

Background: EGFR is an important player in endochondral ossification.
Results: Mice with EGFR deficiency in chondrocytes have delayed secondary ossification center formation due to reduced cartilage degradation, and EGFR regulates MMPs and RANKL expression in chondrocytes partially via Wnt/β-catenin.
Conclusion: EGFR regulates epiphyseal cartilage development.
Significance: The cross-talk between EGFR and Wnt/β-catenin signaling in chondrocytes shed new light on studying cartilage development and diseases.
Keywords: Beta-Catenin, Cartilage Biology, Epidermal Growth Factor Receptor (EGFR), Extracellular Matrix, Matrix Metalloproteinase (MMP), Cartilage Matrix Degradation, Secondary Ossification Center
Abstract
The epidermal growth factor receptor (EGFR) is an essential player in the development of multiple organs during embryonic and postnatal stages. To understand its role in epiphyseal cartilage development, we generated transgenic mice with conditionally inactivated EGFR in chondrocytes. Postnatally, these mice exhibited a normal initiation of cartilage canals at the perichondrium, but the excavation of these canals into the cartilage was strongly suppressed, resulting in a delay in the formation of the secondary ossification center (SOC). This delay was accompanied by normal chondrocyte hypertrophy but decreased mineralization and apoptosis of hypertrophic chondrocytes and reduced osteoclast number at the border of marrow space. Immunohistochemical analyses demonstrated that inactivation of chondrocyte-specific EGFR signaling reduced the amounts of matrix metalloproteinases (MMP9, -13, and -14) and RANKL (receptor activator of NF-κB ligand) in the hypertrophic chondrocytes close to the marrow space and decreased the cartilage matrix degradation in the SOC. Analyses of EGFR downstream signaling pathways in primary epiphyseal chondrocytes revealed that up-regulation of MMP9 and RANKL by EGFR signaling was partially mediated by the canonical Wnt/β-catenin pathway, whereas EGFR-enhanced MMP13 expression was not. Further biochemical studies suggested that EGFR signaling stimulates the phosphorylation of LRP6, increases active β-catenin level, and induces its nuclear translocation. In line with these in vitro studies, deficiency in chondrocyte-specific EGFR activity reduced β-catenin amount in hypertrophic chondrocytes in vivo. In conclusion, our work demonstrates that chondrocyte-specific EGFR signaling is an important regulator of cartilage matrix degradation during SOC formation and epiphyseal cartilage development and that its actions are partially mediated by activating the β-catenin pathway.
Introduction
In mammals, the long bone structure is developed through a process of endochondral ossification in which the skeletal cartilage anlagen are replaced by bone. The anlagen elongate and expand in width by the proliferation of chondrocytes, as well as by the deposition of cartilage matrix. At the embryonic stage, chondrocytes in the central region of the anlagen undergo differentiation into hypertrophic chondrocytes, which eventually become calcified and apoptotic. Angiogenic factors secreted by these cells induce sprouting angiogenesis from the perichondrium. With the vessels come osteoblasts, osteoclasts, and hematopoietic cells, resulting in the formation of the primary ossification center (POC)2 in the center of the diaphysis (1, 2). Shortly after birth, the epiphyseal cartilage is excavated by canals invaginating from the perichondrium. These cartilage canals allow for the arrival of blood vessels and osteoprogenitors to the center of the epiphyseal cartilage. Then the inner ends of the canal fuse and expand by continued excavation to create the marrow space for the secondary ossification center (SOC) (3, 4). The sequential development of the POC and SOC defines the location of the growth plate and the articular cartilage. The former one is responsible for longitudinal bone growth, whereas the latter is an essential component of a synovial joint that provides smooth movement to long bones. Although the formation modes of these two ossification centers share a lot of similar events, such as vascular invasion, chondrocyte hypertrophy, mineralization, apoptosis, and cartilage matrix degradation, they differ in some fundamental aspects. One major difference is that although the aforementioned events are tightly coupled at the chondro-osseous junction between growth plate and primary spongiosa in the POC, cartilage canals with invading vasculature initially penetrate the periarticular cartilage, and this process is uncoupled from the chondrocyte hypertrophy and mineralization in the cartilage center during SOC (5). Currently, the majority of studies have focused on the development of the POC, but the information on the SOC formation is limited. In particular, the systemic and local factors regulating the establishment of the SOC are not clearly elucidated.
The epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor that modulates cell fates for proliferation, differentiation, survival, migration, and adhesion. It plays a critical role during embryonic and postnatal organ development (6), and hence global knock-out of EGFR in mice leads to early lethality due to severe developmental abnormalities in placental, neural, and epithelial tissues (7–9). EGFR ligands include EGF, amphiregulin, and transforming growth factor α (TGFα), which only bind to and signal through EGFR, and heparin-binding EGF, betacellulin, and epiregulin, which bind to both EGFR and ErbB4. Upon ligand binding, EGFR either homodimerizes or heterodimerizes with other members of the EGFR family and relays the extracellular signals into a myriad of intracellular signaling pathways including the Ras-Raf-MAP-kinase and PI3K-Akt pathways (10). Studies over the past several years have identified the EGFR signaling pathway as one of the major modulators of bone metabolism, regulating the proliferation and differentiation of osteoblasts and chondrocytes, osteoclastogenesis, and cancer metastasis to bone (11).
We previously demonstrated that inhibition of EGFR activity by systemic administration of a specific inhibitor, gefitinib, in 1-month-old rats results in a greatly expanded hypertrophic zone in the growth plate due to decreased expression of matrix metalloproteinases (MMPs) in the growth plate and reduced osteoclastogenesis at the chondro-osseous junction (12). To investigate whether this is a chondrocyte-specific event, we specifically inactivated EGFR activity in chondrocytes by generating compound transgenic mice that harbor collagen2a1 promoter-driven Cre (Col2-Cre), a floxed Egfr allele, and an EGFR dominant negative allele, Wa5 (13). A similar approach was used previously to successfully generate preosteoblast/osteoblast-specific Egfr knock-out mice (14). We have reported that these chondrocyte-specific Egfr CKO mice exhibited significantly enlarged hypertrophic zone in the growth plate at P5 (12). In this study, we focused on the postnatal epiphyseal cartilage development in these mice and further investigated the molecular mechanisms by which EGFR signaling regulates cartilage matrix degradation and remodeling. Wnt/β-catenin signaling is involved in many steps of cartilage development, including chondrogenesis, hypertrophic maturation of chondrocytes, and cartilage degradation (15). Notably, recent studies pointed out the involvement of this signaling in SOC development (16). Thus we explored the possible cross-talk between EGFR and Wnt/β-catenin signaling pathways in cartilage. Our results strongly suggest that EGFR signaling in chondrocytes is a novel key regulator of SOC formation during postnatal epiphyseal cartilage development.
EXPERIMENTAL PROCEDURES
Animals
Egfr CKO (Col2-Cre EgfrWa5/f) mice were generated as described previously (12). Briefly, we first bred Col2-Cre with EgfrWa5/+ to obtain Col2-Cre EgfrWa5/+, which were then crossed with Egfrf/f to generate Egfr CKO mice and their Wa5 (EgfrWa5/f) and wild-type (WT, Col2-Cre Egfrf/+, and Egfrf/+) siblings. All animal work performed in this study was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania.
