

Background: Aberrant TNF-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling is observed in inflammation, autoimmune diseases, and cancers.
Results: An integrated computational and experimental study identified small molecule inhibitors of TWEAK-Fn14 interaction.
Conclusion: The TWEAK-Fn14 interaction is tractable and can be inhibited by small molecules.
Significance: This is the first evidence of small molecules targeting TWEAK-Fn14 signaling.
Keywords: Cancer, Cell Signaling, Cell Surface Receptor, Docking, Drug Development, Drug Discovery, Drug Screening, Protein-Protein Interactions
Abstract
Deregulation of the TNF-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling pathway is observed in many diseases, including inflammation, autoimmune diseases, and cancer. Activation of Fn14 signaling by TWEAK binding triggers cell invasion and survival and therefore represents an attractive pathway for therapeutic intervention. Based on structural studies of the TWEAK-binding cysteine-rich domain of Fn14, several homology models of TWEAK were built to investigate plausible modes of TWEAK-Fn14 interaction. Two promising models, centered on different anchoring residues of TWEAK (tyrosine 176 and tryptophan 231), were prioritized using a data-driven strategy. Site-directed mutagenesis of TWEAK at Tyr176, but not Trp231, resulted in the loss of TWEAK binding to Fn14 substantiating Tyr176 as the anchoring residue. Importantly, mutation of TWEAK at Tyr176 did not disrupt TWEAK trimerization but failed to induce Fn14-mediated nuclear factor κ-light chain enhancer of activated B cell (NF-κB) signaling. The validated structural models were utilized in a virtual screen to design a targeted library of small molecules predicted to disrupt the TWEAK-Fn14 interaction. 129 small molecules were screened iteratively, with identification of molecules producing up to 37% inhibition of TWEAK-Fn14 binding. In summary, we present a data-driven in silico study revealing key structural elements of the TWEAK-Fn14 interaction, followed by experimental validation, serving as a guide for the design of small molecule inhibitors of the TWEAK-Fn14 ligand-receptor interaction. Our results validate the TWEAK-Fn14 interaction as a chemically tractable target and provide the foundation for further exploration utilizing chemical biology approaches focusing on validating this system as a therapeutic target in invasive cancers.
Introduction
TWEAK5 is a multifunctional cytokine involved in many cellular activities, including proliferation, migration, differentiation, apoptosis, angiogenesis, and inflammation (1). TWEAK is a type II transmembrane protein that consists of an N-terminal cytoplasmic domain followed by a single transmembrane domain that is separated by a stalk region from the C-terminal tumor necrosis factor (TNF) homology domain (THD) (2, 3). Membrane TWEAK is processed by a protease of the furin family resulting in a soluble ligand containing the THD. The THD functions in ligand trimerization and receptor binding causing TWEAK to signal as a trimerized molecule (4, 5). Importantly, both membrane-bound and -soluble TWEAK (sTWEAK) proteins are fully functional and can mediate similar cellular signaling effects by binding to cellular receptors (6).
TWEAK acts by binding to the Fn14 receptor, the smallest member of the tumor necrosis factor receptor (TNFR) superfamily (1, 7). TWEAK-mediated Fn14 signaling triggers a wide range of physiological activities in cells and tissues, including blood clotting, cell proliferation, cell migration, inflammation, and angiogenesis (8, 9). The Fn14 receptor contains a single cysteine-rich domain (CRD) in the extracellular ligand-binding region and a short cytoplasmic tail possessing a single TNFR-associated factor-binding site (1, 7). Notably, TWEAK is the only known TNF superfamily member that can bind to Fn14. Site-directed mutagenesis has demonstrated that TWEAK binding to the Fn14 CRD requires evolutionarily conserved amino acid residues (Asp45, Lys48, and Met50) and all three of the predicted disulfide bonds (10). Optimal TWEAK-mediated activation of Fn14 is important for promoting productive tissue responses after injury, but excessive TWEAK-Fn14 activation can induce pathological tissue responses, leading to progressive damage and degradation (11). Overexpression of Fn14 has been reported in multiple cancers, including glioblastoma, breast, pancreatic, esophageal, lung, and liver carcinomas (3, 12–16). In glioblastoma, Fn14 mRNA and protein expression are unregulated in migratory cells in vitro and invading cells in vivo (17). Fn14 expression increases with increasing tumor grade with the highest expression observed in glioblastoma multiforme (grade IV). In contrast, the expression of Fn14 is minimal to absent in normal brain tissue. Moreover, TWEAK binding to Fn14 triggers glioma cell invasion and survival (17).
TWEAK-Fn14 signaling plays a key role in various disease states and therefore holds significant therapeutic potential as a novel molecular target for developing anti-cancer and anti-autoimmune therapeutic agents in humans. It has been shown that this interaction plays a pivotal role in various immunological conditions like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, renal injury, ischemic stroke, as well as cardiac dysfunction and failure (18–20). Several studies have confirmed the therapeutic potential of this pathway in human esophageal and pancreatic cancers (21), autoimmune disorders (22), muscle atrophy and injury (23), and chemokine-dependent inflammatory kidney disease (24). The ever increasing knowledge and data on various downstream reactions stimulated by TWEAK-Fn14 interaction has recently been compiled into a complete repository (25). This paves the way for identification of yet unknown components of the signaling pathways.
To date, there are five anti-TNF antibody-based drugs already on the market, and 16 out of ∼22 ligand/receptor pairs under clinical development, constituting one of the most successful classes of biological therapeutics (26). These protein-based therapeutics have some notable disadvantages, including problems associated with drug delivery, stability, and cost. However, very few small molecule inhibitors targeting TNFR family members have been identified. Known small molecule inhibitors for the TNFR family act by disrupting trimerization of their respective ligands, as is the case for TNFα (27) and CD40 (28). Benicchi et al. (29) have also focused on the development of a homogeneous time-resolved fluorescence assay for identification of small molecule inhibitors for the TWEAK-Fn14 interaction and reported the identification of hits at a rate of 0.007%. Currently, the potential therapeutic benefit of inhibiting key nodes of the TWEAK-Fn14 signaling pathway remains unclear and untapped due to the absence of small molecule tools to interrogate this pathway.
In this study, we initiated the discovery of small molecules targeting the TWEAK-Fn14 pathway by determining the molecular basis of the interaction between TWEAK and Fn14 and elucidating key structural elements of this interaction. The ultimate goal of this work is to employ the structural information on TWEAK-Fn14 binding to identify potential inhibitors of this interaction. The importance of the Fn14 CRD has been established utilizing an NMR solution structure of this domain and functional mutation studies (10). To further characterize the TWEAK-Fn14 interaction, six structural models of TWEAK were built based on experimental structures of low homology templates from the TNF superfamily. Protein-protein docking, followed by data-driven prioritization, yielded two promising TWEAK-Fn14 binding hypotheses. Site-directed mutagenesis confirmed one hypothesis providing a structural basis for target-based identification of small molecule inhibitors of the TWEAK-FN14 interaction. Validated models served as a basis for in silico library design. A targeted library of molecules was assembled and screened iteratively, leading to enrichment in activity for compounds with similar scaffolds. These results support the TWEAK-Fn14 interaction as a target of interest for the treatment of cancer, including glioblastoma and other deadly diseases.
EXPERIMENTAL PROCEDURES
Consensus Alignment and Model Building for TWEAK
The templates for homology modeling were selected from the RCSB PDB database (30). Consensus alignment based on three-dimensional structures was performed in MOE to obtain a structure-derived sequence alignment (version 2010.10, Chemical Computing Group Inc.) (31). All template structures were superimposed in three dimensions, with an initial main-chain atom root mean square deviation (r.m.s.d.) of 2.49 Å. In the corresponding sequence alignment, a consensus set of residues was defined based on two criteria as follows: 1) residue identity, retaining those residues with at least 50% of sequence identity per alignment column; and 2) r.m.s.d. of main-chain atoms. The former parameter was kept fixed, and the latter parameter was decremented, starting at the initial value of 2.49 Å and decreasing by increments of 0.5 Å until reaching 1 Å. At each step, the structural superimposition of the proteins was refined using only the consensus residues, and the sequence alignment was subsequently refined to reflect the structural alignment changes. The resulting sequence alignment was utilized as a fixed template to align the sequence of TWEAK. Homology modeling was performed using MOE with the options of disabling C- and N-terminal outgap modeling and enabling automatic disulfide bond detection. A maximum of 10 intermediate models were created. Model refinement was performed at a medium setting for both intermediate and final models. AMBER99 forcefield was used for all energy minimizations, and the GB/VI scoring method was used for model scoring. One final refined model was created per template.
Protein-Protein Docking Simulations
Protein-protein docking simulations were performed using well vetted methodologies implemented in ICM Pro Version 3.7–2b (2012), MolSoft LLC (32). The epitopes were selected on the basis of available biological knowledge of the interacting interfaces from April-TACI (PDB code 1XU1) and April-BCMA (PDB code 1XU2) complexes and knowledge from Fn14 residues required for binding, as established by Winkles and co-workers (10). For the receptor protein, pre-calculated grid maps were generated involving van der Waals, electrostatic, and desolvation terms using ECEPP/3 molecular mechanics force field (33). For the ligand protein, a number of starting conformations were sampled and optimized using a pseudo-Brownian Monte-Carlo-based method, followed by local energy minimizations (32). All the conformations accumulated were merged into a single conformational set compressed by comparison of the atomic coordinates and removal of geometrically similar conformations. The resulting conformations were further optimized by allowing flexibility of the ligand side chains. The interaction energy function uses the internal energy of the ligand and intermolecular energy based on the optimized potential maps. The multiple levels of optimization performed in this approach reduce the possibility of being trapped in local minima. A single docking run was performed for each receptor-ligand complex, and 30–40 poses were obtained for each individual docking run.
Virtual Library Preparation
The peptidomimetic set of ChemDiv (version of May 2011; 13,137 compounds) was prepared using LigPrep version 2.5 in Schrodinger Version 2011 (Schrodinger LLC, New York) by adding hydrogen atoms and calculating protonation states corresponding to pH 7.4. This resulted in generation of 21,682 structures. A conformational search was performed using ConfGen Standard search (default parameters), with ConfGen Version 2.3 in Schrodinger (34), generating a database of 145,995 conformations.
Pharmacophore-based Virtual Screening
Receptor-based pharmacophore generation was performed using Phase version 3.4 implemented in Maestro 9.3 of Schrodinger suite Version 2011 (35). The ligand was defined as the ensemble of TWEAK residues involved in inter-molecular interactions with Fn14. The pharmacophore features were then identified exhaustively for these interacting residues. The features that were not located at the direct protein-protein interaction interface were manually removed. Excluded volumes were included to capture the Fn14 receptor geometry when preparing the pharmacophore model; these were calculated using a scaling factor of 0.9. The conformational ligand database was interrogated for hits matching the generated pharmacophore hypothesis. Pharmacophoric points involving the TWEAK anchoring residue were required, and matching of other pharmacophoric points was set as optional.
Structure-based Virtual Screening
The protein preparation workflow of Maestro 9.3, Version 2011, was employed to prepare the Fn14 receptor by adding missing H-atoms and refining the structure using default parameters. A grid-enclosing box was centered at the centroid of the three binding site residues involved in TWEAK binding as indicated by mutation data from Winkles and co-workers (10), i.e. Asp45, Lys48, and Asp62. Structure-based virtual screening was performed with the conformational ligand database, and the ligands were kept flexible during the docking stage. A three-step docking process was executed as follows. 1) A first parsing was performed by docking of compounds with the fastest HTVS algorithm of Glide (Version 2011, Schrodinger LLC) (36) and scoring of compounds. 2) The top 50% of the virtual hits from step 1 were docked using the standard precision algorithm of Glide and were subsequently scored. 3) Lastly, 10% of the top scoring compounds of step 2 were re-docked using XP algorithm, scored, and considered as hits.
Cell Culture
The human astrocytoma cell line T98G and human HEK293 cells (American Type Culture Collection) were maintained in DMEM (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen) at 37 °C with 5% CO2. For assays involving TWEAK treatment, cells were cultured in reduced serum (0.5% FBS) for 16 h prior to TWEAK stimulation.
