

Background: PARP activation at sites of DNA breaks leads to recruitment of chromatin remodeling enzymes such as ALC1.
Results: TRIM33 associates with ALC1 after DNA damage and regulates its retention at DNA breaks.
Conclusion: TRIM33 has a role in the PARP-dependent DNA damage response pathway.
Significance: The role of TRIM33 in the DNA repair may contribute to its known tumor suppressor function.
Keywords: ATPases, Cancer Biology, Cell Signaling, Chromatin, DNA Damage Response, ALC1, PARP, TRIM33
Abstract
Activation of poly(ADP-ribose) polymerase (PARP) near sites of DNA breaks facilitates recruitment of DNA repair proteins and promotes chromatin relaxation in part through the action of chromatin-remodeling enzyme Amplified in Liver Cancer 1 (ALC1). Through proteomic analysis we find that ALC1 interacts after DNA damage with Tripartite Motif-containing 33 (TRIM33), a multifunctional protein implicated in transcriptional regulation, TGF-β signaling, and tumorigenesis. We demonstrate that TRIM33 is dynamically recruited to DNA damage sites in a PARP1- and ALC1-dependent manner. TRIM33-deficient cells show enhanced sensitivity to DNA damage and prolonged retention of ALC1 at sites of DNA breaks. Conversely, overexpression of TRIM33 alleviates the DNA repair defects conferred by ALC1 overexpression. Thus, TRIM33 plays a role in PARP-dependent DNA damage response and regulates ALC1 activity by promoting its timely removal from sites of DNA damage.
Introduction
Higher-order chromatin structure acts as a major barrier for the detection and repair of DNA damage. Rapid and efficient modification of chromatin facilitates the accessibility of damaged DNA to the DNA repair machinery (1, 2). Breaks in DNA are known to result in rapid activation of the poly(ADP-ribose) (PAR)3 polymerases PARP1 and PARP2, which catalyze the assembly of PAR chains onto chromatin substrates (3–10). PAR modification at damage sites is believed to facilitate DNA repair by attracting PAR-binding DNA repair factors and promoting local chromatin relaxation (11–16).
PARP1/2 activity is required for efficient single-strand break repair, with PARP inhibition causing increased entry of unrepaired breaks into S phase, leading to replication fork stalling and DNA double-strand break equivalents (9, 17, 18). PARP1 and PARP2 are also activated at stalled replication forks, where they play a role in efficient fork restart (19). Upon synthesis by PARP activity, PAR can be rapidly degraded by poly(ADP-ribose) glycohydrolase (PARG). Deletion of the nuclear isoform of PARG leads to DNA repair defects and genomic instability, demonstrating that regulation of PAR production and degradation is critical for efficient DNA repair (20). The rapid assembly and disassembly of PAR and PAR binding proteins at DNA damage sites implies that this process is tightly controlled in the cell and raises the possibility that yet-unknown factors are involved in mediating and regulating these events.
DNA damage-induced PARylation can directly recruit DNA repair proteins such as XRCC1 (X-ray repair cross-complementing protein 1) and APLF (aprataxin- and PNKP-like factor), which contain specific PAR-binding motifs. The chromatin remodeling enzyme Amplified in Liver Cancer 1 (ALC1) is recruited to sites of DNA damage by directly binding PAR through its macro domain. PARP1 activity is thus required to locally target ALC1-dependent nucleosome remodeling, which may facilitate local chromatin relaxation and repair (12, 21, 22, 23). ALC1 is an oncogene that is amplified in some solid tumors, including hepatocellular carcinoma (49). Overexpression of ALC1 leads to altered chromatin structure and increased sensitivity to intercalating agents, such as phleomycin, that induce breaks preferentially at linker DNA (12, 21).
Tripartite motif-containing 33 (TRIM33) is a member of the TIF1 family of transcription regulators, which possess a RING domain, two B-boxes, and a coiled-coil domain at the N terminus as well as the plant homeo domain (PHD) and Bromo domains at the C terminus (Fig. 2A) (24) TRIM33 has been implicated previously in transcriptional regulation during hematopoiesis and interactions with elongation factors (25–28). TRIM33 was also shown to regulate the TGF-β pathway by interacting with both SMAD2/3 and SMAD4 (26, 29–31). TRIM33 helps recruit SMAD2/3 to chromatin via interaction of its PHD and Bromo domains with histone H3 trimethylated at lysine 9 (H3K9me3) and histone H3 acetylated at lysine 18 (H3K18ac), respectively. In embryonic stem cells, binding of the TRIM33 PHD domain to H3K9me3 displaces HP1-g from regions of silenced chromatin, enhancing transcriptional activation ability of the SMAD2/3-SMAD4 complex at target promoter regions (32). Histone binding is also required for TRIM33 ubiquitin ligase activity and its transcriptional repression function (33). TRIM33 may play a dual role in TGF-β signaling, initially enhancing the transcriptional activation function of the SMAD2–3·SMAD4 complex and then promoting the dissociation of this complex from chromatin (32, 33).
FIGURE 2.

TRIM33 participates in DNA damage response. A, Schematics of the TRIM33 constructs showing relevant domains and the mutant constructs used. B, HeLa cells were treated with UV laser scissors and processed for IF using antibodies to γH2AX (green) and TRIM33 (red) at the times indicated. C, quantitation of TRIM33 dynamics at the site of UV laser-induced DNA breaks (mean ± S.D. of at least 20 cells). D, HeLa cells transfected with FLAG-WtTRIM33, TRIM33CA, TRIM33ΔPHD, TRIM33 PHD(AAA), or TRIM33ΔBromo were treated with UV laser scissors and, after 10 min, processed for IF using antibodies to FLAG and γH2AX. E, quantitation of TRIM33 constructs at UV laser-induced DNA breaks (mean ± S.D. of at least 20 cells).
TRIM33 also functions as a tumor suppressor in multiple tissues. Targeted knockout of TRIM33 in liver leads to hepatocellular carcinoma in mice (34), whereas targeted knockout of TRIM33 in hematopoietic precursors leads to myeloproliferative disorders similar to chronic myelomonocytic leukemia (35, 36). Loss of TRIM33 also cooperates with K-ras activation to induce cystic tumors and adenocarcinomas of the pancreas in mice (37). Although the mechanism underlying the tumor suppression function of TRIM33 in these tissues remains unclear, a recent study suggests that this tumor suppressor function is separate from its functions in regulating SMAD4 (38).
Here we identify TRIM33 as an ALC1-interacting protein that is required for efficient DNA repair. We show that TRIM33 is rapidly recruited to sites of DNA breaks in a PAR- and ALC1-dependent manner. We further demonstrate that TRIM33 is required for the timely dissociation of ALC1 from damaged chromatin. Our results raise the possibility that TRIM33 acts to regulate ALC1 activity at DNA lesions. Indeed, we show that increased sensitivity to certain DNA-damaging agents associated with ALC1-overexpressing cells is reversed by concomitant overexpression of wild-type TRIM33. We propose that TRIM33 functions during the PARP-dependent DNA damage response to promote timely removal of ALC1 from damaged chromatin. Thus, TRIM33 regulates ALC1 function in the DNA damage response to facilitate efficient DNA repair.
MATERIALS AND METHODS
Plasmids and Proteins
FLAG-tagged WtTRIM33 and the FLAG-tagged TRIM33CA mutant described previously (29) were obtained from Addgene. The internal deletion FLAG-tagged PHD mutant (TRIM33ΔPHD) and TRIM33 PHD AAA mutants were a gift from the Massague laboratory (32). The internal Bromo domain deletion construct was amplified from FLAG-WtTRIM33 templates using appropriate primers (forward primer, 5′-aactgcgcaggggttacaggaccttcgcac-3′; reverse primer, 5′-gtgcgaaggtcctgtaacccctgcgcagtt-3′) and using a QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The colonies were screened by sequencing. GST-tagged WtTRIM33 (plasmid 15734, Addgene), TRIM33 ShRNA (plasmid 15728, Addgene), and TRIM33 GFP shRNA (plasmid 15721, Addgene) were as described previously (26). The TRIM33 siRNA (Ambion Inc.), FLP-In-FLAG, FLP-In-ALC1, FLP-In-ALC1K77R, and FLP-In-ALC1D723A constructs used have been described previously (12).
