

Background: Hemodynamic force-regulated VWF-GPIbα interaction mediates platelet adhesion during the early stage of hemostatic and thrombotic processes.
Results: The VWF-A1 N-terminal sequence impedes the VWF-GPIbα interaction at low forces but stabilizes it as force increases.
Conclusion: The interplay between force and A1 N-terminal sequence regulates the VWF-GPIbα interaction.
Significance: Force regulation is crucial to the balance of the platelet adhesive function in hemostasis and thrombosis.
Keywords: Kinetics, Platelets, Single Molecule Biophysics, Thrombosis, von Willebrand Factor, Biomembrane Force Probe, Catch Bond, Glycoprotein Ibα
Abstract
Binding of platelet glycoprotein Ibα (GPIbα) to von Willebrand factor (VWF) initiates platelet adhesion to disrupted vascular surface under arterial blood flow. Flow exerts forces on the platelet that are transmitted to VWF-GPIbα bonds, which regulate their dissociation. Mutations in VWF and/or GPIbα may alter the mechanical regulation of platelet adhesion to cause hemostatic defects as found in patients with von Willebrand disease (VWD). Using a biomembrane force probe, we observed biphasic force-decelerated (catch) and force-accelerated (slip) dissociation of GPIbα from VWF. The VWF A1 domain that contains the N-terminal flanking sequence Gln1238–Glu1260 (1238-A1) formed triphasic slip-catch-slip bonds with GPIbα. By comparison, using a short form of A1 that deletes this sequence (1261-A1) abolished the catch bond, destabilizing its binding to GPIbα at high forces. Importantly, shear-dependent platelet rolling velocities on these VWF ligands in a flow chamber system mirrored the force-dependent single-bond lifetimes. Adding the Gln1238–Glu1260 peptide, which interacted with GPIbα and 1261-A1 but not 1238-A1, to whole blood decreased platelet attachment under shear stress. Soluble Gln1238–Glu1260 reduced the lifetimes of GPIbα bonds with VWF and 1238-A1 but rescued the catch bond of GPIbα with 1261-A1. A type 2B VWD 1238-A1 mutation eliminated the catch bond by prolonging lifetimes at low forces, a type 2M VWD 1238-A1 mutation shifted the respective slip-catch and catch-slip transition points to higher forces, whereas a platelet type VWD GPIbα mutation enhanced the bond lifetime in the entire force regime. These data reveal the structural determinants of VWF activation by hemodynamic force of the circulation.
Introduction
During the early stage of hemostatic and thrombotic processes, platelets tether to and translocate or roll on von Willebrand factor (VWF)2 immobilized on disrupted vascular surfaces. This adhesive interaction is primarily mediated by binding of the 45-kDa N-terminal domain of the α subunit of the platelet glycoprotein Ib-IX-V complex (GPIbα) to the A1 domain of VWF (1–3). Remarkably, the VWF-GPIbα interaction has to be delicately balanced because insufficient adhesion cannot stop bleeding to maintain hemostasis, whereas excessive adhesion may result in thrombosis (4). Because such binding occurs under arterial blood flow, the VWF-GPIbα interaction is regulated by hemodynamic force (5–7). Circulating platelets do not bind VWF unless putative conformational changes occur around its A1 domain that exposes the GPIbα binding site (VWF activation) as a result of high hemodynamic force (8). The present view of the force-dependent VWF activation includes two steps: 1) relief of an autoinhibitory mechanism involving interdomain associations within the A1A2A3 tri-domain (9, 10) and between A1 and D′D3 (11) and 2) up-regulation of A1 binding affinity to GPIbα (6, 12–14). However, the biophysical and structural basis of how the VWF-GPIbα interaction is regulated when its hemostatic adhesive function is most needed remains unclear, in part, because of discrepant data.
To elucidate the molecular mechanism regulating VWF-GPIbα interaction, we previously used atomic force microscopy (AFM) and a flow chamber to show that A1-GPIbα and VWF-GPIbα interactions behaved as catch-slip bonds such that increasing force initially prolonged and then shortened bond lifetime (6). However, another study using optical tweezers (OT) observed a different phenomenon at forces lower than those examined by us, reporting an A1-GPIbα bond that could flex between two slip-bond states at ∼10 pN (15). Note that in previous studies, different groups used recombinant sequences with different lengths of the N and C termini outside of the disulfide loop (Cys1272–Cys1458) that comprises the structure of the A1 domain. These sequences can be categorized into two groups: most studies of flow effects on platelet adhesion used A1 constructs that start at ∼1238 (1238-A1) or even beyond 1238 to the N terminus (N-longer A1) (6, 14, 16–22), whereas most structural and kinetic studies used A1 constructs that start at ∼1261 (1261-A1) or even shorter than 1261 at N terminus (N-shorter A1) (15, 23–29). In addition, many lines of evidence suggest that the N-terminal flanking sequence Gln1238–Glu1260 included in the 1238-A1 but excluded in the 1261-A1 constructs may play an important role in the VWF activation and may be responsible for discrepant observations from different studies. It has been shown that a N-shorter A1 (1271–1467) spanning the structurally resolved residues can bind GPIbα but does not support platelet adhesion as efficiently as the N-longer A1 containing the N-terminal flanking sequence at high shear rates (17). Furthermore, it has been shown that a 15-residue N-terminal sequence (1237–1251) can completely inhibit the binding of VWF to GPIbα (30). Recently, another sequence (Gln1238–Glu1260) was suggested to be part of an autoinhibitory mechanism that stabilizes A1A2A3 interdomain associations (31). Still, how this short N-terminal flanking sequence regulates VWF-GPIbα interaction under force remains unclear.
To gain mechanistic insights into the regulatory effects of VWF regions surrounding A1, especially its N-terminal flanking region, on the force-dependent VWF-GPIbα interaction, we used a biomembrane force probe (BFP) to analyze single-bond dissociation of GPIbα from VWF, 1238-A1, and 1261-A1 under forces lower than those previously studied in the absence and presence of the Gln1238–Glu1260 sequence in solution. We showed that GPIbα formed catch-slip bonds with VWF and slip-catch-slip bonds with 1238-A1, which govern platelet rolling on these substrates, but slip-only bonds with 1261-A1. Soluble Gln1238–Glu1260 inhibited whole blood platelet attachment, suppressed the respective GPIbα catch bonds with VWF and 1238-A1, but rescued that with 1261-A1. The 1238-A1 mutation R1450E that exhibits the gain of function (GOF) phenotype of the type 2B VWD prolonged bond lifetime at zero force, whereas the 1238-A1 mutation G1324S that exhibits the loss of function phenotype of the type 2M VWD shifted the respective slip-catch and catch-slip transition points to higher forces. The GPIbα mutation G233V that exhibits the GOF phenotype of platelet type VWD (ptVWD) retained the slip-catch-slip bond feature but greatly prolonged bond lifetime in the entire force regime. These results pinpoint the structural determinants of the VWF-GPIbα catch bond, explain the discrepancy between catch and flex bonds, and elucidate the regulating mechanisms for VWF activation by hemodynamic force of the circulation.
EXPERIMENTAL PROCEDURES
Proteins and Antibodies
The recombinant WT VWF 1238-A1 domain (residues 1238–1471) (16), as well as single-residue mutants R1450E and G1324S, has been described (22). The N-terminally truncated A1 domain (1261-A1) was expressed in bacteria and purified as the 1238-A1 protein. Plasma VWF was from Calbiochem. The N-terminally biotinylated polypeptide Gln1238–Glu1260 (Lp, QEPGGLVVPPTDAPVSPTTLYVE) of the A1 N-terminal flanking region and the scramble polypeptide (Sp, QLPTGVLGEPSDAVPTVYEVTPP) were synthesized by Selleck Chemicals (Houston, TX). Glycocalicin (extracellular domain of GPIbα) was cleaved and purified from outdated platelets as described previously (6). Anti-GPIbα mAb (AK2) was from Abcam (Cambridge, MA). Anti-A1 blocking (5D2) and anti-GPIbα capturing (WM23) mAbs were gifts from Dr. Michael Berndt (Curtin University, Bentley, Australia) and Dr. Renhao Li (Emory University, Atlanta, GA), respectively.
