

Background: Transmembrane domains are critical to the structure and function of bile acid transporters.
Results: Sodium sensitive residues follow distinct α-helical periodicity along TM2.
Conclusion: TM2 forms part of the sodium translocation pathway, which is highly conserved in its putative remote bacterial homologue ASBTNM.
Significance: The proposed sodium translocation mechanism may be universal to other SLC10A family members.
Keywords: Drug Transport, Membrane Proteins, Membrane Transport, Site-directed Mutagenesis, Transporters
Abstract
Human apical sodium-dependent bile acid transporter (hASBT, SLC10A2) is responsible for intestinal reabsorption of bile acids and plays a key role in cholesterol homeostasis. We used a targeted and systematic approach to delineate the role of highly conserved transmembrane helix 2 on the expression and function of hASBT. Cysteine mutation significantly depressed transport activity for >60% of mutants without affecting cell surface localization of the transporter. All mutants were inaccessible toward chemical modification by membrane-impermeant MTSET reagent, strongly suggesting that transmembrane 2 (TM2) plays an indirect role in bile acid substrate translocation. Both bile acid uptake and sodium dependence of TM2 mutants revealed a distinct α-helical periodicity. Kinetic studies with conservative and non-conservative mutants of sodium sensitive residues further underscored the importance of Gln75, Phe76, Met79, Gly83, Leu86, Phe90, and Asp91 in hASBT function. Computational analysis indicated that Asp91 may coordinate with sodium during the transport cycle. Combined, our data propose that a consortium of sodium-sensitive residues along with previously reported residues (Thr134, Leu138, and Thr149) from TM3 may form the sodium binding and translocation pathway. Notably, residues Gln75, Met79, Thr82, and Leu86 from TM2 are highly conserved in TM3 of a putative remote bacterial homologue (ASBTNM), suggesting a universal mechanism for the SLC10A transporter family.
Introduction
hASBT2 (SLC10A2) plays an essential role in the reuptake of bile acids from the intestinal lumen (1, 2). Bile acids are synthesized in the liver from cholesterol, stored in the gall bladder, and excreted into intestinal lumen in the fed state (3), playing a critical role in the digestion and absorption of fat. Hypocholesterolemia associated with idiopathic bile acid malabsorption demonstrates that hASBT is a viable pharmacological target for cholesterol-lowering therapy (4, 5). In addition, hASBT can be used as a potential prodrug target for increasing oral bioavailability of poorly permeable drugs (6, 7).
Bile acid uptake via hASBT is electrogenic (8) using a sodium gradient as the driving force for the transport of bile acids, with a 2:1 Na+:bile acid coupling stoichiometry. Using N-glycosylation scanning and an epitope insertion approach, previous studies from our laboratory have definitively confirmed a 7-transmembrane (TM) topology for this 348-amino acid transporter (see Fig. 1A) (9). Recently, a crystal structure of a putative bacterial homologue of hASBT with ∼24% sequence identity and 10 TMs was reported (10), isolated from Neisseria meningitidis (ASBTNM); although its physiological role in Neisseria, its specificity toward bile acids, and its phylogenetic relationship to mammalian ASBT remain to be established, this structure may aid in the understanding of human SLC10A structure-function relationships.
FIGURE 1.

Secondary structure of hASBT and TM2 sequence alignment among species. A, schematic diagram of the secondary structure of hASBT based on a 7TM topology previously proposed. In this model TM2 ranges from residue Leu73 to Val98. B, sequence of TM2 of hASBT was aligned with SLC10A2 of 10 other species, including sequence of ASBTNM to elucidate the sequence conservation.
