, internalization of mRuby-tagged receptors was observed with ETA-WT and the phosphorylation-deficient mutant ETA-14PD (lacking 14 phosphoacceptor sites) and turned out to be based on a phosphorylation-independent mechanism.
Introduction
The potent vasoconstrictive peptides endothelin-1–3 (1) mediate their multiple physiological effects in various tissues and cell types (2) via activation of the G protein-coupled receptors (GPCRs)2 endothelin receptor A (ETA) and receptor B (3), which couple to Gαq proteins to induce phospholipase C (PLC) activity.
A general property of GPCR signaling is that with continuous or repeated agonist stimulation, GPCR activity wanes. This regulatory process is called desensitization (4). Phosphorylation of activated receptors by GPCR kinases (GRKs) in the third intracellular loop and the C-terminal domain plays a major role as an early regulatory event in GPCR desensitization. This is mediated by increasing the affinity of the phosphorylated receptor for arrestins, which uncouple the receptor from its G protein, initiate receptor internalization, and redirect the signaling to alternative pathways (5, 6). In addition, phosphorylation-independent mechanisms are known to be involved in the regulation of GPCR signaling (7). Phosphorylation-independent GRK-mediated GPCR desensitization may occur by direct binding of the kinase to the receptor (8, 9) or to the activated form of Gαq/11 (10–12) via the RH (regulator of G protein signaling homology) domain of GRK (13).
Stannard et al. (14) identified 15 phosphorylation sites essentially in the C-terminal tail of ETA in human lung fibroblasts by mass spectrometry (Fig. 1). In the same study, palmitoylation of cysteines 383, 385, 386, 387, and 388 in the C-terminal domain was demonstrated. The anchoring of this palmitoylated cysteine cluster to the plasma membrane leads to the development of a fourth intracellular loop (15). Among the identified phosphorylation sites in this study, three Thr residues and seven Ser residues are located in the region distal to the palmitoylation site (amino acids 389–427, subsequently referred to as the C-terminal extremity (CTE)), indicating that this part of the C-terminal tail is a major site of phosphorylation. Within the CTE, two clusters of phospho-Ser and phospho-Thr residues can be distinguished: in the proximal region (amino acids 391–404), six phosphoacceptor sites were identified, whereas in the distal region (amino acids 417–425), four amino acids were shown to be phosphorylated.
FIGURE 1.

Post-translational modifications of human ETA. The primary sequence of human ETA is shown as a snake plot. The plasma membrane is represented by the gray box. Transmembrane amino acids are depicted according to NCBI entry P25101. In this study, mutations of ETA were introduced at known phosphorylation sites, which are represented by black spheres. Palmitoylation sites are marked by zigzag lines. Post-translational modifications are depicted according to Stannard et al. (14). Clustering of phosphoacceptor sites appears in the C-terminal domain of the receptor in the region following the palmitoylation site. This region is referred to as the CTE. Clustered phosphorylation occurs in a distal and a proximal region of the CTE.
As shown by overexpression of dominant-negative GRK mutants, GRK2 is involved in the desensitization of ETA (8, 16). However, the importance of GRK2-mediated receptor phosphorylation is still unclear in this context. Furthermore, because GRK2 does not recognize a well defined consensus sequence but rather shows high specificity toward agonist-occupied GPCRs (17), the location of potential GRK2 phosphorylation sites in ETA is yet unknown.
Therefore, we analyzed the desensitization of a set of different ETA mutants lacking several phosphoacceptor sites essentially in the CTE of the receptor. This was done 1) to gain information about the significance of GRK2 phosphorylation in triggering ETA regulation and 2) to investigate which phosphoacceptor sites, if any, are herein involved.
EXPERIMENTAL PROCEDURES
Construction of Plasmids
The coding sequence of ETA in pCMV-XL5 (SC118901, Origene, Rockville, MD) was amplified by PCR using oligonucleotides 5′-CTCGAGTATTTCCTCAAATTTGCCTCAAGATGGA-3′ and 5′-GCGGCCGCCATAAAAGCTAGCCATGTACTTGAAAGC-3′. The PCR fragment was subcloned into the XhoI and NotI sites of the pLPCX vector (Clontech). The mutant receptor constructs ETA-4PD, ETA-6PD, ETA-8PD, ETA-10PD, ETA-PDZPD, ETA-6E, and ETA-14PD were generated by the exchange of a C-terminal sequence of ETA with a corresponding synthetically generated DNA sequence (ATG:biosynthetics, Merzhausen, Germany) containing the respective point mutations via EcoRI (in the coding sequence of ETA) and NotI. All other constructs were derived from these plasmids by site-directed mutagenesis using the QuikChange Lightning kit (Agilent Technologies, Santa Clara, CA). mRuby (18)-tagged receptor constructs were created by amplification of the respective receptor DNA lacking the stop codon with the use of oligonucleotides 5′-CTCGAGTATTTCCTCAAATTTGCCTCAAGATGGA-3′ and 5′-AAGACCGGTCCGTTCATGCTGTCCTTATGGCTGCTC-3′ (ETA-WT and ETA-PDZPD) or 5′-AAGACCGGTCCGTTCATGGCGTCCTTATGGGCG-3′ (receptor constructs containing S421A and S425A mutations), thereby flanking the PCR product with XhoI and AgeI restriction sites. The amplified products were introduced into a construct similar to the pECFP-N1 vector (Clontech) containing mRuby instead of the enhanced cyan fluorescent protein sequence to obtain ETA constructs C-terminally fused to mRuby with Gly-Pro-Val-Ala-Thr as a linker sequence. Human GRK2 in the pcDNA3 vector was kindly provided by Prof. Martin Lohse (University of Würzburg). Site-directed mutagenesis was performed to obtain GRK2-D110A and GRK2-D110A/K220R. The coding sequence of every construct was verified.