Antibodies
The antibodies used were rabbit anti-TRIM33 (Bethyl Laboratories), mouse anti-ALC1 and mouse anti-XRCC1 antibodies (Abcam), mouse anti γH2AX antibody and mouse anti-P21 antibody (Millipore), rabbit anti-γH2AX antibody (AbD Serotec), mouse anti-PAR antibody (Trevigen), rabbit anti-PARP1 antibody (Enzo Life Sciences), rabbit antibodies against total and phosphorylated Chk2 (Cell Signaling Technology), and mouse anti-tubulin antibody (Sigma). HRP-tagged mouse and rabbit secondary antibodies were purchased from Millipore.
Cell Lines, Cell Culture, and Inhibitors
The stable HEK293-FlP-In ALC1 cell line (12) was grown in regular DMEM supplemented with 150 μg/ml hygromycin B (Invitrogen). Stable U2OS shALC1 and shControl cell lines (12) were grown in DMEM supplemented with 2 μg/ml puromycin (Sigma). Parp1−/− and Parp1+/+ mouse embryonic fibroblasts were maintained in DMEM with 10% FBS and 1% penicillin/streptomycin. Wherever indicated, cells were treated either with PARP inhibitor KU-0058948 (KuDOS Pharmaceuticals) or PARP inhibitor ABT-888 (Abbott Laboratories) at 1 μm final concentration, ATM (Ataxia telangiectasia mutated) inhibitor KU-55933 at a final concentration of 20 μm, and gallotannin (Fluka Biochemika, Buchs, Switzerland) at a concentration of 25 μm.
Laser Microirradiation
Laser microirradiation was carried out as described previously with some modifications (39). To generate subnuclear DNA damage, a laser was focused with LD ×40, NA 0.6 Achroplan objective to yield a spot size of ∼1 μm. The laser output was set to 35% to generate localized damage assisted with PALM Robo software supplied by the manufacturer (P.A.L.M. Microlaser Technologies, Bernried, Germany). Approximately 50 cells were microirradiated in each experiment.
Immunofluorescence Microscopy
Cells were fixed with 4% buffered paraformaldehyde for 10 min, followed by permeabilization with 0.5% Triton X-100. Cells were then incubated for 1 h with the appropriate primary antibodies diluted in 5% goat serum. Cells were then washed and incubated with secondary antibodies coupled with FITC and rhodamine for immunodetection and mounted in Vectashield with DAPI (Vector Laboratories). Images were taken with a ×40 objective using a Nikon Eclipse 80i microscope.
PAR Binding Assay
A PAR binding assay was performed as described previously (12). Proteins were dot-blotted onto a nitrocellulose membrane and blocked with TBST (Tris-buffered saline and Tween 20) buffer supplemented with 5% milk. The nitrocellulose membrane was then incubated with radiolabeled PAR polymer in TBST buffer. The membrane was washed and subjected to autoradiography.
Protein Purification and Mass Spectrometry
Purification of ALC1-associated immunocomplexes was performed as described previously (12). Briefly, stable HEK293T FlP-In FLAG (control) and ALC1 cells were grown in roller bottles, pelleted, washed in PBS, and lysed for 10 min at 4 °C in sucrose buffer (10 mm HEPES (pH 7.9), 0.34 m sucrose, 3 mm CaCl2, 2 mm magnesium acetate, 0.1 mm EDTA, and protease inhibitors) containing 0.5% Nonidet P-40. Nuclei were then pelleted by centrifugation at 3900 × g for 20 min. Residual cytoplasmic contamination was removed by washing with sucrose buffer and subsequent centrifugation at 3900 × g for 20 min. Nuclei were resuspended in nucleoplasmic extraction buffer (20 mm HEPES (pH 7.9), 3 mm EDTA, 10% glycerol, 150 mm potassium acetate, 1.5 mm MgCl2, 1 mm DTT, and protease inhibitors), homogenized, and rotated for 20 min at 4 °C. The chromatin-enriched fraction was pelleted by centrifugation at 13,000 rpm for 30 min. The pellet was resuspended in digestion buffer (150 mm HEPES (pH 7.9), 1.5 mm MgCl2, 150 mm potassium acetate, and protease inhibitors), homogenized, and incubated with benzonase (25 units/μl stock) for 1 h at room temperature. The digested chromatin was cleared by centrifugation at 38,000 × g for 30 min. The soluble chromatin extract was recovered and used for anti-FLAG immunoprecipitation with anti-FLAG M2-agarose. Immunoprecipitated proteins were eluted by 3× FLAG peptide and then precipitated with 10% TCA. Proteins were then trypsinized, purified, and analyzed by LC-MS/MS on an LTQ mass spectrometer (Thermo).
Immunoprecipitation of the TRIM33-associated Complex
Endogenous coimmunoprecipitation using TRIM33 antibody was performed according to the protocol of the manufacturer using a nuclear complex coimmunoprecipitation kit (Active Motif). HeLa cells (8.8 × 106) were washed with ice-cold PBS/phosphatase inhibitors and collected by gentle scraping in the same buffer. Cells were centrifuged and resuspended in hypotonic buffer followed by incubation for 15 min and lysis using detergent. After centrifugation, the nuclear pellet was resuspended in complete digestion buffer with an enzymatic shearing mixture and incubated for 90 min at 4 °C. After centrifugation, supernatants were collected, protein was quantitated, and an equal quantity of protein was mixed with coimmunoprecipitation buffer and precleared with protein A-agarose beads for 2 h. Protein extracts were then incubated overnight at 4 °C with antibody against TRIM33. Protein A-agarose beads were then added and incubated for protein binding for 2 h at 4 °C. Beads were then washed with wash buffer, and proteins were eluted by boiling the beads with 2× Laemmli buffer and loaded onto the SDS-PAGE gels. Resolved proteins were transferred to nitrocellulose membrane and incubated with the appropriate antibody. Aliquots of the extracts were processed directly for Western blotting as an input control.
MTS Assay
To evaluate the effect of TRIM33 on bleomycin sensitivity, 293T cells were transfected with control or TRIM33-specific shRNA. To study the effect of exogenously expressed WtTRIM33 on rescuing the bleomycin sensitivity of TRIM33 knockdown cells, cells were cotransfected with TRIM33 shRNA and either FLAG-WtTRIM33. To study the effect of TRIM33 on rescuing the sensitivity of ALC1-overexpressing cells to bleomycin, FlP-In-WtALC1 cells were transfected with either empty vector or FLAG-tagged WtTRIM33. 48 h after transfection, cells were seeded into 96-well plates (3000 cells/well), treated with the indicated concentrations of DNA-damaging agents, and grown for another 3 days. The relative cell number was measured by incubating cells with Celltiter 96 Aqueous One Solution reagent (Promega) for 3 h and measuring the absorbance at 490 nm. The values were plotted as an average of two different experiments in the case of TRIM33 shRNA-treated cells and three experiments in ALC1 cell lines.
Comet Assay
A comet assay was carried out as described previously (12). FLP-In-FLAG, FLP-In-ALC1, and FLP-In-ALC1 cells transfected with WtTRIM33 were treated with the indicated concentrations of phleomycin. A Comet assay single cell gel electrophoresis kit (R&D Systems) was used to prepare samples according to the instructions of the manufacturer. Approximately 1 × 105/ml cells were combined with molten low-melting agarose at 37 °C at a ratio of 1:10 (v/v). 75 μl of this mix was pipetted onto a comet slide. The slides were left at 4 °C for 10 min, immersed in lysis buffer for 30 min followed by 20-min incubation in alkaline solution, and subjected to electrophoresis at 300 mA for 20 min. Following electrophoresis, the slides were washed in 70% ethanol and left to dry overnight. Samples were then stained with SYBR Green and analyzed with image analysis software (Comet IV, Perceptive Instruments).