Expression Constructs
The coding sequence for the soluble form of TWEAK, designated sTWEAK, encoding amino acids Lys97–His249 was amplified by polymerase chain reaction and ligated in-frame either downstream of a 3× FLAG epitope in p3×FLAG-CMV (Sigma) or upstream of a 3× HA epitope in pcDNA3 (Invitrogen). The sTWEAK Y176D, Y176A, Y176F, and W231G variants were generated using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). All substitutions were verified by DNA sequence analysis. Expression constructs for in vitro transcription were generated using Gateway Technology (Invitrogen). The coding sequences for sTWEAK and sTWEAK variants were first amplified by polymerase chain reaction with oligonucleotide primers containing the appropriate aatB recombination sequences and the aatB-flanked PCR products transferred to the entry vector pDONR 221. Resulting pDONR clones were transferred to the T7-based in vitro transcription expression vector pANT7 (37). All proteins were expressed as epitope-tagged proteins.
Synthesis of sTWEAK and Mutant sTWEAK Proteins
sTWEAK and sTWEAK variant proteins were synthesized using a one-step coupled human in vitro transcription/translation (IVTT) kit (Pierce) or a one-step coupled rabbit IVTT kit (Promega) according to the manufacturers' instructions.
Double Sandwich ELISA
sTWEAK and sTWEAK variant proteins were synthesized as described above. The expression level of each IVTT-synthesized sTWEAK protein was analyzed by Western blot analysis and used in approximately equal amounts in the ELISA. To assay the binding of wild type sTWEAK and sTWEAK variants to Fn14, the human Fc fragment-tagged Fn14 extracellular domain (R&D Systems) was captured in an Immuno 96-microwell white plate by adding 100 μl of 0.025 μg/ml Fn14-Fc to wells coated with goat anti-human Fcγ fragment-specific monoclonal antibody. After capture, the wells were washed three times with Dulbecco's phosphate-buffered saline containing 0.05% Tween 20. Unbound sites were blocked by addition of 100 μl of blocking solution containing 0.05% Tween 20, 1% BSA, and 3% normal goat serum in Dulbecco's PBS for 1 h at room temperature. 0.01 μl of sTWEAK or sTWEAK variant made with the human IVTT kit or 2 μl of sTWEAK or sTWEAK variant made with the rabbit IVTT kit was diluted in 100 μl of sample diluent (Dulbecco's PBS + 1% BSA + 0.005% Tween 20) and then added to the wells for 2 h followed by addition of 100 μl of 50 ng/ml biotinylated TWEAK detection antibody (R & D Systems) in sample diluent. Following incubation for additional 2 h at room temperature, wells were washed three times with Dulbecco's PBS containing 0.05% Tween 20, and bound biotinylated TWEAK antibody was detected by incubation with an HRP-conjugated streptavidin. The total luminescent signal was obtained using Femto ELISA kit (Pierce) and compared with the standard curve signal obtained from the binding of 0–4000 pg/ml recombinant TWEAK (PeproTech) to Fn14-Fc. Using 5-parameter logistic curve fitting for standard curve analysis (Sigmaplot 11.0, Systat Software Inc.), binding of sTWEAK or TWEAK variant to Fn14-Fc was determined. The data represent that observed for at least four replicate assays.
The small molecule screening was performed using the ELISA described above with minor modifications. Briefly, after capturing Fn14-Fc in the microwell plate, 80 μl of drug solution in sample diluent was added to desired wells and incubated for 2 h at room temperature. Subsequently, 20 μl of 2500 pg/ml (5×) TWEAK was added to each well to achieve a final TWEAK concentration of 500 pg/ml and incubated for an additional 2 h at room temperature. Bound TWEAK was detected as described in the protocol above. All small molecule inhibitors were screened at a 25 μm final concentration (final DMSO concentration of 0.125%) in duplicate. Cycloheximide, a nonspecific small molecule, at 25 μm concentration was used as negative control. The anti-Fn14 antibody ITEM-4, added at 2.5 μg/ml, was used as a positive control for complete blockade of TWEAK binding. Reduction in TWEAK binding due to compound addition or controls was calculated by using standard curve (separate standard curve was obtained for every screening plate).
Nondenaturing/Native Gel Electrophoresis
Native gel electrophoresis kit and reagents were purchased from Invitrogen, and electrophoresis and Western blotting were performed according to the manufacturer's protocol. Briefly, IVTT protein lysates (1 μl) were mixed with 1 μl of 10% n-dodecyl β-d-maltoside, 0.5 μl of 5% NativePAGETM G-250 additive, 2.5 μl of 4× NativePAGETM sample buffer (4×), and deionized water to make the total volume to 10 μl. Electrophoresis was performed for 2 h at 16 mA at room temperature using NativePAGETM Novex® 4–16% BisTris gels. Calibration was achieved by separation of NativeMarkTM protein standards of known molecular masses. After gel electrophoresis, proteins were transferred to PVDF membrane for immunoblotting with an anti-FLAG antibody (Sigma).
NF-κB Luciferase Reporter Assay
The capacity of sTWEAK or sTWEAK variants to activate Fn14 signaling was evaluated using engineered reporter cell lines that express luciferase upon NF-κB activation. Two reporter cell lines were utilized for these experiments. HEK293 NF-κB luciferase (courtesy of Dr. Jeff Winkles) were generated by transfecting HEK293 cells with a reporter plasmid containing five copies of a consensus NF-κB-binding site upstream of a minimal CMV promoter driving expression of firefly luciferase. The second cell line, designated HEK293 NF-κB luciferase Fn14 FL, was generated by stably transfecting the HEK293 NF-κB luciferase cell line with full-length Fn14. To assay the binding of sTWEAK or sTWEAK variants to Fn14, reporter cells were seeded in tissue culture-treated white 96-well plates at 1 × 104 cells/well in 80 μl Opti-MEM media (Invitrogen) and incubated for 48 h at 37 °C. After 48 h incubation, 20 μl of 5× purified recombinant TWEAK (PeproTech.; 150 ng/ml) in 1 mg/ml BSA in PBS was added to each well and incubated for 5 h at 37 °C as positive control. Similarly, equivalent amounts of sTWEAK or sTWEAK variant prepared via IVTT as determined using ELISA described above was added in 20 μl. An IVTT solution lacking a cDNA template in 1 mg/ml BSA in PBS and 1 mg/ml BSA in PBS alone was used as an additional control. At the end of a 5-h incubation, the luminescent signal was determined using Bright-Glo assay kit (Promega, Madison, WI) according to the manufacturer's instructions.
Small molecules that demonstrated ≥15% inhibition of TWEAK binding to Fn14 in the ELISA screen were validated using the cell-based NF-κB luciferase reporter assay with minor modifications. Briefly, Fn14-NF-κB-Luc reporter cells (Fn14 overexpressing HEK293 NF-κB luciferase cells) were seeded in tissue culture-treated white 96-well plates at 1 × 104 cells/well in 80 μl of Opti-MEM media (Invitrogen) and incubated for 48 h at 37 °C. After 48 h of incubation, 10 μl of the drug solution (200 μm) in DMSO was added to the designated wells at a final concentration of 20 μm. After 1 h of drug incubation at 37 °C, 10 μl of 10× purified recombinant TWEAK (PeproTech; 300 ng/ml) in 1 mg/ml BSA in PBS was added to each well and incubated for 5 h at 37 °C. DMSO alone was used as a negative control, and anti-TWEAK antibody suspended in DMSO was used as a positive control for the assay. Luminescent signal was determined using Bright-Glo assay kit (Promega, Madison, WI) according to the manufacturer's instructions and normalized to negative control. A counter-screen assay was carried out using TNFα to stimulate NF-κB activity in NF-κB-Luc reporter cells (HEK293 NF-κB luciferase cells). The counter-screen assay was performed similar to the drug screening assay described above, except 10 μl of 10× purified recombinant TNFα (R & D Scientific; 300 ng/ml) in 1 mg/ml BSA in PBS was added to each well instead of TWEAK for NF-κB activation. Small molecules, which showed inhibition of the luciferase signal following TWEAK stimulation but not after TNFα stimulation, were further validated by performing a dose-response analysis. The selected small molecule inhibitor was tested at concentrations ranging from 0.75 to 250 μm for its ability to suppress TWEAK-induced NF-κB activity in Fn14-overexpressing HEK293 NF-κB luciferase cells compared with its ability to suppress TNFα-induced NF-κB activity in HEK293 NF-κB luciferase cells. IC50 values for the dose-response curve were determined using the curve fitting functionality of GraphPad Prism software.
Molecular Interaction Measurement by Surface Plasmon Resonance Assay
The binding affinity of L524-0366 to TWEAK and to Fn14-Fc was determined on a BIAcore T100 optical biosensor (GE Healthcare) at 25 °C at the Arizona Proteomics Consortium (University of Arizona, Tucson). Recombinant human TWEAK (20 μg/ml in 10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.05% Tween 20) or recombinant human Fn14-Fc (20 μg/ml in 10 mm NaOAc, pH 4.0) was covalently coupled to separate flow cells on a BIAcore CM5 sensor chip using standard amine coupling chemistry as per manufacturer's protocol. Final immobilization levels were 6000 relative units for TWEAK and 13,000 relative units for Fn14-Fc. The first flow path of the chip was treated with the same coupling and blocking reagents without protein and was used as a reference for each binding cycle. Functionality of the TWEAK and the Fn14-Fc sensor surfaces were verified by injecting Fn14-Fc and TWEAK over them respectively. Serial dilutions of L524-0366 or cycloheximide (control) from 0 to 50 μm were made in running buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 1% DMSO). The compounds were injected over the TWEAK and Fn14-Fc sensor surfaces for 60 s at a flow rate of 45 μl/min. Running buffer was injected for 5 min at a flow rate of 45 μl/min to dissociate bound drug molecules from the sensor surface. The fluidics were washed with 50% DMSO after each sample injection, and to minimize sample carryover, a buffer wash step was included after every binding cycle. A DMSO calibration curve was used to correct for any bulk responses due to mismatches between sample and running buffer (38). Equilibrium dissociation constants for the small molecules were calculated by fitting the double reference subtracted data to Req = ((CRmax)/(KD + C)) + RI, where RI is the bulk refractive index contribution.
Cell Migration Assay
The effect of pharmacological inhibition of TWEAK/Fn14 signaling on glioma cell migration was analyzed using a modified Boyden chamber (Neuroprobe, Cabin John, MD) as described previously (39, 40). Each well contains an 8-μm pore size Nucleopore filter that had been coated with 50 μg/ml PureCol® (bovine collagen) (Advanced Biomatrix, Poway, CA). T98G glioma cells were treated with selected drug compound for 1 h and then seeded at 4.8 × 104 cells in 100 μl of DMEM with 0.1% bovine serum assay medium to the top well of the chamber. TWEAK was added to the lower wells of the chamber using DMEM with 0.1% bovine serum albumin as assay medium. After incubation for 5 h at 37 °C, nonmigrated cells were scraped off the upper side of the filter, and filters were stained with 4′,6-diamidino-2-phenylindole (DAPI). Nuclei of migrated cells were counted in five high power fields with a ×20 objective. Values were assessed in triplicate.
Cytotoxicity Assay
Cytotoxic effects of drugs on glioma cells were analyzed by quantifying the ATP, an indicator of metabolically active cells. Briefly, glioma cells were seeded in tissue culture-treated white 96-well plates at 3 × 103 cells/well in 80 μl of Opti-MEM media (Invitrogen) and incubated for 24 h at 37 °C. After 24 h of incubation, 20 μl of 5× drug solution at required concentration in Opti-MEM was added to each well and incubated for 72 h at 37 °C. The Opti-MEM/DMSO mixture without drug was used as the negative control and 20 μm staurosporine was used as the positive control. At the end of 72 h of incubation, the number of viable cells were quantified by using CellTiter-Glo assay kit (Promega, Madison, WI) according to the manufacturer's instructions. The luminescence signal measured was normalized to negative control to determine % cell viability.