Cell Culture and Transfection
HEK-293 cells were cultured in DMEM supplemented with 10% FBS (Invitrogen), penicillin (100 units/ml), and streptomycin (100 μg/ml) and incubated at 37 °C with 5% CO2 in a humidified incubator. Transfection of trypsinized cells was performed 24 h before the beginning of the experiments using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Unless stated otherwise, we used 400 ng of receptor plasmid DNA/cm2 of culture surface. Cotransfection of GRK2 was performed using 5 ng of GRK2 plasmid DNA/cm2 of culture surface to obtain a molar ratio of 1:75 GRK2 to ETA plasmid DNA. For high level cotransfection experiments, the amount of GRK2 plasmid DNA was increased by 5-fold. For knockdown experiments, 75 nm validated Silencer® Select siRNA (Ambion AM51331) or negative control siRNA (Ambion AM4611) was transfected along with 200 ng of receptor plasmid DNA/cm2 of culture surface 48 h before the beginning of the experiment.
Western Blotting
HEK-293 cells were lysed in PBS containing 0.2% Triton X-100 and protease inhibitor mixture (Sigma-Aldrich P2714). After shaking for 30 min at 4 °C, the lysate was cleared by centrifugation at 20,000 × g for 15 min. Protein content was determined using bicinchoninic acid with BSA as a standard. 7 μg of protein was separated on a 12% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. GRK2 was detected using an anti-GRK2/3 antibody (1:10,000 dilution; Upstate 05-465) and an HRP-coupled anti-mouse IgG antibody (1:2000 dilution; BD Pharmingen 554002). Densitometric analysis of chemiluminescence was performed using a MultiImage light cabinet (Alpha Innotech Corp., San Leandro, CA).
Membrane Preparation
2.85 × 106 HEK-293 cells were transiently transfected with receptor plasmid DNA in a 6-well plate. After 24 h, cells were washed with PBS containing protease inhibitor mixture and transferred to 1.5-ml reaction tubes. The additional procedure was performed according to Elshourbagy et al. (19).
Radioligand Binding
4 μg of membrane preparation was mixed with 10–1000 pm 125I-ET-1 and incubated in a total volume of 120 μl of binding buffer (10 mm Tris (pH 7.4) and 154 mm NaCl). After binding at 37 °C for 4.5 h, 10 ml of binding buffer was added, and the samples were filtered through glass-fiber filters presoaked in 0.1% (w/v) BSA. After three washes with 10 ml of binding buffer, the activity of the filters was quantified in a γ-counter.
Determination of Cell Surface Receptors with 125I-ET-1
1.14 × 106 HEK-293 cells in 12-well plates were transiently transfected with plasmids encoding receptor variants. After 24 h, cells were washed with binding buffer (Hanks' balanced salt solution with 20 mm HEPES (pH 7.4), 0.2% BSA, and 0.1% glucose) and incubated with 470 μl of 100 pm 125I-ET-1 (23 μCi; PerkinElmer Life Sciences) in binding buffer for 3 h at 10 °C. Cells were transferred to glass-fiber filters and washed three times with PBS. The activity of 125I was quantified in a γ-counter.
Determination of PLC Activity
Accumulation of myo-inositol 1-phosphate (IP1) was determined using the IP-One kit (Cisbio Bioassays, Codolet, France) according to the manufacturer's protocol. 30,000 HEK-293 cells were transfected with plasmids encoding receptor and, in the case of cotransfection, GRK2 variants in a white 384-well plate (Greiner Bio-One, Kremsmünster, Austria). Stimulation was performed with 100 nm ET-1 at 37 °C in the presence of 50 mm LiCl to inhibit myo-inositol 1-phosphatase. Time-resolved fluorescence signals were measured using an Infinite M1000 plate reader (Tecan, Männedorf, Switzerland).
Confocal Microscopy
Confocal microscopy experiments were performed with transiently transfected HEK-293 cells in a Nunc Lab-Tek Chamber SlideTM system on a Zeiss LSM 780 microscope using a 63× water immersion objective. For analysis of receptor internalization, HiLyte FluorTM 488-labeled ET-1 (ET-1-HiLyte; Eurogentec, Seraing, Belgium) was added to the cells expressing the mRuby-tagged receptor to a final concentration of 100 nm. Images were captured immediately after agonist administration (∼1 min) and after 15, 30, 45, and 60 min. HiLyte FluorTM 488 was excited with the 488-nm line of an argon laser, and emission was detected at a range of 493–551 nm. For excitation of mRuby, a DPSS laser (561 nm) was used. Fluorescence intensities were recorded from 569 to 652 nm. In both cases, a 488/561 nm main beam splitter was used. Signal amplification was constant for all measurements.
Data Analysis
Data were analyzed using GraphPad Prism Version 6 software (GraphPad Software, La Jolla, CA). Mean values from individual treatment groups were statistically analyzed by one-way analysis of variance (ANOVA) with subsequent Bonferroni correction. Direct comparison of two treatment groups was performed using Student's t test.
RESULTS
Human ETA was analyzed with respect to the involvement of phosphorylation sites in receptor desensitization and internalization. Therefore, various constructs of ETA lacking phosphoacceptor sites were generated (Fig. 2). Binding of 125I-ET-1 to HEK-293 cells transiently transfected with the corresponding constructs demonstrated comparable surface expression of ETA-WT and all mutant receptors used in this study (Fig. 3). Furthermore, the binding characteristics of the maximally mutated variant ETA-14PD did not differ significantly from those of ETA-WT (Table 1). Because of the nearly irreversible binding of ET-1 to ETA (20, 21), pre-stimulation with ET-1 to induce receptor desensitization was not possible. Ca2+ flux experiments revealed desensitization of ETA-mediated Ca2+ release (data not shown), but they are not suited for monitoring specific ETA desensitization because the inositol trisphosphate receptor itself is rapidly desensitized (22). Therefore, in this study, impairment of ETA desensitization was analyzed by measuring inositol production. An augmented inositol accumulation during continuous ET-1 stimulus was interpreted as a desensitization defect.
FIGURE 2.

ETA variants used in this study. Shown are the amino acid sequences of the WT and mutant ETA CTEs. The numbers above indicate the amino acid positions in the receptor protein. Palmitoylation occurs at cysteine residues indicated in black (14). Phosphorylated amino acids in ETA-WT (14) are underlined; mutant amino acids are highlighted in boldface. Additional substitutions of ETA-14PD (S289A, S295A, and S382A) located upstream of the palmitoylation site are not shown.
FIGURE 3.