Western Blot Analysis
Nuclear extracts were prepared using an Active Motif kit following the instructions of the manufacturer. Equal amounts of protein were resolved on SDS-PAGE, and Western blotting was carried out using the indicated antibodies. Densitometric measurements of bands on Western blots were carried out using Adobe Photoshop software. Western blot images were inverted, and mean intensity was measured. This was then normalized to the background intensity, and normalized values were plotted as mean ± S.E.
RESULTS
TRIM33 Interacts with ALC1 upon Induction of DNA Damage
Previous studies have shown that ALC1 is rapidly recruited to DNA damage sites via a macro domain-dependent interaction with PAR. The retention of ALC1 on damaged chromatin is short-lived, with a half-life of 2.5 min (12, 21).The rapid association/disassociation kinetics of ALC1 from damage sites implies that its chromatin association is subject to strict regulatory control. To gain insight into the regulation of ALC1 during the DNA damage response, we sought to identify proteins that interact with ALC1 upon DNA damage. HEK293 control cells and cells expressing FLAG-tagged wild-type ALC1 (12) were either mock-treated or treated with bleomycin and subjected to immunoprecipitation using FLAG beads. Immunoprecipitates were then analyzed by LC-MS/MS to identify potential interacting proteins. In agreement with previous reports, peptides for PARP1, APLF, H2B, Ku70, Ku80, and DNA-PKcs were identified in ALC1 immunoprecipitates both before and after DNA damage but not in controls (12). Intriguingly, peptides for TRIM33 were also found in ALC1 immunoprecipitates but only from the bleomycin-treated samples (Fig. 1A). This observation raised the possibility that TRIM33 interacts with ALC1 upon induction of DNA damage. To confirm these findings, HeLa cells were mock-treated or treated with either 300 μm bleomycin for 1 h or 3 mm hydroxyurea for 3 h and then subjected to immunoprecipitation using antibody against endogenous TRIM33 and processed for Western blotting using antibodies specific for ALC1 and TRIM33. As shown in Fig. 1B, ALC1 is found in the TRIM33 immunoprecipitation only after induction of DNA damage with either bleomycin or hydroxyurea, indicating that TRIM33 and ALC1 interact in a DNA damage-dependent manner. Analysis of the cell extracts demonstrated that TRIM33 was present in undamaged cells and that its total protein level is not increased upon DNA damage.
FIGURE 1.
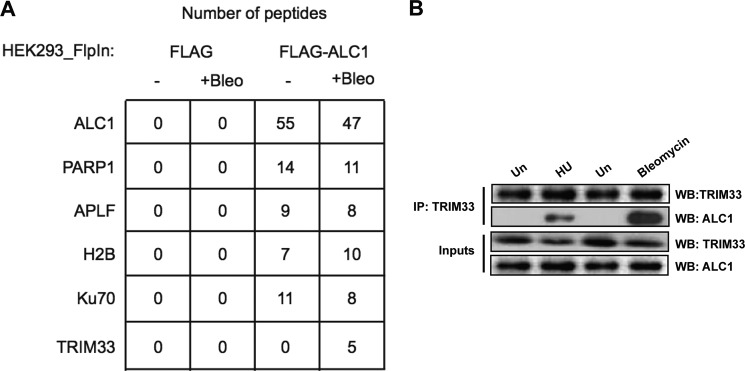
TRIM33 interacts with ALC1 upon induction of DNA damage. A, HEK293 cells expressing either vector or FLAG-ALC1 were treated with either vehicle or bleomycin, subjected to immunoprecipitation using FLAG beads, and then processed by LC-MS/MS. The relative peptide counts for each condition are shown for ALC1, PARP1, APLF, histone H2B, Ku70, and TRIM33. As shown, TRIM33 peptides were identified only in the bleomycin-treated samples. B, endogenous interaction of TRIM33 and ALC1 upon hydroxyurea (HU) and bleomycin treatment. DNase-treated nuclear extracts from untreated (Un) cells or cells treated with HU (3 mm, 3 h) or bleomycin (300uM, 1 h) were immunoprecipitated with anti-TRIM33 antibody. Immunoprecipitates were processed for Western blotting (WB) using antibodies to TRIM33 and ALC1 (top two panels, IP). Aliquots of nuclear extract were also directly processed for Western blotting with these antibodies (bottom two panels, Inputs).
TRIM33 Localizes to DNA Breaks
Given that TRIM33 interacts with ALC1 in response to DNA damage, we next sought to determine whether TRIM33 is also recruited to DNA damage acutely induced by UV laser scissors in HeLa cells sensitized with iododeoxyuridine. Endogenous TRIM33 rapidly localized to DNA damage, as seen by its colocalization with γH2AX, a marker of DNA strand breaks (Fig. 2, A–C). Unlike continuing exposure to agents such as bleomycin that induce ongoing DNA damage, laser scissors induce acute, transient DNA damage at a defined time point, making it amenable to high-resolution time course analysis. TRIM33 recruitment to DNA damage sites was rapid and short-lived. TRIM33 was detected within 5 min of damage induction and disassociated from DNA lesions between 15 and 20 min (Fig. 2, B and C).
To determine whether TRIM33 also localizes to sites of replication stress, HeLa cells were treated with either 0 or 3 mm hydroxyurea for 3 h, and localization of TRIM33 and γH2AX were evaluated by immunofluorescence. Hydroxyurea treatment led to induction of nuclear foci of TRIM33 that colocalized with γH2AX (data not shown). To determine which domains of TRIM33 contribute to its localization to DNA breaks, HeLa cells were transfected with FLAG-tagged constructs encoding either WtTRIM33 or a series of mutant constructs. These included a RING domain mutant (TRIM33CA) that has two cysteine-to-alanine mutations at amino acids 125 and 128, internal deletion mutants of the histone-binding PHD (TRIM33ΔPHD) or Bromo domain (TRIM33ΔBromo) (32), and the mutation of highly conserved residues in the PHD domain (TRIM33-PHD(AAA)) (40) (Fig. 2A). The TRIM33CA mutant has been shown to lack ubiquitin ligase activity (41), and TRIM33-PHD(AAA) has been shown to be unable to bind methylated histone residues (40). Cells were then subjected to UV laser scissor-induced DNA damage, and TRIM33 localization was monitored by immunofluorescence (IF). The WtTRIM33 and TRIM33CA mutants both localized rapidly to DNA breaks. In contrast, deletion of either the PHD or Bromo domain abrogates TRIM33 localization to sites of laser scissor-induced DNA breaks. TRIM33 PHD(AAA) also has greatly reduced localization to DNA breaks (Fig. 2, A, D, and E). Thus, the chromatin-binding PHD and Bromo domains are critical for robust localization of TRIM33 to sites of DNA breaks.
TRIM33 Knockdown Sensitizes Cells to DNA-damaging Agents and Activates Cell Cycle Checkpoints
To investigate a potential role for TRIM33 in the DNA damage response, we next examined the effect of depleting TRIM33 on the sensitivity of cells to DNA-damaging agents. HeLa cells were transfected with control sh/siRNA, with either of two different TRIM33 shRNAs, or one TRIM33 siRNA, and after 48 h, the cells were treated with different concentrations of bleomycin or hydroxyurea. TRIM33 knockdown enhanced sensitivity to both bleomycin (Fig. 3A) and hydroxyurea treatment (data not shown). Introduction of shRNA-resistant WtTRIM33 could rescue the bleomycin sensitivity of TRIM33 depletion (Fig. 3B). These results indicate that TRIM33 plays a role in DNA damage response.
FIGURE 3.