Platelets and Cells
Abiding protocols approved by the respective institutional review boards of the Georgia Institute of Technology and Baylor College of Medicine, 10 ml of blood were collected from healthy donors for isolation of platelets and red blood cells (RBCs) and for direct use in flow chamber experiments after mixed with Lp or Sp. RBCs were washed, biotinylated, and stored for experiments for several weeks as described previously (32). Platelet-rich plasma was prepared in a 1:10 citrate buffer (170 mm sodium citrate and 83 mm citric acid in distilled water). To prevent unwanted platelet activation, 2 μm prostaglandin E1 was added to the citrate buffer. Blood was centrifuged at 150 × g for 15 min at room temperature. Platelet-rich plasma was then centrifuged at 900 × g for 10 min. The platelet pellet was resuspended in the HEPES-Tyrode buffer (134 mm NaCl, 12 mm NaHCO3, 2.9 mm KCl, 0.34 mm sodium phosphate monobasic, 5 mm HEPES, and 5 mm glucose, 1% BSA, pH 7.4) as the washed platelets. CHOαβ9 cells transfected to express WT or G233V GPIbα have been described previously (33).
Platelet and CHOαβ9 Cell Lysates
Platelets (2 × 109) were pelleted and resuspended in a 5-ml lysis buffer (125 mm NaCl, 50 mm Tris, pH 8.0, 1% Triton X-100) at 4 °C for 30 min with stirring. To avoid the unwanted shedding of GPIbα, the final medium included 5 mm EDTA, 2 mm phenylmethylsulfonyl fluoride, and 1× protease inhibitor (#78437; Pierce). After 16,000 × g centrifugation at 4 °C for 20 min, the platelet lysates in the supernatant were collected through a filter (50 K; Millipore, Billerica, MA). CHOαβ9 cells transfected to express WT or G233V GPIbα were lysed similarly.
Functionalization of Glass Beads
VWF, A1, glycocalicin, and capturing mAb WM23 were precoupled covalently with maleimide-PEG3500-N-hydroxysuccinimide (molecular mass, ∼3500 Da; JenKem Technology, Plano, TX). Modified proteins were mixed with streptavidin-maleimide (Sigma-Aldrich), linked to 3-μm (diameter) silanized beads (Fig. 1, B and D, left for VWF or A1 and right for glycocalicin or WM23) and washed, as described previously (32). To immobilize full-length GPIbα on surfaces, beads precoated with WM23 were incubated with platelet or CHOαβ9 cell lysates in a rotor for 2 h at room temperature and washed (Fig. 1B, right). After resuspending in PBS with 1% human serum albumin, beads were ready for immediate use in BFP experiments. To coat Lp or Sp on beads, streptavidin-precoupled beads were prepared as above. Biotin-labeled Lp or Sp was coupled to streptavidin beads (Fig. 1B, left) by a 2-h incubation at room temperature. After that, the same washing steps were used before BFP experiments.
FIGURE 1.

BFP force clamp assay. A and C, BFP photomicrograph. A micropipette-aspirated RBC with a bead (left, termed “probe”) attached to the apex formed a pico-force sensor, as depicted by a spring. It was aligned with another bead (A) or a platelet (C) (right, termed “target”) aspirated by an apposing micropipette. B and D, BFP functionalization with purified molecules. A1 or full-length VWF was covalently coupled to the probe (left). GPIbα was captured from lysates of platelets or CHOαb9 cells by mAb WM23, which was covalently precoated on the target (B, right). Glycocalicin was covalently coupled to the target (D, right). Streptavidin was also covalently coupled to the probe for attachment to biotinylated RBC, as well as coupling the polypeptide Gln1238–Glu1260 (Lp) or scramble polypeptide (Sp). E and F, force versus time traces from two representative test cycles. A target was driven to approach a probe (<1.1 s, ∼0 pN), contacted for 0.5 s (1.1–1.6 s, ∼15 pN), retracted, and ended the cycle if no adhesion was detected (>1.6 s, ∼0 pN) (E) or held at a preset force until dissociation if adhesion was detected (1.6–2 s, ∼12 pN, indicated in red), after which the target was retracted to the starting position (>2 s, ∼0 pN) (F). Lifetime was measured from the point when the clamped force (12 pN) was reached (1.26 s) to the point when the bond dissociated (2 s), signified by a force drop to zero. G, binding specificity. Adhesion frequencies between targets coated with platelet lysate GPIbα, glycocalicin, or platelet GPIbα and probes coated without (−, black columns) or with (+, white columns) 1238-A1 or VWF, in the absence or presence of 50 μg/ml anti-A1 (5D2, gray columns) or anti-GPIbα (AK2, hatched columns) blocking mAb. Each probe-target pair was tested repeatedly for 100 approach-contact-retract cycles to estimate an adhesion frequency. Five probe-target pairs were tested to obtain mean ± S.E. ***, p < 0.001 as assessed by unpaired, two-tailed Student's t test.
BFP Experiments
Our home-built BFP has been described (34). A personal computer installed with custom-written LabViewTM programs were used for image analysis and piezoelectric translator control, which ran in repeated cycles to measure adhesion frequency as well as bond lifetime (force clamp assay at non-zero forces and thermal fluctuation assay at the zero force) (32, 34–36). In each cycle, the target was driven to approach and contact the probe with a 20-pN compressive force for a certain contact time (0.5 s for glycocalicin, 0.2 s for GPIbα from platelet or CHOαβ9 cell lysates and for platelet GPIbα) that allowed for bond formation and then retracted for adhesion detection. During the retraction phase, an adhesion event was signified by tensile force (Fig. 1F), but no tensile force was detected in a no-adhesion event (Fig. 1E). For the adhesion frequency assay, adhesion and no-adhesion events were enumerated to calculate an adhesion frequency in 100 repeated cycles for each probe and target pair. Because the adhesion frequency Pa depends on the site densities of receptors (mr) and ligands (ml) on the contact area, to compare the binding affinity of Lp to different receptor candidates, the effective affinities per receptor density (Fig. 2D) were derived via formula −ln(1 − Pa)/mr (37). In the force clamp assay, the target was held at a desired force to wait for bond dissociation and returned to the original position to complete the cycle. Lifetime was measured from the instant when the force reached the desired level to the instant of bond dissociation (Fig. 1F, red trace) (34, 35). In the thermal fluctuation assay, the target was held at zero force to allow contact with the probe via thermal fluctuation. Bond formation and dissociation were identified from the respective reduction and resumption of the probe thermal fluctuation. Lifetime was measured from the moment of bond formation to the moment of bond dissociation as described previously (32).
FIGURE 2.

The role of Gln1238–Glu1260 on GPIbα interactions with VWF and 1238-A1. A, force-dependent of lifetime of platelet lysate GPIbα bonds with the VWF and two forms of A1. Lifetimes (mean ± S.E. of >20 measurements per point) were measured by the thermal fluctuation assay (closed symbols) at zero force and by the force clamp assay at nonzero forces (open symbols). B, wall shear stress-dependent rolling velocity (mean ± S.E. of ∼20 platelets per point) on VWF and two forms of A1. C, platelets attach differently to 1238- and 1261-A1 coated surfaces. Washed platelets were perfused over a surface coated with 1238- or 1261-A1 at 8 dyn/cm2 wall shear stress. Representative snapshots were shown. D, adhesion frequency Pa (left y axis, closed bars, mean ± S.E. of >3 probe-target pairs each contacting 100 times per bar) between targets bearing 1238-A1, 1261-A1, VWF, or platelet lysate GPIbα and probes bearing Gln1238–Glu1260 (Lp, black columns) or a control scramble polypeptide (Sp, gray columns). NS, not significant; **, p < 0.01; ***, p < 0.001, assessed by unpaired, two-tailed Student's t test. The right y axis variable (open bars) represents a metric of the propensity of Lp binding to its counter-molecule whose variable site density (mr, measured separately by flow cytometry) has been accounted for via the formula −ln(1 − Pa)/mr to allow for comparison of Lp binding to 1238-A1, 1261-A1, VWF, and GPIbα (37). E, ELISA showing specific binding of polypeptide Lp in a purified system. Data (mean ± S.E. of triplicates, two separate experiments) show binding of lysate GPIbα, 1261-A1, and VWF-A2 (as a negative control) to immobilized polypeptide Lp (black columns) or Sp (gray columns) (0.5 mm). F–H, effect of Gln1238–Glu1260 on platelet adhesion under shear. Whole blood mixed with 0.5 mm Lp (left column) or Sp (right column) was perfused over a surface coated with collagen (F), VWF (G), or 1238-A1 (H) at 1,500 s−1 wall shear rate. After a 2-min perfusion, the platelets were washed with PBS and observed microscopically. All photomicrographs are representatives of separate experiments using the same blood donor.