Surface expression of ETA-WT and mutants. HEK-293 cells were transiently transfected with constructs of ETA-WT or mutants. Transfection of empty vector was performed as a control. Total binding of 125I-ET-1 was determined as described under “Experimental Procedures.” The binding of mutant receptors was normalized to the binding of ETA-WT. The means ± S.D. of four independent experiments were compared with ETA-WT using one-way ANOVA with Bonferroni correction. *, p < 0.001.
TABLE 1.
Radioligand binding characteristics of ETA-WT and ETA-14PD
125I-ET-1 binding to membrane preparations of HEK-293 cells expressing the WT or mutant receptor was performed to obtain the equilibrium dissociation constants (Kd) of the two receptor variants and the maximal binding (Bmax) of membranes from cells expressing receptors. The data represent the mean ± S.E. of six experiments. There were no significant differences in 125I-ET-1 binding of ETA-WT and ETA-14PD.
| Kd | Bmax | |
|---|---|---|
| pm | pmol/mg protein | |
| ETA-WT | 287.2 ± 80.5 | 2.33 ± 1.08 |
| ETA-14PD | 435.0 ± 106.3 | 2.00 ± 1.06 |
15 phosphorylation sites, including Tyr-184, were previously identified in human ETA (Fig. 1) (14). Tyr-184 is part of the (E/D)RY motif, one of the most highly conserved sequences in the class A family of GPCRs. This motif is involved in the adjustment of conformational states of receptors (23). Because the exchange of Tyr-184 resulted in decreased PLC activity (Fig. 4), we excluded this amino acid from further investigations. The phosphorylation-deficient mutant ETA-14PD is depleted of all other phosphorylation sites described by Stannard et al. (14). Their involvement in the desensitization of ETA was analyzed by investigating the PLC activity in HEK-293 cells overexpressing ETA-14PD under continuous ET-1 stimulation (Fig. 4). This mutant showed a significant increase in IP1 accumulation, reflecting a decreased desensitization compared with ETA-WT. We concluded that proper regulation of ETA correlates with the ability of the receptor to become phosphorylated. Next, we tried to identify which of the phosphorylation sites are involved in receptor regulation. We first analyzed ETA-10PD, depleted of all 10 phosphoacceptor sites within the CTE. With this mutant receptor, an impaired desensitization was observed (Fig. 4), showing similar augmented IP1 accumulation as with ETA-14PD. Hence, phosphorylation sites located somewhere in the CTE of ETA play a pivotal role in its desensitization. To address the impact of each single Ser and Thr residue located in the CTE, 10 mutants were generated in which these amino acids were individually exchanged with Ala. No influence on agonist-stimulated receptor desensitization was detected (Fig. 4). Thus, none of the Ser or Thr residues located in the CTE of the receptor is indispensable for ETA regulation. Accordingly, only the combined deletion of several phosphoacceptor sites at once influences the desensitization of ETA-10PD. Therefore, we created a series of ETA multiple mutants including two to nine Ala substitutions. We started with the deletion of the two most distally located phosphorylation sites and removed additional Ser and Thr residues successively from the C terminus heading toward the palmitoylation site (Fig. 4). Desensitization was not affected by deletion of two to four phosphorylation sites in the distal region but was attenuated by the additional replacement of Ser-404 (ETA-5PD) and was further impaired by removing Thr-403 (ETA-6PD) to a maximum extent. Desensitization of ETA-6PD was indistinguishable from that obtained with ETA-14PD or ETA-10PD. In addition, no further impact on receptor regulation was detected with ETA-7PD, ETA-8PD, or ETA-9PD.
FIGURE 4.
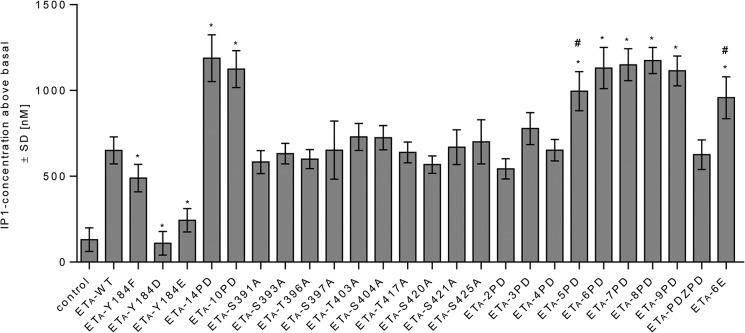
Inositol production of ETA-WT and mutants. HEK-293 cells were transiently transfected with constructs of ETA-WT or mutants as indicated. Cells were incubated for 2 h in the absence (basal) or presence of 100 nm ET-1. IP1 accumulation was quantified as described under “Experimental Procedures.” Values are presented as signal above the basal level. An impairment of receptor desensitization was observed upon enhanced IP1 accumulation compared with the WT-induced signal. Data are expressed as the means ± S.D. of three independent experiments, each performed in at least quadruplicate, and were compared with ETA-WT using one-way ANOVA with Bonferroni correction (*, p < 0.001). Furthermore, t test analysis (#, p < 0.01) was performed to compare the signals of ETA-5PD and ETA-6E with the signal of ETA-6PD.
From these experiments, it appears that phosphorylation of the distal region of the CTE is not involved in ETA desensitization (or is involved only if Thr-403 and Ser-404 are present). To investigate the involvement of distal phosphoacceptor sites in ETA desensitization, we analyzed the second messenger accumulation of the mutant ETA-PDZPD, lacking all phosphoacceptor sites in the proximal region. Remarkably, phosphorylation sites in the distal part of the C terminus were sufficient to trigger the desensitization process as indicated by the WT-like signal of this mutant receptor.
We conclude that phosphorylation of ETA initiating its desensitization is located at the CTE and is not restricted to one cluster of Ser/Thr residues. Rather, phosphorylation may occur redundantly at either proximal (ETA-4PD) or distal (ETA-PDZPD) phosphoacceptor sites.