Our laboratory has carried out a systematic analysis to unveil residues or regions that are important for the expression, function, and stability of hASBT. For example, mutations on ∼70% TM1 residues resulted in impaired function (11), with residue Leu30 critical for both bile acid and sodium binding. The large hydrophilic region lining the cytosolic half of TM3 may be partially involved in substrate exit during bile acid translocation (12). Consistent with the orientation of TM4 in our three-dimensional model (13), experimental data ruled out the direct involvement of TM4 in sodium translocation; instead, the high solvent accessibility of the cytosolic half of the transmembrane domain indicates an important role of TM4 in substrate transport (14). Conversely, TM5 may not be directly involved in substrate translocation, yet the presence of a helix-helix interacting motif, GXXXG, suggests that it may play a role in structural stability of the transporter (15). Functional flexibility enabled by residues Pro234, Gly237, and Gly241 allow TM6 to form a “conformational switch” for substrate turnover (16). Both experimental data and computational modeling suggest a pivotal role for TM7 in substrate translocation (17, 18). Overall, these studies have provided a roadmap for interpreting hASBT structure and function.
The present study analyzes the role of TM2 in the expression, function, and/or stability of hASBT using the substituted cysteine accessibility method (SCAM). Each individual residues of TM2 was mutated to cysteine, and the effect of these mutations on hASBT surface expression, function, sodium sensitivity, and aqueous accessibility were assessed. Conservative and non-conservative mutations were generated on functionally impaired and sodium-sensitive residues to further elucidate their functional importance/requirement.
EXPERIMENTAL PROCEDURES
Materials
Radiolabeled [3H]taurocholic acid (TCA; 1 mCi/mmol) was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO), bovine serum albumin and TCA were from Sigma, [2-(trimethylammonium)ethyl] methanethiosulfonate (MTSET) reagent was from Toronto Research Chemicals, Inc. (North York, ON, Canada), EDTA-free protease inhibitor mixture tablets were from Roche Applied Science, Ez-linkTM Sulfo-NHS-SS-Biotin was from Pierce, and cell culture media and supplies were from Invitrogen. All other reagents and chemicals were of highest purity available commercially.
Cell Culture and Transient Transfection
The monkey kidney fibroblast cell line, COS-1 (ATCC CRL-1650), was maintained in Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal calf serum, 4.5 g/liter glucose, 0.1 mm nonessential amino acid at 37 °C in a humidified atmosphere with 5% CO2. Transfections were carried out as previously described (19). Briefly, cells were seeded at a density of 6 × 104 cells/ml on 12- or 24-well plates (Costar, Corning, NY). The next day, after reaching ∼70–90% confluence, cells were transfected with TurboFect transfection agent (Thermo Scientific). 40–48 h after the transfection, cells were used for uptake and surface protein labeling studies.
Site-directed Mutagenesis
Mutagenesis primers were custom-synthesized and purchased from Sigma. To generate mutations on each individual residue of TM2, C270A or wild type (WT) plasmid of hASBT in pCMV5 vector was used as the template. Our previous studies demonstrated that the C270A mutant is functionally active and has resistance to thiol modification (20). The mutations were generated by QuikChange site-directed mutagenesis. Plasmids were purified using kits from Qiagen (Valencia, CA). The sequences of the mutants were verified by DNA sequencing at the University of Maryland Medical School sequencing facility.
TCA Uptake Study
Uptake studies were carried out as previously described (11). Briefly, COS-1 cells were transiently transfected with pCMV5 plasmid containing C270A mutant, TM2 mutants, or WT_ASBT DNA. 48 h after transfection, cells were washed twice with phosphate-buffered saline (PBS) and incubated in 200 μl of modified Hanks' balanced salt solution containing 5.0 μm cold TCA spiked with 1 μCi/ml [3H]TCA at 37 °C. The uptake study was carried out for 12 min to ensure steady-state kinetics. Uptake was halted by washing the wells with ice-cold PBS (pH 7.4) containing 0.5 mm TCA. Cells were lysed by incubation in 350 μl of 1 n NaOH for 2 h followed by neutralizing with 50 μl of concentrated HCl. 250 μl of sample from each well were subjected to analysis by scintillation counting using and LS6500 liquid scintillation counter (Beckman Coulter, Inc., Fullerton, CA). Uptake activity was normalized to total protein in each well, which was quantified using Bradford protein assay (Bio-Rad). The final uptake activity was expressed as pmol of [3H]TCA/min/mg of protein.