Flow Chamber Assays
Whole blood perfusion was performed as described previously (9, 38). Briefly, whole blood mixed with 0.5 mm of Lp or Sp was perfused through a parallel plate flow chamber (Glycotech, Gaithersburg, MD) at 1,500 s−1 shear rate over the chamber floor coated with collagen (100 μg/ml) or VWF (100 μg/ml) by physical absorption. After a 2-min perfusion, the surface was washed with PBS, and several frames of attached platelets were recorded. Tethered platelets were observed with a phase contrast objective (40×) and recorded by video microscopy.
Platelet rolling velocity was measured as described previously (6). Briefly, washed platelets were perfused at various wall shear stresses over 1238-A1 or 1261-A1 (100 μg/ml) absorbed on the flow chamber floor. Mean rolling velocities were measured over a 0.1-s interval by video microscopy with frame by frame analysis by the Nikon NIS-Elements software. At each wall shear stress, ∼20 velocities of rolling platelets were measured and averaged. The data are presented as means ± S.E. The washed platelet tethering was snapshot with a phase contrast objective (20×) during the experiments.
Direct Binding Assay by ELISA
This assay was performed similarly to that described previously (10, 31). Microtiter wells were coated with lysate GPIbα, A1, or A2. Biotin-labeled Lp or Sp (0.5 mm) were incubated with above putative receptor-coated wells for 1 h at 37 °C. After washing, the wells were incubated with 1:200 dilution of neutravidin-horseradish peroxidase conjugate (Pierce) 1 h at 37 °C. The wells were again washed, and the substrate (o-phenylenediamine; Sigma) was added. The reaction was stopped with 0.025 ml of 2 n H2SO4, and the plates were read at 490-nm absorbance. Net binding was determined by subtracting absorbance values from wells added only BSA from the total binding values obtained in wells added the corresponding polypeptides. VWF-A2 as the isotype control in this assay has been described previously (9).
Analysis of Lifetime Distributions with a Two-state Model
At each force bin, the survival frequency as a function of bond lifetime tb was calculated as the fraction of events with lifetime >tb. In our previous study (6), 1238-A1-GPIbα bonds were assumed to dissociate from a single state so that the pooled bond lifetimes were analyzed by a single exponential distribution (Equation 1) after treating a small fraction (<10%) of long lifetimes (>2 s) as outliers.
Taking the log linearizes the right-hand side of Equation 1, the semi-log survival frequency versus bond lifetime plot is predicted to appear linear. The negative slope of the line would be used as an estimate for off rate koff. However, if long lifetime events were included, the ln(survival frequency) versus tb plots were no longer linear (see Fig. 8). To analyze such data, we extended the single-state model to a two-state model that assumes a slow dissociation state coexisting with a fast dissociation state (35). The survival frequency data for each force bin were fitted by Equation 2, which superimposes two exponential decays,
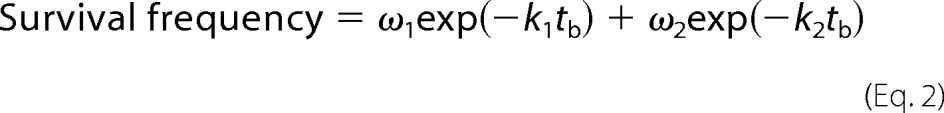 |
where ki and ωi are the off rate and the associated fraction of the ith state (i = 1, 2), which satisfy the constraints ω1 + ω2 = 1 and k1 < k2, respectively.
FIGURE 8.

Comparison with previous results. A, plots of lifetime versus force of GPIbα bonds with 1238-A1 (black square) or 1261-A1 (red square) measured by BFP in this study (mean ± S.E. of >20 measurements per point), with 1238-A1 measured by AFM in our previous study (6) (orange diamond), and with 1261-A1 measured by OT in a previous study (15) (blue circle and triangle). B, comparison of 1261-A1 lifetime distributions measured by BFP in this study (black symbols) and by OT in the previous study (15) (blue symbols) at two very close force bins (squares and circles). C–E, lifetime distributions measured by BFP in this study (black) and those measured by AFM in our previous study (red) were compared at three representative forces: 5 pN for the low force regime where the low slip bond was observed in this study but not in our previous study (C), 22 pN for the intermediate force regime where the catch bond was observed in both of our studies (D), and 50 pN for high force regime where the high slip bond was observed (E) in both of our studies.
RESULTS
We used a BFP to measure adhesion frequency and bond lifetime between VWF, A1 domains, and polypeptides coated on a glass bead (Fig. 1, B and D, left, Probe) attached to the apex of a micropipette-aspirated RBC (Fig. 1, A and C, left) and GPIbα or glycocalicin immobilized on another glass bead (Fig. 1, B and D, right) or platelet aspirated by an apposing micropipette (Fig. 1, A and C, right, Target). For better molecular orientation with purified protein, GPIbα was captured from platelet lysates by WM23, a mAb with the epitope in the macroglycopeptide region (Fig. 1B). After a controlled contact, the BFP detected the absence (Fig. 1E) or the presence (Fig. 1F) of adhesion from the force signal upon target retraction, calculated adhesion frequency from repeated tests (Fig. 1G), and measured bond lifetime under constant force (Fig. 1F).
The binding specificity was established as GPIbα-bearing targets (lysate GPIbα, glycocalicin, or platelet GPIbα) adhered at significantly higher frequencies to probes coated with VWF ligands (1238-A1, 1261-A1, or VWF) than those not coated with VWF ligands or blocked by anti-A1 or anti-GPIbα mAb (Fig. 1G). The severely shortened lifetimes of the rare adhesions in the presence of blocking antibodies suggest that the remaining adhesion events are mostly nonspecific (data not shown).
GPIbα Dissociates from 1238-A1 as a Triphasic Slip-Catch-Slip Bond, 1261-A1 as a Monophasic Slip-only Bond, and Full-length VWF as a Biphasic Catch-Slip Bond
Using AFM in a previous study (6), we observed that 1238-A1-glycocalicin dissociated as a biphasic catch-slip bond at forces of >15 pN. A recent study using OT reported that the 1261-A1-GPIbα dissociation behaved as a “flex bond” at forces of <20 pN that switched between two slip-bonds with 20-fold different off rates (15). To investigate this apparent discrepancy, we measured single-bond lifetimes in the full force regime by BFP using both 1238- and 1261-A1 domains. The former includes the Gln1238–Glu1260 sequence, whereas the latter excludes it. We observed a very similar catch-slip bond for 1238-A1 dissociation from GPIbα (captured from platelet lysates by WM23) in the same force regime as that of the previous AFM study (6) (Fig. 2A, blue circle). At forces of <15 pN, however, an additional slip bond was observed, qualitatively similar to the slip bond observed in the OT study (15). In addition to force clamp experiments, we used a thermal fluctuation assay to directly measure the bond lifetime at zero force (32). The value of 5.4 ± 1.6 s so measured matched the zero force extrapolation of the slip bond trend in the low force regime (Fig. 2A, blue circle). Thus, the force-dependent single 1238-A1–GPIbα bond lifetime exhibits a triphasic pattern transitioning from a slip bond at 0–16 pN to a catch bond at 16–25 pN, followed by another slip bond at >25 pN.