As presented above, desensitization of ETA takes place without phosphorylation of distal Ser and Thr residues (ETA-4PD) but is impaired by deleting further phosphorylation sites in the proximal part (ETA-5PD and ETA-6PD). Subsequently, we investigated whether Thr-403 and Ser-404 play a particular role in ETA-4PD desensitization or if instead the amount of phosphoacceptor sites at arbitrary positions in the proximal region is sufficient for ETA-4PD regulation, regardless of their precise location. This was done by the insertion of extra Ala substitutions into the proximal region of ETA-4PD. It turned out that each phosphorylation site deleted in this experiment is involved in the desensitization of ETA-4PD because an increased IP1 accumulation was found for all investigated mutants (Fig. 5), but not for ETA-4PD (Fig. 4). However, the complete presence of the three phosphorylation sites Ser-397, Thr-403, and Ser-404 appears to play a more important role compared with the complete presence of Ser-391, Ser-393, and Thr-396. The absence of one of the latter phosphorylation sites reduced the ability for desensitization to a lesser extent. The simultaneous deletion of two phosphoacceptor sites belonging to the highlighted triplet (Ser-397, Thr-403, and Ser-404) even reduced the desensitization competence to a maximum degree as shown with the mutant ETA-6PD containing the mutations T403A and S404A in the proximal region (Fig. 5).
FIGURE 5.

Desensitization of mutants derived from ETA-4PD. HEK-293 cells were transiently transfected with constructs of ETA-WT or mutants as indicated. Cells were incubated for 2 h in the absence (basal) or presence of 100 nm ET-1. IP1 accumulation was quantified as described under “Experimental Procedures,” and values above the basal level were normalized to the value above the basal level of ETA-6PD. Bars represent the mean ± S.D. of two independent experiments, each performed at least in triplicate. Data were compared with the values of ETA-6PD (*, p < 0.001), ETA-5PD (#, p < 0.01), or ETA-WT ($, p < 0.001) using one-way ANOVA with Bonferroni correction.
In summary, every Ser and Thr phosphorylation site located in the proximal part of the CTE is obligatory for proper receptor regulation in the absence of distal phosphoacceptor sites. However, the lack of Ser-397, Thr-403, and Ser-404 influences desensitization to a greater degree compared with the lack of Ser-391, Ser-393, and Thr-396.
To verify that the desensitization incompetence of ETA-6PD results from its lack of phosphorylation, we generated a mutant containing six substitutions with Glu instead of Ala to mimic a phosphorylated receptor. Indeed, the IP1 accumulation of this mutant (ETA-6E) was significantly reduced compared with the ETA-6PD signal (Fig. 4), indicating that the increased signal of ETA-6PD does not result from the absence of Ser and Thr residues but rather from the phosphorylation deficiency of this mutant. However, the signal of ETA-6E was not reduced to an extent comparable to that of the WT receptor.
To assess the influence of GRK2 on the desensitization of ETA-WT and ETA-6PD, we investigated the IP1 accumulation in HEK-293 cells coexpressing receptor and kinase (Fig. 6). For the cotransfection we used GRK2-WT and mutants GRK2-D110A and GRK2-D110A/K220R. The point mutation D110A (11, 24) in the RH domain of GRK2 influences binding of the kinase to Gαq/11 (12), whereas the mutation K220R in the catalytic domain results in kinase-deficient GRK2 (25). As verified by Western blot experiments, protein expression of GRK2 was not different between the WT and mutants in HEK-293 cells (Fig. 6A).
FIGURE 6.

GRK2 involvement in ETA desensitization. A, for expression control, HEK-293 cells were transiently transfected with ETA-WT and with the indicated GRK2 construct. Control cells were transfected exclusively with ETA-WT. After 24 h, cells were lysed, and the expression of GRK2/3 was investigated in a Western blot experiment using a GRK2/3-specific antibody (upper) and by densitometric analysis (lower). Bars represent the mean ± S.D. of three independent experiments. No significant differences in GRK2 expression could be observed. B, HEK-293 cells were transiently transfected with constructs of ETA-WT or ETA-6PD. Cotransfection was performed with 5 ng of plasmid DNA/cm2 of culture surface coding for WT or mutant GRK2 as indicated. No GRK2 cotransfection was performed with control cells. The cells were stimulated with 100 nm ET-1 for 30 min. IP1 accumulation was quantified as described under “Experimental Procedures.” Data were collected in triplicate. From each data point, the basal value obtained with non-stimulated control cells was subtracted to obtain PLC activity above the basal level (PLCnet). Inhibition of PLC activity is given in percent (means ± S.D.) and calculated as follows: 100 × (PLCnet of control cells − PLCnet of GRK-transfected cells)/(PLCnet of control cells). Results were compared with the values obtained with control cells using one-way ANOVA and Bonferroni correction. C, High level cotransfection using 25 ng of plasmid DNA/cm2 of culture surface encoding GRK2 variants. The experimental conditions and data processing are analogous to those described for B. D, knockdown of GRK2 using siRNA. HEK-293 cells were transfected with ETA-WT and control siRNA or GRK2-specific siRNA. After cell lysis, the expression of GRK2/3 was investigated in a Western blot experiment (upper) and by densitometric analysis (lower). Bars represent the mean ± S.D. of three independent experiments. Statistical analysis was performed using Student's t test. E, HEK-293 cells were transfected with ETA-WT or ETA-6PD and 75 nm control siRNA (control cells) or GRK2-specific siRNA. After 48 h, the cells were stimulated with 100 nm ET-1 for 30 min. IP1 accumulation was quantified as described under “Experimental Procedures.” From each data point, the basal value obtained with non-stimulated control cells was subtracted to obtain PLCnet. Bars represent the additional signal intensity obtained by treatment with GRK2-specific siRNA compared with control cells given in percent (means ± S.D.) and calculated as follows: 100 × ((PLCnet/PLCnet of control cells) − 1). Data were collected in triplicate. Statistical analysis was performed using Student's t test. *, p < 0.001; **, p < 0.01.
Coexpression of GRK2-WT along with ETA or ETA-6PD decreased the second messenger response of both receptors. However, the PLC activity of ETA-WT was inhibited by ∼80%, whereas the second messenger response of ETA-6PD was influenced to a lesser degree (∼35%), suggesting that receptor phosphorylation is involved in signal inhibition. Accordingly, GRK2-D110A (which does not bind Gαq/11) inhibited activity of ETA-WT by 45% but had no significant impact on ETA-6PD signaling. Finally, the mutant GRK2-D110A/K220R (lacking kinase activity in addition) did not influence ETA-WT or ETA-6PD receptor activity. Thus, desensitization of ETA-WT is mediated by two GRK2-dependent mechanisms: binding to Gαq/11 and receptor phosphorylation. However, in the case of ETA-6PD, kinase-dependent desensitization is disabled, which proves the impact of phosphorylation sites in the ETA CTE for receptor regulation.