Histologic Analyses, Immunohistochemistry, and in Situ Hybridization
Mouse hind long bones were harvested at P5, P9, P12, and P18 for histological analyses. We used at least four animals per age per group. Tibiae were fixed in 10% neutral buffered formalin overnight, washed under tap water, and then decalcified in 0.5 m EDTA (pH 7.4) for 1 week prior to processing and embedding in paraffin. A series of 6 μm-thick sections was cut through the entire bone and adhered to Superfrost Plus slides at three sections per slide. One out of every eight slides was stained with hematoxylin and eosin (H&E) to identify the location of the cartilage canal and SOC, which were usually in the center of the bone. The neighboring slices were then subjected to staining as described below. For safranin O staining, sections were stained with hematoxylin followed by 0.001% (w/v) fast green and 0.1% (w/v) safranin O. TUNEL staining was carried out using the ApopTag Plus peroxidase in situ apoptosis detection kit (EMD Millipore, Billerica, MA) according to the manufacturer's directions. For TRAP staining, sections were stained with a leukocyte acid phosphatase kit (Sigma-Aldrich). TRAP-positive cells at the junction between marrow space of SOC and adjacent hypertrophic chondrocytes were counted, and the length of the junction was measured under a microscope at 100× magnification. Standard immunohistochemistry was performed with following primary antibodies: anti-EGFR (Abcam), anti-Runx2, anti-RANKL (Santa Cruz Biotechnology), anti-MMP9, anti-MMP13, anti-MMP14 (Abcam), anti-Col2–3/4C (MMP-13-generated type 2 collagen cleavage fragment), and anti-CGGVDIPEN (MMP-generated aggrecan cleavage fragment. Both were gifts from Dr. John S Mort), anti-β-catenin (BD Biosciences). To quantify the percentage of antigen-positive cells, we randomly chose three fields per section containing mostly hypertrophic chondrocytes adjacent to the marrow space of SOC and counted antigen-positive and total hypertrophic chondrocytes within 5- (for MMP9) and 15- (for all of the rest of the antigens) cell distance to the marrow border at ×100 magnification (n = 3 mice/group). In situ hybridization of paraffin sections was performed as described previously (17).
To study epiphyseal morphology and calcification, tibiae were dissected, fixed in 70% ethanol, processed for methyl methacrylate embedding, and cut into 5-mm longitudinal sections using a Reichert-Jung Polycut-S motorized microtome (Reichert). Consecutive sections were processed for Goldner's trichrome and von Kossa staining.
Primary Chondrocytes, Real Time RT-PCR, and Western Blotting
Primary mouse (18) and rat (12) epiphyseal chondrocytes from newborn pups were harvested and cultured as described previously. Cells were plated at a density of 4 × 104/cm2 in 12-well plates in chondrogenic medium (DMEM/F12 medium with 5% fetal bovine serum, 50 μg/ml l-ascorbic acid, 1% glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin). Four days later, when cells reached 90% confluence, cultures were given fresh medium overnight before adding 50 ng/ml recombinant human TGFα (PeproTech) in the absence or presence of pathway-specific inhibitors, such as U0126 (20 μm), SB203580 (10 μm), wortmannin (3 μm), JNK II (20 μm, EMD Millipore), BIO (0.5 μg/ml), IWR-endo (10 μm, Cayman Chemicals), LiCl (20 mm, Sigma-Aldrich), and DKK1 (500 ng/ml, R&D Systems). RNA and protein were harvested at the indicated time points for qRT-PCR and Western blot, respectively. The rat primers used for qRT-PCR were reported previously (12). The mouse primer sequences are: β-actin (forward, 5′-TCCTCCTGAGCGCAAGTACTCT-3′; reverse, 5′-CGGACTCATCGTACTCCTGCTT-3′), Mmp9 (forward, 5′-CCACATCGAACTTCGACACTGA-3′; reverse, 5′-TGATCTAAGCCCAGTGCATGG-3′), Mmp13 (forward: 5′-TGACCTCCACAGTTGACAGG-3′; reverse, 5′-ATCAGGCACTCCACATCTTGG-3′), and Rankl (forward, 5′-TGCAGGAGGATGAAACAAGC-3′; reverse, 5′-CCATGAGCCTTCCATCATAGC-3′). The following antibodies were used for immunoblotting: anti-MMP9, anti-TATA-binding protein (Abcam), anti-MMP13, anti-active β-catenin (EMD Millipore), anti-β-catenin (BD Biosciences), anti-phospho-LRP6, anti-LRP6, anti-phospho-p38 (Cell Signaling Technology), anti-phospho-ERK, and anti-β-actin (Santa Cruz Biotechnology) antibodies.
Cell Fractionation for Western Blotting
Cells were washed twice with ice-cold PBS and harvested by scraping with a rubber policeman in lysis buffer (10 mm HEPES, pH 7.5, 10 mm KCl, 1.5 mm MgCl2, 0.5 mm DTT, 5 mm EDTA, 1 mm Na3VO4, 1 mm PMSF, and 50 mm β-glycerophosphate). After incubation on ice for 15 min, cells were homogenized by passing through a 22.5-gauge needle 20 times. The homogenate was centrifuged at 3,500 rpm for 5 min to sediment the nuclei. This supernatant was then centrifuged at 16,000 × g for 20 min, and the subsequent supernatant was harvested as the non-nuclear fraction. The nuclear pellet was washed three times with lysis buffer to remove contamination with cytoplasmic membranes. To extract nuclear proteins, isolated nuclei were resuspended in lysis buffer and sonicated briefly. Nuclear lysates were collected after centrifugation.
Immunofluorescence Analysis
Primary chondrocytes grown on glass coverslips were fixed in 4% (w/v) paraformaldehyde for 10 min and then permeabilized in 0.2% (w/v) Triton X-100 for 5 min and 2 mg/ml BSA for 10 min. The cells were incubated sequentially with anti-β-catenin antibody (BD Biosciences) at 4 °C overnight and Alexa Fluor 488-labeled goat anti-mouse IgG antibody (Invitrogen) at room temperature for 1 h. The coverslips were then placed on slides with mounting medium (VECTASHIELD-DAPI, Vector Laboratories) and examined under a fluorescence microscope (Nikon Eclipse 90i). Five different fields were randomly imaged and counted for total cell number (DAPI staining) and the number of cells with nuclear staining of β-catenin.
Statistical Analysis
All data were analyzed by independent Student's t test assuming equal variances in each group or two-way analysis of variance with a Bonferroni's post test. For cell culture experiments, all results are derived from experiments being repeated independently at least three times. A value of p < 0.05 was considered significant. All data are expressed as mean ± S.E.
RESULTS
Chondrocyte-specific EGFR Activity Is Required for SOC Formation
We first performed immunohistochemistry to examine whether epiphyseal cartilage expresses EGFR. As shown in Fig. 1A, panel a, the majority of epiphyseal chondrocytes, except most of the periarticular and perichondrial cells, showed EGFR-positive staining, implying that EGFR might play a role in epiphyseal cartilage development. Egfr CKO (Col2-Cre EgfrWa5/f) mice were viable and born at the expected Mendelian ratio. They had comparable birth weight with WT (Col2-Cre Egfr+/f and Egfr+/f) and Wa5 (EgfrWa5/f) siblings (data not shown). To confirm the reduced EGFR activity, we cultured epiphyseal chondrocytes from these mice and tested their EGF responsiveness. As shown in Fig. 1B, EGF treatment stimulated a strong and sustained increase in the amounts of phospho-ERK1/2 in cells derived from WT mice. This EGF-stimulated ERK phosphorylation was only slightly decreased in chondrocytes from Wa5 mice but was dramatically diminished in chondrocytes from Egfr CKO mice both at basal level and upon EGF stimulation. Similar observations were also found with another EGFR ligand, TGFα (data not shown). These results suggest that the EGFR activity in chondrocytes from these mice follows this order: WT>Wa5≫Egfr CKO.
FIGURE 1.