Protein Membrane Expression
Transiently transfected COS-1 cells in 12-well plates were first washed with ice-cold PBS followed by incubating with 1.2 mg/ml Sulfo-NHS-SS-Biotin in PBS for 30 min at 4 °C with gentle rocking. The reaction was quenched with 1 m Tris buffer (pH 8.0) followed by washing with Tris-buffered saline (TBS). Cells were lysed in 200 μl of lysis buffer (50 mm Tris buffer (pH 7.4), 150 mm NaCl, 1% Triton X-100, 1× sigma protease inhibitor mixture) for 30 min at 4 °C. Cell lysates were centrifuged at 10,000 × g for 2 min. 30 μl of the supernatant was used for total protein expression assessment. The remaining supernatants were mixed with 250 μl of neutravidin resin (Pierce) and rocked overnight at 4 °C. The supernatant was removed after centrifugation at 2500 × g for 2 min. The resins were mixed with 2× sample buffer after washing twice with TBS and lysis buffer and heated for 10 min at 95 °C. The suspension was centrifuged at 5000 × g for 5 min, and the supernatants were subjected to SDS-PAGE and Western blot analysis.
For SDS-PAGE and immunoblotting studies, the total protein or the surface-labeled proteins were separated on 12.5% SDS-polyacrylamide gel (Bio-Rad) and transferred onto a polyvinylidene difluoride membrane (Bio-Rad). Blots were probed with rabbit anti-ASBT primary antibody (Santa Cruz Biotechnology; 1:1000 dilutions). The absence of a 90-kDa band for calnexin (mouse anti-calnexin; Sigma; 1:1000 dilutions) and a 140-kDa band for cadherin (mouse anti-cadherin; Abcam; 1:1000 dilutions) was used as an indicator for surface and total expression of hASBT. Goat anti-rabbit IgG (H+L) Dylight 800 conjugated and goat anti-mouse IgG (H+L) Dylight 680 conjugated (Thermoscientific, 1:10,000 dilutions) were used as the secondary antibodies. An Odyssey imaging system (LI-COR, Lincoln, NE) was used to visualize hASBT and control bands. The anti-ASBT primary antibody detects both the glycosylated (∼41 kDa) and unglycosylated (∼38 kDa) ASBT proteins.
Sodium Activation and Kinetics
To assess Na+ sensitivity of individual mutants, [3H]TCA uptake was measured at equilibrative (12 mm; equilibrium with cytosolic Na+) and physiological (137 mm) Na+ concentrations. The results are expressed as a ratio of uptake at 12 mm over 137 mm Na+ to determine sodium sensitivity of each mutant. A ratio less than 1 indicates greater necessity for physiological sodium concentration for proper function. TCA uptake kinetics or Na+ activation kinetics were measured at TCA concentrations ranging from 0 to 200 μm and Na+ concentrations ranging from 0 to 137 mm.
Solvent Accessibility Studies
The possible involvement of the TM2 residues in substrate translocation was analyzed on the cysteine mutants by assessing thiol accessibility of these mutants to positively charged, membrane impermeable MTSET. COS-1 cells transiently transfected with TM2 mutants were preincubated with MTSET for 10 min at room temperature and washed twice with Hanks' balanced salt solution followed by [3H]TCA uptake at 37 °C for 12 min. MTSET working solution was freshly prepared to ensure stability. Control wells were treated with buffer in the absence of MTSET reagent and run parallel with the MTSET-treated cells.