The PLC activity of ETA-WT and ETA-6PD along with the high level cotransfection of GRK2 constructs (Fig. 6C) resembles the results described above, albeit all signals are decreased. However, enhanced desensitization cannot be explained only by an augmentation of both aforementioned GRK2 effects because expression of GRK2-D110A and GRK2-D110A/K220R affected ETA-6PD- and ETA-WT-mediated signaling, respectively, to a significant degree.
Congruent with the data obtained by GRK2 overexpression, siRNA-mediated down-regulation of endogenously expressed GRK2 resulted in augmented PLC activity of both receptors (Fig. 6, D and E), verifying a role of GRK2 in ETA desensitization. However, the impact of GRK2 silencing on the signal mediated by ETA-6PD (with disabled kinase-dependent desensitization) was less pronounced.
To investigate the role of phosphorylation sites in the internalization of ETA, we fused the red fluorescent protein mRuby to the C termini of the receptors (18). HEK-293 cells transfected with ETA-WT-mRuby or the phosphorylation-deficient mutant ETA-14PD-mRuby were incubated with fluorescent ET-1-HiLyte. Binding of ET-1-HiLyte to cells expressing receptor constructs could be observed immediately after the addition of the ligand (Fig. 7). However, non-transfected cells showed no detectable binding of the fluorescent agonist (data not shown). After 15, 30, 45, and 60 min, cells expressing ETA-WT-mRuby and ETA-14PD-mRuby showed an increasing number of vesicles containing ET-1-HiLyte, indicating internalization of agonist-bound receptors (Fig. 7). No impact of mutations on the internalization was detected. We conclude that the mechanism of ETA internalization is independent of receptor phosphorylation in HEK-293 cells.
FIGURE 7.

Internalization of ETA-WT and ETA-14PD. HEK-293 cells expressing ETA-WT-mRuby (A) or ETA-14PD-mRuby (B) were loaded with 100 nm ET-1-HiLyte and studied for internalization of the membrane-bound ligand by confocal laser scanning microscopy. The cells were observed over a period of 60 min. Scale bars = 10 nm. C, quantification of receptor internalization at the indicated time points after the addition of the fluorescent agonist was performed using ImageJ software. The summarized ET-1-HiLyte signal intensity of intracellular vesicles was background-subtracted and normalized to the total cellular fluorescence intensity to account for possible photobleaching. The value was then multiplied by 100 to obtain the percent of total cellular ET-1-HiLyte fluorescence intensity. The mRuby signal served as a control and was not incorporated into the calculation. Data points represent the mean ± S.E. of at least 25 cells for each construct. No significant differences were observed between ETA-WT and ETA-14PD.
DISCUSSION
For endothelin receptors, the mechanism of desensitization, especially the role of receptor phosphorylation, is not unambiguously resolved. In a study performed by Freedman et al. (8), GRK2 overexpression in HEK-293 cells led to an increase of ETA phosphorylation. Furthermore, agonist-induced receptor phosphorylation, as well as agonist-induced reduction of ETA-mediated GTPase activity, was abolished by inhibition of GRK2 activity caused by coexpression of the dominant-negative mutant GRK2-K220R. Congruent with these observations, overexpression of GRK2-D110A/K220R and introduction of siRNA targeting endogenous GRK2 both attenuated desensitization of endogenous ETA in mesenteric arterial smooth muscle cells of adult rats (16). These reports propose that ETA regulation is mediated by GRK2 phosphorylation of the receptor.
However, there are data suggesting that this canonical model for GPCR desensitization does not suffice to explain ETA desensitization. Because GRK2-K220R, although lacking phosphotransferase activity, retains the ability to suppress phosphoinositide hydrolysis (8), it was suggested that GRK-mediated ETA phosphorylation is not relevant for decreasing of receptor signaling.
In this study, we have demonstrated a defective regulation of phosphorylation-deficient ETA mutants (Fig. 4). This suggests that receptor phosphorylation contributes to ETA desensitization. Furthermore, the involvement of phosphorylation-independent desensitization as proposed by Freedman et al. (8) was verified in our study by transfection of GRK2 constructs lacking kinase activity and the ability to bind Gαq/11 proteins (Fig. 6B). Both phosphorylation-dependent and -independent regulation of PLC activity were shown to be GRK2-mediated.
Physical association of GRK2 with the receptor may be a complementary regulatory action of the kinase (8). In our results obtained with high level co-transfection of GRK2 variants, inhibition of ETA-WT- and ETA-6PD-mediated signaling was induced even with the use of GRK2-D110A/K220R or GRK2-D110A, respectively (Fig. 6C), most likely attributable to the latter mechanism.
The increase in PLC activity mediated by silencing of endogenously expressed GRK2 underlines the existence of both 1) a phosphorylation-independent desensitization mechanism by the enhancement of ETA-6PD signaling and 2) a phosphorylation-dependent regulatory component by the additional signal increase in the case of ETA-WT.
Little is known about the localization of phosphoacceptor sites involved in GRK2-mediated desensitization of endothelin receptors. Clustering of Ser and Thr residues within the extreme C terminus is a common feature of many GPCRs. For rhodopsin (26), PAR2 (protease-activated receptor-2) (27), and the β2-adrenergic receptor (28) among other GPCRs, GRK-mediated phosphorylation of Ser and Thr residues is located within the C-terminal tail subsequent to the palmitoylation site. These phosphoacceptor sites are an important determinant for arrestin interaction, receptor desensitization, or both. The CTE of ETA contains a high density of phosphorylatable residues, and all 10 of them are indeed phosphorylated in human lung fibroblasts (14). In this study, we have shown that the combined deletion of these 10 phosphoacceptor sites in ETA results in an impaired desensitization most likely due to the lack of GRK2-mediated phosphorylation.