The development of SOC in the proximal epiphysis of the tibia is delayed in the chondrocyte-specific EGFR-deficient mice. A, immunohistochemistry of EGFR in the epiphyseal cartilage of P9 WT mice reveals that most chondrocytes express EGFR (a). Black arrows point to EGFR negative periarticular region. Negative control (b) shows immunostaining with omission of the primary antibody. B, primary epiphyseal chondrocytes derived from Egfr CKO mice exhibit decreased ERK phosphorylation (p-ERK) in response to EGF when compared with those from WT and Wa5 mice as shown by immunoblots. Cells were harvested at the indicated times after 50 ng/ml EGF treatment. C, safranin O-stained sections from WT, Wa5, and Egfr CKO mice at the age of 5 (a–c), 9 (d–i), 12 (j–l), and 18 (p–r) days show the SOC and growth plate development. Double-headed arrows indicate the hypertrophic zone. Cartilage canals and hypertrophic chondrocytes in the SOC are indicated by yellow arrowheads and yellow arrows, respectively. g–i, magnified images from d–f, respectively. m–o, darkfield images of in situ hybridization with probes to col2a1 at the SOC in mice at the age of 12 days. D, consecutive sections from methyl methacrylate-embedded samples, one stained with Goldner's trichrome (a–c and g–l) and the other stained with von Kossa (d–f and m–o), reveal matrix calcification in the hypertrophic cartilage (black arrows) in the SOC from mice at the age of 9 (a–f) and 12 (g–o) days. j–l, magnified images from g–i, respectively. The yellow arrowhead in i points to a cartilage canal.
At P5, the proximal tibial epiphysis of all mice was composed exclusively of cartilaginous tissue (Fig. 1C, panels a–c). Consistent with our previous study (12), we observed a moderate increase in the length of the hypertrophic zone in Wa5 mice and a much more remarkable increase in Egfr CKO mice, suggesting that EGFR expression in chondrocytes is responsible for the growth plate development. At P9, cartilage canals began to form by invaginations of the perichondrium and to invade the surrounding cartilage matrix in both WT and Wa5 mice (Fig. 1C, panels d, e, g, and h, arrowheads). In contrast, we observed that cartilage canals initiated at the perichondrium, but they were unable to penetrate further into the cartilage (Fig. 1C, panels f and i). At P12, canals excavated into the center of the cartilage generating the marrow space in both WT and Wa5 mice. When compared with WT, Wa5 mice had a slightly smaller marrow space and less cartilage-to-bone conversion in the marrow space (Fig. 1C, panels j and k, and D, panels g, h, j, k, m, and n). However, canals in Egfr CKO mice just started to extend and excavated a small marrow space into the hypertrophic cartilage (Fig. 1D, panel i). The SOC eventually formed in Egfr CKO mice with smaller marrow space and less bone matrix at P18 (Fig. 1C, panels p–r). Similar observations were also noticed in the distal femoral epiphysis (data not shown). Taken together, these data demonstrate that EGFR signaling in chondrocytes is critical for the excavation of cartilage canal and marrow space during SOC formation in long bones.
Analyses of the Effects of Chondrocyte-specific EGFR Deficiency on Chondrocyte Hypertrophy, Mineralization, and Apoptosis
We next examined the cellular mechanisms underlying the delay in SOC formation resulting from EGFR deficiency in chondrocytes. As shown in Fig. 1C, panels d–l, the morphological appearance of chondrocytes within the epiphyseal cartilage revealed that chondrocyte hypertrophy developed at the same extent among WT, Wa5, and Egfr CKO mice. In line with this result, the future SOC site, where the chondrocytes proceeded toward hypertrophy and were surrounded by col2a1-expressing chondrocytes, was similar in size among these mice (Fig. 1C, panels m–o). However, matrix mineralization at the center of cartilage initially occurred in WT and Wa5 mice at P9 but was completely absent in Egfr CKO mice (Fig. 1D, panels d–f). At P12, the mineralized cartilage area remained much smaller in Egfr CKO mice when compared with others (Fig. 1D, m–o). These results clearly indicate that deficiency of EGFR activity in chondrocytes does not affect the hypertrophy of chondrocytes but suppresses their mineralization. Therefore, the delayed SOC formation in the Egfr CKO mice cannot be due to the abnormality of the initial chondrocyte differentiation.
Runx2 is an important transcription factor required for chondrocyte maturation and highly expressed in chondrocytes proceeding toward terminal differentiation (19), which is characterized by matrix mineralization and expression of Mmp13 (20). Immunostaining of Runx2 revealed that fewer hypertrophic chondrocytes adjacent to the marrow space of SOC in Egfr CKO mice expressed Runx2 when compared with those in WT and Wa5 mice (Fig. 2A, panels a–c, and B), suggesting that terminal differentiation is suppressed in these chondrocytes. Furthermore, TUNEL staining at the same area revealed 40 and 88% decreases of apoptotic hypertrophic chondrocytes in Wa5 and Egfr CKO mice, respectively, when compared with WT mice (Fig. 2A, panels d–f, and C). Hence, we conclude that EGFR signaling stimulates terminal differentiation, matrix mineralization, and apoptosis in epiphyseal chondrocytes.
FIGURE 2.
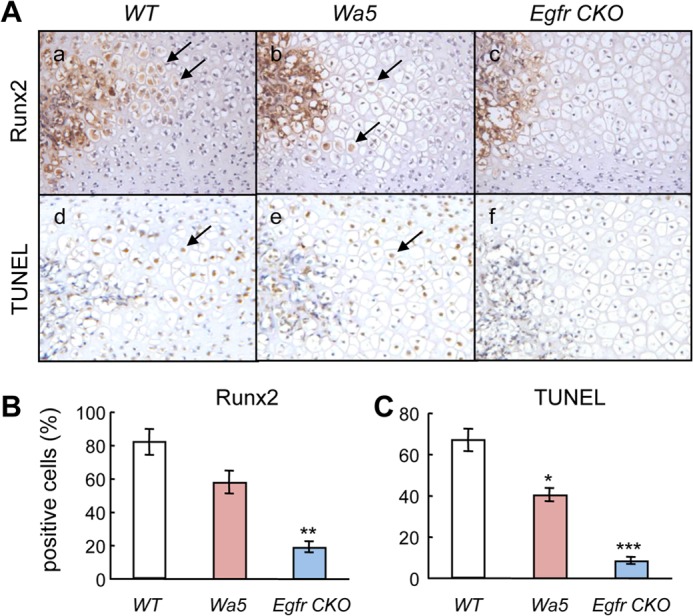
EGFR deficiency in chondrocytes suppresses the terminal differentiation and apoptosis of hypertrophic chondrocytes adjacent to the marrow space in the SOC. A, immunohistochemistry of Runx2 protein (a–c) and TUNEL (d–f) staining in sections of proximal epiphysis of tibia from 12-day-old WT, Wa5, and Egfr CKO mice. Arrows indicate positively stained hypertrophic chondrocytes. B and C, quantification of Runx2- (B) and TUNEL- (C) positive chondrocytes. n = 3 mice per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus WT.
Deficiency in Chondrocyte-specific EGFR Activity Impedes the Expression of MMPs and Extracellular Matrix (ECM) Degradation
The aforementioned cellular events that are regulated by EGFR signaling are tightly coupled with each other and together with cartilage ECM degradation and bone replacement. MMPs are members of a family of zinc-dependent proteolytic enzymes. Some of them, including MMP9, -13, and -14, are expressed at high levels in bone and cartilage and are essential for the cleavage of cartilage ECM and remodeling of cartilage into bone (21). To investigate whether these MMPs mediate the effect of EGFR signaling on the formation of the SOC, the localization and amounts of these proteins were examined by immunohistochemistry. We observed that MMP9 was mainly expressed inside the marrow space and its adjacent thin layer of chondrocytes in both WT and Wa5 mice (Fig. 3A, panels a and b). Although the expression inside the marrow space remained the same within all three types of mice, MMP9 expression in the chondrocytes was much less in Egfr CKO mice (Fig. 3A, panel c, and B). Expression of both MMP13 and MMP14 was much broader within the hypertrophic chondrocyte zone in WT and Wa5 mice (Fig. 3A, panels d, e, g, and h), and their staining intensities were significantly less in Egfr CKO mice (Fig. 3A, panels f and i, C, and D). In correlation with decreases in amounts of MMPs, the MMP-cleaved aggrecan (Fig. 3A, panels j–l, and E) and type II collagen (Fig. 3A, panels m–o, and F) products were detected at lower levels in the cartilage of Egfr CKO mice when compared with WT and Wa5 mice.