RESULTS AND DISCUSSION
Fn14 Receptor Selection
An NMR structure of the Fn14 CRD is available in PDB (code 2RPJ) (41). All 20 models captured in the structure were visually inspected to assess areas of structural flexibility in the putative receptor-binding site. This analysis revealed a highly conserved core region (Ala34–Ala69) with very few flexible side chains. This rigid core includes the residues Asp45, Lys48, and Asp62 previously identified as required for TWEAK binding by Winkles and co-workers (10) and delineating the protein-protein binding interface. Importantly, the side chain of Arg58, located in close proximity of the putative protein-protein interface, presents a high degree of flexibility. We therefore hypothesized that Arg58 could potentially act as a switch that opens the binding groove. Models 1 and 17 (Fig. 1) capture two extreme closed and open geometries, respectively. In model 17, the side chain of Arg58 points toward the solvent that reveals a potential binding site on the surface of the Fn14 CRD. Conversely, in model 1, the side chain of Arg58 obscures that potential binding site. Both configurations of the receptor were considered as initial receptor models for protein-protein docking, with the understanding that the open configuration captured in model 17 is likely to be more favorable than the closed configuration captured in model 1. Thus, we compared 20 NMR models of Fn14 for side chain flexibility, and two were selected for further consideration.
FIGURE 1.
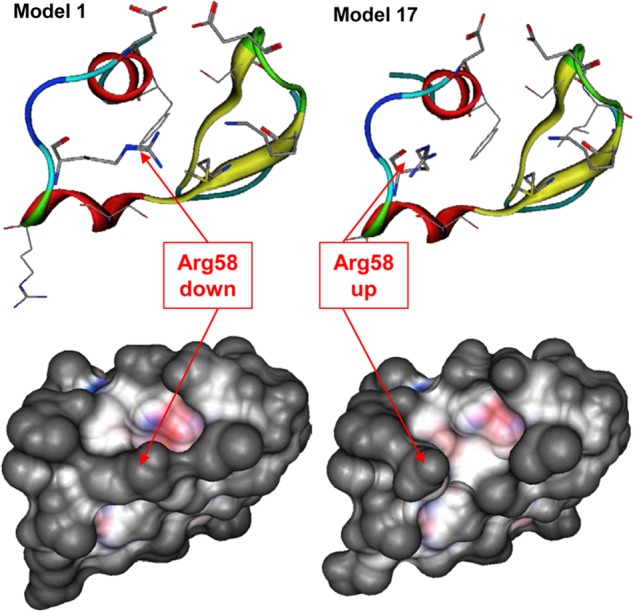
Flexibility of the Arg58 side chain in the Fn14 CRD. The position of Arg58 side chain in models 1 and 17 of the Fn14 CRD NMR structure defines closed and open states of the Fn14-binding site. Top views, the Fn14 CRD main chain is represented as a ribbon and key side chains are shown as sticks. Bottom views, the van der Waals surface of both Fn14 CRD models are represented, illustrating the changes in geometries associated with the different Arg58 side chain positions.
Homology Modeling of TWEAK
Homology modeling of TWEAK was undertaken in the absence of an experimental crystal structure. The C-terminal extracellular domain of TWEAK was predicted to have a β-pleated sheet structure based on structures of other members of the TNF superfamily (42). Several other members of the TNF superfamily are characterized by experimental structures in PDB, but the low sequence identity with TWEAK limits their selection as a direct template for homology modeling. We overcame this limitation by using consensus-based structural overlay of the template structures available to derive a quality multiple sequence alignment and by employing this alignment for building homology models (Fig. 2). The advantage of preferring structural alignment over sequence alignment is due to the fact that structural conservation predominates sequence conservation and is closer to function (43). Six members of TNF superfamily associated with an experimental structure were used as template structures. These include TNF ligand superfamily member 11 (PDB code 1S55), TNF-β (PDB code 1TNR), TNF ligand superfamily member 13 (PDB code 1XU2), TNFα (PDB code 2E7A), TNF superfamily ligand TL1A (PDB code 2RJL), and TNF ligand superfamily member 9 (PDB code 2X29). Because the residue identity of TWEAK with these templates is very low (12.8–17.9%) (Table 1), we first obtained a structure-derived sequence alignment of the six template proteins, which was then used to align the sequence of TWEAK. TWEAK sequence aligned onto that of each individual template protein was considered the starting point of an extensive homology modeling campaign, ultimately leading to six TWEAK homology models as described under “Experimental Procedures.”
FIGURE 2.
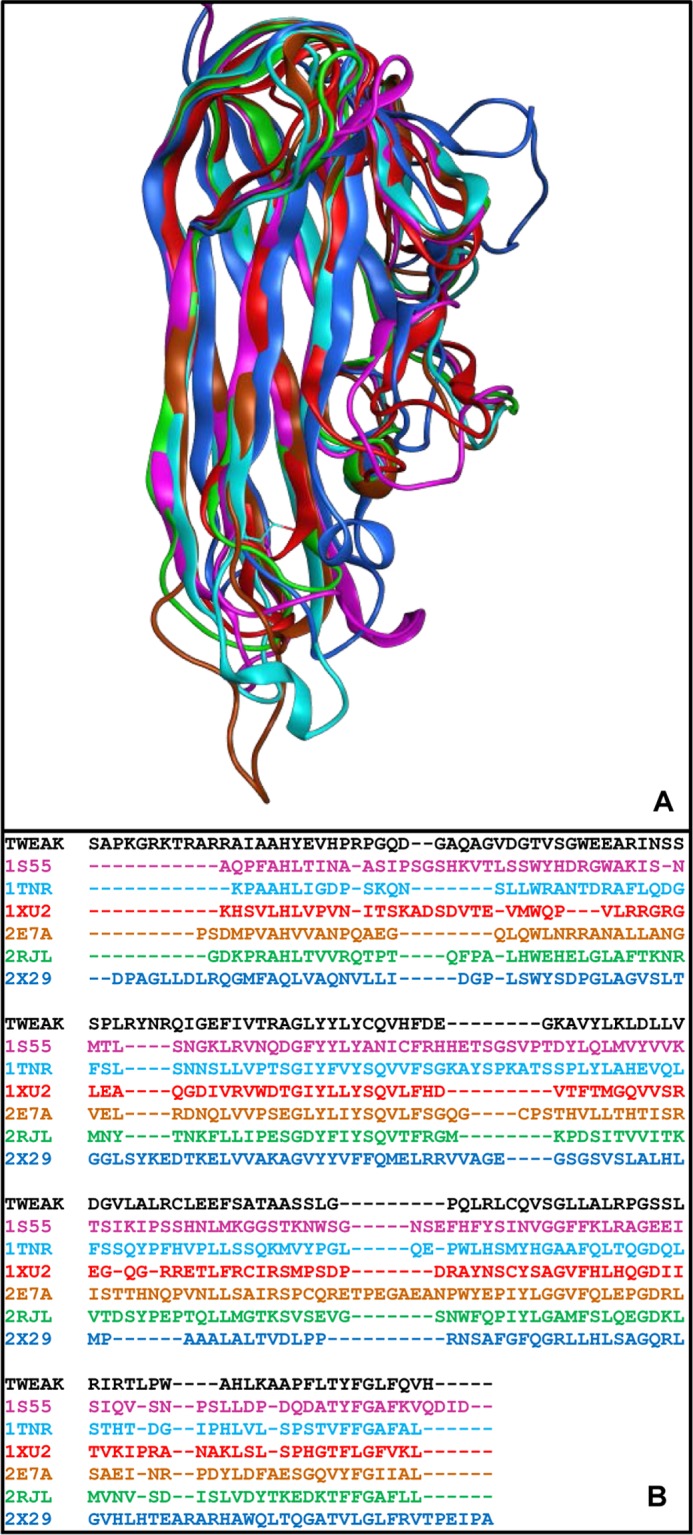
Homology modeling of TWEAK. A, consensus structural alignment performed in MOE (Chemical Computing Group) for six members of TNF superfamily. B, TWEAK sequence (black) aligned to the consensus structure-derived sequence alignment of the six template TNF ligands. The sequence alignment of the six template ligands, identified by their PDB code and color-coded based on the chain color in A, was derived from the structure alignment. The TWEAK sequence was aligned onto the multiple alignment of the six templates.
TABLE 1.
Similarity matrix of the percentages of sequence identity for the six template structures (identified by their PDB code) and TWEAK, based on the structure-derived sequence alignment generated by structural consensus in MOE
| Protein | TWEAK | 1S55 | 1TNR | 1XU2 | 2E7A | 2RJL | 2X29 |
|---|---|---|---|---|---|---|---|
| TWEAK | 16.7 | 19.4 | 15.3 | 18.0 | 14.2 | 16.7 | |
| 1S55 | 16.7 | 22.7 | 16.8 | 22.0 | 28.4 | 17.9 | |
| 1TNR | 17.9 | 21.2 | 21.9 | 32.0 | 32.6 | 14.7 | |
| 1XU2 | 13.5 | 14.7 | 20.8 | 22.7 | 17.0 | 10.9 | |
| 2E7A | 17.3 | 21.2 | 33.3 | 24.8 | 29.1 | 13.5 | |
| 2RJL | 12.8 | 25.6 | 31.9 | 17.5 | 27.3 | 15.4 | |
| 2X29 | 16.7 | 17.9 | 16.0 | 12.4 | 14.0 | 17.0 |
TWEAK-Fn14 Binding Mode Prediction via Protein-Protein Docking
Prior to docking of the TWEAK protein to the Fn14 CRD, benchmarking was performed to parameterize the algorithms and verify the predictive ability of the ICM-Pro algorithms for TNFR and their ligands. For that purpose, we used the crystal structure of the April-BCMA complex (PDB code 1XU2) (44) as a model system. In a first step, the ligand (BCMA) was translated away from the complex and rotated, but the conformation was unchanged. The ICM-Pro protein-protein docking algorithm identified multiple poses, with the lowest energy solution corresponding to the geometry of the crystal structure. In a second step, the ligand was moved away from the binding site, and the side chains of the interfacing residues were randomized to evaluate the ability to identify a complex in the ensemble of solutions that approaches the experimental complex. We found that the 7th best solution rank-ordered by interaction energy was consistent with the actual binding mode. In light of these outcomes, we concluded that the ICM-Pro algorithm, as parameterized, was suitable to identify plausible poses of TNF ligands bound to their receptors in an ensemble of low energy solutions.
On this basis, protein-protein docking calculations were performed for two Fn14 structures and six TWEAK homology models to predict TWEAK-Fn14 binding modes. A total of 12 protein-protein docking simulations were performed, producing hundreds of poses. Initial binding epitopes required to start the protein-protein docking calculation were defined by similarity to the known interacting interfaces from April-TACI and April-BCMA complexes. Each simulation generated between 30 and 40 ligand-receptor poses. TWEAK-Fn14 candidate solutions were analyzed and prioritized following a data-driven decision-making process as summarized in Fig. 3. 1) Poses with the predicted TWEAK-anchoring residue lying at the putative TWEAK trimerization interface were excluded. 2) Poses coherent with mutation data by Winkles and co-workers (10) were retained. 3) Candidate solutions with the TWEAK-predicted anchoring residue being Phe, Trp, or Tyr were prioritized as they have been reported to be some of the most frequently observed anchoring residues in protein-protein interactions (45). Following this process, a total of four models with a Tyr anchor and three models with a Trp anchor were identified from all protein-protein docking runs. 4) The remaining candidate complexes were individually examined using the internet-based webserver AnchorQuery (46) to further analyze the anatomies of the predicted protein-protein interaction interfaces. The program leverages the concept of amino acid residues as anchors that bury a large amount of solvent-accessible surface area at the protein-protein interface, and it calculates the changes in a solvent-accessible surface area upon binding of each side chain at the interface. This information is utilized to rank the residues, with the top ranking ones being likely the anchoring residue of the interaction based on solvent-accessible surface area loss upon complex formation. We retained those solutions for which the anchoring residues identified in step 3 ranked first in this methodology. In summary, this process identified four models with two putative TWEAK residues, Tyr176 and Trp231, anchoring the ligand to Fn14 CRD (Fig. 4).
FIGURE 3.