However, individual phosphorylation of each site is dispensable for receptor desensitization as shown with single mutant receptors. Thus, ETA regulation is not based on a key phosphorylation of a specific priming site initiating a hierarchical receptor phosphorylation as seen for other GPCRs, such as rhodopsin (29), the complement 5a receptor (30), and the δ-opioid receptor (31). Our data instead suggest a comparable unspecific phosphorylation of Ser and Thr residues located within the CTE of ETA. This fits the fact that GRKs do not recognize a defined consensus sequence of specific receptors but bind specifically to activated receptors. Upon activation of GPCRs, a pocket in the cytoplasmic receptor surface is shaped (32), supporting GRK binding. Once docked, the kinase is activated and is able to phosphorylate any phosphoacceptor site located nearby in an unspecific fashion (17). The redundancy of phosphorylation of either proximal or distal phosphoacceptor sites in the regulation of ETA leads to the conclusion that the desensitization process is initiated by a certain number of phosphorylated amino acids, regardless of their precise location. Similarly, in the m2 muscarinic acetylcholine receptor, two redundant clusters of phosphoacceptor sites were reported to be involved in receptor internalization (33). However, arrestin binding (34) and desensitization (33) were shown to be dependent on the phosphorylation of one particular cluster.
For desensitization, however, the phosphorylation of ETA is not entirely interchangeable. Phosphorylation of Ser-397, Thr-403, and Ser-404 (despite not being indispensable) seems to have a more important role in receptor desensitization than phosphorylation of Ser-391, Ser-393, and Thr-396. This may be attributable to the different locations of phospho groups within the tertiary structure of the cytoplasmic domain, which are unequally accessible for arrestin.
For internalization studies, we used ETA constructs C-terminally fused to a fluorescent protein. This tag does not alter the internalization characteristics of ETA (35). We showed that internalization of ETA is independent of phosphoacceptor sites (at least of phosphoacceptor sites proven in Ref. 14). Similar results were obtained using a truncated ETA mutant with deletion of the CTE, including 10 phosphoacceptor sites (36), in CHO cells. In concert with these findings, GRK2 overexpression failed to affect ETA internalization in CHO cells (35).
Arrestin binding is essential for internalization of GPCRs (37, 38). Especially for ETA expressed in CHO (35) and HEK-293T (39) cells, arrestin-dependent receptor internalization was demonstrated. It was claimed for a long time that receptor phosphorylation is essential for arrestin binding (40). In this context, a mutant of the human P2Y1 receptor lacking Ser-352 and Thr-358 in the distal portion of the C-terminal tail failed to induce the translocation of arrestin and consequently receptor internalization in HEK-293 cells (41). This contrasts with our data suggesting a phosphorylation-independent internalization mechanism of ETA.
However, the arrestin-GPCR interaction seems to reflect a more complex process than just binding or not. Binding to a non-phosphorylated receptor may induce an arrestin conformation different from that resulting from the binding to a phosphorylated receptor. This can result in different downstream effects. Such differential arrestin binding was shown, for example, for PAR2. Here, a stable arrestin interaction was lost upon a truncation in the C-terminal tail impairing prolonged ERK2 activation, whereas receptor internalization mediated by a more unstable arrestin association was not influenced (27).
GPCR desensitization and internalization likewise may have different requirements for arrestin conformations resulting from different arrestin receptor associations. Consequently, both events are not necessarily correlated in phosphorylation-deficient GPCR mutants. This is the case, for example, for the somatostatin subtype 2A receptor. A Thr-deficient mutant of this receptor inhibited internalization but did not affect desensitization (42). Recently, results analogous to ours were obtained with BLT1 (B4 leukotriene receptor-1) in rat basophilic leukemia RBL-2H3 cells (43). In this study, arrestin-mediated internalization of the receptor was not influenced by conversion of phosphoacceptor sites in the C-terminal domain to Ala residues, whereas desensitization of the same construct was reduced as shown by enhanced G protein-mediated activities. These and other (e.g. Refs. 27, 30, 44, and 45) results agree with our finding that phosphorylation of GPCRs is not an absolute requirement for arrestin binding or internalization. The phosphorylation-independent interaction of a ligand-occupied receptor with arrestin may be mediated through the activation sensor of arrestin (5).
The flexibility of arrestins is a prerequisite for our current view on these multifunctional adaptor proteins. For instance, binding of different arrestin-biased agonists to GPCRs leads to the establishment of distinct phosphorylation patterns of the same receptor by different GRKs leading to the adoption of different arrestin conformations and functional outcomes (46).
Our investigations using the phospho-mimic mutant ETA-6E revealed a partly reduced desensitization. This demonstrates that negative charges are not sufficient to promote proper arrestin binding required for ETA desensitization. One would anticipate that mutant ETA-6E would show low signal intensity and behave as a constitutive desensitized receptor, as is the case for BLT1. Here, a mutant containing Asp/Glu insertions mimicking the phosphorylation of substituted Ser/Thr behaved as a constitutive desensitized receptor (43). On the other hand, for the β2-adrenergic receptor, a reduction of GRK-mediated desensitization was observed upon the substitution of Asp for putative GRK sites (47). This shows that signaling of different GPCRs can be individually influenced by the introduction of phospho-mimic mutations. In the case of ETA-6E, the phospho-mimic does not equal real receptor phosphorylation with respect to the ability to induce receptor internalization.
In this study, we have shown that ETA desensitization is a complex process involving phosphorylation-dependent and -independent regulatory mechanisms. The participation of diverse desensitization mechanisms presumably allows cell-specific fine-tuning of endothelin signaling. Understanding the flexible nature of endothelin receptor regulation may help in the development of drugs targeting tissue-specific receptor desensitization, e.g. for the treatment of pulmonary hypertension.
Acknowledgments
We thank Prof. Gerd Ulrich Nienhaus (Karlsruhe Institute of Technology) for providing the mRuby construct; Prof. Hanns Hatt (Ruhr University Bochum) for providing the opportunity to measure Ca2+ fluxes; Dr. Andreas Brodehl, Prof. Martin Lohse, and Prof. Karl-Josef Dietz (University of Bielefeld) for helpful discussions; and Désirée Gerdes, Birte Bohms, and Ramona Cebulla for excellent technical assistance.