FIGURE 3.

Blocking EGFR activity in chondrocytes reduces the amounts of MMPs and cartilage matrix degradation in the hypertrophic chondrocytes adjacent to the marrow space in the SOC. A, immunohistochemistry of MMP9 (a–c), 13 (d–f), 14 (g–i), MMP-cleaved aggrecan (j–l), and type II collagen (Col-II) (m–o) products in sections of proximal epiphysis of tibia from 12-day-old WT, Wa5, and Egfr CKO mice. Arrows indicate positively stained hypertrophic chondrocytes. B–F, the percentages of hypertrophic chondrocytes positive for the above proteins were quantified and shown to the right of their immunohistochemistry images. n = 3 mice per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus WT.
EGFR Signaling Stimulates Osteoclastogenesis at the Borders of Bone Marrow Space
Osteoclasts at the chondro-osseous junction play an important role in endochondral ossification by secreting various proteases to digest cartilage matrix. At the SOC, TRAP staining identified abundant osteoclasts within the marrow space and at the border of the marrow space. Although the Wa5 mutation did not affect the overall number and distribution pattern of osteoclasts, chondrogenic knockdown of EGFR activity greatly reduced the osteoclast number at the border of the marrow space by 61% (Fig. 4, A and B). A similar phenomenon was also observed at the junction between the growth plate and metaphyseal bone (data not shown). RANKL is a major regulator of osteoclast formation and differentiation (22). Immunohistochemistry showed that the amount of RANKL in the hypertrophic chondrocytes adjacent to the marrow space in the SOC was significantly less in Egfr CKO mice when compared with WT and Wa5 mice (Fig. 4, C and D). These results indicate that one approach for chondrocytes to regulate cartilage degradation during SOC formation is through expressing RANKL to stimulate osteoclastogenesis at the edge of the marrow space and that this mechanism is tightly controlled by EGFR signaling.
FIGURE 4.

Osteoclast differentiation at the border of marrow space in the SOC is delayed in the Egfr CKO mice. A, TRAP staining (red) of the epiphyseal tibial sections reveals that there were fewer osteoclasts at the junction between the marrow space and chondrocytes in the SOC of Egfr CKO mice. B, the number of TRAP+ cells along the junction was quantified microscopically. n = 3–5 mice per group. **, p < 0.01 versus WT. C, immunostaining of RANKL protein in the hypertrophic chondrocytes adjacent to the marrow cavity in the SOC. Top panels are high magnification images from bottom panels. D, the percentages of RANKL+ hypertrophic chondrocytes were quantified. ***, p < 0.001 versus WT.
EGFR Signaling Regulates MMP9 and RANKL Expression via MEK/ERK, p38 Kinase, and β-Catenin Pathways
The above transgenic mouse data indicate that delay of SOC formation in Egfr CKO mice is closely linked to the inhibition of cartilage matrix degradation and osteoclastogenesis. In line with these in vivo findings, we demonstrated that in vitro TGFα treatment increase the expression of Mmp9 and -13 and Rankl in WT mouse primary chondrocytes but not in those derived from Egfr CKO mice (Fig. 5A). To elucidate the underlying signaling pathway mechanisms, we next performed a series of in vitro assays using rat primary epiphyseal chondrocytes because of a much higher yield of chondrocytes from rats when compared with mice. Consistent with the above mouse data and our previous study (12), both EGFR ligands, TGFα (Fig. 5B), and EGF (data not shown), up-regulated the mRNA levels of Mmp9 and -13 and Rankl in rat primary chondrocytes, with a stronger effect with TGFα-treated samples. Pathway-specific inhibitor assays revealed that MEK1 inhibitor U0126 and p38 kinase inhibitor SB203580 were able to abolish the TGFα-induced Mmp9 and Rankl expression, whereas PI3K inhibitor wortmannin and JNK inhibitor JNK II had no such effect. On the contrary, TGFα-stimulated expression of Mmp13 was only suppressed by U0126 (Fig. 5B). These changes on MMP9 and -13 were further confirmed at the protein level (Fig. 5C). Note that TGFα did phosphorylate ERK and p38 kinases in chondrocytes via an EGFR-dependent manner (Fig. 5D). These findings prove that ERK and p38 pathways are downstream targets of EGFR required for its regulation of Mmp9 and Rankl gene expression in chondrocytes.
FIGURE 5.

EGFR signaling stimulates the expression of Mmp9 and -13 and Rankl through β-catenin-dependent and -independent pathways. A, TGFα stimulates the expression of Mmp9 and -13 and Rankl in mouse chondrocytes through the EGFR pathway. Mouse primary epiphyseal chondrocytes from either WT or Egfr CKO mice were treated with TGFα for 48 h followed by qRT-PCR analysis. ***, p < 0.001 versus vehicle-treated WT cells. B and C, rat primary epiphyseal chondrocytes were pretreated with either control (DMSO, 0.1% v/v) or pathway-specific inhibitors for 30 min followed by TGFα treatment for 48 h. The total RNA and protein were prepared for qRT-PCR analysis (B) and immunoblotting (C), respectively. con, DMSO; SB, SB203580; IWR, IWR-endo; wort, wortmannin; JNK, JNK II. #, p < 0.01; &, p < 0.001 versus DMSO- and vehicle-treated cells; *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus DMSO- and TGFα-treated cells. a, proMMP9 (92 kDa); b, proMMP13 (54 kDa). D, TGFα stimulates the phosphorylation of ERK (p-ERK) and p38 kinase (p-p38) in chondrocytes through activating EGFR. Rat primary epiphyseal chondrocytes were treated with DMSO or EGFR inhibitor gefitinib (gef) for 30 min followed by 15 min of TGFα treatment. Cell lysates were collected for Western blotting for phosphorylated and total ERK and p38 kinase. E and F, activation of the β-catenin pathway stimulates the expression of Mmp9 (E) and Rankl (F) in chondrocytes. Rat primary epiphyseal chondrocytes were treated with Wnt3a (100 ng/ml), LiCl (20 mm), or BIO (0.5 μg/ml) for 48 h. RNA was harvested for qRT-PCR analysis of Mmp9 and Rankl mRNA levels. *, p < 0.05, **, p < 0.01, ***, p < 0.001 versus control (con).
Interestingly, a Wnt pathway inhibitor, IWR-endo, partially suppressed the induction of Mmp9 by TGFα, reducing its mRNA -fold change from 12.8- to 3.4-fold and further reducing its protein level (Fig. 5, B and C). Similar inhibitor results were also observed with TGFα-induced up-regulation of Rankl, but not MMP13 (Fig. 5B). Activators of the canonical Wnt pathway, such as Wnt3a, LiCl, or BIO, strongly enhanced the expression of Mmp9 (Fig. 5E) and Rankl (Fig. 5F), suggesting that these two genes are indeed Wnt pathway targets. Taken together, these results indicate that the catabolic actions of EGFR on cartilage ECM could be partially mediated by cross-talking with the β-catenin signaling pathway.