In sharp contrast, the 1261-A1–GPIbα interaction was a slip-only bond whose lifetime decreased monotonically with increasing force (Fig. 2A, red triangle). Below 15 pN, the lifetime versus force curves of the two A1s coincided. Between 16 and 25 pN, however, the lifetime curve for 1238-A1 increased, whereas that for 1261-A1 decreased. Beyond 25 pN, the lifetimes of GPIbα bonds with both A1 domains decreased with increasing force, but the bond lifetimes were significantly shorter for 1261-A1 than 1238-A1. In addition, without force-induced bond strengthening, few 1261-A1 bonds survived the target retraction to reach >40 pN forces for lifetime measurement (Fig. 2A). These results indicate that the Gln1238–Glu1260 sequence in cis plays a key role in stabilizing the A1-GPIbα interaction under force, because truncating this sequence eliminated the A1 catch bond with GPIbα.
To determine how well the GPIbα binding properties of the A1 domain represent those of VWF, we measured the force-dependent lifetime of single GPIbα bond with full-length VWF using BFP thermal fluctuation and force clamp assays. Gratifyingly, the lifetime versus force curve of the VWF–GPIbα bond (Fig. 2A, black diamond) was very similar to that of the 1238-A1–GPIbα bond (Fig. 2A, blue circle) at the catch-slip bond force regime. At lower forces (<16 pN), however, the GPIbα bond with VWF continued to behave as a catch bond, sharply contrasting to those with both A1s. At zero force, the GPIbα bond lifetime was ∼80-fold lower with VWF than A1 (0.067 versus 5.4 s). These data suggest an autoinhibitory mechanism involving VWF domains surrounding A1 that reduced the bond stability with GPIbα. However, this inhibition can be relieved as VWF was progressively activated by increasing force such that the GPIbα bond with 1238-A1 achieved its full stability as that with VWF, giving rise to the VWF–GPIbα catch bond at forces <16 pN. These data also caution us not to overinterpret and generalize binding results obtained using A1 domains before confirmation with full-length VWF.
Platelet Rolling Velocities on 1238-A1, 1261-A1, and VWF Were Governed by Their Respective Slip-Catch-Slip, Slip-only, and Catch-Slip Bonds with GPIbα
Previously, we demonstrated an inverse relationship between rolling velocity and bond lifetime—the longer the bond lifetime, the slower the rolling velocity (6, 39)—that provided definitive evidence that flow-enhanced rolling is caused by catch bonds (40). To corroborate the single-bond lifetime results in Fig. 2A, we measured platelet rolling on surfaces coated with either of the two A1 domains or VWF over a range of wall shear stresses in a flow chamber. The velocities of washed platelets rolling on these substrates showed stress-dependent patterns mirroring the corresponding patterns of bond lifetimes: triphasic for 1238-A1, monophasic for 1261-A1, and biphasic for VWF (Fig. 2B). At shear stress >1 dyn/cm2, 1261-A1 no longer supported platelet rolling, preventing measurements beyond that point. This is evident from the snapshots taken at 8 dyn/cm2 that show 1238-A1 supported platelet tethering and rolling at high shear, whereas 1261-A1 did not (Fig. 2C).
Polypeptide Gln1238–Glu1260 Binds GPIbα and 1261-A1 and Inhibits Platelet Adhesion
It has been reported that the VWF-derived polypeptide Leu1232–Asp1261 acts as a putative GPIbα binding site and can inhibit VWF binding to platelet (30). In addition, it was recently suggested that this A1 N-terminal sequence may bind A1 noncovalently (31). We therefore measured direct binding of BFP probes bearing a similar polypeptide Gln1238–Glu1260 (Lp) to BFP targets coated with GPIbα, VWF, 1238-A1, or 1261-A1. Specific binding of Lp to GPIbα, VWF, and 1261-A1, but not 1238-A1, was observed as the adhesion frequencies to the former three molecules (but not the last molecule) were abrogated when Lp was replaced by a control scramble polypeptide (Sp) (Fig. 2D, closed bars). To compare the propensities of Lp binding to these molecules, the specific adhesion frequencies (Pa, obtained by subtracting the adhesion frequency of Sp from that of Lp) were converted to effective binding affinities per unit receptor site density (−ln(1 − Pa)/mr) by normalizing the average number of adhesion bonds (−ln(1 − Pa)) by the respective site densities of 1238-A1, 1261-A1, VWF, and GPIbα (mr). Although we did not measure the site density of Lp, the same batch of Lp-bearing beads were used to analyze binding of all four receptors. Hence the respective effective affinities per unit receptor density should be equal to the effective two-dimensional affinities of Lp for 1238-A1, 1261-A1, VWF, and GPIbα multiplied by the same constant of Lp site density. The values for the latter three molecules were ∼11-, 4.7-, and 17-fold higher than that for 1238-A1, respectively (Fig. 2D, open bars). Compared with 1261-A1 and GPIbα, the VWF binding to Lp was much weaker, possibly because this binding was mediated by the VWF structure outside 1238-A1. Additional population experiments by ELISA (Fig. 2E) confirmed the direct interactions of Lp to GPIbα and 1261-A1 by single-bond experiments. Thus, VWF has at least two binding sites for GPIbα: one resides within 1261-A1 and the other involves Gln1238–Glu1260. This sequence also binds noncovalently to 1261-A1 and other VWF domains in addition to forming a covalent peptide bond with 1261-A1 in the full-length VWF structure.
To investigate the functional role of Lp in platelet adhesion under shear, we perfused whole blood mixed with soluble Lp or Sp over a collagen-coated (Fig. 2F), VWF-coated (Fig. 2G), or 1238-A1-coated (Fig. 2H) surface in a flow chamber. These experiments mimic the hemostatic scenario at disrupted vascular sites where plasma VWF deposits on exposed subendothelum to capture circulating platelets (41). Many more attached platelets were observed when whole blood was mixed with Sp (Fig. 2, F–H, right panels) than with Lp (Fig. 2, F–H, left panels), confirming the role of Gln1238–Glu1260 to inhibit the binding between platelet and VWF (30).
Soluble Gln1238–Glu1260 Reduces GPIbα Bond Lifetimes with VWF and 1238-A1 but Rescues the Catch Bond with 1261-A1
Because Lp binds GPIbα much better than VWF (Fig. 2D), its inhibitory effect on platelet adhesion (Fig. 2, F–H) may be explained by its binding to GPIbα (Fig. 2, D and E), which may interfere, at least in part, with the VWF–GPIbα interaction that mediates platelet adhesion. To test this hypothesis, we used BFP to directly measure the effect of Lp on force-dependent VWF–GPIbα bond lifetime. Supporting our hypothesis, VWF–GPIbα bond lifetime was shortened by the addition of soluble Lp (Fig. 3A), but not Sp (compare Figs. 2A and 3A), over the full force regime.
FIGURE 3.
The effect of soluble Gln1238–Glu1260 on GPIbα interactions with VWF and two forms of A1. A–C, plots of lifetime versus force of platelet lysate GPIbα bonds with VWF (A), 1238-A1 (B), or 1261-A1 (C) in the presence of soluble Lp or Sp (10 μg/ml or 4.2 μm). Lifetimes (mean ± S.E. of >20 measurements per point) were measured by the thermal fluctuation assay (closed symbols) at zero force and by the force clamp assay at nonzero forces (open symbols).
To further elucidate the mechanism for the A1 N-terminal flanking region to regulate platelet binding, we measured lifetimes of GPIbα bonds with 1238-A1 and 1261-A1 in the presence of soluble Lp or Sp. As expected, the respective lifetime versus force curves of GPIbα interactions with 1238-A1 and 1261-A1 were indistinguishable in the absence (Fig. 2A) and presence (Fig. 3, B and C) of Sp, for the control sequence should have no effect. In the presence of Lp, however, the lifetime curves of the 1238-A1 (Fig. 3B) downshifted but still retained their triphasic profile. More interestingly, the monophasic lifetime curve of 1261-A1 became triphasic, first decreasing, then increasing, and again decreasing with increasing force (Fig. 3C). In fact, in the presence of Lp, the 1261-A1 and 1238-A1 curves were close to each other but distinct from the VWF curve. Thus, soluble Gln1238–Glu1260 suppresses GPIbα binding to VWF and 1238-A1 but rescues the GPIbα catch bond with 1261-A1. The former suggests that Lp competes for GPIbα binding with VWF and 1238-A1, whereas the latter suggests that a possible 1261-A1–Lp–GPIbα sandwich bond may restore the catch bond behavior. These data show that the Gln1238–Glu1260 sequence plays a similar role in trans as it does in cis in stabilizing the A1–GPIbα bond against increasing force. The inability for the recent OT study (15) to observe GPIbα catch bond may be explained by their use of 1261-A1 in the fusion construct.