This work was supported by Actelion Pharmaceuticals (Allschwil, Switzerland) and the Erich and Hanna Klessmann Foundation (Gütersloh, Germany) (to F. G. and H. M.).
- GPCR
- G protein-coupled receptor
- ETA
- endothelin receptor A
- PLC
- phospholipase C
- GRK
- GPCR kinase
- CTE
- C-terminal extremity
- IP1
- myo-inositol 1-phosphate
- ANOVA
- analysis of variance.
REFERENCES
- 1. Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y., Yazaki Y., Goto K., Masaki T. (1988) A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332, 411–415 [DOI] [PubMed] [Google Scholar]
- 2. Masaki T. (1993) Endothelins: homeostatic and compensatory actions in the circulatory and endocrine systems. Endocr. Rev. 14, 256–268 [DOI] [PubMed] [Google Scholar]
- 3. Sakurai T., Yanagisawa M., Masaki T. (1992) Molecular characterization of endothelin receptors. Trends Pharmacol. Sci. 13, 103–108 [DOI] [PubMed] [Google Scholar]
- 4. Lefkowitz R. J. (1993) G protein-coupled receptor kinases. Cell 74, 409–412 [DOI] [PubMed] [Google Scholar]
- 5. Gurevich V. V., Gurevich E. V. (2006) The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol. Ther. 110, 465–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 7. Pao C. S., Benovic J. L. (2002) Phosphorylation-independent desensitization of G protein-coupled receptors? Sci. STKE 2002, pe42. [DOI] [PubMed] [Google Scholar]
- 8. Freedman N. J., Ament A. S., Oppermann M., Stoffel R. H., Exum S. T., Lefkowitz R. J. (1997) Phosphorylation and desensitization of human endothelin A and B receptors. Evidence for G protein-coupled receptor kinase specificity. J. Biol. Chem. 272, 17734–17743 [DOI] [PubMed] [Google Scholar]
- 9. Dhami G. K., Anborgh P. H., Dale L. B., Sterne-Marr R., Ferguson S. S. G. (2002) Phosphorylation-independent regulation of metabotropic glutamate receptor signaling by G protein-coupled receptor kinase 2. J. Biol. Chem. 277, 25266–25272 [DOI] [PubMed] [Google Scholar]
- 10. Carman C. V., Parent J. L., Day P. W., Pronin A. N., Sternweis P. M., Wedegaertner P. B., Gilman A. G., Benovic J. L., Kozasa T. (1999) Selective regulation of Gαq/11 by an RGS domain in the G protein-coupled receptor kinase, GRK2. J. Biol. Chem. 274, 34483–34492 [DOI] [PubMed] [Google Scholar]
- 11. Sterne-Marr R., Tesmer J. J. G., Day P. W., Stracquatanio R. P., Cilente J.-A. E., O'Connor K. E., Pronin A. N., Benovic J. L., Wedegaertner P. B. (2003) G protein-coupled receptor kinase 2/Gαq/11 interaction. A novel surface on a regulator of G protein signaling homology domain for binding Gα subunits. J. Biol. Chem. 278, 6050–6058 [DOI] [PubMed] [Google Scholar]
- 12. Tesmer V. M., Kawano T., Shankaranarayanan A., Kozasa T., Tesmer J. J. G. (2005) Snapshot of activated G proteins at the membrane: the Gαq-GRK2-Gβγ complex. Science 310, 1686–1690 [DOI] [PubMed] [Google Scholar]
- 13. Huang C.-c., Tesmer J. J. G. (2011) Recognition in the face of diversity: interactions of heterotrimeric G proteins and G protein-coupled receptor (GPCR) kinases with activated GPCRs. J. Biol. Chem. 286, 7715–7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stannard C., Lehenkari P., Godovac-Zimmermann J. (2003) Functional diversity of endothelin pathways in human lung fibroblasts may be based on structural diversity of the endothelin receptors. Biochemistry 42, 13909–13918 [DOI] [PubMed] [Google Scholar]
- 15. Moench S. J., Moreland J., Stewart D. H., Dewey T. G. (1994) Fluorescence studies of the location and membrane accessibility of the palmitoylation sites of rhodopsin. Biochemistry 33, 5791–5796 [DOI] [PubMed] [Google Scholar]
- 16. Morris G. E., Nelson C. P., Standen N. B., Challiss R. A. J., Willets J. M. (2010) Endothelin signalling in arterial smooth muscle is tightly regulated by G protein-coupled receptor kinase 2. Cardiovasc. Res. 85, 424–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gurevich E. V., Tesmer J. J. G., Mushegian A., Gurevich V. V. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133, 40–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kredel S., Oswald F., Nienhaus K., Deuschle K., Röcker C., Wolff M., Heilker R., Nienhaus G. U., Wiedenmann J. (2009) mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS ONE 4, e4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elshourbagy N. A., Korman D. R., Wu H. L., Sylvester D. R., Lee J. A., Nuthalaganti P., Bergsma D. J., Kumar C. S., Nambi P. (1993) Molecular characterization and regulation of the human endothelin receptors. J. Biol. Chem. 268, 3873–3879 [PubMed] [Google Scholar]
- 20. Waggoner W. G., Genova S. L., Rash V. A. (1992) Kinetic analyses demonstrate that the equilibrium assumption does not apply to [125I]endothelin-1 binding data. Life Sci. 51, 1869–1876 [DOI] [PubMed] [Google Scholar]
- 21. Wang J., Chiou W. J., Gagne G. D., Wu-Wong J. R. (2000) Internalization of type-A endothelin receptor. J. Cardiovasc. Pharmacol. 36, S61–S65 [DOI] [PubMed] [Google Scholar]
- 22. Hajnóczky G., Thomas A. P. (1994) The inositol trisphosphate calcium channel is inactivated by inositol trisphosphate. Nature 370, 474–477 [DOI] [PubMed] [Google Scholar]
- 23. Rovati G. E., Capra V., Neubig R. R. (2007) The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol. Pharmacol. 71, 959–964 [DOI] [PubMed] [Google Scholar]
- 24. Willets J. M., Challiss R. A. J., Nahorski S. R. (2003) Non-visual GRKs: are we seeing the whole picture? Trends Pharmacol. Sci. 24, 626–633 [DOI] [PubMed] [Google Scholar]