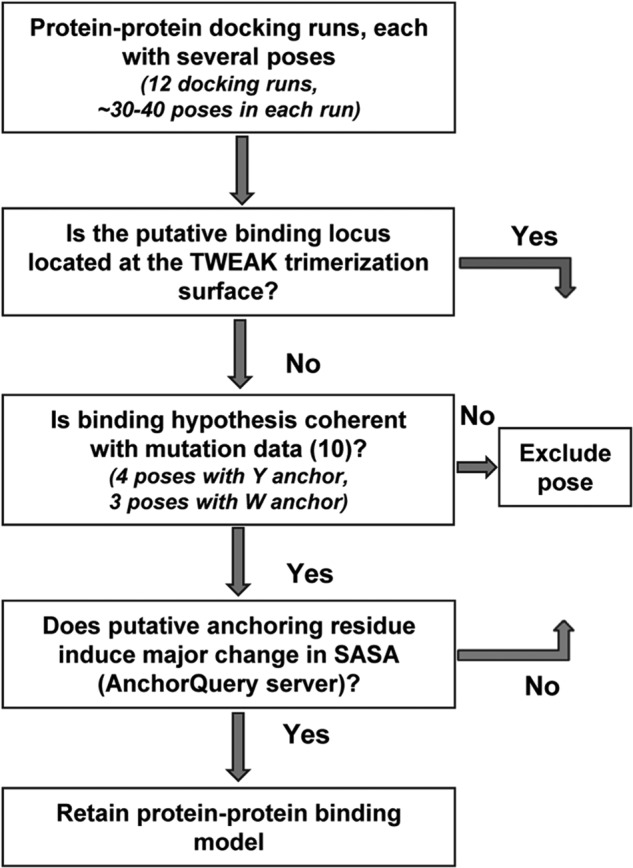
Data-driven decision-making workflow for prioritization of protein-protein docking results. Protein-protein docking of six TWEAK models was performed with two Fn14 models, leading to 12 protein-protein docking runs and hundreds of poses. Poses where the putative binding locus is located at the TWEAK trimerization interface were not considered as valid. Poses with the mutational validated residues Asp45, Lys48, Met50, and Asp62 present at the binding interface were retained. Finally, the AnchorQuery server was used to rank-order the anchoring residues, and those models with Phe, Tyr, or Trp resulting in solvent-accessible surface area loss upon complex formation were retained.
FIGURE 4.

Prioritized binding models for Fn14-TWEAK. A and B, three-dimensional representations of the two TWEAK-Fn14 prioritized models with TWEAK Tyr176 serving as an anchor residue to bind Fn14 CRD. C and D, three-dimensional representations of the two prioritized models with TWEAK Trp231 serving as an anchor to bind Fn14 CRD. A van der Waals surface is overlaid on the Fn14 CRD in all panels.
Experimental Confirmation of Binding by TWEAK Mutagenesis
Binding mode prediction via protein-protein docking identified TWEAK Tyr176 or Trp231 as plausible anchoring amino acid residues mediating the TWEAK-Fn14 interaction. To experimentally validate models generated from protein-protein docking calculation and to determine which predicted residue is critical for TWEAK binding to the Fn14, we performed a double sandwich ELISA to analyze the binding of sTWEAK and sTWEAK variants to Fn14. Failure of any sTWEAK variant to bind to Fn14 would be indicated by reduction in the chemiluminescent signal. Immunoblot analysis using anti-HA antibody indicated that sTWEAK HA, sTWEAK Y176D HA, and sTWEAK W231G HA were synthesized equivalently using the rabbit IVTT system and were used for the ELISA (Fig. 5, top). In the ELISA, substitution of amino acid Tyr176 significantly reduced TWEAK binding to Fn14, and substitution of amino acid Trp231 significantly increased the TWEAK binding to the Fn14 (Fig. 5, bottom), suggesting that Tyr176 is a critical amino acid residue for binding of TWEAK to Fn14. Based on these results, all further experiments utilized sTWEAK with substitutions at residue Tyr176.
FIGURE 5.
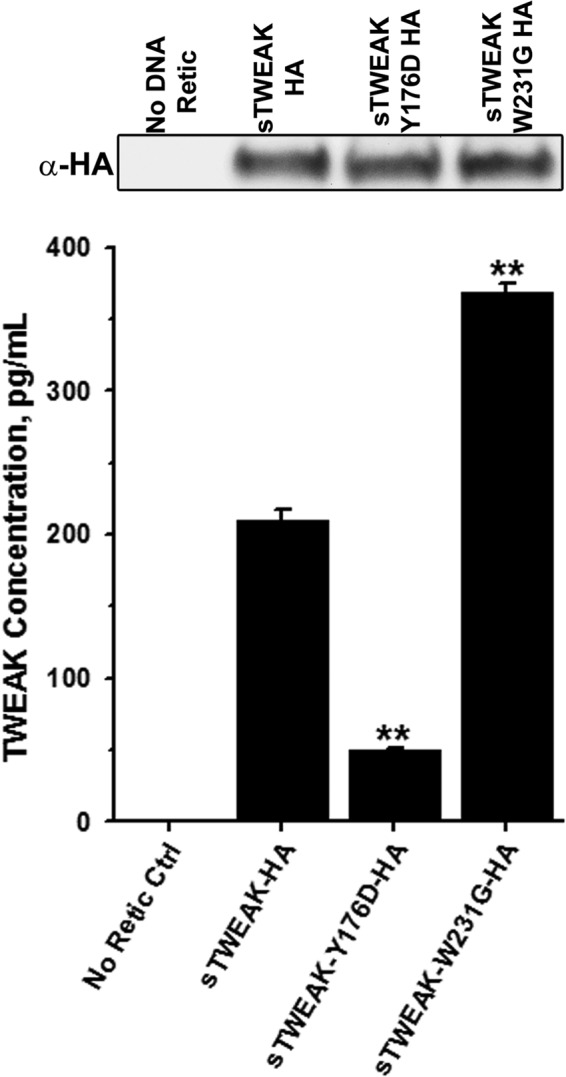
Substitution of TWEAK Tyr176 disrupts the Fn14-TWEAK interaction. Top, immunoblot analysis with anti-HA antibody showing equal expression of sTWEAK, sTWEAK Y176D, or sTWEAK W231G in IVTT lysates. No DNA, IVTT without added cDNA template. Bottom, double-sandwich ELISA showing binding of sTWEAK, or indicated sTWEAK variants to Fn14 (**, p < 0.001). Concentrations of sTWEAK and sTWEAK variants were determined using standard curve obtained with recombinant TWEAK. Ctrl, control.
Nondenaturing/Native Gel Electrophoresis
The results of the ELISA indicated that substitution of TWEAK Tyr176 disrupted TWEAK binding to Fn14. To examine whether substitution of Tyr176 caused significant changes in the secondary, tertiary, or quaternary structure of TWEAK, we performed nondenaturing gel electrophoresis comparing sTWEAK and sTWEAK Tyr176 variants. As shown in Fig. 6, sTWEAK, sTWEAK Y176A, and sTWEAK Y176F exhibited similar bands between 66 and 146 kDa when immunoblotted with an anti-FLAG antibody, indicative of sTWEAK trimer. IVTT lysate generated with a cDNA for GFP was used as a control and did not show any specific anti-FLAG staining. These results demonstrate that the conservative substitutions of Y176A or Y176F did not appear to cause significant changes in structure and surface charge relative to sTWEAK, suggesting that the sTWEAK Tyr176 variants also exist in a homotrimeric state similar to wild type sTWEAK (19, 47).
FIGURE 6.
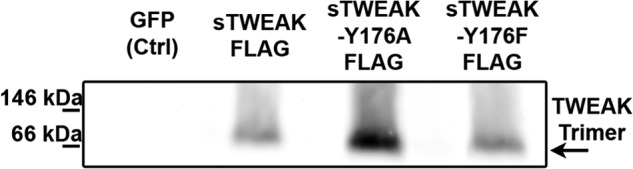
Substitution of TWEAK Tyr176 does not significantly affect TWEAK structure or surface charge. IVTT lysates containing FLAG epitope-tagged sTWEAK, sTWEAK Y176A, or sTWEAK Y176F were resolved by native gel electrophoresis. IVTT lysate with GFP cDNA as template was run as negative control. Lysates were immunoblotted with anti-FLAG monoclonal antibody.
Luciferase Induction Assay
The ELISA indicated that substitution of Tyr176 significantly reduced sTWEAK binding to Fn14. Furthermore, native gel electrophoresis further indicated that conservative substitutions of Tyr176 did not cause obvious alterations in sTWEAK structure and surface charge. However, neither of these assays can accurately predict the effect of the sTWEAK variants on Fn14 cellular signaling. TWEAK binding to the Fn14 receptor results in NF-κB phosphorylation (48). Therefore, we examined the ability of sTWEAK and the sTWEAK Tyr176 variants to initiate Fn14 signaling using cells expressing an NF-κB luciferase reporter. Stimulation of HEK293 cells expressing the NF-κB luciferase reporter with sTWEAK Y176A or sTWEAK Y176F resulted in luciferase expression that was 87 and 100% less, respectively, than cells stimulated with sTWEAK (Fig. 7A). Cells treated with IVTT lysate without cDNA template or with recombinant TWEAK served as controls. Immunoblotting of IVTT lysates (Fig. 8) ensured equivalent amounts of sTWEAK and sTWEAK Tyr176 variants were added to the cells. Additionally, induction of luciferase expression in HEK293 NF-κB luciferase cells that stably overexpress full-length Fn14 was 98% less following stimulation with sTWEAK Y176A or sTWEAK Y176F relative to cells stimulated with sTWEAK (Fig. 7B). Together, these results substantiate the results of the ELISA and indicate that substitution of TWEAK Tyr176 abrogates TWEAK binding to cellular Fn14 and Fn14 signaling. These results are consistent with those published very recently by Pellegrini et al. (49) in a structural biology study focused on the structural characterization of the Fn14-TWEAK binding interface in two different species to investigate the evolution of structural conservation in the cysteine-rich domains of the TNF receptor family.
FIGURE 7.
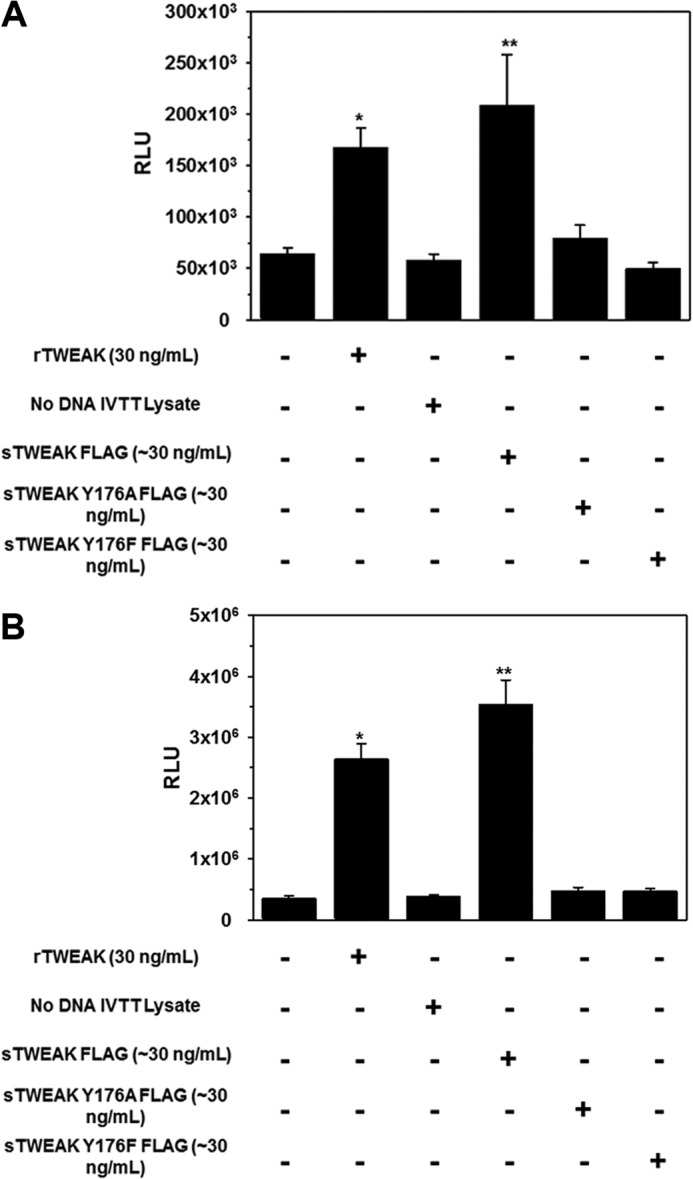
Substitution of TWEAK Tyr176 inhibits Fn14-mediated NF-κB activation. A, HEK293 expressing an NF-κB luciferase reporter were treated with rTWEAK or IVTT-synthesized sTWEAK, sTWEAK Y176A, or sTWEAK Y176F. After 5 h of incubation, luciferase generation was detected with Bright-Glo reagent. *, p < 0.05; **, p < 0.01. B, HEK293 NF-κB luciferase cells overexpressing full-length Fn14 were treated with rTWEAK or IVTT synthesized sTWEAK, sTWEAK Y176A, or sTWEAK Y176F. After 5 h of incubation, luciferase generation was detected with Bright-Glo reagent. RLU, relative luminescence units. *, p < 0.005; **, p < 0.001.