Effects of Different Forms of WT GPIbα
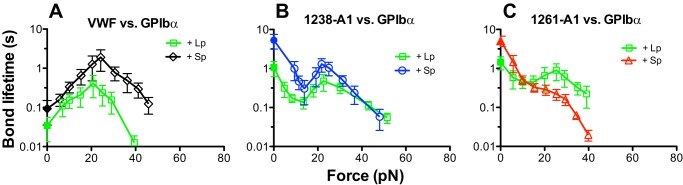
In addition to studying different force regimes and using different A1 domains, another possible cause of the discrepancies between our previous AFM data (6) and the OT data from another study (15) may be the use of different forms of GPIbα. To test this possibility, we compared lysate GPIbα from platelets or CHOαβ9 cells captured by WM23 mAb on target beads (Fig. 1B) and glycocalicin covalently linked to target beads (Fig. 1D) with native GPIbα expressed on platelets (Fig. 1C). The lifetime curves of 1238-A1 bonds with GPIbα captured from lysates of platelets (Fig. 4A, black square) or CHOαβ9 cells (33) (Fig. 4B, black diamond) or with glycocalicin (Fig. 4C, black circle) are indistinguishable from 1238-A1–platelet curve, displaying the same triphasic force dependence (Fig. 4, red square). These data indicate that the 1238-A1–GPIbα bond dissociation characteristic is determined by the extracellular domain of GPIbα and not affected by the different post-translational modifications of GPIbα in platelets and CHOαβ9 cells or how GPIbα is immobilized on surfaces.
FIGURE 4.
Force-dependent lifetimes of 1238-A1 bonds with different GPIbα preparations. Plots of lifetime versus force of 1238-A1 bonds with GPIbα from lysates of platelets (A, black squares) or CHOαβ9 cells (B, black diamonds) captured on targets or with glycocalicin (C, black circles) coated on targets. The curve of 1238-A1 bonds with GPIbα expressed on platelets (red squares) were used as the standard to compare the properties of purified proteins. The previous data of 1238-A1 interaction with platelet lysate GPIbα from Fig. 2A were replotted for comparison in A. Lifetimes (mean ± S.E. of >20 measurements per point) were measured by the thermal fluctuation assay (closed symbols) at zero force and by the force clamp assay at nonzero forces (open symbols).
Effects of VWD Mutants
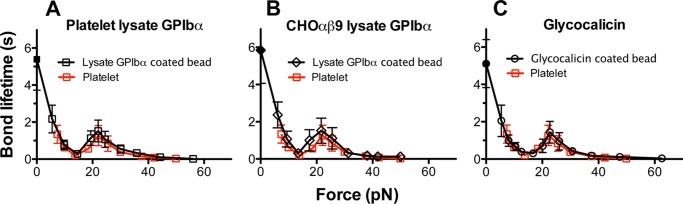
The putative conformational change resulting in VWF activation may be induced or affected by VWF or GPIbα mutations naturally occurring in patients with VWD. Our previous AFM study demonstrated that a type 2B VWD mutation, 1238-A1R1450E, eliminated the catch bond found in the 1238-A1–glycocalicin interaction by prolonging the lifetimes at low forces, resulting in an up and left shifts of the lifetime versus force curve relative to the WT curve (6). In contrast, a type 2M VWD mutant 1238-A1G1324S shortened lifetimes at low forces and right-shifted the biphasic catch-slip bond curve toward higher forces (12). The present BFP study extended the lifetime versus force curves for these mutants to lower forces. We found that glycocalicin interaction with 1238-A1R1450E displayed a monophasic slip-only bond (Fig. 5A, black), but that with 1238-A1G1324S showed a right-shifted triphasic slip-catch-slip bond (Fig. 5B, black), qualitatively similar to that with the WT 1238-A1 (Fig. 5, gray). These data confirm the previous AFM results at forces >20 pN and add a new slip-bond regime at low forces for 1238-A1G1324S. The extended results also included lifetimes at zero force measured via the thermal fluctuation assay, which are 13.9 ± 2.7 and 4 ± 1.1 s for R1450E (Fig. 5A, black closed square) and G1324S (Fig. 5B, black closed square), respectively. The former value is much longer, whereas the latter value is slightly shorter than the zero force lifetime of 5.1 ± 1.2 s for the WT 1238-A1 (Fig. 5, gray closed square), explaining the GOF phenotype for 1238-A1R1450E and the loss of function phenotype for 1238-A1G1324S. We also studied a ptVWD mutant GPIbα (G233V) captured from lysates of G233V CHOαβ9 cells. Consistent with its GOF phenotype in other assays (42–44), the lifetime versus force curve of 1238-A1 bonds with GPIbαG233V (Fig. 5C, black) was significantly upshifted relative to that with WT GPIbα (Fig. 5C, gray). The zero force lifetime measured by the thermal fluctuation assay was 20.5 ± 7.4 s, ∼4-fold longer than the WT value of 5.8 ± 1.8 s (p < 0.01 by Student's t test).
FIGURE 5.
Force-dependent lifetimes of 1238-A1-GPIbα bonds with VWD mutations. A and B, plots of lifetime versus force of glycocalicin bonds with type 2B VWD mutant 1238-A1R1450E (A) and type 2M VWD mutant 1238-A1G1324S (B) (black squares). The previous data of 1238-A1-glycocalicin interaction from Fig. 4C were replotted for comparison (gray squares). C, plots of lifetime versus force of 1238-A1 bonds with WT (gray squares) or G233V (black squares) GPIbα from CHOαβ9 cell lysates. Lifetimes (mean ± S.E. of >20 measurements per point) were measured by the thermal fluctuation assay (closed symbols) at zero force and by the force clamp assay at nonzero forces (open symbols).
Characterization of A1-GPIbα Dissociation by a Two-state Model
In addition to the force-dependent average of lifetimes, their distributions also demonstrated that 1238-A1–GPIbα interaction dissociated as a slip-catch-slip bond (Fig. 6, A–C), but 1261-A1–GPIbα interaction dissociated as a slip-only bond (Fig. 6, D–F). For the first order irreversible dissociation of a single monomeric bond from a single state along a single pathway in a single step, lifetime at each force bin should follow a single-exponential distribution, appearing linearly in the semi-log survival frequency versus lifetime plot, with the negative slope equal to the off rate. However, the semi-log lifetime distributions at many force bins (Fig. 6) appear as two line segments connected at ∼2 s, invalidating the assumption underlying the simple kinetics model that predicts a single exponential decay (Equation 1). We found that such double exponentially distributed lifetimes are similar to those of the bonds between α5β1 integrin and fibronectin (45, 46) but distinct from those of the bonds between P- or L-selectin and P-selectin glycoprotein ligand 1 or other ligands, whose lifetimes are distributed as single exponentials (47, 48).
FIGURE 6.

Bond lifetime distributions. Survival frequencies, calculated as the fraction of measurements (including both short and long lifetime events) with a bond lifetime >tb, were plotted versus tb for each force bin in each of three regimes: low (A and D), intermediate (B and E), and high (C and F) forces, for GPIbα bonds with 1238-A1 (A–C) and 1261-A1 (D–F). These forces corresponded to the low slip bond, the catch bond, and the high slip bond regime for 1238-A1 interactions, but 1261-A1 interactions only showed slip bonds in the entire force regime. The arrows indicate the directions of force increase.