TRIM33 knockdown results in DNA damage sensitivity. A, HeLa cells treated with control shRNA, TRIM33 shRNA1, TRIM33 shRNA2, and TRIM33 siRNA were exposed to increasing concentrations of bleomycin. Relative cell counts measured by MTS assay, normalized to no treatment, were performed on day 3 and were plotted. p < 0.005 for control versus TRIM33 shRNA or siRNA. Relative expression of TRIM33 and tubulin is shown. B, HeLa cells treated with control shRNA, TRIM33 shRNA, or TRIM33 shRNA cells complemented with WtTRIM33 were exposed to increasing concentrations of bleomycin, and relative cell counts were measured as above. The Western blot analysis shows levels of TRIM33 and tubulin. C, whole cell extracts from control and TRIM33 sh2-treated cells were processed for Western blotting using antibodies to the indicated proteins. D, TRIM33 localization to DNA damage is not dependent on ATM, ATR, or DNA-dependent protein kinase (DNApk). HeLa cells treated with vehicle or ATM inhibitor (ATMi) (KU-55933), GM18366 (ATR mutant), and M059J (DNApk−/−) cells were subject to laser scissor-induced DNA damage. Cells were fixed after 10 min and processed for IF using antibodies to γH2AX (green) and TRIM33 (red). E, quantitation of TRIM33 at sites of DNA damage is shown. Each data point is the mean ± S.D. of at least 20 cells.
Cells treated with TRIM33 shRNA also exhibited evidence of spontaneous unrepaired DNA damage, evident from the elevated levels of γH2AX (Fig. 3C). Furthermore, TRIM33 shRNA-treated cells also exhibited hallmarks of damage-induced checkpoint activation, including increased levels of p21 and enhanced phosphorylation of CHK2 on threonine 68 when compared with cells treated with control shRNA (Fig. 3C). Collectively, these results reveal that TRIM33 knockdown results in increased sensitivity to DNA-damaging agents, accumulation of spontaneous DNA damage, and activation of DNA damage-induced checkpoints.
TRIM33 Localization to DNA Breaks Is Dependent upon PARP Activity
TRIM33 localization to sites of DNA breaks was intact in Ataxia telangiectasia- and Rad3-related (ATR)-deficient (GM18366) and DNA-PKcs-deficient (M059J) cells and was unaffected in HeLa cells treated with an ATM inhibitor, KU-55933 (42) (Fig. 3, D and E). The level of γH2AX at the UV laser scissor stripes was reduced but still detectable in these cells, as has been reported previously (10). These data demonstrate that recruitment of TRIM33 to sites of DNA damage is independent of ATM/ATR/DNA-PKcs activation.
Given that PAR formation is required for the recruitment of some DNA repair proteins, including ALC1, to the sites of DNA damage (6, 12, 21, 43), we next examined the role of PAR in TRIM33 recruitment. The effect of inhibiting PAR formation on the localization of TRIM33 to UV laser scissor-induced DNA breaks was evaluated by treating cells with 1 μm PARP inhibitor ABT-888 (44). PARP inhibitor treatment abolished the induction of PAR polymers at sites of laser scissor-induced DNA breaks and greatly reduced the localization of TRIM33 to these sites when compared with mock-treated cells (Fig. 4, A and B). Furthermore, recruitment of TRIM33 to UV laser scissor-induced DNA damage was also greatly reduced in Parp1−/− mouse embryonic fibroblasts when compared with Parp1+/+ mouse embryonic fibroblasts (Fig. 4, C and D). PAR polymers are normally rapidly degraded by the action of PARG (45, 46). Treatment of HeLa cells with a PARG inhibitor, gallotannin, led to prolonged retention of both PAR and TRIM33 at sites of DNA breaks (data not shown).
FIGURE 4.

Localization of TRIM33 to DNA breaks is dependent upon PARP and ALC1. A, PAR (top two panels) and TRIM33 (bottom two panels) were localized by IF in untreated (Un) and in cells pretreated with 1 μm PARPi (Pi) ABT-888 for 1 h. B, quantitation of PAR and TRIM33 colocalization with γH2AX at sites of laser scissors. *, p < 0.005. C, Parp1+/+ or Parp1−/− mouse embryonic fibroblasts were treated with laser scissors, and γH2AX and TRIM33 were localized by IF. Images are shown at identical magnification. D, quantitation of PAR and TRIM33 colocalization with γH2AX at sites of laser scissors. *, p < 0.005. E, APLF, WtALC1, C1 (ALC1 macro domain only), and TRIM33 proteins were dot-blotted onto a nitrocellulose membrane and incubated with 32P-labeled PAR. F, TRIM33 localization to DNA breaks is ALC1-dependent. U2OS cells stably expressing control sh, ALC1sh, or cells expressing ALC1 sh were reconstituted with WT ALC1, KR (kinase dead) and DA (PAR binding mutant) were analyzed. All cells were subjected to UV laser scissor-induced DNA breaks. After 10 min, cell were fixed, and IF was performed using antibodies to γH2AX and TRIM33. G, Western blot analyses showing levels of ALC1 and TRIM33 in U2OS cells expressing control and ALC1 shRNA and different constructs of ALC1 in ALC1sh cells. H, quantitation of relative intensity of TRIM33 at sites of DNA damage. Each data point is mean ± S.D. of at least 20 cells. *, p < 0.005.
PARP1 is also known to be activated at sites of replication stress where it is believed to play a role in replication fork restart (19). Treatment of HeLa cells with PARP inhibitors also reduced TRIM33 focus formation in response to hydroxyurea treatment, with only 19% of the TRIM33 foci colocalizing with γH2AX foci compared with 74% observed in controls (data not shown). Together, these results demonstrate that the recruitment of TRIM33 to sites of DNA breaks and stalled replication forks is PAR-dependent.
TRIM33 Recruitment to DNA Damage Is Dependent on ALC1
The DNA repair proteins APLF and ALC1 are recruited and bind directly to sites of active PAR synthesis via their PAR-binding PBZ (PAR-binding zinc finger) and macro domains, respectively (12, 47, 48). Because TRIM33 is rapidly recruited to sites of DNA damage in a PAR-dependent manner, we sought to determine whether TRIM33 also binds directly to PAR. Purified recombinant FLAG-tagged WtTRIM33, WtALC1, C1 (macro domain) fragment of ALC1, and APLF were spotted onto nitrocellulose, and their ability to bind 32P-radiolabeled PAR was measured. Both the WT and C1 (macro domain) region of ALC1 exhibited a strong interaction with labeled PAR, as demonstrated previously (12, 21). However, TRIM33 failed to bind PAR directly (Fig. 4E), suggesting that PAR-dependent recruitment of TRIM33 to DNA damage is not via direct binding to PAR and may involve some intermediary factor.
Given that TRIM33 and ALC1 associate in response to DNA damage, we investigated whether the recruitment of TRIM33 to DNA damage is dependent on ALC1. The localization of TRIM33 to sites of laser scissor-induced DNA breaks was therefore examined in U2OS cells expressing either control shRNA or ALC1-shRNA. TRIM33 recruitment to UV laser-induced DNA damage was greatly reduced in ALC1 shRNA-expressing cells (Fig. 4, F, G, and H). Thus, TRIM33 recruitment to DNA breaks is dependent on the presence of ALC1.
We further investigated which regions of ALC1 are required for TRIM33 localization to DNA breaks. Cells stably expressing ALC1sh were reconstituted with WtALC1, ALC1-K77R (ATPase dead), or the ALC1-D723A macro domain mutant, which is unable to interact with PAR and fails to localize to DNA breaks. Cells were then subjected to UV laser-induced DNA breaks, and TRIM33 localization was observed by IF using antibodies against endogenous TRIM33. Reconstitution of ALC1sh cells with either WtALC1 or ALC1-K77R rescued TRIM33 localization to DNA breaks (Fig. 4, F, G, and H). However, reconstitution with the ALC1-D723A macro domain mutant failed to rescue TRIM33 localization to DNA damage. This result suggests that PAR binding of ALC1, but not its catalytic activity, is required for its function in localizing TRIM33 to DNA breaks.
The Interaction of TRIM33 with ALC1 Is PARP-dependent
To determine whether the DNA-damage induced interaction of ALC1 and TRIM33 is dependent upon PAR synthesis, endogenous coimmunoprecipitations were performed in DNase-treated nuclear extracts. HeLa cells were either mock-treated or treated with PARP inhibitor (PARPi) and then exposed to 0 gray, 10 gray IR, or 100 J/m2 UV light (Fig. 5A). These treatments were chosen because we could follow the dynamics of interaction after an acute episode of DNA damage. Although an interaction between ALC1 and TRIM33 was not detected in untreated cells, a robust interaction was evident 5 min after either IR or UV light treatment, which diminished after 10 min. The interaction of TRIM33 and ALC1 after IR and UV light treatment was significantly reduced in cells pretreated with PARP inhibitor ABT-888 (PARPi) (Fig. 5, A and B). These results suggest that TRIM33 and ALC1 interact in response to DNA damage and that this is partly dependent on active PAR synthesis.