- 25. Kong G., Penn R., Benovic J. L. (1994) A β-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the β2-adrenergic receptor. J. Biol. Chem. 269, 13084–13087 [PubMed] [Google Scholar]
- 26. Zhang L., Sports C. D., Osawa S., Weiss E. R. (1997) Rhodopsin phosphorylation sites and their role in arrestin binding. J. Biol. Chem. 272, 14762–14768 [DOI] [PubMed] [Google Scholar]
- 27. Stalheim L., Ding Y., Gullapalli A., Paing M. M., Wolfe B. L., Morris D. R., Trejo J. (2005) Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol. Pharmacol. 67, 78–87 [DOI] [PubMed] [Google Scholar]
- 28. Seibold A., Williams B., Huang Z. F., Friedman J., Moore R. H., Knoll B. J., Clark R. B. (2000) Localization of the sites mediating desensitization of the β2-adrenergic receptor by the GRK pathway. Mol. Pharmacol. 58, 1162–1173 [DOI] [PubMed] [Google Scholar]
- 29. Ohguro H., Palczewski K., Ericsson L. H., Walsh K. A., Johnson R. S. (1993) Sequential phosphorylation of rhodopsin at multiple sites. Biochemistry 32, 5718–5724 [DOI] [PubMed] [Google Scholar]
- 30. Christophe T., Rabiet M. J., Tardif M., Milcent M. D., Boulay F. (2000) Human complement 5a (C5a) anaphylatoxin receptor (CD88) phosphorylation sites and their specific role in receptor phosphorylation and attenuation of G protein-mediated responses. Desensitization of C5a receptor controls superoxide production but not receptor sequestration in HL-60 cells. J. Biol. Chem. 275, 1656–1664 [DOI] [PubMed] [Google Scholar]
- 31. Kouhen O. M., Wang G., Solberg J., Erickson L. J., Law P. Y., Loh H. H. (2000) Hierarchical phosphorylation of δ-opioid receptor regulates agonist-induced receptor desensitization and internalization. J. Biol. Chem. 275, 36659–36664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rasmussen S. G. F., Choi H.-J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pals-Rylaarsdam R., Hosey M. M. (1997) Two homologous phosphorylation domains differentially contribute to desensitization and internalization of the m2 muscarinic acetylcholine receptor. J. Biol. Chem. 272, 14152–14158 [DOI] [PubMed] [Google Scholar]
- 34. Pals-Rylaarsdam R., Gurevich V. V., Lee K. B., Ptasienski J. A., Benovic J. L., Hosey M. M. (1997) Internalization of the m2 muscarinic acetylcholine receptor. Arrestin-independent and -dependent pathways. J. Biol. Chem. 272, 23682–23689 [DOI] [PubMed] [Google Scholar]
- 35. Bremnes T., Paasche J. D., Mehlum A., Sandberg C., Bremnes B., Attramadal H. (2000) Regulation and intracellular trafficking pathways of the endothelin receptors. J. Biol. Chem. 275, 17596–17604 [DOI] [PubMed] [Google Scholar]
- 36. Paasche J. D., Attramadal T., Sandberg C., Johansen H. K., Attramadal H. (2001) Mechanisms of endothelin receptor subtype-specific targeting to distinct intracellular trafficking pathways. J. Biol. Chem. 276, 34041–34050 [DOI] [PubMed] [Google Scholar]
- 37. Ferguson S. S. (2001) Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 53, 1–24 [PubMed] [Google Scholar]
- 38. Luttrell L. M., Lefkowitz R. J. (2002) The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 39. Hamdan F. F., Rochdi M. D., Breton B., Fessart D., Michaud D. E., Charest P. G., Laporte S. A., Bouvier M. (2007) Unraveling G protein-coupled receptor endocytosis pathways using real-time monitoring of agonist-promoted interaction between beta-arrestins and AP-2. J. Biol. Chem. 282, 29089–29100 [DOI] [PubMed] [Google Scholar]
- 40. Gurevich V. V., Gurevich E. V. (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 25, 105–111 [DOI] [PubMed] [Google Scholar]
- 41. Reiner S., Ziegler N., Leon C., Lorenz K., von Hayn K., Gachet C., Lohse M. J., Hoffmann C. (2009) β-Arrestin-2 interaction and internalization of the human P2Y1 receptor are dependent on C-terminal phosphorylation sites. Mol. Pharmacol. 76, 1162–1171 [DOI] [PubMed] [Google Scholar]
- 42. Liu Q., Dewi D. A., Liu W., Bee M. S., Schonbrunn A. (2008) Distinct phosphorylation sites in the SST2A somatostatin receptor control internalization, desensitization, and arrestin binding. Mol. Pharmacol. 73, 292–304 [DOI] [PubMed] [Google Scholar]
- 43. Jala V. R., Shao W.-H., Haribabu B. (2005) Phosphorylation-independent β-arrestin translocation and internalization of leukotriene B4 receptors. J. Biol. Chem. 280, 4880–4887 [DOI] [PubMed] [Google Scholar]
- 44. Richardson M. D., Balius A. M., Yamaguchi K., Freilich E. R., Barak L. S., Kwatra M. M. (2003) Human substance P receptor lacking the C-terminal domain remains competent to desensitize and internalize. J. Neurochem. 84, 854–863 [DOI] [PubMed] [Google Scholar]
- 45. Klenk C., Vetter T., Zürn A., Vilardaga J.-P., Friedman P. A., Wang B., Lohse M. J. (2010) Formation of a ternary complex among NHERF1, β-arrestin, and parathyroid hormone receptor. J. Biol. Chem. 285, 30355–30362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reiter E., Ahn S., Shukla A. K., Lefkowitz R. J. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vaughan D. J., Millman E. E., Godines V., Friedman J., Tran T. M., Dai W., Knoll B. J., Clark R. B., Moore R. H. (2006) Role of the G protein-coupled receptor kinase site serine cluster in β2-adrenergic receptor internalization, desensitization, and β-arrestin translocation. J. Biol. Chem. 281, 7684–7692 [DOI] [PubMed] [Google Scholar]