Assuming that two subpopulations of A1-GPIbα bonds coexist and dissociate at a slow (k1) and a fast (k2) off rate, respectively, we analyzed the lifetime distributions using a two-state model (Equation 2) to obtain the best fit force-dependent off rates and their associated fractions of these subpopulations of bonds of GPIbα with 1238-A1 (Fig. 7, A and B) and with 1261-A1 (Fig. 7, C and D). For GPIbα dissociation from both A1s, the off rate of the long-lived bond (k1) was 1 order of magnitude smaller than that of the short-lived bond (k2) at zero force (Fig. 7, A and C), which is consistent with the results obtained by the recent OT study at low forces (15). Because the fractions of the long-lived (ω1) and short-lived (ω2) bonds were similar (Fig. 7, B and D), the long-lived bonds dominated the average lifetime. Although the relative fractions were stable around the catch bond force regime, overall, force gradually increased the fraction of short-lived bonds at the expense of the long-lived bonds. The fast off rate k2 increased monotonically as the force increased. By comparison, the slow off rate k1 first increased, then decreased, and again increased with force, accounting for the triphasic slip-catch-slip bond behavior. Surprisingly, the two states merged into one at force above 60 pN for 1238-A1 (Fig. 7A), but at much lower force ∼30 pN for 1261-A1 (Fig. 7C). This analysis confirms that losing the Gln1238–Glu1260 sequence makes forced dissociation of GPIbα from 1261-A1 much faster than 1238-A1.
FIGURE 7.

Analysis of two-state kinetics. A two-state model was fit to the bond lifetime distribution at each force bin of GPIbα dissociation from 1238-A1 (A and B) or 1261-A1 (C and D) to evaluate model parameters at that force, which are plotted versus force. The kinetic parameters are: the slow (k1) and fast (k2) off rates of dissociation from the two states of different stabilities (A and C) and the fractions (ω1 and ω2, ω1 + ω2 = 1) of bonds associated with the corresponding short-lived (high off rate) and long-lived (low off rate) states (B and D). The error bars represent ± 95% confident interval of the best fit value.
Comparison with Previous Measurements by Different Methods
We replotted the present BFP lifetime versus force curves in Fig. 8A to compare with previous data obtained by AFM using 1238-A1 (6) and OT using 1261-A1 (15). At low forces, the OT measurements determined two states that dissociated at distinct rates (Fig. 8A, blue circle and blue triangle): one state at low forces and the other engaging at ∼10 pN with a 20-fold longer lifetime and a greater force resistance. The authors termed this behavior “flex bond” (15). For a direct comparison, we overlaid the 1261-A1 lifetime distributions measured by the BFP (Fig. 8B, black symbols) on those measured the OT at ∼10 pN (Fig. 8B, blue symbols). The BFP measurement at ∼10 pN also showed two states, although only the long-lived state was consistent with the OT data. However, we observed two states at forces between 0 and 60 pN for 1238-A1 (Fig. 7B) and between 0 and 30 pN for 1261-A1 (Fig. 7D) instead of just a single state below 9.55 pN that flexed to another state above 10.05 pN (15). The effect of force measured by the BFP was gradual; 1-pN force change ∼10 pN did not drastically change the two-state characteristics of the A1-GPIbα bond (Fig. 8B, black symbols).
The second and third phases of the triphasic bond lifetime versus force curve in the present BFP study are qualitatively similar to the biphasic lifetime curve observed in our previous AFM study in the same force range and with the same 1238-A1 construct but have quantitatively longer lifetimes (6). This is due to an improvement—a counter-drifting system—to the BFP used in the present study, which enabled us to measure longer bond lifetimes. By comparison, bond lifetimes of >2 s were excluded as possible outliers in our previous AFM experiments (6), for they might be affected by instrument drift and/or resulted from multiple bonds. This can be seen by overlaying the lifetime distributions of the present BFP study with those of the previous AFM study measured at three representative force bins. Because of the 2-s cutoff in the lifetime measurement, the AFM data only match the short-lived state portion of the BFP data in the first slip regime (5 pN; Fig. 8C) and the catch regime (22 pN; Fig. 8D) but miss the long-lived state of the BFP data, explaining the shorter average lifetimes of the AFM data than the BFP data (Fig. 8A). By comparison, the AFM and BFP data match well in the second slip bond regime (55 pN; Fig. 8E) where the short-lived state dominates over the long-lived state (Fig. 7B), thus preventing the 2-s cutoff used in the AFM study from biasing the data.
DISCUSSION
The present study investigated the structural determinants of force-induced activation of VWF binding to GPIbα. We found that the 1238-A1–GPIbα could fully account for VWF–GPIbα at forces >20 pN, which behaved as a catch-slip bond, in agreement with our previous results (6). As force decreased, the VWF-GPIbα continued to behave as a catch bond even when force became zero, whereas the 1238-A1-GPIbα turned into a slip bond with ∼80-fold longer lifetime at zero force (Fig. 2A). Similar triphasic force-dependent kinetics has been observed by us in the dissociations of sLex-bearing microspheres and carbohydrate ligand-expressing cells from E-selectin (49). The 1238-A1-GPIbα slip bond at low force suggests that VWF structural elements surrounding the 1238-A1 impede its binding to GPIbα, which may be relieved by force. In sharp contrast, the 1261-A1-GPIbα bond behaved as a slip-only bond in the entire force range and hence did not capture the dissociation characteristics of the VWF-GPIbα bond despite the resemblance with the 1238-A1-GPIbα bond at force <15 pN (Fig. 2A).
The distinct GPIbα dissociation (Fig. 2A) from and platelet rolling (Fig. 2, B and C) on 1238-A1 and 1261-A1 highlight the regulatory role of the A1 N-terminal flanking region. The addition of Lp to whole blood suppressed platelet attachment to immobilized collagen (Fig. 2F), VWF (Fig. 2G), and 1238-A1 (Fig. 2H) under flow, which may be explained, at least in part, by the competitive binding of this polypeptide to GPIbα (Fig. 2, D and E). This may reduce the VWF-GPIbα complex interface, thereby shortening bond lifetime (Fig. 3A). Interestingly, soluble Lp weakens the catch bond for 1238-A1 by downshifting the lifetime curve (Fig. 3B), whereas it rescues the catch bond for 1261-A1 by prolonging the lifetime of its bond with GPIbα at intermediate forces (Fig. 3C). The former can be explained by an inhibitory role of Lp, which was shown previously (30) and confirmed in this study (Fig. 2). Note that the zero force lifetimes for both 1238- and 1261-A1s were reduced by soluble Lp (Fig. 3, B and C), suggesting that Lp exerts its inhibitory effect at the low force regime and increasing force relieves the inhibitory effect. The data also suggest a similar function of the Gln1238–Glu1260 sequence in stabilizing the A1-GPIbα interaction at forces >16 pN regardless of whether it exists in cis (Fig. 2A, 1238-A1) or in trans (Fig. 3B, 1261-A1 + Lp) to A1. This may be explained by the interaction of Lp with 1261-A1 and GPIbα (Fig. 2, D and E), which may provide an additional cross-link by sandwiching Lp between 1261-A1 and GPIbα to strengthen their binding interface.
We extended our previous results on two 1238-A1 mutations by showing that the type 2B VWD mutant R1450E eliminated the catch bond by prolonging bond lifetime at low forces (Fig. 5A) and type 2M VWD mutant G1324S right-shifted the bond lifetime versus force curve (Fig. 5B). In addition, we analyzed 1238-A1 dissociation from the ptVWD mutant GPIbαG233V for the first time and found that this mutation significantly prolonged single-bond lifetime at all forces tested (Fig. 5C). Importantly, thermal fluctuation experiments showed that the bond lifetime at zero force for 1238-A1R1450E was ∼2-fold longer than WT (p < 0.05), that for 1238-A1G1324S was 26% shorter than WT (p < 0.05), and that for GPIbαG233V was ∼4-fold longer than WT (p < 0.01) (Fig. 5). These results are consistent with their respective GOF and loss of function phenotypes found in other assays, explaining the spontaneous binding of VWF to platelet for type 2B VWD (50, 51) and ptVWD (42–44), as well as the ineffective binding of VWF to platelet for type 2M VWD (22, 52) observed in vitro.