EGFR/MEK/ERK Signaling Phosphorylates LRP6, Stabilizes β-Catenin, and Promotes Its Nuclear Translocation
To understand the interaction between EGFR and β-catenin signaling pathways, we treated primary chondrocytes with TGFα for 48 h. We did not observe any changes in β-catenin mRNA by qRT-PCR (data not shown) or total β-catenin amount by Western blot (Fig. 6A). However, we found that TGFα increased the level of active (nonphosphorylated) β-catenin and that this increase was abolished by inhibitors for EGFR (gefitinib), MEK/ERK (U0126), and p38 kinase (SB203580) pathways (Fig. 6A).
FIGURE 6.

TGFα activates β-catenin and stimulates its nuclear translocation in chondrocytes. A, rat primary chondrocyte cultures were pretreated with the indicated inhibitors followed by 48 h of TGFα treatment and subjected to immunoblot analyses of the active form of β-catenin (active), total β-catenin (total), or β-actin as internal control (actin). SB, SB203580. B and C, immunofluorescence was performed with rat primary chondrocytes to detect the nuclear accumulation of β-catenin after 24 h of TGFα treatment in the absence and presence of pathway-specific inhibitors. Representative images were shown in B. The percentages of cells with β-catenin nuclear staining were quantified in C. ***, p < 0.001 versus vehicle-treated control (con). gef, gefitinib. D, the nuclear and cytosolic fractions of rat primary chondrocytes treated with TGFα and MAPK inhibitors were processed for immunoblotting of β-catenin and its internal nuclear (TATA-binding protein (TBP)) and cytosolic (β-actin) controls. E, a similar immunofluorescence experiment was performed with mouse primary chondrocytes derived from WT and Egfr CKO mice. **, p < 0.01 versus vehicle-treated WT cells.
The active form of β-catenin translocates from the cytoplasm to the nucleus and acts as a co-transcription factor to regulate Wnt target gene expression. An immunofluorescence experiment showed that TGFα stimulated the accumulation of β-catenin in the nucleus, resulting in a 2.7-fold increase in the percentage of chondrocytes displaying strong β-catenin nuclear staining (Fig. 6, B and C). Consistent with the immunoblot results, inhibitors for EGFR, MEK/ERK, and p38 kinase also abolished the nuclear translocation of β-catenin induced by TGFα (Fig. 6, B and C). Indeed, the nuclear β-catenin amount greatly increased after TGFα treatment, and this increase was diminished by suppressing either MEK/ERK or p38 kinase pathways (Fig. 6D). Similar nuclear accumulation of β-catenin was observed in WT chondrocytes but not in those derived from Egfr CKO mice (Fig. 6E).
To investigate how EGFR signaling activates and stabilizes β-catenin, we treated primary chondrocytes with TGFα with or without IWR-endo for 48 h. IWR-endo stabilizes the destruction complex for β-catenin and promotes its proteasomal degradation. Interestingly, pretreatment of IWR-endo abolished TGFα-induced accumulation of active β-catenin (Fig. 7A), implying that EGFR signaling regulates Wnt/β-catenin signaling upstream of the glycogen synthase kinase 3 (GSK3) destruction complex. A recent study demonstrated that MAPKs, such as ERK1/2, p38 kinase, and JNK1, are able to phosphorylate LRP6, the co-receptor for Wnts, at the conserved PPP(S/T)P motif (23). Phosphorylated LRP6 is known to interfere with the function of the destruction complex for β-catenin, thereby enabling the accumulation of active β-catenin. Indeed, TGFα treatment rapidly phosphorylated LRP6, which was abrogated by inhibiting either EGFR or MEK/ERK but not p38 kinase (Fig. 7B). In addition, treatment with DKK1, an inhibitor for LRP6 (24), abolished the TGFα-induced LRP6 phosphorylation (Fig. 7C) and β-catenin nuclear translocation (Fig. 7D). Furthermore, DKK1 attenuated the up-regulation of Mmp9 and Rankl mRNA by TGFα (Fig. 7E). Consistent with previous data (Fig. 5B) that the β-catenin pathway does not participate in the regulation of Mmp13 by EGFR, pretreatment of DKK1 did not affect the stimulation of Mmp13 by TGFα (Fig. 7E). At last, we found that activating EGFR and β-catenin signaling by TGFα and BIO, respectively, synergistically up-regulated Mmp9 and Rankl expression in chondrocytes (Fig. 7F). Collectively, these data reveal a novel signaling mechanism by which an EGFR ligand can regulate β-catenin signaling and genes related to cartilage ECM degradation through Lrp6 phosphorylation in chondrocytes.
FIGURE 7.

TGFα activates β-catenin in chondrocytes through LRP6 phosphorylation. A, primary chondrocyte cultures were pretreated with IWR-endo for 30 min followed by TGFα treatment for 48 h and subjected to immunoblot for active β-catenin (active), total β-catenin (total), and β-actin as internal control (actin). B and C, primary chondrocyte cultures were pretreated with gefitinib (gef), U0126, SB203580 (SB) (B) or DKK1 (C) for 30 min followed by TGFα for 15 min and subjected to immunoblot for phosphorylated and total LRP6. con, control. D, DKK1 abrogates the TGFα-induced nuclear translocation of β-catenin in chondrocytes as demonstrated by immunofluorescence analysis using anti-β-catenin antibody. **, p < 0.01 versus control. E, qRT-PCR revealed that TGFα-induced Mmp9 and RANKL expression but not MMP13 expression was partially abolished by DKK1. *, p < 0.05 versus TGFα-treated control. F, qRT-PCR revealed that 48 h of TGFα (50 ng/ml) and BIO (0.5 μg/ml) treatment synergistically increased Mmp9 and RANKL expression. *, p < 0.05, **, p < 0.01, ***, p < 0.001 versus vehicle-treated control; &, p < 0.001 versus TGFα alone or BIO alone.
EGFR Signaling Regulates β-Catenin Amount in Cartilage
Next, we studied whether suppressing EGFR activity affects the β-catenin pathway in vivo. Immunohistochemistry of tibial epiphyseal sections indicated that the protein levels of β-catenin were much lower in the hypertrophic chondrocytes either adjacent to the marrow space in the SOC or in the growth plate in Egfr CKO mice than in those located at the corresponding areas in WT mice (Fig. 8A), suggesting that EGFR signaling is required for stabilization of β-catenin in cartilage.
FIGURE 8.
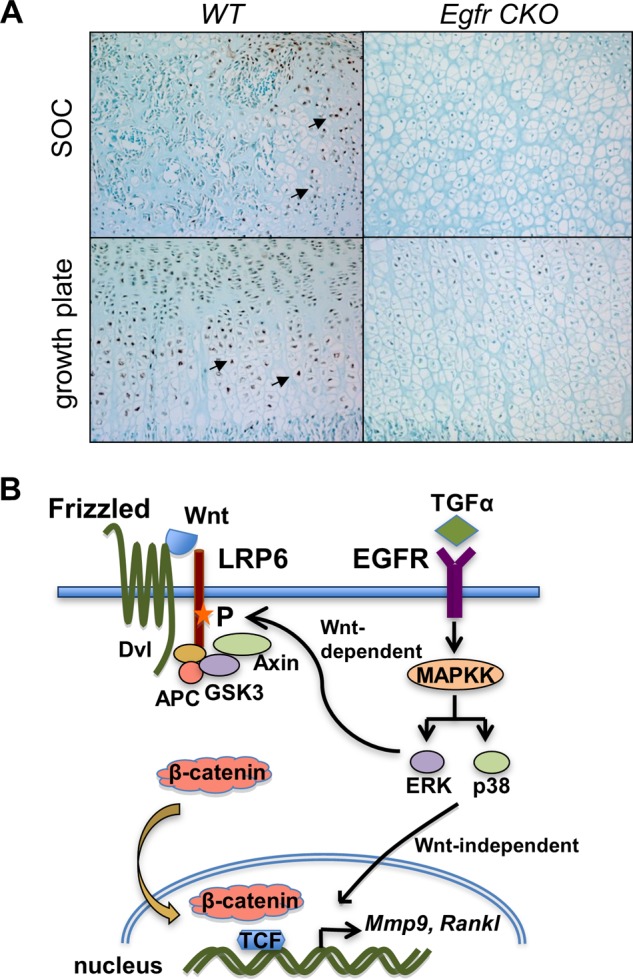
EGFR signaling increases β-catenin amount in hypertrophic chondrocytes in vivo. A, immunohistochemical staining of β-catenin in hypertrophic chondrocytes located in either SOC or growth plate in the proximal tibiae of 12-day-old WT and Egfr CKO mice. Arrows indicate β-catenin-positive cells. B, a model depicting how EGFR signaling stimulates the expression of Mmp9 and RANKL via β-catenin-dependent and -independent pathways in chondrocytes. P, phosphorylation.