FIGURE 5.

TRIM33 dynamically interacts with ALC1 in a PARP-dependent manner. A, HeLa cells were untreated (Un) or treated with IR or UV light, with or without PARP inhibitor (PARPi), and DNase-treated nuclear extracts were prepared at 5 and 10 min. TRIM33 IP was performed, followed by Western blotting (WB) using the indicated antibodies. IP, immunoprecipitation; No Ab, no antibody. B, quantitation of ALC1 interaction with TRIM33. The plot shows the ratio of the signal of ALC1 coimmunoprecipitation to ALC1 input. C, The FLAG WtALC1, ALC1K77R, and ALC1D723A mutants were expressed in 293 cells and subjected to UV irradiation. Protein extracts were prepared after 5 min and immunoprecipitated with anti-FLAG antibodies, followed by Western blotting using antibody against TRIM33 and FLAG. D, PAR-bound ALC1 does not bind TRIM33 in vitro. WtALC1, KR (ATPase dead) and DA (PAR binding mutant) ALC1 mutants and TRIM33 proteins were dot-blotted onto a nitrocellulose membrane and incubated with PAR, washed, and then incubated with purified TRIM33. Membranes were then processed for Western blot analysis with antibodies to PAR (top panel) or antibody to TRIM33 (bottom panel).
To determine whether the PAR binding activity of ALC1 or its ATPase activity is required for its interaction with TRIM33, cells were transfected with FLAG-tagged constructs encoding either WtALC1, the ATPase dead mutant, ALC1-K77R, or ALC1-D723A macro domain mutant that cannot bind to PAR (12). The cells were subjected to UV damage, cell extracts were collected after 5 min post-damage, immunoprecipitated with anti-FLAG beads, and processed for Western blot analyses using antibodies to TRIM33. Both the WtALC1 and ALC1-K77R mutant interact with TRIM33 after UV damage. However, the macro domain mutant ALC1-D723A, which does not localize to DNA breaks, fails to interact with TRIM33 (Fig. 5C). These data are consistent with the IF data presented in Fig. 4, and together, they suggest that the interaction of TRIM33 and ALC1 requires PARP-dependent localization of ALC1 to DNA breaks but that it is not dependent upon its ATPase activity.
To determine whether PAR binding of ALC1 is sufficient to induce interaction with TRIM33, PAR-bound ALC1 immobilized on nitrocellulose was incubated with purified TRIM33, and, after washing, analyzed by immunoblotting with antibodies to TRIM33. No interaction of PAR-bound ALC1 with TRIM33 was observed using this approach (Fig. 5D). This suggests that PAR binding of ALC1 is not, by itself, sufficient to induce interaction of ALC1 with TRIM33 in vitro.
TRIM33 Knockdown Results in Prolonged Accumulation of ALC1 at Sites of DNA Damage
Previous studies have shown that ALC1 transiently localizes to laser scissor-induced DNA damage, appearing within seconds of induction and disassociating from the damage site within 10–20 min. Mutant forms of ALC1 that retain the macro domain but are inactive for chromatin remodeling show prolonged retention and persistence of XRCC1 on damaged chromatin (12). To investigate the effect of TRIM33 on the dynamics of ALC1 recruitment to damage sites, we depleted TRIM33 by shRNA in HeLa cells (Fig. 6, A–C). In control shRNA-treated cells, ALC1 was rapidly recruited to sites of laser scissor-induced damage but was undetectable at these sites 45 min after damage (Fig. 6, A and C). In contrast, treatment of cells with TRIM33 shRNA resulted in prolonged retention of ALC1 (Fig. 6, A–C) at sites of laser scissor-induced DNA damage, with ALC1 evident at damage sites 45 min after treatment. Consistent with a prior report, the prolonged retention of ALC1 was also accompanied by prolonged retention of XRCC1 at sites of DNA damage (data not shown) (12).
FIGURE 6.

TRIM33 knockdown induces delayed dissociation of ALC1 but does not affect PAR dissociation from sites of DNA damage. A, HeLa cells expressing either control shRNA, TRIM33 shRNA, TRIM33shRNA + WtTRIM33, or the TRIM33shRNA + TRIM33CA mutant were subjected to UV laser scissors and stained for γH2AX (green) and ALC1 (red) at the time points indicated. B, Western blot analysis showing levels of TRIM33 in cells treated with control shRNA (lane 1), TRIM33 shRNA (lane 2), TRIM33sh RNA + WtTRIM33 (lane 3), and TRIM33sh RNA + TRIM33CA (lane 4). C, quantification of ALC1 localization to DNA damage in TRIM33 knockdown, WtTRIM33, or TRIM33CA reconstituted cells. Each data point is mean ± S.D. of at least 20 cells. D, TRIM33 knockdown does not affect ALC1 protein levels. Shown is a Western blot analysis showing levels of ALC1, TRIM33, and tubulin in control sh (1), TRIM33sh and TRIM33sh (2) cells reconstituted with either TRIM33Wt (3) or TRIM33CA (4). Protein extracts were collected from untreated or UV-treated cells, as indicated, and probed by Western blot analysis using the antibodies indicated. E, HeLa cells expressing either control shRNA or TRIM33 sh2 RNA were treated with laser scissors and stained by IF for γH2AX (green) and PAR (red). F, quantification of PAR intensity was performed 5 and 45 min after induction of DNA damage. Data are plotted as the mean ± S.D. of at least 20 cells. *, p < 0.005 for control versus TRIM33 shRNA-treated cells. G, Western blot analysis showing TRIM33 knockdown. HeLa cells transfected with control or TRIM33 shRNA and proteins levels were detected by Western blot analysis by probing with antibodies against TRIM33 and tubulin.
Of note, TRIM33 knockdown has no effect on protein levels of ALC1 after DNA damage, as analyzed by Western blot analysis (Fig. 6D), suggesting that TRIM33 does not influence ALC1 protein stability. TRIM33 knockdown has no effect on the dynamics of PAR at the UV laser-induced DNA breaks. In both control and TRIM33 knockdown cells, PAR rapidly localized to DNA breaks at 5 min but was not present at breaks after 45 min. This suggests that, in the absence of TRIM33, ALC1 remains at breaks even when PAR is no longer present (Fig. 6, E, F, and G).
To examine the impact of TRIM33 on ALC1 recruitment and retention at damage sites, we complemented TRIM33 knockdown cells with shRNA-resistant wild-type TRIM33 or the RING domain TRIM33CA mutant. Importantly, the prolonged retention of ALC1 at damage sites observed in TRIM33 knockdown cells was corrected by introduction of the shRNA-resistant WtTRIM33 construct but not by the TRIM33CA RING domain mutant (Fig. 6, A–C). Collectively, these data suggest that TRIM33 is required for timely dissociation of ALC1 from sites of damaged DNA and that this function requires an intact RING domain.
The DNA Repair Phenotype Associated with ALC1 Overexpression Is Reversed by TRIM33 Overexpression
ALC1 dissociation was delayed in TRIM33-depleted cells. To determine whether ALC1 overexpression leads to a similar effect, we evaluated the dynamics of ALC1 overexpression on its localization to UV laser scissor-induced DNA breaks. Cells overexpressing WtALC1 (FLP-In-ALC1) show prolonged retention of ALC1 at DNA breaks (Fig. 7A, B, and C). Overexpression of WtTRIM33 restores the ALC1 dynamics, and ALC1 is no longer detectable at sites of breaks after 45 min. These data suggest that proper stoichiometry between TRIM33 and ALC1 is essential for timely dissociation of ALC1 from sites of DNA breaks (Fig. 7, A, B, and C).
FIGURE 7.