Molecular Dynamic Simulation
The structural model of the hASBT dimer was developed using an elaborate homology modeling scheme followed by refinement using both implicit and explicit protein-membrane molecular dynamics (MD) simulations as has been described in detail elsewhere.3 The hASBT dimer model showed excellent correlation with experimental substituted cysteine accessibility method profiles. In this study we performed additional analysis on the hASBT dimer trajectory specifically to probe the interactions with sodium ions. Briefly, bilayer protein-membrane simulations on the hASBT dimer were performed using the biomolecular simulation program CHARMM (22) and NAMD (23). The CHARMM22 protein force field (24) with CMAP (dihedral correction map (25)) modified the TIP3P water model (26), and the CHARMM lipid force field was used for the in silico modeling. A mixed 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine/cholesterol lipid bilayer was used to model the membrane (27). The electrostatic interactions were treated via the particle mesh Ewald method with a real-space cutoff of 12 Å, a κ value of 0.34 Å−1, and a sixth-order spline (28). Nonbond interaction lists were updated heuristically out to 16 Å with a force switch smoothing function from 10 to 12 Å used for the Lennard-Jones interactions. SHAKE algorithm was used to constrain all covalent bonds involving hydrogen atoms (29). All the bilayer calculations were performed at 303.15 K. A salt (NaCl) concentration of 0.137 m was used. A Langevin coupling coefficient of 1 ps−1 with a temperature bath of 303.15 K was applied to all atoms to achieve constant temperature. A piston oscillation period of 200 fs and a barostat damping time scale of 100 fs were used to maintain a piston pressure of 1 atm. A time step of 2 fs was used. After initial equilibration in CHARMM (using gradually decreasing restraints) 60-ns production runs were performed using NAMD Version 2.7b1. The final system size was 98.39 × 98.39 × 98.39 Å3. Additional details of the simulation and the systems setup have been discussed elsewhere.3
Data Analysis
Studies were carried out at least three times in triplicate, and bars represent the S.E. for n ≥3. GraphPad Prism 5.0 (GraphPad Software, San Diego, CA) was used for data analysis using one-way analysis of variance with Dunnett's post hoc test to determine statistical significance.
RESULTS
Cysteine Scanning of TM2
According to a 7TM topology model (Fig. 1A), residues spanning Leu73 to Val98 form the TM2 helix. High sequence conservation in TM2 was observed among various species (Fig. 1B). To elucidate the role of TM2 on hASBT expression, function, and stability, each individual residue of TM2 (except Cys74) was mutated to cysteine. The mutations were generated using the hASBT C270A scaffold, which is largely insensitive to thiol-specific chemical reagents. Consequently, the expression and function of the mutants were compared with that of the C270A.
Transport Activity and Membrane Expression of TM2 Mutants
Each of the TM2 mutants was transiently transfected into COS-1 cells, which have extremely low background expression of ASBT. The cell surface localization of these mutants was determined with the membrane-impermeable agent, sulfo-NHS-SS-Biotin followed by immunoblotting and densitometric analysis of the developed bands. Total cellular and membrane-localized (surface) expression of these mutants was compared with the expression of the C270A scaffold.
The total expression of each TM2 mutants is comparable to the expression of the C270A control, except P94C, L95C, and V98C (see Fig. 3A); these three mutants had detectable levels of total cellular hASBT expression, albeit with low cell surface localization. Membrane localization of the majority of TM2 mutants is similar to that of C270A (Fig. 2B). Interestingly, mutations made to the residues in the center of the TM2 helix showed relatively higher cell surface expression than C270A, especially T82C. The surface expression of each mutant was first semiquantified by normalizing to cadherin, a plasma membrane expression marker; then [3H]TCA uptakes were normalized to quantified cell surface hASBT expression (Fig. 2C). Significant changes in TCA uptake rates were observed for the majority (64%) of the mutants compared with the C270A control, suggesting a functional role for these residues. Uptake activity of P80C and P94C mutations were almost abolished. A severe loss of activity (<10% activity remaining) for Q75C, F76C, M79C, P80C, G83C, P94C, and Q96C mutations, yet measurable cell surface transporter expression, suggests that the reduction in activities did not result from a loss of protein at the plasma membrane. These residues may play a significant role in bile acid substrate or Na+ translocation.
FIGURE 3.