DISCUSSION
In the present study, we have provided results strongly supporting that chondrocyte-specific EGFR signaling plays an essential role in stimulation of cartilage ECM degradation in postnatal SOC formation in long bones via activating the expression of Mmps and Rankl in hypertrophic chondrocytes. Whether this signaling pathway plays roles in regulating the development of bones other than long bones, such as craniofacial bones and vertebral column, is still unknown and needs further detailed investigation. To enhance the reduction of EGFR activity in chondrocytes, we added a dominant negative Wa5 allele to the Col2-Cre system and generated compound Egfr CKO mice. Because Wa5 itself exhibited only slightly decreased EGFR activity and minor changes in epiphyseal SOC formation, it is likely that the strong phenotypes observed with Egfr CKO mice are largely chondrocyte-dependent. However, we cannot completely exclude the possibility of involvement of other types of cells, such as synovial cells, where Col2-Cre can also be induced (25).
Our data demonstrate that the excavation of cartilage canals through hypertrophic chondrocytes is greatly inhibited in Egfr CKO mice due to reduced Mmps and Rankl expression. This event is also tightly coupled with delays in the terminal differentiation, mineralization, and apoptosis of chondrocytes within epiphyseal cartilage. We previously identified a similar mechanism in growth plate development using EGFR inhibitor-treated young rats (12). Our findings that EGFR pathway uses similar approaches to regulate both SOC and growth plate development suggest that these two events, although occurring at a different time and location, share the same local factors and signaling pathways for degrading the cartilage matrix. TGFα is very likely to be the endogenous ligand for EGFR activation because its null mice at the early postnatal stage display similar growth plate and SOC phenotypes (26). In addition, we provided strong evidence showing that EGFR regulates the expression of those genes through both β-catenin-dependent and β-catenin-independent pathways.
Cartilage ECM has two major components: the collagen fibrils (type II and type X) and the proteoglycan aggrecan. To convert cartilage matrix into bone matrix during endochondral ossification, these components need to be degraded first by proteases secreted by surrounding cells, including chondrocytes, osteoclasts, and blood vessel-associated cells, to make space for osteoblasts to deposit bone matrix, mainly containing type I collagen. Hence, cartilage ECM degradation is a necessary step for both POC and SOC formation. Suppressing MMP activity by broad spectrum MMP inhibitors strikingly enlarges the hypertrophic zone within the growth plate (27) and a complete inhibition of SOC formation (28). Analyzing individual Mmp knock-out mice identified that both MMP9 and MMP13 are crucial for degrading type II collagen and aggrecan at the chondro-osseous junction in the growth plate (29–31). Although Mmp9 and -13 single knock-out mice were not observed to display any abnormalities in SOC formation, the double knock-out mice had severely delayed SOC formation with no bone structures in the epiphyseal cartilage at 2 weeks of age (30), indicating that both of their activities are required for degrading the cartilage ECM during SOC progression. By contrast, Mmp14 null mice did not form cartilage canals (32, 33). Their SOCs were eventually formed through an alternative mechanism bypassing the cartilage canal step. All these MMPs were found to be expressed abundantly at the blind ends of canals and at the expanding borders of the marrow space in the epiphyseal region during SOC development (34). Our studies demonstrate that EGFR signaling in chondrocytes supports cell autonomous action for these MMPs, particularly those synthesized by hypertrophic chondrocytes adjacent to the marrow space. It is possible that EGFR signaling could achieve this autonomous effect through regulating proteases other than the MMPs studied here. However, our pilot experiments showed no stimulation of adamts-4 and -5 and cathepsin K in the chondrocyte culture (data not shown).
The conversion of cartilage matrix to bone matrix at the growth plate requires the activity of osteoclasts. Mice that are unable to generate osteoclasts developed enlarged hypertrophic zones in their growth plate (35). A recent study suggested that the expression of TRAP, an osteoclast marker, is dispensable for epiphyseal excavation and establishment of the SOC (36). Our data provide a positive correlation between decreased number of osteoclasts adjacent to chondrocytes and delayed SOC formation resulting from EGFR deficiency. Emerging evidence suggests that hypertrophic chondrocytes are one of the major sources of RANKL controlling mineralized cartilage resorption at the growth plate (37). In supporting this notion, we found that Rankl expression in the hypertrophic chondrocytes next to the marrow cavity correlates well with the number of osteoclasts at the junction, implying that chondrocytes at both POC and SOC use a cell non-autonomous mechanism to regulate cartilage resorption by modulating osteoclastogenesis.
As summarized in the graphic shown in Fig. 8B, our data suggest that EGFR ligands activate MAPK pathways, mainly the phosphorylation of ERK and p38 kinases. Although only ERK activation is essential for EGFR-induced Mmp13 expression, both pathways are indispensable for the up-regulation of Mmp9 and Rankl genes. ERK phosphorylation, but not p38 kinase phosphorylation, leads to the phosphorylation of LRP6 at Ser-1490 within one of the five conserved PPP(S/T)P motifs present in the intracellular domain of LRP6. This phosphorylation of LRP6, which is also considered to mediate Wnt-induced β-catenin activation, recruits the GSK3-APC-Axin complex to the cell membrane, thus stabilizing β-catenin and promoting its nuclear translocation to stimulate the transcription of Mmp9 and Rankl genes. Both ERK and p38 phosphorylation are important for β-catenin entering the nucleus. However, how p38 kinase causes β-catenin translocation remains to be determined. Inhibiting the Wnt/β-catenin pathway by either the LRP6 inhibitor DKK1 or IWR-endo only partially abolished the up-regulation of Mmp9 and Rankl by EGFR, suggesting that β-catenin-independent pathways also contribute to this regulation.
Both EGFR and Wnt/β-catenin signaling pathways are important for tumorigenesis. In recent years, the cross-talks between these two pathways have been well documented in cancer cells. In the majority of studies, they synergistically induce tumor formation and progression (38). On the one hand, it has been shown in mammary cells that Wnt binding to its receptor, Frizzled (Fz), transactivates EGFR by MMP-mediated release of soluble EGFR ligands and subsequently stimulates MAPK activation (39, 40). On the other hand, EGFR signaling mainly influences the Wnt pathway at the β-catenin level through a variety of approaches, including directly and indirectly modifying phosphorylation, stabilization, and transcriptional activity of β-catenin (41–45). A recent study found that MAPKs, including ERK1, p38α, and JNK1, directly phosphorylate LRP6 at the PPP(S/T)P motif and consequently enhance β-catenin transcriptional activity (23).