TRIM33 rescues ALC1 overexpression sensitivity to DNA-damaging agents. A, ALC1 overexpression results in delayed dissociation of ALC1 from DNA damage. FLP-In ALC1 and FLP-In ALC1 + WtTRIM33 cells were exposed to UV laser scissor-induced DNA damage, and cells were fixed at the indicated time points. Cells were stained by IF for γH2AX (green) and ALC1 (red). B, Western blot analysis showing levels of TRIM33 and ALC1 in ALC1-overexpressing cells. C, quantification of ALC1 intensity was performed at the indicated time points after induction of DNA damage. Data are plotted as the mean ± S.D. of at least 20 cells. D, effect of TRIM33 on sensitivity of ALC1-overexpressing cells to bleomycin. U2OS cells stably expressing either empty vector (FLP-In-FLAG), WtALC1 (FLP-In-ALC1), WtALC1 (FLP-In-ALC1) + WtTRIM33, and WtALC1 (FLP-In-ALC1) + TRIM33CA were treated with increasing concentrations of bleomycin for 3 days. Relative cell counts at day 3 were measured by MTS assay and were plotted normalized to untreated cells. Data are mean ± S.D. of three experiments. E, induction of DNA breaks in cells (n > 200 cells) of indicated genotypes after treatment with increasing amounts of phleomycin were measured by alkaline comet assay. Data are plotted as the mean ± S.D. of at least three experiments.
ALC1 is amplified and overexpressed in certain cancers, suggesting that it may function as an oncogene (49). Overexpression of ALC1 leads to chromatin relaxation and sensitivity of cells to the DNA-damaging agent phleomycin, which induces breaks preferentially in linker DNA (12). Our data raise the possibility that the relative levels of ALC1 and TRIM33 may be important for the regulation of ALC1 activity. To directly investigate the effect of TRIM33 expression on the phenotype associated with ALC1 overexpression, cells stably overexpressing either an empty vector (FLP-In-FLAG) or WtALC1 (FLP-In-ALC1) were analyzed for sensitivity to bleomycin. Confirming prior reports, overexpression of Wt ALC1 confers increased sensitivity to bleomycin (Fig. 7D) (12). Overexpression of WtTRIM33 greatly reduced the sensitivity of ALC1-overexpressing cells to bleomycin, with these cells now showing similar sensitivity as vector-expressing cells. However, overexpression of the RING domain mutant TRIM33CA failed to rescue the bleomycin sensitivity of ALC1-overxpressing cells (Fig. 7D).
The effect of TRIM33 overexpression on induction of DNA breaks by phleomycin in ALC1-overexpressing cells was also evaluated using the comet assay. Consistent with prior reports, phleomycin exposure produces longer tail moments in ALC1-overexpressing cells compared with control cells, suggesting that ALC1 overexpression promotes chromatin relaxation and increased accessibility of linker DNA to phleomycin (12). Overexpression of WtTRIM33, but not the TRIM33CA mutant, counteracted the effect of ALC1 overexpression on induction of DNA breaks induced by phleomycin treatment, as measured by comet tail moments (Fig. 7E). This is consistent with our prior results, and collectively these data demonstrate that elevated TRIM33 expression can counteract the phenotype of ALC1 overexpression and that this requires an intact RING domain.
DISCUSSION
PARP activation at sites of DNA breaks leads to local changes in chromatin structure required for efficient DNA repair. A key insight into how PARylation impacts chromatin structure came from the finding that PAR-binding proteins such as ALC1 are recruited to sites of DNA breaks and participate in chromatin remodeling and DNA repair (12, 21, 23). Here, we implicate TRIM33 in the PAR-induced DNA damage response through its interaction with ALC1. This assertion is supported by the following observations. 1) TRIM33 is rapidly recruited to DNA damage sites in a PAR- and ALC1-dependent fashion; 2) TRIM33 interacts with ALC1 in response to DNA damage; 3) TRIM33 knockdown in cells confers DNA damage sensitivity and delays disassociation of ALC1 from damaged chromatin; and 4) overexpression of TRIM33, but not the TRIM33-CA mutant, alleviates the DNA repair phenotype resulting from ALC1 overexpression.
Our data suggest that TRIM33 does not directly interact with PAR but that its enrichment at damage sites is dependent upon interaction with ALC1. Recruitment also depends upon the presence of an intact Bromo domain and an intact PHD domain in TRIM33, suggesting an important role for the interaction with modified histone residues (32, 33). The interaction of TRIM33 with ALC1 is highly dynamic and is evident within minutes after acute, transient DNA damage but declines rapidly. Although we cannot rule out a possibility of a low level of interaction between TRIM33 and ALC1 in undamaged cells, this interaction is clearly enhanced by DNA damage with kinetics that parallel production of PAR. Indeed, the interaction between TRIM33 and ALC1 is reduced by PARP inhibitor treatment or by point mutations in the macro domain of ALC1 that abolishes PAR binding, suggesting that PAR binding by ALC1 is important for its interaction with TRIM33. However, PAR binding alone might not be sufficient to induce interaction of ALC1 by TRIM33 because PAR-bound ALC1 could not directly interact with TRIM33 by a far-western approach (Fig. 5D). It is possible that binding of the TRIM33 PHD-Bromo domain with histones, which is required for recruitment of TRIM33 to DNA breaks, may also be required for optimal interaction of ALC1 and TRIM33. It is also possible that other proteins recruited to DNA breaks may facilitate or mediate the interaction between ALC1 and TRIM33.
Loss of TRIM33 leads to increased base-line H2AX phosphorylation and activation of cell cycle checkpoints, suggesting that it may be required for efficient DNA repair. TRIM33 loss also leads to increased sensitivity to DNA-damaging agents such as bleomycin. Of note, loss of TRIM33 leads to somewhat slower growth (data not shown). However, the sensitivity to DNA-damaging agents remains proportionately increased. This is similar to other DNA repair proteins whose loss can lead to both decreased proliferation and increased sensitivity to DNA-damaging agents. However, we cannot rule out that the role of TRIM33 in transcriptional regulation may contribute to these phenotypes.
TRIM33 appears to regulate the dynamics of ALC1 retention at sites of DNA breaks. ALC1 is normally recruited rapidly to sites of DNA damage but dissociates quickly. Loss of TRIM33 leads to prolonged retention of ALC1 at sites of DNA breaks. Furthermore, reintroduction of WtTRIM33, but not the RING domain mutant TRIM33, was found to rescue the effect of TRIM33 knockdown on ALC1 retention. Our observations are consistent with a model in which, upon DNA damage, TRIM33 interacts with ALC1 and promotes the timely removal of ALC1 from damaged chromatin.
We surmise that overexpression of ALC1, a putative oncogene, disrupts the normal stoichiometry of ALC1 and TRIM33, leading to dysregulated ALC1 activity, which confers promiscuous chromatin relaxation and enhanced sensitivity to bleomycin. Concomitant overexpression of WtTRIM33 was found to counteract the DNA repair phenotype and enhanced susceptibility to phleomycin-induced DNA breaks evident in ALC1-overexpressing cells. Conversely, knockdown of TRIM33 leads to a phenotype similar to that induced by ALC1 overexpression, including increased sensitivity of cells to bleomycin. Thus, loss of TRIM33 may be, in part, functionally analogous to ALC1 overexpression, with both leading to abnormal ALC1 retention at sites of DNA breaks. This effect may also contribute to the tumor suppressor function of TRIM33. The functional impact of abnormal ALC1 retention at DNA breaks on the DNA repair process needs to be further characterized. TRIM33 likely has additional ALC1-independent functions that contribute to the phenotype associated with TRIM33 loss.
TRIM33 has been implicated in the regulation of the TGF-β pathway, where it interacts with and regulates the SMAD3-SMAD4 complex and its chromatin association (29, 41). TRIM33 also interacts with FACT1 and other members of the transcriptional elongation complex and plays a key role in transcriptional regulation during development (27). It is not currently clear whether the transcription and DNA repair roles of TRIM33 are separate functions or are related mechanistically. It is, however, intriguing to note that both the SMAD pathway and the FACT1 complex are regulated by PARP activity, with both SMAD3 and FACT1 reported as substrates for PARylation by PARP1 (50, 51). Because PARP1 functions in both transcription and DNA repair, it is possible that TRIM33 may act downstream of PARP1 activation in several distinct cellular contexts where it interacts with specific target proteins: SMAD4 linked to transcription and ALC1 associated with DNA repair.