Probing solvent accessibility of TM2 residues. COS-1 cells transiently transfected withTM2 cysteine mutants were preincubated in buffer for 10 min at room temperature with or without 1 mm MTSET, then assayed for [3H]TCA uptake. Uptake results were first expressed as a ratio of uptake with MTSET pretreatment over uptake without MTSET pretreatment, then the final results were expressed as the percentage of C270A. Bars represent the S.E. of three experiments with triplicates each time.
FIGURE 2.

[3H]TCA uptake by TM2 mutants and transporter expression. A, COS-1 cells were transfected with TM2 mutants, pCMV5 and C270A controls as described under “Experimental Procedures.” 40–48 h after transfection, [3H]TCA uptake was carried out for 12 min before cells were washed 3 times and lysed in 1 n NaOH, and the lysates were subjected to radioactive counter analysis. TCA uptake rates were calculated as pmol [3H]TCA/min/mg of protein. The final results were expressed as the percentage of the uptake rate of the C270A control. B, total (whole cell homogenate) and cell surface expression of each cysteine mutant were subjected to 12.5% polyacrylamide gel analysis followed by transferring to PVDF membrane. hASBT bands were confirmed with anti-ASBT antibody (1:1000 dilution). Cadherin was used as a cell surface expression marker; calnexin was used as an ER protein marker. C, uptake for each mutant was normalized to surface expression level of hASBT mutants and expressed as a percentage of Control (C270A). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Solvent Accessibility via Sulfhydryl Modification
To explore whether TM2 residues are involved in substrate binding and/or translocation, a membrane-impermeable, sulfhydryl-reactive labeling agent, MTSET, was used to probe solvent accessibility. Residues involved in bile acid translocation are likely exposed to the aqueous phase; therefore, cysteine mutation of this residue would make it available for thiol modification by MTSET. Once the residue is chemically modified, substrate binding or translocation will be impeded. No significant change in TCA uptake rate was observed for any of the TM2 mutants upon chemical modification (Fig. 3), indicating that TM2 residues may not be directly involved in bile acid binding and/or translocation. Control experiments with WT_ASBT revealed significant changes in TCA uptake rate upon MTSET modification, confirming previous findings (19).
Na+ Sensitivity of TM2 Residues
hASBT relies on the physiological sodium gradient to transport bile acids, with an approximate 2:1 Na+ to bile acid co-transport stoichiometry (8). To identify residues of TM2 that may be involved in sodium interactions, TCA uptake was studied at equilibrating (12 mm) and physiological (137 mm) sodium concentrations. Previous studies have demonstrated minimal sodium sensitivity for the C270A control (16, 17, 19, 30). Therefore, the ratio of TCA uptake at 12 mm versus 137 mm Na+ for each mutant was compared with that of the C270A control. Several TM2 mutants exhibited sensitivity toward changes in Na+ concentration following a clear pattern of α-helical periodicity, i.e. mutants of every third or fourth residue were significantly affected (Fig. 4); Q75C, M79C, G83C, and L86C showed a significant decrease, whereas I92C and Q96C exhibited a significant increase in TCA uptake. Interestingly, residues of adjacent TM3 (13) were also found to be Na+-sensitive and may participate in sodium binding and translocation (12). Based on their close proximity, it is tempting to speculate that TM2 and TM3 may cooperate during sodium translocation.
FIGURE 4.