FIGURE 8.
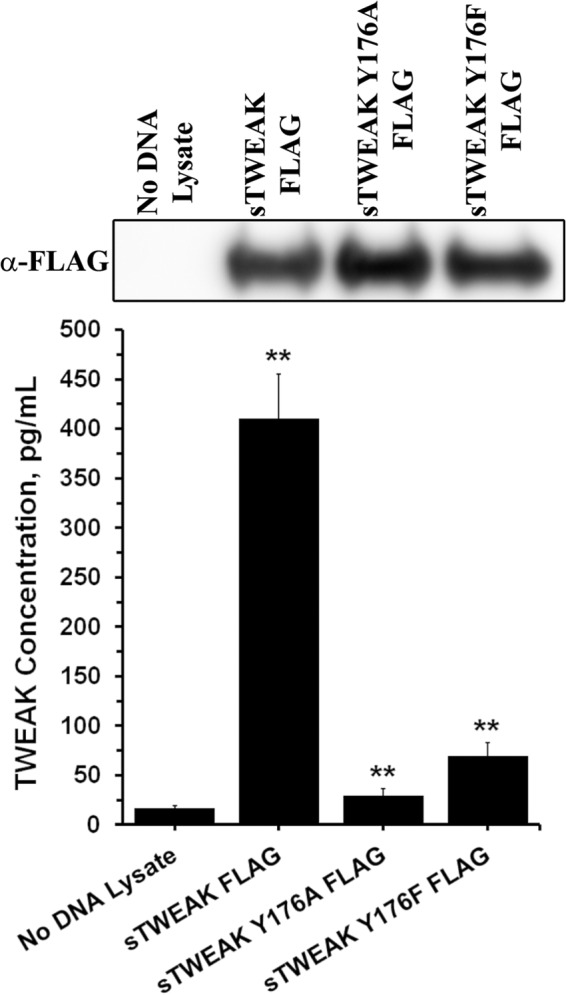
Substitution of TWEAK Tyr176 disrupts Fn14-TWEAK interaction. Top, immunoblot analysis of IVTT lysates using anti-FLAG antibody showing equal expression of sTWEAK and sTWEAK Tyr176 variants. Bottom, double sandwich ELISA depicting binding of sTWEAK and TWEAK Tyr176 variants to Fn14. sTWEAK shows significantly higher binding as compared with no DNA lysate control, and TWEAK Tyr176 variants show significantly lower binding as compared with sTWEAK (**, p < 0.001).
In Silico Identification of Small Molecule Inhibitors of TWEAK-Fn14 Interaction
The work presented above predicted plausible binding modes of the TWEAK-Fn14 association involving TWEAK residue Tyr176, and it was validated experimentally. These data provided a structural basis to enable further examination of the chemical tractability of the system with the goal to determine whether the characterized TWEAK-Fn14 interaction would be of utility as a therapeutic target for small molecule discovery. To pursue that goal, the structural TWEAK-Fn14 complexes predicted by protein-protein docking involving the Tyr176 anchoring residue of TWEAK to Fn14 were used as starting point for virtual screening of a library of commercially available small molecules. The peptidomimetic set of ChemDiv (13,137 compounds, Version 05.2011) was selected for this study and pre-processed as described under “Experimental Procedures.” A two-pronged virtual screening workflow intersecting the results of a ligand-biased pharmacophore-based and an unbiased structure-based approach was followed, as illustrated in Fig. 9. In the pharmacophore-based virtual screening approach, the two TWEAK-Fn14 complexes predicted by protein-protein docking (Tyr176 anchor models) served as a structural basis to generate two distinct pharmacophore hypotheses, respectively, composed of 10 and 13 sites (Table 2). Virtual hits were defined as those compounds matching the required pharmacophoric sites of the model and matching three of the optional sites. The hit lists from both models were combined, leading to a set of 7,308 compounds. In the parallel structure-based approach, a sequential high throughput docking workflow was followed in three steps, as described under “Experimental Procedures.” The best NMR model (model 1) was prepared and optimized, leading to a change in receptor side-chain orientations. This optimized geometry of the Fn14 CRD was utilized as a receptor for structure-based virtual screening, to eliminate potential bias from protein-protein docking. The set of solutions included 498 compounds, after removal of redundancies. The intersection of this hit list, with that obtained via pharmacophore-based virtual screening, produced an ensemble of 296 compounds considered as virtual hits. Among these, 60 were readily available in our internal compound collection and were advanced to screening in the ELISA.
FIGURE 9.
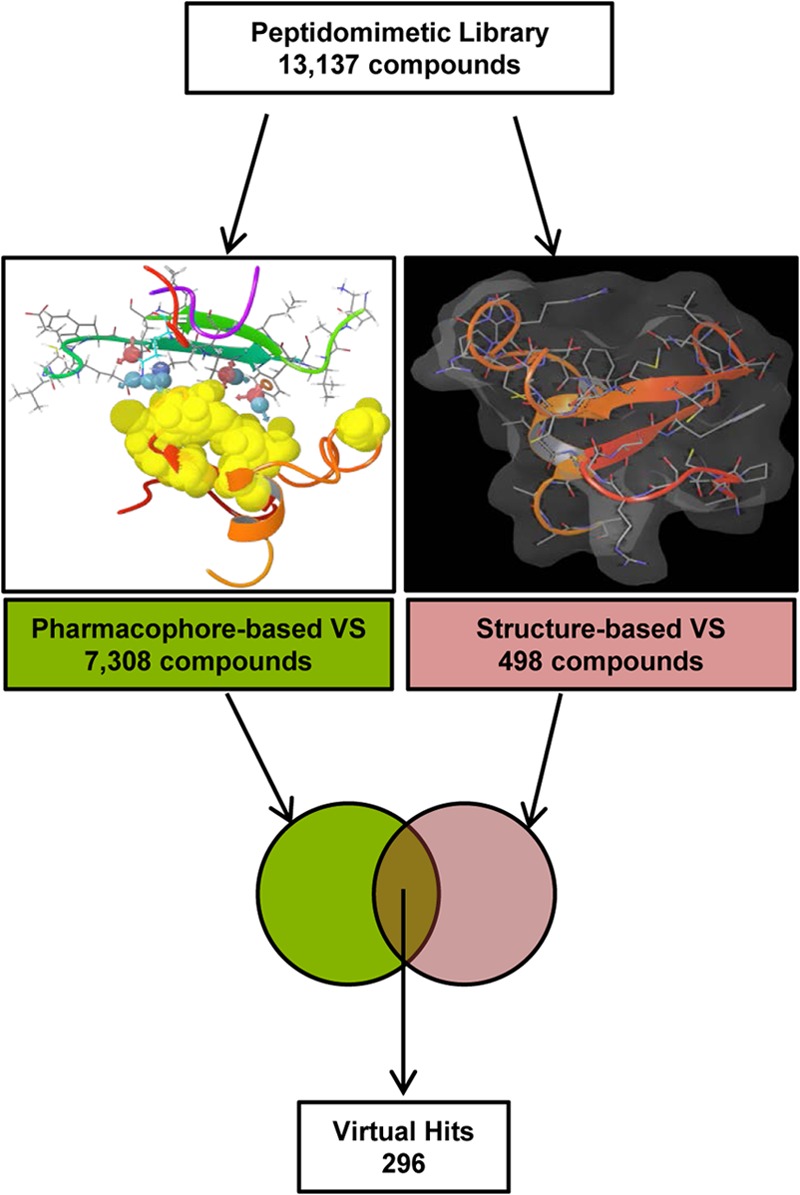
Virtual screening workflow for in silico peptidomimetic compound selection. Two parallel virtual screening (VS) strategies were implemented to identify virtual hits from a vendor compound library of peptidomimetic molecules (ChemDiv, 13,137 compounds). The pharmacophore-based virtual screening provided a biased approach utilizing the protein-protein complexes identified in Fig. 4, A and B. The structure-based virtual screening utilized an optimized Fn14 CRD model to identify candidate compounds from the initial library. Both result sets were intersected, and compounds identified by both methodologies were considered as virtual hits and selected for further testing.
TABLE 2.
Pharmacophore sites for the two TWEAK-Fn14 models validated after protein-protein docking
Sites corresponding to the anchoring residues are required as a pharmacophore match, whereas match with other sites is optional.
| TWEAK-Fn14 model | TWEAK residues | Pharmacophoric sitea |
|---|---|---|
| Tyr-176 model 1 | Tyr176 (anchor) | R, A, D (required) |
| Gln123 | D, D, A | |
| Lys178 | P, D | |
| Thr198 | A,D | |
| Tyr-176 model 2 | Tyr176 (anchor) | R, A, D (required) |
| Lys173 | P, D, D | |
| Ala174 | A, D | |
| Arg227 | H, P | |
| Trp231 | R, R, D |
a Pharmacophoric site abbreviations are as follows: D, hydrogen bond donor; A, hydrogen bond acceptor; H, hydrophobic; P, positive ion; R, aromatic ring.