Our findings have potential clinical implications because TRIM33 is mutated, translocated, and has decreased expression in several human cancers, including hepatocellular cancer, pancreatic cancer, and chronic myelomonocytic leukemia (34–36, 38, 52, 53). Tissue-specific TRIM33 knockout in mouse liver leads to hepatocellular carcinoma (34). Because amplification of ALC1 and knockout of TRIM33 are both implicated in the pathogenesis of hepatocellular cancer (49), this observation lends support to the potential antagonistic role of TRIM33 and ALC1. Decreased TRIM33 expression is also seen in a subset of human pancreatic cancers, and a tissue-specific knockout of TRIM33 is known to cooperate with KRAS mutation in the development of adenocarcinomas of the pancreas in mice (37). A recent study also demonstrates that pancreas-specific TRIM33 knockout is not epistatic with SMAD4 knockout in the development of Kras-associated pancreatic cancer, suggesting that the role of TRIM33 as a pancreatic tumor suppressor may be independent from its effect on SMAD4 (38). Our findings raise the possibility that the DNA repair defects associated with TRIM33 loss may contribute to tumorigenesis. Moreover, treatment of tumors exhibiting loss of TRIM33 function could be designed to exploit the DNA repair defect present in these cells.
Acknowledgments
We thank Bing Xia and Zhiyuan Shen for valuable discussions, the Cancer Therapy and Evaluation Program at the NCI, National Institutes of Health for ABT-888, and Joan Massague for the TRIM33 ΔPHD and TRIM33 PHD(AAA) constructs.
This work was supported, in whole or in part, by the National Institutes of Health (to S. G.). This work was also supported by the New Jersey Commission for Cancer Research (to A. K., J. O., and S. G.), by the Department of Defense (to J. O. and S. G.), and by the Rutgers Cancer Institute of New Jersey (CINJ) Foundation (to S. G.). The laboratory of S. J. B. was funded by Cancer Research UK.
- PAR
- poly(ADP-ribose)
- PARP
- poly(ADP-ribose) polymerase
- PARG
- poly(ADP-ribose) glycohydrolase
- PHD
- plant homeo domain
- IF
- immunofluorescence.
REFERENCES
- 1. Downs J. A., Nussenzweig M. C., Nussenzweig A. (2007) Chromatin dynamics and the preservation of genetic information. Nature 447, 951–958 [DOI] [PubMed] [Google Scholar]
- 2. Goodarzi A. A., Noon A. T., Jeggo P. A. (2009) The impact of heterochromatin on DSB repair. Biochem. Soc. Trans. 37, 569–576 [DOI] [PubMed] [Google Scholar]
- 3. Satoh M. S., Lindahl T. (1992) Role of poly(ADP-ribose) formation in DNA repair. Nature 356, 356–358 [DOI] [PubMed] [Google Scholar]
- 4. Lindahl T., Satoh M. S., Poirier G. G., Klungland A. (1995) Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 20, 405–411 [DOI] [PubMed] [Google Scholar]
- 5. Kim M. Y., Zhang T., Kraus W. L. (2005) Poly(ADP-ribosyl)ation by PARP-1. “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 19, 1951–1967 [DOI] [PubMed] [Google Scholar]
- 6. Timinszky G., Till S., Hassa P. O., Hothorn M., Kustatscher G., Nijmeijer B., Colombelli J., Altmeyer M., Stelzer E. H., Scheffzek K., Hottiger M. O., Ladurner A. G. (2009) A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16, 923–929 [DOI] [PubMed] [Google Scholar]
- 7. Jackson S. P., Bartek J. (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kubota Y., Takanami T., Higashitani A., Horiuchi S. (2009) Localization of X-ray cross complementing gene 1 protein in the nuclear matrix is controlled by casein kinase II-dependent phosphorylation in response to oxidative damage. DNA Repair 8, 953–960 [DOI] [PubMed] [Google Scholar]
- 9. Ciccia A., Elledge S. J. (2010) The DNA damage response. Making it safe to play with knives. Mol. Cell 40, 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Polo S. E., Kaidi A., Baskcomb L., Galanty Y., Jackson S. P. (2010) Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 29, 3130–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hakmé A., Wong H. K., Dantzer F., Schreiber V. (2008) The expanding field of poly(ADP-ribosyl)ation reactions. “Protein Modifications. Beyond the Usual Suspects” Review Series. EMBO Rep. 9, 1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahel D., Horejsí Z., Wiechens N., Polo S. E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S. C., Jackson S. P., Owen-Hughes T., Boulton S. J. (2009) Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325, 1240–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahel I., Ahel D., Matsusaka T., Clark A. J., Pines J., Boulton S. J., West S. C. (2008) Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451, 81–85 [DOI] [PubMed] [Google Scholar]
- 14. Hassa P. O., Haenni S. S., Elser M., Hottiger M. O. (2006) Nuclear ADP-ribosylation reactions in mammalian cells. Where are we today and where are we going? Microbiol Mol. Biol. Rev. 70, 789–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malanga M., Althaus F. R. (2005) The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem. Cell Biol. 83, 354–364 [DOI] [PubMed] [Google Scholar]
- 16. Realini C. A., Althaus F. R. (1992) Histone shuttling by poly(ADP-ribosylation). J. Biol. Chem. 267, 18858–18865 [PubMed] [Google Scholar]
- 17. El-Khamisy S. F., Masutani M., Suzuki H., Caldecott K. W. (2003) A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31, 5526–5533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin S. A., Lord C. J., Ashworth A. (2008) DNA repair deficiency as a therapeutic target in cancer. Curr. Opin. Genet. Dev. 18, 80–86 [DOI] [PubMed] [Google Scholar]
- 19. Bryant H. E., Petermann E., Schultz N., Jemth A. S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., Helleday T. (2009) PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Min W., Cortes U., Herceg Z., Tong W. M., Wang Z. Q. (2010) Deletion of the nuclear isoform of poly(ADP-ribose) glycohydrolase (PARG) reveals its function in DNA repair, genomic stability and tumorigenesis. Carcinogenesis 31, 2058–2065 [DOI] [PubMed] [Google Scholar]
- 21. Gottschalk A. J., Timinszky G., Kong S. E., Jin J., Cai Y., Swanson S. K., Washburn M. P., Florens L., Ladurner A. G., Conaway J. W., Conaway R. C. (2009) Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U.S.A. 106, 13770–13774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pines A., Vrouwe M. G., Marteijn J. A., Typas D., Luijsterburg M. S., Cansoy M., Hensbergen P., Deelder A., de Groot A., Matsumoto S., Sugasawa K., Thoma N., Vermeulen W., Vrieling H., Mullenders L. (2012) PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 199, 235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gottschalk A. J., Trivedi R. D., Conaway J. W., Conaway R. C. (2012) Activation of the SNF2 family ATPase ALC1 by poly(ADP-ribose) in a stable ALC1·PARP1·nucleosome intermediate. J. Biol. Chem. 287, 43527–43532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venturini L., You J., Stadler M., Galien R., Lallemand V., Koken M. H., Mattei M. G., Ganser A., Chambon P., Losson R., de Thé H. (1999) TIF1γ, a novel member of the transcriptional intermediary factor 1 family. Oncogene 18, 1209–1217 [DOI] [PubMed] [Google Scholar]
- 25. Peng H., Feldman I., Rauscher F. J., 3rd. (2002) Hetero-oligomerization among the TIF family of RBCC/TRIM domain-containing nuclear cofactors. A potential mechanism for regulating the switch between coactivation and corepression. J. Mol. Biol. 320, 629–644 [DOI] [PubMed] [Google Scholar]