Sodium sensitivity of TM2 residues. 48 h after COS-1 cells were transfected with TM2 mutant, C270A, and pCMV5 plasmids, [3H]TCA uptake was carried out with 5 μm TCA under equilibrating (12 mm) and physiological (137 mm) sodium concentration. The results were calculated as the ratio of equilibrating over saturating sodium concentration. The final results were expressed as the percentage of C270A. Bars represent the S.E. of three experiments with triplicates each time. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Sodium and TCA Kinetics of Select TM2 Mutants
To further delineate the underlying mechanism for the observed changes in TCA uptake rates and sodium sensitivities, a subset of residues were selected for TCA and Na+ kinetic analysis. Only Q75C and G83C showed a significant change in TCA affinity (KTCA) compared with C270A control (Table 1). Because both mutants have minimal TCA uptake compared with control (<10%, Fig. 1), it is difficult to draw conclusions from kinetic parameters alone. The other mutations (F76C, M79C, G83C, D91C, I92C, and Q96C) did not affect bile acid binding affinity (KTCA) but demonstrated significantly diminished maximal turnover rates (Jmax), suggesting that these mutations affected hASBT structure or Na+ coupling rates. Thus, Na+ kinetics for this subset of residues was further evaluated. M79C and L86C showed a significant increase in KNa+ as well as a significant decrease in Jmax compared with that of C270A control (Table 2). Interestingly, mutants L73C, D91C, I92C, and Q96C demonstrated a significant increase in sodium affinity that coincided with a significant decrease in maximal TCA transport rate (Jmax). We hypothesize that the concomitant changes in sodium affinity and maximal transport rates (Table 2) may, at least in part, provide evidence for either direct or indirect involvement of these residues in sodium translocation.
TABLE 1.
TCA kinetics constants for selected TM2 mutants
COS-1 cells were transiently transfected with several selected TM2 mutants as described under “Experimental Procedures.” Uptake was measured at the TCA concentrations ranging from 0 to 200 μm. Experiments were carried out three times in triplicate. Kinetic data were analyzed using GraphPad Prism 5.0 software.
| Mutations | TCA kinetics |
|
|---|---|---|
| KTCA | Jmax | |
| mm | pmol /min·mg protein | |
| C270A | 17.80 ± 2.365 | 2623 ± 106.1 |
| Q75C | 1097.0 ± 688.0a | 584.9 ± 319.6a |
| F76C | 24.05 ± 5.58 | 80.72 ± 4.79a |
| M79C | 60.45 ± 11.35 | 157.9 ± 12.48a |
| G83C | 478.8 ± 199.4b | 676.1 ± 213.6a |
| D91C | 17.75 ± 1.83 | 442.1 ± 13.89a |
| I92C | 13.38 ± 2.92 | 479.5 ± 29.37a |
| Q96C | 18.6 ± 3.58 | 64.01 ± 3.79a |
a p < 0.001.
b p < 0.05.
TABLE 2.
Sodium kinetics parameters for selected TM2 mutants
COS-1 cells were transiently transfected with several sodium sensitive TM2 mutants as described under “Experimental Procedures.” Uptake was measured at the Na+ ion concentration ranging from 0 to 200 mm. Experiments were carried out three times with triplicates each time. Kinetic data were analyzed by Graphpad Prism 5.0 software.
| Mutations | Na+ kinetics |
|
|---|---|---|
| KNa+ | Jmax | |
| mm | pmol/min·mg protein | |
| C270A | 14.22 ± 3.03 | 544.3 ± 33.08 |
| L73C | 6.52 ± 1.52 | 375.7 ± 19.09a |
| G77C | 16.01 ± 2.99 | 529.9 ± 29.2 |
| M79C | 162.9 ± 35.59a | 26.25 ± 6.43a |
| L86C | 32.9 ± 5.65 | 328.5 ± 20.16a |
| F90C | 18.51 ± 6.06 | 215.5 ± 22.15a |
| D91C | 5.197 ± 1.98 | 68.8 ± 5.20a |
| I92C | 4.50 ± 1.32 | 195.8 ± 11.43a |
| Q96C | 3.30 ± 2.16 | 27.07 ± 3.11a |
a p < 0.001.