However, all those studies were performed within tumor cell lines. To our knowledge, we are the first to report the interaction of EGFR and β-catenin signaling pathways in chondrocytes both in vitro and in vivo. It should be pointed out that the level of β-catenin activation by EGFR signaling in primary chondrocytes is not as high as that by bona fide Wnt ligands. Although Wnt3a or BIO is able to induce nuclear translocation of β-catenin in almost 100% of primary chondrocytes, we only observed about 10–24% of chondrocytes with positive nuclear β-catenin staining. Moreover, in contrast with the results shown in tumor cells that EGFR signaling increases Topflash activity, we did not observe a similar increase in primary chondrocytes transfected with Topflash plasmid (data not shown), suggesting that in normal cells, EGFR only modifies a fraction of β-catenin available for transducing Wnt signaling. Nevertheless, this moderate regulation is sufficient for regulating the expression of genes related to cartilage degradation. Another explanation for the low efficiency of EGFR-induced β-catenin nuclear translocation could be that primary epiphyseal chondrocytes do not completely represent hypertrophic chondrocytes in vivo. Indeed, our data analyzing epiphyseal cartilage tissue revealed that EGFR activity is required to maintain the total β-catenin level within chondrocytes.
Wnt/β-catenin signaling is important for chondrocyte differentiation and hypertrophy (16, 46–49). Our previous studies showed that activation of β-catenin in mature chondrocytes stimulates hypertrophy, matrix mineralization, and Mmp9 and -13 expression (50). This is consistent with another in vivo study indicating that deletion of β-catenin causes delayed cartilage mineralization and reduced Mmp13 expression during POC formation at the embryonic stage (49). Moreover, Dao et al. (16) recently demonstrated that the cartilage-specific gain of function of β-catenin by using a Col2-Cre system promotes cartilage canal formation and enhances Mmp9, -13, and -14 expression, resulting in an early occurrence of the SOC, and that cartilage-specific loss of function of β-catenin has the opposite effects. Interestingly, we found that activating β-catenin and EGFR pathways had synergistic actions on Mmp9 and RANKL expression. Taken together, these results strongly suggest both overlapping and non-overlapping roles between EGFR and Wnt/β-catenin signaling in the regulation of cartilage ECM degradation. Further investigation into the cross-talk between these two important signaling pathways during endochondral ossification will shed new light on the skeletal development and management of cartilage-related diseases.
Acknowledgments
We thank Dr. Frank Beier at the University of Western Ontario and Dr. Maurizio Pacifici at the Children's Hospital of Philadelphia for providing critical comments and technical advice on this project and Dr. John S. Mort at Shriners Hospital for Children (Montreal, Canada) for kindly providing anti-MMP-13-generated type 2 collagen cleavage fragment and anti-MMP-generated aggrecan cleavage fragment antibodies.
This work was supported, in whole or in part, by National Institutes of Health Grants AR060991 (to L. Q.) and AR062908 (to M. E. I.) and Penn Center for Musculoskeletal Disorders Grant P30AR050950. This study was also supported by American Society for Bone and Mineral Research (ASBMR) Research Career Enhancement Award (to L. Q.).
- POC
- primary ossification center
- SOC
- secondary ossification center
- EGFR
- epidermal growth factor receptor
- RANKL
- receptor activator of NF-κB ligand
- MMP
- matrix metalloproteinase
- TRAP
- tartrate-resistant acid phosphatase
- CKO
- conditional knock-out
- qRT-PCR
- quantitative RT-PCR
- ECM
- extracellular matrix
- DMSO
- dimethyl sulfoxide
- P
- postnatal day
- BIO
- 6-bromoindirubin-3′-oxime.
REFERENCES
- 1. Kronenberg H. M. (2003) Developmental regulation of the growth plate. Nature 423, 332–336 [DOI] [PubMed] [Google Scholar]
- 2. Mackie E. J., Tatarczuch L., Mirams M. (2011) The skeleton: a multi-functional complex organ: the growth plate chondrocyte and endochondral ossification. J. Endocrinol 211, 109–121 [DOI] [PubMed] [Google Scholar]
- 3. Blumer M. J., Longato S., Fritsch H. (2008) Structure, formation and role of cartilage canals in the developing bone. Ann. Anat. 190, 305–315 [DOI] [PubMed] [Google Scholar]
- 4. Lee E. R., Lamplugh L., Davoli M. A., Beauchemin A., Chan K., Mort J. S., Leblond C. P. (2001) Enzymes active in the areas undergoing cartilage resorption during the development of the secondary ossification center in the tibiae of rats ages 0–21 days: I. Two groups of proteinases cleave the core protein of aggrecan. Dev. Dyn. 222, 52–70 [DOI] [PubMed] [Google Scholar]
- 5. Alvarez J., Costales L., López-Muñiz A., López J. M. (2005) Chondrocytes are released as viable cells during cartilage resorption associated with the formation of intrachondral canals in the rat tibial epiphysis. Cell Tissue Res. 320, 501–507 [DOI] [PubMed] [Google Scholar]
- 6. Sibilia M., Kroismayr R., Lichtenberger B. M., Natarajan A., Hecking M., Holcmann M. (2007) The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 75, 770–787 [DOI] [PubMed] [Google Scholar]
- 7. Miettinen P. J., Berger J. E., Meneses J., Phung Y., Pedersen R. A., Werb Z., Derynck R. (1995) Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376, 337–341 [DOI] [PubMed] [Google Scholar]
- 8. Sibilia M., Wagner E. F. (1995) Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 269, 234–238 [DOI] [PubMed] [Google Scholar]
- 9. Threadgill D. W., Dlugosz A. A., Hansen L. A., Tennenbaum T., Lichti U., Yee D., LaMantia C., Mourton T., Herrup K., Harris R. C., et al. (1995) Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 269, 230–234 [DOI] [PubMed] [Google Scholar]
- 10. Pines G., Köstler W. J., Yarden Y. (2010) Oncogenic mutant forms of EGFR: lessons in signal transduction and targets for cancer therapy. FEBS Lett. 584, 2699–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schneider M. R., Sibilia M., Erben R. G. (2009) The EGFR network in bone biology and pathology. Trends Endocrinol Metab. 20, 517–524 [DOI] [PubMed] [Google Scholar]
- 12. Zhang X., Siclari V. A., Lan S., Zhu J., Koyama E., Dupuis H. L., Enomoto-Iwamoto M., Beier F., Qin L. (2011) The critical role of the epidermal growth factor receptor in endochondral ossification. J. Bone Miner Res. 26, 2622–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee D., Cross S. H., Strunk K. E., Morgan J. E., Bailey C. L., Jackson I. J., Threadgill D. W. (2004) Wa5 is a novel ENU-induced antimorphic allele of the epidermal growth factor receptor. Mamm Genome. 15, 525–536 [DOI] [PubMed] [Google Scholar]
- 14. Zhang X., Tamasi J., Lu X., Zhu J., Chen H., Tian X., Lee T. C., Threadgill D. W., Kream B. E., Kang Y., Partridge N. C., Qin L. (2011) Epidermal growth factor receptor plays an anabolic role in bone metabolism in vivo. J. Bone Miner Res. 26, 1022–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chun J. S., Oh H., Yang S., Park M. (2008) Wnt signaling in cartilage development and degeneration. BMB Rep. 41, 485–494 [DOI] [PubMed] [Google Scholar]