ELISA and Expansion on Activities
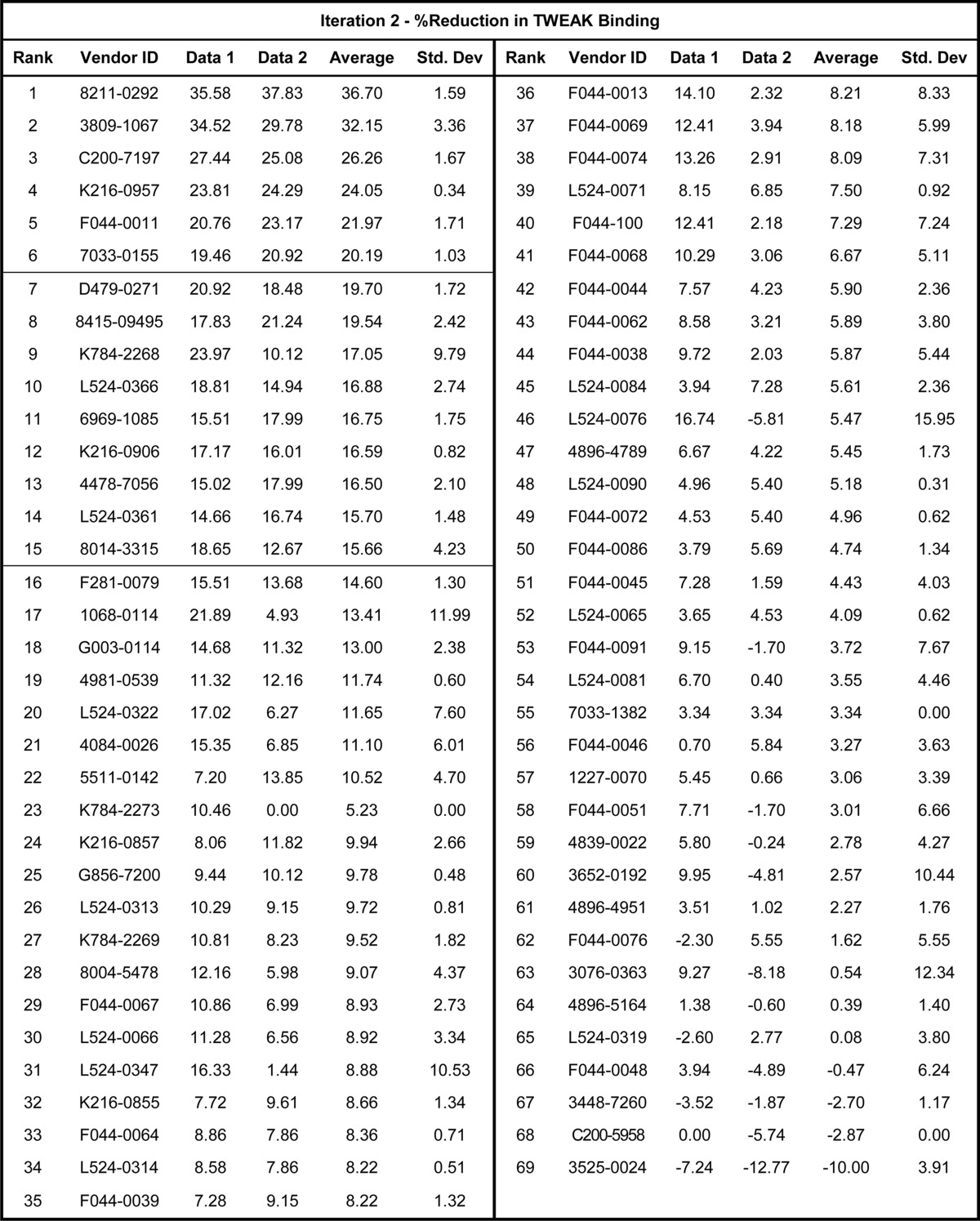
The 60 compounds selected by virtual screen were obtained from the internal compound collection and assayed in the ELISA screen as described under “Experimental Procedures.” These compounds demonstrated variable inhibitory activity of the Fn14-TWEAK interaction, with individual data points ranging from 0 to 26%. The data set capturing the reduction in Fn14-TWEAK binding demonstrated by each compound was rank-ordered by decreasing average inhibition and summarized in Table 3. As shown in Fig. 10A, 4 compounds from the supplier ChemDiv with compound identifiers G873-0032, F151-0435, D715-0890, and J004-1091 showed an average inhibition in TWEAK-Fn14 binding over 15%. Compounds with similar scaffolds and single point activities above 15% were also identified (G873-0031 and D715-0114). Finally, compounds with high repeatability and moderate activities slightly under 15% of activity were identified in the top 10 compounds and were also retained for further consideration (D715-2673, F044-0043, and F044-0075). Indeed, two of these molecules present similar scaffolds (F044-0043 and F044-0075), and a third one, D715-2673, shares similarity to two other compounds discussed above (D715-0890 and D715-0114). To confirm the mild activities observed in these five scaffold classes in the first screening iteration, these five chemical spaces were expanded. Common core scaffolds were identified and deconstructed into smaller substructures. These core substructures served as a query to perform a substructure search in the ChemDiv on-line catalogue. Compounds matching those queries were further triaged visually to remove those compounds with flags for reactivity, and a total of 69 compounds was procured. These compounds were plated as described under “Experimental Procedures” and were evaluated for Fn14-TWEAK inhibition in a second screening iteration using the ELISA. The results, summarized in Table 4, clearly show a significant increase in activity with individual measurements reaching up to 37.8% inhibition of TWEAK-Fn14 interaction. 15 out of 69 compounds showed ≥15% and 6 out 69 compounds showed ≥20% inhibition of TWEAK binding to Fn14 (Fig. 10B). Compounds 8211-0292 and 3809-1067 reached 36.7 and 32.1% inhibition in TWEAK binding to Fn14, respectively. Interestingly, five of the six most inhibitory compounds came from scaffold expansion of the core substructures of compound F151-0435. These results indicate the chemical tractability of the TWEAK-Fn14 target of interest.
TABLE 3.
Bioassay data of the first screening iteration expressed as the percentage of reduction in Fn14-TWEAK binding
Compounds are rank-ordered and identified using the supplier ID (ChemDiv). Zero values indicate missing data.
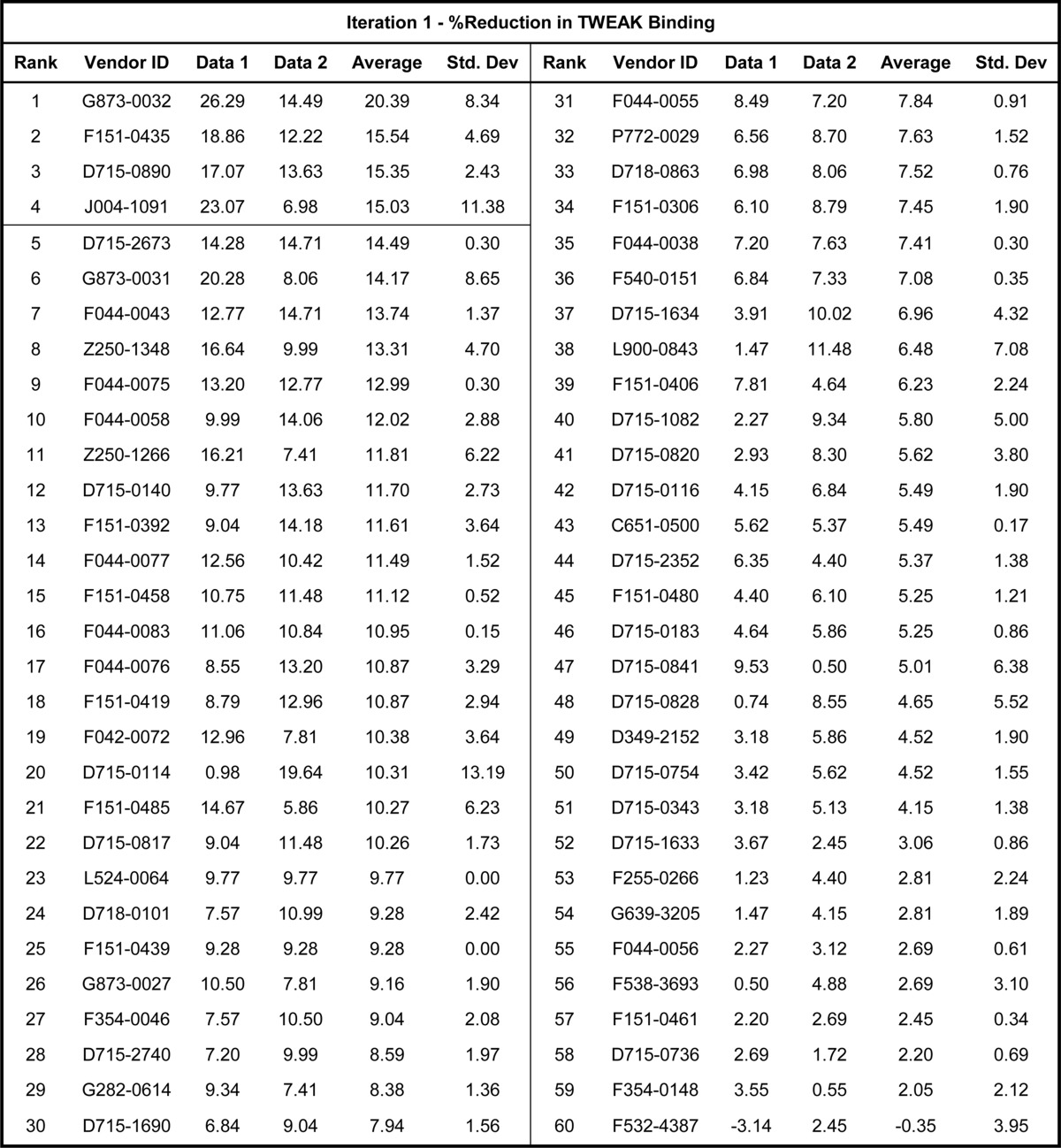
FIGURE 10.
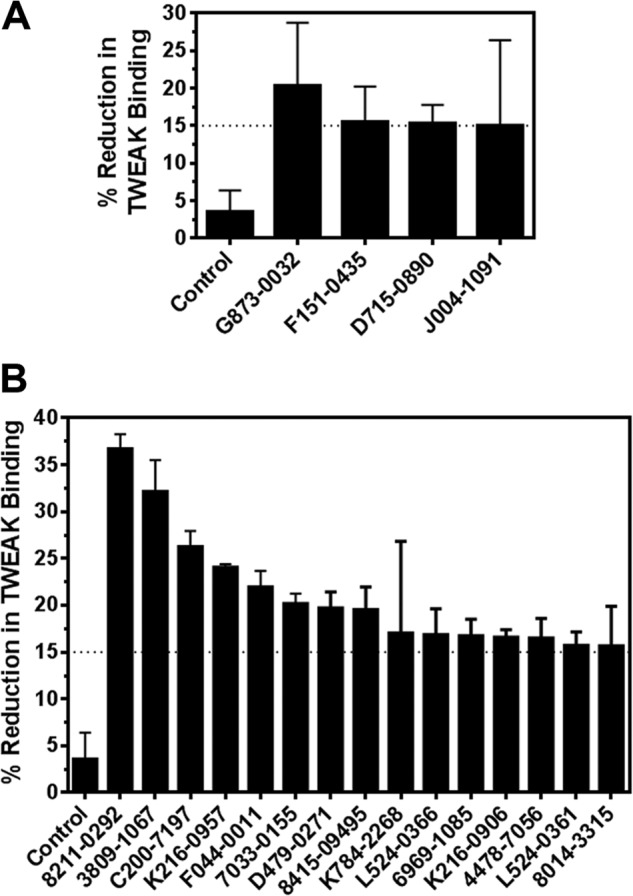
Average inhibitory activities of TWEAK binding to Fn14. A, average inhibitory activities for compounds that demonstrated ≥15% reduction in Fn14-TWEAK binding in the ELISA during the first round of screening (4 of 60 compounds). B, average inhibitory activities for compounds that demonstrated ≥15% reduction in Fn14-TWEAK binding in the ELISA during the follow-up round of screening (15 of 69 compounds).
TABLE 4.
Bioassay data of the second screening iteration expressed as the percentage of reduction in Fn14-TWEAK binding
Compounds are rank-ordered and identified using the supplier ID (ChemDiv). Zero values indicate missing data.
Cell-based Luciferase Induction Assay and Validation of Activities
Compounds that demonstrated ≥15% inhibition of TWEAK binding to Fn14 in the ELISA were selected for further validation using a cell-based NF-κB luciferase induction assay. A total of 19 compounds were selected and screened for their capacity to inhibit cellular NF-κB-stimulated luciferase activity in NF-κB-Luc and Fn14-NF-κB-Luc cells stimulated with either TWEAK or TNFα. One compound, L524-0366, demonstrated significant inhibition of TWEAK-induced NF-κB-driven luciferase activity in Fn14-NF-κB-Luc cells but only minor inhibition of TNFα-induced NF-κB-driven luciferase activity in NF-κB-Luc cells (Fig. 11A). In addition, L524-0366 showed specific dose-dependent inhibition of TWEAK-Fn14-stimulated luciferase induction (Fig. 11B). L524-0366 showed ∼5-fold higher inhibitory activity against TWEAK-Fn14 signaling (IC50 of 7.8 μm for Fn14-NF-κB-Luc cells stimulated with TWEAK as compared with IC50 of 31.03 μm for NF-κB-Luc cells stimulated with TNFα). Other compounds that exhibited inhibitory activity in the ELISA either did not demonstrate specific inhibition of TWEAK-Fn14-stimulated luciferase induction relative to TNFα-stimulated luciferase induction or exerted significant cellular toxicity.
FIGURE 11.