Mutation of Critical Residues on a Wild Type Background
To exclude the possibility that the C270A scaffold affects the functional changes observed above, conservative and non-conservative mutations were made against a WT_ASBT background to residues with significantly affected TCA uptake rates (Fig. 5). When Gln75 was mutated to Asp/Glu, TCA uptake was almost abolished. Similarly, a non-conservative mutation Q75A resulted in loss of function, indicating that both the size and physicochemical properties of the Gln75 side chain are critical for hASBT function. Non-conservative mutation F76A severely dampened hASBT function, whereas the more conservative mutations, F76Y and F76W, preserved >10% bile acid uptake activity. These data suggest that the phenyl moiety on the Phe76 side chain may play an important role in hASBT function, perhaps by providing a scaffold for π-charge interactions with Na+. A complete loss of function for the P80A and P94A mutations was expected as proline mutations in transmembrane domains may lead to structural instability (31) by disrupting the hinge movement Pro residues provide to α-helices (32). The conservative and non-conservative mutations Q96N and Q96A resulted in complete loss of function; however, Q96E did not affect TCA uptake compared with control, indicating that the size of the side chain is critical and that charged moieties are tolerated at this position. Gln96 is located at the border between plasma membrane and extracellular space and may provide a role in stabilizing membrane localization.
FIGURE 5.

[3H]TCA uptake activity and cell-surface expression of conservative and non-conservative TM2 mutants. A, conservative and non-conservative mutants of selected TM2 residues were generated using WT_ASBT as the scaffold. [3H]TCA uptake was expressed as a percentage of the WT_ASBT transporter, in pmol of [3H]TCA/min/mg of protein. Bars represent the S.E. of three separate experiments with triplicates each time. B, 40–48 h after COS-1 cells transiently transfected with each mutant or WT_ASBT were treated with sulfo-NHS-SS-Biotin. Samples were subjected to 12.5% polyacrylamide gel analysis followed by Western blot analysis. Blots were probed with goat anti-ASBT antibody (1:1000 dilutions) followed by Donkey anti-goat secondary antibody. Cadherin was used as a cell surface expression marker.
Asp91 Is Na+-sensitive
Asp91 is the sole residue in TM2 with a polar and possibly charged side chain. Cysteine substitution of Asp91 and its adjacent residues Phe90 and Ile92 resulted in significantly decreased TCA uptake rates (Fig. 2C), suggesting that this region is critical to hASBT function. Solvent accessibility data ruled out the direct involvement of this region in bile acid substrate translocation (Fig. 3), prompting the hypothesis that altered sodium interactions might be responsible for the observed change in TCA uptake. I92C resulted in a significantly increased sodium affinity (KNa+ = 4.5 ± 1.32 mm) compared with the C270A control (KNa+ = 14.22 ± 3.03 mm). However, the increased sodium binding significantly reduced TCA uptake activity for the mutant, indicating that either Na+ translocation was hindered or that tighter binding of Na+ to the mutant transporter did not result in a conformational change that promotes bile acid binding and, consequently, translocation.
Next, conservative and non-conservative mutations were made to Asp91 using WT_ASBT as a scaffold. D91A resulted in a total loss of function, whereas conservative mutations, D91E and D91N, preserved ∼20% activity (Fig. 6A). Asp91 is not fully conserved across species, and rASBT and mASBT feature a glycine in position 91. Here, a D91G mutation preserved ∼15% uptake activity of hASBT. Mutations at Asp91 neither significantly altered the total expression or the cell surface presence of the mutant protein (Fig. 6B). To further evaluate the role of Asp91 in hASBT function, we determined Na+ kinetics of these mutants (Fig. 7). The Na+ binding affinity for D91E and D91N was similar to that of the WT transporter with similar Hill coefficients (Table 3), yet mutations resulted in a significant decrease in transporter activity. Interestingly, the Hill coefficient of D91G was nearly 1, suggesting a possible loss of one of the two sodium binding sites or translocation pathways.
FIGURE 6.

[3H]TCA uptake activity and expression of Asp91 mutants. A, COS-1 cells were transiently transfected with Asp91 mutants. 40–48 h after transfection [3H]TCA uptake was carried out for 12 min at 37 °C. The final uptake for each mutant was calculated as a percentage of the uptake by the WT_ASBT and expressed as pmol of [3H]TCA/min/mg of protein. Bars represent the S.E. of three separate experiments with triplicates each time. B, 48 h after transfection with Asp91 mutants, cells were treated with sulfo-NHS-SS-Biotin. Cell lysates were separated on 4–15% SDS-PAGE followed by Western blot analysis.