To reconcile the discrepant results between our previous AFM study (6) and a recent OT study (15), we investigated several differences between the two studies. We showed that 1238-A1 dissociated from glycocalicin (used in our previous work (6)) indistinguishably from platelet GPIbα or GPIbα from lysates of platelets or CHOαβ9 cells (Fig. 4). The present BFP data qualitatively agreed with our previous AFM data at forces >20 pN and the OT data at forces <20 pN (Fig. 8), indicating that the different force ranges used by the two studies may contribute to the discrepancies, whereas the different measurement techniques used were not the cause of the discrepancies. However, the different A1 constructs have been identified as the main cause of the discrepancies because the present BFP measurements qualitatively reproduced the same force-dependent lifetimes of GPIbα bonds with 1238-A1 as the previous AFM measurements (6) and with 1261-A1 as the recent OT measurements (15) in their respective force ranges. We note that considerable variations in their ability to support platelet attachment between the N-longer and N-shorter A1s have long been reported. It has been shown that an N-longer A1 interacted with platelets at 50–6,000 s−1 shear rates indistinguishably from full-length VWF, whereas an N-shorter A1 exhibited progressive loss of activity with increasing shear above 1,500 s−1 and became essentially inactive at 6,000 s−1. Our data reaffirm the importance of the VWF-GPIbα catch bond by its direct observation with full-length VWF and by elucidation of its different force-dependent dissociation characteristics from the two A1 domains. More significantly, both the present and our previous studies showed that the VWF-GPIbα catch bond governed flow-enhanced platelet rolling on VWF (Fig. 2B). Although we also observed a slip bond at forces of <20 pN resembling a flex bond (Fig. 8B) as reported by the OT study (15), this behavior differs from that of full-length VWF. Thus, our work has elucidated the regulation of VWF activation and catch bond formation with GPIbα by the A1 N-terminal flanking region.
Recently, a protein thermodynamic study showed that the cis-binding of Lp to A1 stabilizes the overall 1238-A1 structure (53). However, their flow chamber data showed that platelets roll slower on 1261-A1 than 1238-A1 under a range of shear stresses and suggested that 1261-A1 would also adapt a catch bond as 1238-A1 does. A possible explanation for this discrepancy is that two binding states exist for 1261-A1. Clarification of this issue is one of the goals of future studies.
Acknowledgments
We thank Michael Berndt for providing anti-A1 mAb (5D2); Renhao Li for providing anti-GPIbα mAb (WM23); Carol Sun for the purification of glycocalicin; and Jizhong Lou, Zhenhai Li, Wei Chen, Kaitao Li, Ning Tan, Veronika I. Zarnitsyna, and Rodger P. McEver for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants HL091020 (to C. Z.), HL072886 (to M. A. C.), and HL71895 (to J.-F. D.).
- VWF
- von Willebrand factor
- AFM
- atomic force microscopy
- BFP
- biomembrane force probe
- Lp
- polypeptide Gln1238–Glu1260
- OT
- optical tweezers
- ptVWD
- platelet-type VWD
- Sp
- scramble polypeptide
- VWD
- von Willebrand disease
- GPIbα
- Glycoprotein Ibα
- GOF
- gain of function
- RBC
- red blood cell.
REFERENCES
- 1. Ruggeri Z. M. (2003) Von Willebrand factor, platelets and endothelial cell interactions. J. Thromb. Haemost. 1, 1335–1342 [DOI] [PubMed] [Google Scholar]
- 2. Andrews R. K., Berndt M. C. (2004) Platelet physiology and thrombosis. Thrombosis Research 114, 447–453 [DOI] [PubMed] [Google Scholar]
- 3. Ruggeri Z. M., Mendolicchio G. L. (2007) Adhesion mechanisms in platelet function. Circ. Res. 100, 1673–1685 [DOI] [PubMed] [Google Scholar]
- 4. Lenting P. J., Pegon J. N., Groot E., de Groot P. G. (2010) Regulation of von Willebrand factor-platelet interactions. Thromb. Haemost. 104, 449–455 [DOI] [PubMed] [Google Scholar]
- 5. Mazzucato M., Santomaso A., Canu P., Ruggeri Z. M., De Marco L. (2007) Flow dynamics and haemostasis. Ann. Ist. Super. Sanita 43, 130–138 [PubMed] [Google Scholar]
- 6. Yago T., Lou J., Wu T., Yang J., Miner J. J., Coburn L., López J. A., Cruz M. A., Dong J.-F., McIntire L. V., McEver R. P., Zhu C. (2008) Platelet glycoprotein Ibα forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J. Clin. Invest. 118, 3195–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jackson S. P., Nesbitt W. S., Westein E. (2009) Dynamics of platelet thrombus formation. J. Thromb. Haemost. 7, 17–20 [DOI] [PubMed] [Google Scholar]
- 8. Shankaran H., Alexandridis P., Neelamegham S. (2003) Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood 101, 2637–2645 [DOI] [PubMed] [Google Scholar]
- 9. Martin C., Morales L. D., Cruz M. A. (2007) Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibα. J. Thromb. Haemost. 5, 1363–1370 [DOI] [PubMed] [Google Scholar]
- 10. Auton M., Sowa K. E., Smith S. M., Sedlák E., Vijayan K. V., Cruz M. A. (2010) Destabilization of the A1 domain in von Willebrand factor dissociates the A1A2A3 tri-domain and provokes spontaneous binding to glycoprotein Ibα and platelet activation under shear stress. J. Biol. Chem. 285, 22831–22839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ulrichts H., Udvardy M., Lenting P. J., Pareyn I., Vandeputte N., Vanhoorelbeke K., Deckmyn H. (2006) Shielding of the A1 domain by the D′D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J. Biol. Chem. 281, 4699–4707 [DOI] [PubMed] [Google Scholar]
- 12. Auton M., Sedlák E., Marek J., Wu T., Zhu C., Cruz M. (2009) Changes in thermodynamic stability of von Willebrand factor differentially affect the force-dependent binding to platelet GPIbα. Biophys. J. 97, 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Auton M., Zhu C., Cruz M. A. (2010) The mechanism of VWF-mediated platelet GPIbα binding. Biophys. J. 99, 1192–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coburn L. A., Damaraju V. S., Dozic S., Eskin S. G., Cruz M. A., McIntire L. V. (2011) GPIbα-vWF rolling under shear stress shows differences between type 2B and 2M von Willebrand disease. Biophys. J. 100, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J., Zhang C.-Z., Zhang X., Springer T. A. (2010) A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature 466, 992–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cruz M. A., Handin R. I. (1993) The interaction of the von Willebrand factor-A1 domain with platelet glycoprotein Ib/IX. J. Biol. Chem. 268, 21238–21245 [PubMed] [Google Scholar]
- 17. Miyata S., Ruggeri Z. M. (1999) Distinct structural attributes regulating von Willebrand factor A1 domain interaction with platelet glycoprotein Ibα under flow. J. Biol. Chem. 274, 6586–6593 [DOI] [PubMed] [Google Scholar]
- 18. Celikel R., Ruggeri Z. M., Varughese K. I. (2000) von Willebrand factor conformation and adhesive function is modulated by an internalized water molecule. Nat. Struct. Biol. 7, 881–884 [DOI] [PubMed] [Google Scholar]
- 19. Cruz M. A., Diacovo T. G., Emsley J., Liddington R., Handin R. I. (2000) Mapping the glycoprotein Ib-binding site in the von willebrand factor A1 domain. J. Biol. Chem. 275, 19098–19105 [DOI] [PubMed] [Google Scholar]
- 20. Doggett T. A., Girdhar G., Lawshé A., Schmidtke D. W., Laurenzi I. J., Diamond S. L., Diacovo T. G. (2002) Selectin-like kinetics and biomechanics promote rapid platelet adhesion in flow. The GPIbα-vWF tether bond. Biophys. J. 83, 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arya M., Anvari B., Romo G. M., Cruz M. A., Dong J.-F., McIntire L. V., Moake J. L., López J. A. (2002) Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex. Studies using optical tweezers. Blood 99, 3971–3977 [DOI] [PubMed] [Google Scholar]