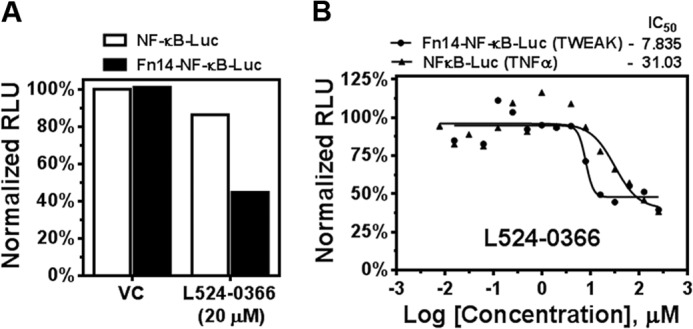
L524-0366 specifically inhibits TWEAK-Fn14-mediated NF-κB activation. A, NF-kB Luc reporter cells treated with TNFα or Fn14-NF-kB-Luc reporter cells treated with TWEAK were incubated with vehicle (VC) or L524-0366. NF-κB-driven luminescent signal was determined using Bright-Glo assay. B, dose-response curve of inhibitory activity of L524-0366 in Fn14-NF-κB-Luc and NF-κB-Luc cells following TWEAK and TNFα stimulation, respectively. RLU, relative luminescence units.
Surface Plasmon Resonance Assay Validates the Direct Interaction of L524-0366 with Fn14
To define the molecular basis of how L524-0366 inhibits the TWEAK-Fn14 signaling cascade, the interaction of L524-0366 with TWEAK and Fn14 was analyzed using surface plasmon resonance assay. The binding of cycloheximide to Fn14 or TWEAK was used as a control. Although cycloheximide did not show significant binding to either the Fn14 or TWEAK surface (Fig. 12, A and B), L524-0366 bound specifically to the Fn14 surface with a KD of 7.12 μm (Fig. 12C) but not to the TWEAK surface (Fig. 12D). These results are coherent with the structure-guided strategy presented above that targeted Fn14 CRD as the receptor for this study. The functionality of the TWEAK and Fn14 sensor surfaces was determined by observing the binding of TWEAK to the Fn14 surface and binding of Fn14-Fc to the TWEAK surface (data not shown).
FIGURE 12.
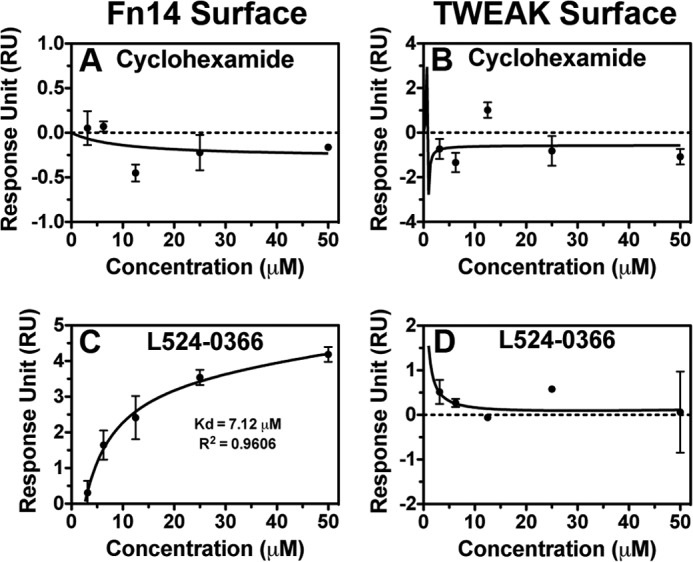
L524-0366 specifically binds to Fn14. A and B, serial dilutions of cycloheximide from 0 to 50 μm were injected over Fn14 and TWEAK sensor surfaces, and its binding affinity to Fn14 and TWEAK was measured. Values are mean ± S.D. of duplicate measurements. C and D, serial dilutions of L524-0366 from 0 to 50 μm were injected over Fn14 and TWEAK sensor surfaces, and its binding affinity to Fn14 and TWEAK was measured. Values are mean ± S.D. of duplicate measurements.
Validation of TWEAK-Fn14 Interaction Inhibitor in Phenotypic Assay
The capacity of L524-0366 to bind to Fn14 and inhibit TWEAK-Fn14 signaling suggests that it could functionally inhibit TWEAK-Fn14-driven cell migration. Therefore, we examined the capacity of L524-0366 to inhibit TWEAK-induced glioma cell migration. Although TWEAK significantly stimulated T98G glioma cell migration, L524-0366 (10 μm) completely suppressed TWEAK-induced T98G cell migration (Fig. 13A). Notably, addition of L524-0366 did not demonstrate any cytotoxicity up to 50 μm (Fig. 13B). Therefore, the observed decrease in glioma cell migration is not due to compound toxicity. Collectively, these results indicate the chemical tractability of the TWEAK-Fn14 target of interest and set the stage for subsequent medicinal chemistry efforts aimed at the identification and optimization of lead compounds capable of disrupting the interaction of Fn14 and TWEAK.
FIGURE 13.
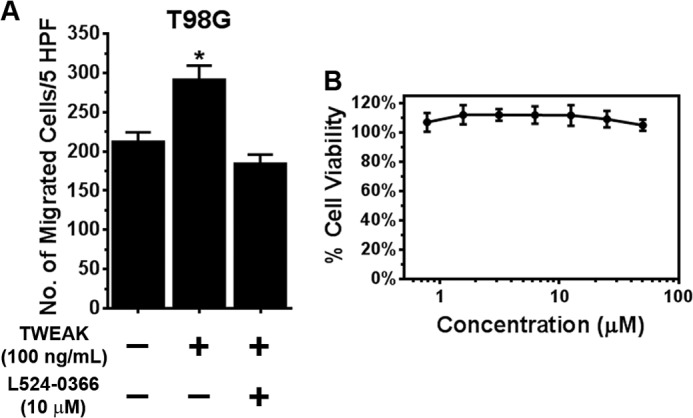
L524-0366 inhibits TWEAK-induced glioma cell migration. A, T98G glioma cells were added to the top well of a modified transwell chamber pre-coated with collagen. TWEAK (100 ng/ml) or TWEAK and PP2 (10 μm) was added to the lower wells, and the number of cells invaded to the bottom chamber quantitated after 5 h. Values are mean ± S.D. of triplicate measurements (*, p < 0.05). HPF, high power field. B, cytotoxic effect of L524-0366 on T98G glioma cells was assessed by quantifying metabolic activity of the cells after the drug treatment. Glioma cells were seeded in 96-well plates, and after 24 h of incubation, either vehicle (DMSO) or L524-0366 at the indicated concentration was added to each well. After 72 h of incubation, viability of the cells was measured using CellTiter-Glo assay kit. Values are mean ± S.D. of six separate measurements.
In summary, this work elucidated key structural elements of the TWEAK-Fn14 binding interaction using in silico protein modeling and protein-protein docking, followed by experimental validation in vitro. Six homology models of the TWEAK cytokine were built and docked to two of the NMR models of the Fn14 CRD selected as receptors. A data-driven workflow was followed to parse the results, leading to two trends in binding hypotheses. Recently, Lammens et al. (50) published an experimental crystal structure of human TWEAK (PDB code 4HT1; resolution, 2.50 Å) in complex with the Fab fragment of a neutralizing antibody. To validate our structural models of TWEAK, the three-dimensional structures of TWEAK homology models occurring in the protein-protein docking solutions with Tyr176 as the anchor residue were overlaid with the TWEAK experimental structure (PDB code 4HT1). For that purpose, the consensus alignment strategy was used as described under “Experimental Procedures.” The initial overlays of our first and second models to experimental TWEAK structure are characterized by r.m.s.d. of 3.40 and 2.25 Å, respectively. Further analysis of our second model by consensus analysis revealed that 78% of the protein was correctly predicted, with main-chain atoms of consensus residues having an r.m.s.d. of 1.12 Å. As would be anticipated, a higher agreement is observed in the β-pleated sheet structure of the protein and a lower agreement in unstructured loop areas. We also observed that the secondary structure of strand E was correctly predicted but misaligned due to low sequence conservation to the template structures, to a longer D strand in template structures, leading to inaccurate prediction of the D–E loop. Additionally, Lammens et al. (50) proposed an interaction model of TWEAK and Fn14 interpolated by structural overlay of TWEAK and Fn14 CRD structures, respectively, to cytokines and TNF receptor CRDs of other complexes with experimental co-crystallized structures. By doing so, they pinpointed eight residues of TWEAK predicted to participate in the interaction with Fn14 CRD as derived from the overlays. Our models correctly positioned six of those residues, including Tyr176. Despite the inaccuracies in positioning strand E discussed above, our two models were overall found to be in good agreement with the experimental 4HT1 structure, especially given that the two models were generated based on low homology templates, with sequence identities of 19.4 and 14.2%, respectively (templates, 1TNR and 2RJL, see Table 1). This emphasizes the usefulness of structure-derived sequence alignment strategies for homology modeling and consensus alignment of structures and sequences. To predict the TWEAK-Fn14 interface, we preferred an approach utilizing protein-protein docking over structural overlays, because the Fn14 CRD fold was reported to be the first A1–C2-type CRD that could bind to the known target (41). The results identified TWEAK Tyr176 as an important anchor residue in the interaction of TWEAK with the Fn14 CRD. Leveraging this functionally validated information, we demonstrated that the predicted TWEAK-Fn14 interaction interface structural models could guide the virtual selection of small molecule inhibitors that disrupt the TWEAK-Fn14 interaction. By doing so, 60 compounds were identified, of which four were confirmed to inhibit TWEAK binding to Fn14 by ≥15% relative to control. These inhibitory activities were confirmed and increased in a second iteration of screening of 69 compounds selected by expanding the chemical spaces of active scaffolds identified in the initial screening iteration. A higher rate of activities was observed in this second screening as well as an overall increase in the average inhibitory activities, with a particular enrichment of actives around structures with similarity to one of the initial hits. Compounds that demonstrated ≥15% inhibition in TWEAK binding to Fn14 were further validated using a cell-based functional screen that evaluates the ability of compounds to inhibit TWEAK-Fn14 signaling. One compound (L524-0366) was confirmed to be a specific dose-dependent inhibitor of TWEAK-Fn14 interaction and found to confer its activity by binding specifically to Fn14. Finally, L524-0366 demonstrated functional activity and completely suppressed TWEAK-induced glioma cell migration without any potential cytotoxic effects. These results represent a significant step toward proving that the TWEAK-Fn14 interaction is chemically tractable and can serve as a foundation for further exploration utilizing chemical biology approaches focusing on functional validation of this interaction as a therapeutic target of interest in invasive cancers.
Acknowledgments
We thank Dr. Gerald M. Maggiora for fruitful discussions and Dr. Jeffrey Winkles (University of Maryland) for the HEK293-Fn14-NFκB luciferase cells. Surface plasmon resonance data were acquired by the Arizona Proteomics Consortium supported by National Institutes of Health Grant ES06694 from NIEHS to the Southwest Environmental Health Sciences Center and Grant CA023074 from NCI to the Arizona Cancer Center, and by the BIO5 Institute of the University of Arizona.
This work was supported, in whole or in part, by National Institutes of Health Grants P50 CA108961 (to J. C. L.) and R01 CA130940 (to N. L. T.) and by National Institute of Mental Health Grant RC2MH090878 (to N. M.). This work was also supported by the Ben and Catherine Ivy Foundation (to M. E. B.) and by the Helios Education Foundation (to G. T.).