- 26. He W., Dorn D. C., Erdjument-Bromage H., Tempst P., Moore M. A., Massagué J. (2006) Hematopoiesis controlled by distinct TIF1Rγ and Smad4 branches of the TGFβ pathway. Cell 125, 929–941 [DOI] [PubMed] [Google Scholar]
- 27. Bai X., Kim J., Yang Z., Jurynec M. J., Akie T. E., Lee J., LeBlanc J., Sessa A., Jiang H., DiBiase A., Zhou Y., Grunwald D. J., Lin S., Cantor A. B., Orkin S. H., Zon L. I. (2010) TIF1γ controls erythroid cell fate by regulating transcription elongation. Cell 142, 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Howard P. W., Ransom D. G., Maurer R. A. (2010) Transcription intermediary factor 1γ decreases protein expression of the transcriptional cofactor, LIM-domain-binding 1. Biochem. Biophys. Res. Commun. 396, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dupont S., Mamidi A., Cordenonsi M., Montagner M., Zacchigna L., Adorno M., Martello G., Stinchfield M. J., Soligo S., Morsut L., Inui M., Moro S., Modena N., Argenton F., Newfeld S. J., Piccolo S. (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 136, 123–135 [DOI] [PubMed] [Google Scholar]
- 30. Morsut L., Yan K. P., Enzo E., Aragona M., Soligo S. M., Wendling O., Mark M., Khetchoumian K., Bressan G., Chambon P., Dupont S., Losson R., Piccolo S. (2010) Negative control of Smad activity by ectodermin/Tif1γ patterns the mammalian embryo. Development 137, 2571–2578 [DOI] [PubMed] [Google Scholar]
- 31. Doisne J. M., Bartholin L., Yan K. P., Garcia C. N., Duarte N., Le Luduec J. B., Vincent D., Cyprian F., Horvat B., Martel S., Rimokh R., Losson R., Benlagha K., Marie J. C. (2009) iNKT cell development is orchestrated by different branches of TGF-β signaling. J. Exp. Med. 206, 1365–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xi Q., Wang Z., Zaromytidou A. I., Zhang X. H., Chow-Tsang L. F., Liu J. X., Kim H., Barlas A., Manova-Todorova K., Kaartinen V., Studer L., Mark W., Patel D. J., Massagué J. (2011) A poised chromatin platform for TGF-β access to master regulators. Cell 147, 1511–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Agricola E., Randall R. A., Gaarenstroom T., Dupont S., Hill C. S. (2011) Recruitment of TIF1γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 43, 85–96 [DOI] [PubMed] [Google Scholar]
- 34. Herquel B., Ouararhni K., Khetchoumian K., Ignat M., Teletin M., Mark M., Béchade G., Van Dorsselaer A., Sanglier-Cianférani S., Hamiche A., Cammas F., Davidson I., Losson R. (2011) Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc. Natl. Acad. Sci. U.S.A. 108, 8212–8217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aucagne R., Droin N., Paggetti J., Lagrange B., Largeot A., Hammann A., Bataille A., Martin L., Yan K. P., Fenaux P., Losson R., Solary E., Bastie J. N., Delva L. (2011) Transcription intermediary factor 1γ is a tumor suppressor in mouse and human chronic myelomonocytic leukemia. J. Clin. Invest. 121, 2361–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aucagne R., Droin N., Solary E., Bastie J. N., Delva L. (2011) TIF1γ: a tumor suppressor gene in chronic myelomonocytic leukemia. Med. Sci. 27, 696–698 [DOI] [PubMed] [Google Scholar]
- 37. Vincent D. F., Yan K. P., Treilleux I., Gay F., Arfi V., Kaniewski B., Marie J. C., Lepinasse F., Martel S., Goddard-Leon S., Iovanna J. L., Dubus P., Garcia S., Puisieux A., Rimokh R., Bardeesy N., Scoazec J. Y., Losson R., Bartholin L. (2009) Inactivation of TIF1γ cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 5, e1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vincent D. F., Gout J., Chuvin N., Arfi V., Pommier R. M., Bertolino P., Jonckheere N., Ripoche D., Kaniewski B., Martel S., Langlois J. B., Goddard-Léon S., Colombe A., Janier M., Van Seuningen I., Losson R., Valcourt U., Treilleux I., Dubus P., Bardeesy N., Bartholin L. (2012) Tif1γ suppresses murine pancreatic tumoral transformation by a Smad4-independent pathway. Am. J. Pathol. 180, 2214–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ziv Y., Bielopolski D., Galanty Y., Lukas C., Taya Y., Schultz D. C., Lukas J., Bekker-Jensen S., Bartek J., Shiloh Y. (2006) Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 8, 870–876 [DOI] [PubMed] [Google Scholar]
- 40. Wang G. G., Song J., Wang Z., Dormann H. L., Casadio F., Li H., Luo J. L., Patel D. J., Allis C. D. (2009) Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dupont S., Zacchigna L., Cordenonsi M., Soligo S., Adorno M., Rugge M., Piccolo S. (2005) Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 121, 87–99 [DOI] [PubMed] [Google Scholar]
- 42. Hickson I., Zhao Y., Richardson C. J., Green S. J., Martin N. M., Orr A. I., Reaper P. M., Jackson S. P., Curtin N. J., Smith G. C. (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64, 9152–9159 [DOI] [PubMed] [Google Scholar]
- 43. Haince J. F., McDonald D., Rodrigue A., Déry U., Masson J. Y., Hendzel M. J., Poirier G. G. (2008) PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283, 1197–1208 [DOI] [PubMed] [Google Scholar]
- 44. Palma J. P., Rodriguez L. E., Bontcheva-Diaz V. D., Bouska J. J., Bukofzer G., Colon-Lopez M., Guan R., Jarvis K., Johnson E. F., Klinghofer V., Liu X., Olson A., Saltarelli M. J., Shi Y., Stavropoulos J. A., Zhu G. D., Penning T. D., Luo Y., Giranda V. L., Rosenberg S. H., Frost D. J., Donawho C. K. (2008) The PARP inhibitor, ABT-888 potentiates temozolomide. Correlation with drug levels and reduction in PARP activity in vivo. Anticancer Res. 28, 2625–2635 [PubMed] [Google Scholar]
- 45. Blenn C., Althaus F. R., Malanga M. (2006) Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochem. J. 396, 419–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rapizzi E., Fossati S., Moroni F., Chiarugi A. (2004) Inhibition of poly(ADP-ribose) glycohydrolase by gallotannin selectively up-regulates expression of proinflammatory genes. Mol. Pharmacol. 66, 890–898 [DOI] [PubMed] [Google Scholar]
- 47. Iles N., Rulten S., El-Khamisy S. F., Caldecott K. W. (2007) APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell Biol. 27, 3793–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rulten S. L., Cortes-Ledesma F., Guo L., Iles N. J., Caldecott K. W. (2008) APLF (C2orf13) is a novel component of poly(ADP-ribose) signaling in mammalian cells. Mol. Cell Biol. 28, 4620–4628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma N. F., Hu L., Fung J. M., Xie D., Zheng B. J., Chen L., Tang D. J., Fu L., Wu Z., Chen M., Fang Y., Guan X. Y. (2008) Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 47, 503–510 [DOI] [PubMed] [Google Scholar]
- 50. Heo K., Kim H., Choi S. H., Choi J., Kim K., Gu J., Lieber M. R., Yang A. S., An W. (2008) FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell 30, 86–97 [DOI] [PubMed] [Google Scholar]
- 51. Lönn P., van der Heide L. P., Dahl M., Hellman U., Heldin C. H., Moustakas A. (2010) PARP-1 attenuates Smad-mediated transcription. Mol. Cell 40, 521–532 [DOI] [PubMed] [Google Scholar]
- 52. Klugbauer S., Rabes H. M. (1999) The transcription coactivator HTIF1 and a related protein are fused to the RET receptor tyrosine kinase in childhood papillary thyroid carcinomas. Oncogene 18, 4388–4393 [DOI] [PubMed] [Google Scholar]
- 53. Herquel B., Ouararhni K., Davidson I. (2011) The TIF1α-related TRIM cofactors couple chromatin modifications to transcriptional regulation, signaling and tumor suppression. Transcription 2, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]