- 22. Morales L. D., Martin C., Cruz M. A. (2006) The interaction of von Willebrand factor-A1 domain with collagen. Mutation G1324S (type 2M von Willebrand disease) impairs the conformational change in A1 domain induced by collagen. J. Thromb. Haemost. 4, 417–425 [DOI] [PubMed] [Google Scholar]
- 23. Emsley J., Cruz M., Handin R., Liddington R. (1998) Crystal structure of the von Willebrand factor A1 domain and implications for the binding of platelet glycoprotein Ib. J. Biol. Chem. 273, 10396–10401 [DOI] [PubMed] [Google Scholar]
- 24. Celikel R., Varughese K. I., Madhusudan, Yoshioka A., Ware J., Ruggeri Z. M. (1998) Crystal structure of the von Willebrand factor A1 domain in complex with the function blocking NMC-4 Fab. Nat. Struct. Biol. 5, 189–194 [DOI] [PubMed] [Google Scholar]
- 25. Miura S., Li C. Q., Cao Z., Wang H., Wardell M. R., Sadler J. E. (2000) Interaction of von Willebrand factor domain A1 with platelet glycoprotein Ibα-(1–289). Slow intrinsic binding kinetics mediate rapid platelet adhesion. J. Biol. Chem. 275, 7539–7546 [DOI] [PubMed] [Google Scholar]
- 26. Uff S., Clemetson J. M., Harrison T., Clemetson K. J., Emsley J. (2002) Crystal structure of the platelet glycoprotein Ibα N-terminal domain reveals an unmasking mechanism for receptor activation. J. Biol. Chem. 277, 35657–35663 [DOI] [PubMed] [Google Scholar]
- 27. Huizinga E. G., Tsuji S., Romijn R. A., Schiphorst M. E., de Groot P. G., Sixma J. J., Gros P. (2002) Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science 297, 1176–1179 [DOI] [PubMed] [Google Scholar]
- 28. Dumas J. J., Kumar R., McDonagh T., Sullivan F., Stahl M. L., Somers W. S., Mosyak L. (2004) Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Ibα complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J. Biol. Chem. 279, 23327–23334 [DOI] [PubMed] [Google Scholar]
- 29. Fukuda K., Doggett T., Laurenzi I. J., Liddington R. C., Diacovo T. G. (2005) The snake venom protein botrocetin acts as a biological brace to promote dysfunctional platelet aggregation. Nat. Struct. Mol. Biol. 12, 152–159 [DOI] [PubMed] [Google Scholar]
- 30. Mohri H., Fujimura Y., Shima M., Yoshioka A., Houghten R. A., Ruggeri Z. M., Zimmerman T. S. (1988) Structure of the von Willebrand factor domain interacting with glycoprotein Ib. J. Biol. Chem. 263, 17901–17904 [PubMed] [Google Scholar]
- 31. Auton M., Sowa K. E., Behymer M., Cruz M. A. (2012) N-terminal flanking region of A1 domain in von Willebrand factor stabilizes structure of A1A2A3 complex and modulates platelet activation under shear stress. J. Biol. Chem. 287, 14579–14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen W., Evans E. A., McEver R. P., Zhu C. (2008) Monitoring receptor-ligand interactions between surfaces by thermal fluctuations. Biophys. J. 94, 694–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dong J., Schade A. J., Romo G. M., Andrews R. K., Gao S., McIntire L. V., López J. A. (2000) Novel gain-of-function mutations of platelet glycoprotein Ibα by valine mutagenesis in the Cys209-Cys248 disulfide loop. Functional analysis under statis and dynamic conditions. J. Biol. Chem. 275, 27663–27670 [DOI] [PubMed] [Google Scholar]
- 34. Chen W., Lou J., Evans E. A., Zhu C. (2012) Observing force-regulated conformational changes and ligand dissociation from a single integrin on cells. J. Cell Biol. 199, 497–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen W., Lou J., Zhu C. (2010) Forcing switch from short- to intermediate- and long-lived states of the A domain generates LFA-1/ICAM-1 catch bonds. J. Biol. Chem. 285, 35967–35978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang J., Zarnitsyna V. I., Liu B., Edwards L. J., Jiang N., Evavold B. D., Zhu C. (2010) The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 464, 932–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chesla S. E., Selvaraj P., Zhu C. (1998) Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 75, 1553–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cruz M. A., Chen J., Whitelock J. L., Morales L. D., López J. A. (2005) The platelet glycoprotein Ib-von Willebrand factor interaction activates the collagen receptor α2β1 to bind collagen. Activation-dependent conformational change of the α2-I domain. Blood 105, 1986–1991 [DOI] [PubMed] [Google Scholar]
- 39. Yago T., Wu J., Wey C. D., Klopocki A. G., Zhu C., McEver R. P. (2004) Catch bonds govern adhesion through L-selectin at threshold shear. J. Cell Biol. 166, 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu C., Yago T., Lou J., Zarnitsyna V. I., McEver R. P. (2008) Mechanisms for flow-enhanced cell adhesion. Ann. Biomed. Eng. 36, 604–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruggeri Z. M., Orje J. N., Habermann R., Federici A. B., Reininger A. J. (2006) Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood 108, 1903–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marchese P., Saldívar E., Ware J., Ruggeri Z. (1999) Adhesive properties of the isolated amino-terminal domain of platelet glycoprotein Ibα in a flow field. Proc. Natl. Acad. Sci. U.S.A. 96, 7837–7842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Doggett T. A. (2003) Alterations in the intrinsic properties of the GPIb-VWF tether bond define the kinetics of the platelet-type von Willebrand disease mutation, Gly233Val. Blood 102, 152–160 [DOI] [PubMed] [Google Scholar]
- 44. Kumar R. A., Dong J.-F., Thaggard J. A., Cruz M. A., López J. A., McIntire L. V. (2003) Kinetics of GPIbα-vWF-A1 tether bond under flow. Effect of GPIbα mutations on the association and dissociation rates. Biophys. J. 85, 4099–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kong F., García A. J., Mould A. P., Humphries M. J., Zhu C. (2009) Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 185, 1275–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kong F., Li Z., Parks W. M., Dumbauld D. W., García A. J., Mould A. P., Humphries M. J., Zhu C. (2013) Cyclic mechanical reinforcement of integrin-ligand interactions. Mol. Cell 49, 1060–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marshall B. T., Long M., Piper J. W., Yago T., McEver R. P., Zhu C. (2003) Direct observation of catch bonds involving cell-adhesion molecules. Nature 423, 190–193 [DOI] [PubMed] [Google Scholar]
- 48. Sarangapani K. K., Yago T., Klopocki A. G., Lawrence M. B., Fieger C. B., Rosen S. D., McEver R. P., Zhu C. (2004) Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J. Biol. Chem. 279, 2291–2298 [DOI] [PubMed] [Google Scholar]
- 49. Wayman A. M., Chen W., McEver R. P., Zhu C. (2010) Triphasic force dependence of E-selectin/ligand dissociation governs cell rolling under flow. Biophys. J. 99, 1166–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsushita T., Sadler J. E. (1995) Identification of amino acid residues essential for von Willebrand factor binding to platelet glycoprotein Ib. Charged-to-alanine scanning mutagenesis of the A1 domain of human von Willebrand factor. J. Biol. Chem. 270, 13406–13414 [DOI] [PubMed] [Google Scholar]
- 51. Ruggeri Z. M. (2004) Type IIB von Willebrand disease. A paradox explains how von Willebrand factor works. J. Thromb. Haemost. 2, 2–6 [DOI] [PubMed] [Google Scholar]
- 52. Rabinowitz I., Tuley E. A., Mancuso D. J., Randi A. M., Firkin B. G., Howard M. A., Sadler J. E. (1992) von Willebrand disease type B. A missense mutation selectively abolishes ristocetin-induced von Willebrand factor binding to platelet glycoprotein Ib. Proc. Natl. Acad. Sci. U.S.A. 89, 9846–9849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tischer A., Cruz M. A., Auton M. (2013) The linker between the D3 and A1 domains of vWF suppresses A1-GPIbα catch bonds by site-specific binding to the A1 domain. Protein Sci. 22, 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]