- TWEAK
- TNF-like weak inducer of apoptosis
- Fn14
- fibroblast growth factor-inducible 14
- THD
- TNF homology domain
- sTWEAK
- soluble TWEAK
- TNFR
- TNF receptor
- CRD
- cysteine-rich domain
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank
- IVTT
- in vitro transcription/translation
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Wiley S. R., Winkles J. A. (2003) TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 14, 241–249 [DOI] [PubMed] [Google Scholar]
- 2. Chicheportiche Y., Bourdon P. R., Xu H., Hsu Y. M., Scott H., Hession C., Garcia I., Browning J. L. (1997) TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 272, 32401–32410 [DOI] [PubMed] [Google Scholar]
- 3. Feng S. L., Guo Y., Factor V. M., Thorgeirsson S. S., Bell D. W., Testa J. R., Peifley K. A., Winkles J. A. (2000) The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am. J. Pathol. 156, 1253–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kölzsch J. (1990) Clinical aspects and therapy of frequent viral diseases of the skin. Z. Arztl. Fortbild. 84, 1199–1202 [PubMed] [Google Scholar]
- 5. Kolfschoten G. M., Pradet-Balade B., Hahne M., Medema J. P. (2003) TWE-PRIL; a fusion protein of TWEAK and APRIL. Biochem. Pharmacol. 66, 1427–1432 [DOI] [PubMed] [Google Scholar]
- 6. Brown S. A., Ghosh A., Winkles J. A. (2010) Full-length, membrane-anchored TWEAK can function as a juxtacrine signaling molecule and activate the NF-κB pathway. J. Biol. Chem. 285, 17432–17441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meighan-Mantha R. L., Hsu D. K., Guo Y., Brown S. A., Feng S. L., Peifley K. A., Alberts G. F., Copeland N. G., Gilbert D. J., Jenkins N. A., Richards C. M., Winkles J. A. (1999) The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J. Biol. Chem. 274, 33166–33176 [DOI] [PubMed] [Google Scholar]
- 8. Harada N., Nakayama M., Nakano H., Fukuchi Y., Yagita H., Okumura K. (2002) Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 299, 488–493 [DOI] [PubMed] [Google Scholar]
- 9. Polek T. C., Talpaz M., Darnay B. G., Spivak-Kroizman T. (2003) TWEAK mediates signal transduction and differentiation of RAW264.7 cells in the absence of Fn14/TweakR. Evidence for a second TWEAK receptor. J. Biol. Chem. 278, 32317–32323 [DOI] [PubMed] [Google Scholar]
- 10. Brown S. A., Hanscom H. N., Vu H., Brew S. A., Winkles J. A. (2006) TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem. J. 397, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burkly L. C., Michaelson J. S., Zheng T. S. (2011) TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol. Rev. 244, 99–114 [DOI] [PubMed] [Google Scholar]
- 12. Han H., Bearss D. J., Browne L. W., Calaluce R., Nagle R. B., Von Hoff D. D. (2002) Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 62, 2890–2896 [PubMed] [Google Scholar]
- 13. Tran N. L., McDonough W. S., Donohue P. J., Winkles J. A., Berens T. J., Ross K. R., Hoelzinger D. B., Beaudry C., Coons S. W., Berens M. E. (2003) The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am. J. Pathol. 162, 1313–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watts G. S., Tran N. L., Berens M. E., Bhattacharyya A. K., Nelson M. A., Montgomery E. A., Sampliner R. E. (2007) Identification of Fn14/TWEAK receptor as a potential therapeutic target in esophageal adenocarcinoma. Int. J. Cancer 121, 2132–2139 [DOI] [PubMed] [Google Scholar]
- 15. Willis A. L., Tran N. L., Chatigny J. M., Charlton N., Vu H., Brown S. A., Black M. A., McDonough W. S., Fortin S. P., Niska J. R., Winkles J. A., Cunliffe H. E. (2008) The fibroblast growth factor-inducible 14 receptor is highly expressed in HER2-positive breast tumors and regulates breast cancer cell invasive capacity. Mol. Cancer Res. 6, 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Whitsett T. G., Cheng E., Inge L., Asrani K., Jameson N. M., Hostetter G., Weiss G. J., Kingsley C. B., Loftus J. C., Bremner R., Tran N. L., Winkles J. A. (2012) Elevated expression of Fn14 in non-small cell lung cancer correlates with activated EGFR and promotes tumor cell migration and invasion. Am. J. Pathol. 181, 111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tran N. L., McDonough W. S., Savitch B. A., Fortin S. P., Winkles J. A., Symons M., Nakada M., Cunliffe H. E., Hostetter G., Hoelzinger D. B., Rennert J. L., Michaelson J. S., Burkly L. C., Lipinski C. A., Loftus J. C., Mariani L., Berens M. E. (2006) Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-κB and correlate with poor patient outcome. Cancer Res. 66, 9535–9542 [DOI] [PubMed] [Google Scholar]
- 18. Burkly L. C., Michaelson J. S., Hahm K., Jakubowski A., Zheng T. S. (2007) TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine 40, 1–16 [DOI] [PubMed] [Google Scholar]
- 19. Winkles J. A. (2008) The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 7, 411–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jain M., Jakubowski A., Cui L., Shi J., Su L., Bauer M., Guan J., Lim C. C., Naito Y., Thompson J. S., Sam F., Ambrose C., Parr M., Crowell T., Lincecum J. M., Wang M. Z., Hsu Y. M., Zheng T. S., Michaelson J. S., Liao R., Burkly L. C. (2009) A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation 119, 2058–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoriki R., Akashi S., Sho M., Nomi T., Yamato I., Hotta K., Takayama T., Matsumoto S., Wakatsuki K., Migita K., Yagita H., Nakajima Y. (2011) Therapeutic potential of the TWEAK/Fn14 pathway in intractable gastrointestinal cancer. Exp. Ther. Med. 2, 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng T. S., Burkly L. C. (2008) No end in site: TWEAK/Fn14 activation and autoimmunity associated-end-organ pathologies. J. Leukocyte Biol. 84, 338–347 [DOI] [PubMed] [Google Scholar]
- 23. Bhatnagar S., Kumar A. (2012) The TWEAK-Fn14 system: breaking the silence of cytokine-induced skeletal muscle wasting. Curr. Mol. Med. 12, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Campbell S., Burkly L. C., Gao H. X., Berman J. W., Su L., Browning B., Zheng T., Schiffer L., Michaelson J. S., Putterman C. (2006) Proinflammatory effects of TWEAK/Fn14 interactions in glomerular mesangial cells. J. Immunol. 176, 1889–1898 [DOI] [PubMed] [Google Scholar]
- 25. Bhattacharjee M., Raju R., Radhakrishnan A., Nanjappa V., Muthusamy B., Singh K., Kuppusamy D., Lingala B. T., Pan A., Mathur P. P., Harsha H. C., Prasad T. S., Atkins G. J., Pandey A., Chatterjee A. (2012) A bioinformatics resource for TWEAK-Fn14 signaling pathway. J. Signal Transduct. 2012:376470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tansey M. G., Szymkowski D. E. (2009) The TNF superfamily in 2009: new pathways, new indications, and new drugs. Drug Discov. Today 14, 1082–1088 [DOI] [PubMed] [Google Scholar]
- 27. He M. M., Smith A. S., Oslob J. D., Flanagan W. M., Braisted A. C., Whitty A., Cancilla M. T., Wang J., Lugovskoy A. A., Yoburn J. C., Fung A. D., Farrington G., Eldredge J. K., Day E. S., Cruz L. A., Cachero T. G., Miller S. K., Friedman J. E., Choong I. C., Cunningham B. C. (2005) Small-molecule inhibition of TNF-α. Science 310, 1022–1025 [DOI] [PubMed] [Google Scholar]
- 28. Silvian L. F., Friedman J. E., Strauch K., Cachero T. G., Day E. S., Qian F., Cunningham B., Fung A., Sun L., Shipps G. W., Su L., Zheng Z., Kumaravel G., Whitty A. (2011) Small molecule inhibition of the TNF family cytokine CD40 ligand through a subunit fracture mechanism. ACS Chem. Biol. 6, 636–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benicchi T., Iozzi S., Svahn A., Axelsson H., Mori E., Bernocco S., Cappelli F., Caramelli C., Fanti P., Genesio E., Maccari L., Markova N., Micco I., Porcari V., Schultz J., Fecke W. (2012) A homogeneous HTRF assay for the identification of inhibitors of the TWEAK-Fn14 protein interaction. J. Biomol. Screen. 17, 933–945 [DOI] [PubMed] [Google Scholar]
- 30. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Molecular Operating Environment (2010) Molecular Operating Environment, Version 2010.10, Chemical Computing Group Inc., Montreal, Quebec, Canada [Google Scholar]
- 32. Abagyan R., Totrov M., Kuznetsov D. (1994) ICM–A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 [Google Scholar]
- 33. Nemethy G., Gibson K. D., Palmer K. A., Yoon C. N., Paterlini G., Zagari A., Rumsey S., Scheraga H. A. (1992) Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J. Phys. Chem. 96, 6472–6484 [Google Scholar]
- 34. Watts K. S., Dalal P., Murphy R. B., Sherman W., Friesner R. A., Shelley J. C. (2010) ConfGen: a conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 50, 534–546 [DOI] [PubMed] [Google Scholar]
- 35. Dixon S. L., Smondyrev A. M., Rao S. N. (2006) PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 67, 370–372 [DOI] [PubMed] [Google Scholar]
- 36. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 37. Ramachandran N., Hainsworth E., Bhullar B., Eisenstein S., Rosen B., Lau A. Y., Walter J. C., LaBaer J. (2004) Self-assembling protein microarrays. Science 305, 86–90 [DOI] [PubMed] [Google Scholar]
- 38. Frostell-Karlsson A., Remaeus A., Roos H., Andersson K., Borg P., Hämäläinen M., Karlsson R. (2000) Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J. Med. Chem. 43, 1986–1992 [DOI] [PubMed] [Google Scholar]
- 39. Lamszus K., Schmidt N. O., Jin L., Laterra J., Zagzag D., Way D., Witte M., Weinand M., Goldberg I. D., Westphal M., Rosen E. M. (1998) Scatter factor promotes motility of human glioma and neuromicrovascular endothelial cells. Int. J. Cancer 75, 19–28 [DOI] [PubMed] [Google Scholar]
- 40. Brockmann M. A., Ulbricht U., Gruner K., Fillbrandt R., Westphal M., Lamszus K. (2003) Glioblastoma and cerebral microvascular endothelial cell migration in response to tumor-associated growth factors. Neurosurgery 52, 1391–1399 [DOI] [PubMed] [Google Scholar]
- 41. He F., Dang W., Saito K., Watanabe S., Kobayashi N., Güntert P., Kigawa T., Tanaka A., Muto Y., Yokoyama S. (2009) Solution structure of the cysteine-rich domain in Fn14, a member of the tumor necrosis factor receptor superfamily. Protein Sci. 18, 650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Banner D. W., D'Arcy A., Janes W., Gentz R., Schoenfeld H. J., Broger C., Loetscher H., Lesslauer W. (1993) Crystal structure of the soluble human 55 kD TNF receptor-human TNFβ complex: implications for TNF receptor activation. Cell 73, 431–445 [DOI] [PubMed] [Google Scholar]
- 43. Chothia C., Lesk A. M. (1986) The relation between the divergence of sequence and structure in proteins. EMBO J. 5, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hymowitz S. G., Patel D. R., Wallweber H. J., Runyon S., Yan M., Yin J., Shriver S. K., Gordon N. C., Pan B., Skelton N. J., Kelley R. F., Starovasnik M. A. (2005) Structures of APRIL-receptor complexes: like BCMA, TACI employs only a single cysteine-rich domain for high affinity ligand binding. J. Biol. Chem. 280, 7218–7227 [DOI] [PubMed] [Google Scholar]
- 45. Bogan A. A., Thorn K. S. (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280, 1–9 [DOI] [PubMed] [Google Scholar]
- 46. Meireles L. M., Dömling A. S., Camacho C. J. (2010) ANCHOR: a web server and database for analysis of protein-protein interaction binding pockets for drug discovery. Nucleic Acids Res. 38, W407–W411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schneider P., Schwenzer R., Haas E., Mühlenbeck F., Schubert G., Scheurich P., Tschopp J., Wajant H. (1999) TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur. J. Immunol. 29, 1785–1792 [DOI] [PubMed] [Google Scholar]
- 48. Tran N. L., McDonough W. S., Savitch B. A., Sawyer T. F., Winkles J. A., Berens M. E. (2005) The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFκB pathway activation and BCL-XL/BCL-W expression. J. Biol. Chem. 280, 3483–3492 [DOI] [PubMed] [Google Scholar]
- 49. Pellegrini M., Willen L., Perroud M., Krushinskie D., Strauch K., Cuervo H., Day E. S., Schneider P., Zheng T. S. (2013) Structure of the extracellular domains of human and Xenopus Fn14: implications in the evolution of TWEAK and Fn14 interactions. FEBS J. 280, 1818–1829 [DOI] [PubMed] [Google Scholar]
- 50. Lammens A., Baehner M., Kohnert U., Niewoehner J., von Proff L., Schraeml M., Lammens K., Hopfner K. P. (2013) Crystal structure of human TWEAK in complex with the Fab fragment of a neutralizing antibody reveals insights into receptor binding. PLoS One 8, e62697. [DOI] [PMC free article] [PubMed] [Google Scholar]