- 16. Dao D. Y., Jonason J. H., Zhang Y., Hsu W., Chen D., Hilton M. J., O'Keefe R. J. (2012) Cartilage-specific β-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J. Bone Miner Res. 27, 1680–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koyama E., Young B., Nagayama M., Shibukawa Y., Enomoto-Iwamoto M., Iwamoto M., Maeda Y., Lanske B., Song B., Serra R., Pacifici M. (2007) Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134, 2159–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yasuhara R., Ohta Y., Yuasa T., Kondo N., Hoang T., Addya S., Fortina P., Pacifici M., Iwamoto M., Enomoto-Iwamoto M. (2011) Roles of β-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab. Invest. 91, 1739–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ueta C., Iwamoto M., Kanatani N., Yoshida C., Liu Y., Enomoto-Iwamoto M., Ohmori T., Enomoto H., Nakata K., Takada K., Kurisu K., Komori T. (2001) Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell Biol. 153, 87–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wuelling M., Vortkamp A. (2011) Chondrocyte proliferation and differentiation. Endocr Dev. 21, 1–11 [DOI] [PubMed] [Google Scholar]
- 21. Krane S. M., Inada M. (2008) Matrix metalloproteinases and bone. Bone. 43, 7–18 [DOI] [PubMed] [Google Scholar]
- 22. Edwards J. R., Mundy G. R. (2011) Advances in osteoclast biology: old findings and new insights from mouse models. Nat. Rev. Rheumatol. 7, 235–243 [DOI] [PubMed] [Google Scholar]
- 23. Červenka I., Wolf J., Mašek J., Krejci P., Wilcox W. R., Kozubík A., Schulte G., Gutkind J. S., Bryja V. (2011) Mitogen-activated protein kinases promote WNT/β-catenin signaling via phosphorylation of LRP6. Mol. Cell. Biol. 31, 179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bafico A., Liu G., Yaniv A., Gazit A., Aaronson S. A. (2001) Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat. Cell Biol. 3, 683–686 [DOI] [PubMed] [Google Scholar]
- 25. Fosang A. J., Golub S. B., East C. J., Rogerson F. M. (2013) Abundant LacZ activity in the absence of Cre expression in the normal and inflamed synovium of adult Col2a1-Cre; ROSA26RLacZ reporter mice. Osteoarthritis Cartilage. 21, 401–404 [DOI] [PubMed] [Google Scholar]
- 26. Usmani S. E., Pest M. A., Kim G., Ohora S. N., Qin L., Beier F. (2012) Transforming growth factor α controls the transition from hypertrophic cartilage to bone during endochondral bone growth. Bone. 51, 131–141 [DOI] [PubMed] [Google Scholar]
- 27. Renkiewicz R., Qiu L., Lesch C., Sun X., Devalaraja R., Cody T., Kaldjian E., Welgus H., Baragi V. (2003) Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 48, 1742–1749 [DOI] [PubMed] [Google Scholar]
- 28. Ortega N., Behonick D. J., Werb Z. (2004) Matrix remodeling during endochondral ossification. Trends Cell Biol. 14, 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inada M., Wang Y., Byrne M. H., Rahman M. U., Miyaura C., López-Otín C., Krane S. M. (2004) Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Natl. Acad. Sci. U.S.A. 101, 17192–17197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stickens D., Behonick D. J., Ortega N., Heyer B., Hartenstein B., Yu Y., Fosang A. J., Schorpp-Kistner M., Angel P., Werb Z. (2004) Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 131, 5883–5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vu T. H., Shipley J. M., Bergers G., Berger J. E., Helms J. A., Hanahan D., Shapiro S. D., Senior R. M., Werb Z. (1998) MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holmbeck K., Bianco P., Caterina J., Yamada S., Kromer M., Kuznetsov S. A., Mankani M., Robey P. G., Poole A. R., Pidoux I., Ward J. M., Birkedal-Hansen H. (1999) MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 99, 81–92 [DOI] [PubMed] [Google Scholar]
- 33. Zhou Z., Apte S. S., Soininen R., Cao R., Baaklini G. Y., Rauser R. W., Wang J., Cao Y., Tryggvason K. (2000) Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. U.S.A. 97, 4052–4057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alvarez J., Costales L., Serra R., Balbín M., López J. M. (2005) Expression patterns of matrix metalloproteinases and vascular endothelial growth factor during epiphyseal ossification. J. Bone Miner Res. 20, 1011–1021 [DOI] [PubMed] [Google Scholar]
- 35. Ortega N., Wang K., Ferrara N., Werb Z., Vu T. H. (2010) Complementary interplay between matrix metalloproteinase-9, vascular endothelial growth factor and osteoclast function drives endochondral bone formation. Dis. Model. Mech. 3, 224–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blumer M. J., Hausott B., Schwarzer C., Hayman A. R., Stempel J., Fritsch H. (2012) Role of tartrate-resistant acid phosphatase (TRAP) in long bone development. Mech. Dev. 129, 162–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu T., Li C. (2010) Convergence between Wnt-β-catenin and EGFR signaling in cancer. Mol. Cancer 9, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Civenni G., Holbro T., Hynes N. E. (2003) Wnt1 and Wnt5a induce cyclin D1 expression through ErbB1 transactivation in HC11 mammary epithelial cells. EMBO Rep. 4, 166–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schlange T., Matsuda Y., Lienhard S., Huber A., Hynes N. E. (2007) Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Res. 9, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ji H., Wang J., Nika H., Hawke D., Keezer S., Ge Q., Fang B., Fang X., Fang D., Litchfield D. W., Aldape K., Lu Z. (2009) EGF-induced ERK activation promotes CK2-mediated disassociation of α-catenin from β-catenin and transactivation of β-catenin. Mol. Cell 36, 547–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krejci P., Aklian A., Kaucka M., Sevcikova E., Prochazkova J., Masek J. K., Mikolka P., Pospisilova T., Spoustova T., Weis M., Paznekas W. A., Wolf J. H., Gutkind J. S., Wilcox W. R., Kozubik A., Jabs E. W., Bryja V., Salazar L., Vesela I., Balek L. (2012) Receptor tyrosine kinases activate canonical WNT/β-catenin signaling via MAP kinase/LRP6 pathway and direct β-catenin phosphorylation. PLoS One 7, e35826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee C. H., Hung H. W., Hung P. H., Shieh Y. S. (2010) Epidermal growth factor receptor regulates β-catenin location, stability, and transcriptional activity in oral cancer. Mol. Cancer 9, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu Z., Ghosh S., Wang Z., Hunter T. (2003) Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of β-catenin, and enhanced tumor cell invasion. Cancer Cell 4, 499–515 [DOI] [PubMed] [Google Scholar]
- 45. Yang W., Xia Y., Ji H., Zheng Y., Liang J., Huang W., Gao X., Aldape K., Lu Z. (2011) Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 480, 118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akiyama H., Lyons J. P., Mori-Akiyama Y., Yang X., Zhang R., Zhang Z., Deng J. M., Taketo M. M., Nakamura T., Behringer R. R., McCrea P. D., de Crombrugghe B. (2004) Interactions between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev. 18, 1072–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Day T. F., Guo X., Garrett-Beal L., Yang Y. (2005) Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 [DOI] [PubMed] [Google Scholar]
- 48. Enomoto-Iwamoto M., Kitagaki J., Koyama E., Tamamura Y., Wu C., Kanatani N., Koike T., Okada H., Komori T., Yoneda T., Church V., Francis-West P. H., Kurisu K., Nohno T., Pacifici M., Iwamoto M. (2002) The Wnt antagonist Frzb-1 regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev. Biol. 251, 142–156 [DOI] [PubMed] [Google Scholar]
- 49. Guo X., Mak K. K., Taketo M. M., Yang Y. (2009) The Wnt/β-catenin pathway interacts differentially with PTHrP signaling to control chondrocyte hypertrophy and final maturation. PLoS One. 4, e6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamamura Y., Otani T., Kanatani N., Koyama E., Kitagaki J., Komori T., Yamada Y., Costantini F., Wakisaka S., Pacifici M., Iwamoto M., Enomoto-Iwamoto M. (2005) Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J. Biol. Chem. 280, 19185–19195 [DOI] [PubMed] [Google Scholar]