FIGURE 7.

Na+ kinetics of Asp91 mutants. COS-1 cells transfected with WT_ASBT and mutants of Asp91 were incubated with 5 μm cold TCA spiked with 1 μCi/ml [3H]TCA under sodium concentrations ranging from 0 to 137 mm at 37 °C for 12 min. Changes in sodium concentration were compensated with choline chloride.
TABLE 3.
Na+ kinetics parameters of the conservative and non-conservative mutants of Asp91
Conservative and non-conservative mutants of Asp91 were generated using WT_ASBT as the scaffold. COS-1 cells were transiently transfected with these mutants as described under “Experimental Procedures.” Uptake was measured at the Na+ concentrations ranging from 0 to 200 mm. Experiments were carried out three times in triplicate. Kinetic data were analyzed with GraphPad Prism 5.0 software.
| Hill coefficient | Jmax | |
|---|---|---|
| pmol/min/mg protein | ||
| WT_ASBT | 1.66 | 333.4 |
| D91E | 1.77 | 50.7 |
| D91N | 1.51 | 38.8 |
| D91A | 0.48 | 4.1 |
| D91G | 1.04 | 19.1 |
TM2/TM3 May Form a Na+ Translocation Pathway
To gain insight into the structural arrangement of the residues involved in sodium binding, an analysis was performed on the explicit membrane molecular dynamics (MD) simulations of the hASBT dimer. The structural model of the hASBT dimer was developed using an elaborate homology modeling scheme followed by refinement using both implicit and explicit protein-membrane MD simulations.3 The functional significance of the dimer model was evaluated by comparing it with mutagenesis results. Cross-linking experiments verified the presence of the functional dimer predicted by in silico modeling.3 In this study we perform additional analysis on the trajectory obtained from dimer MD simulations. The MD simulation was able to capture two sodium binding sites, one in each monomer. High sodium distribution was observed around residue Asp91 (Fig. 8). This observation correlated well with the experimental results. In addition, the sodium-sensitive residues Gln75/Phe76, Met79, Thr82, Leu86, and Phe90/Asp91 in TM2 are in line and are facing previously reported sodium-sensitive residues, Thr134, Leu138, and Thr149, from TM3 (Fig. 9). Therefore, a consortium of these residues may form the sodium translocation pathway.
FIGURE 8.
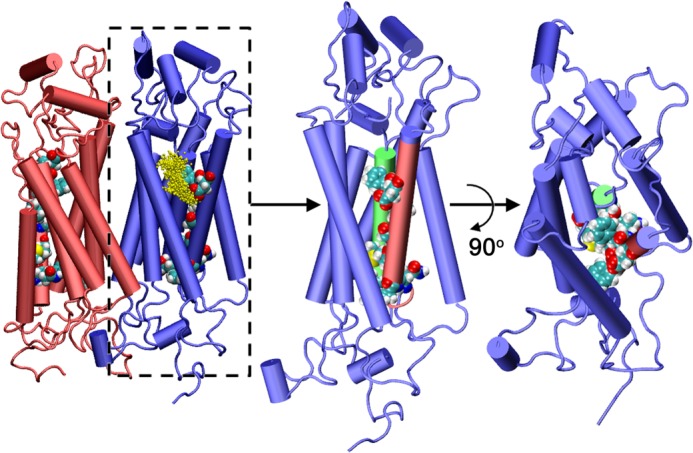
Representative structure of hASBT dimeric assembly obtained from explicit MD simulations. The lipid bilayer and solvent environment have been omitted for clarity. Sodium distribution in one of the monomers captured from the MD simulations is also shown. The side view and top view of one the monomer is presented to highlight the arrangement of the amino acids involved in the proposed TM2-TM3 translocation pathway. Representation: protein, new schematic representation; sodium ions, CPK representation; amino acids in TM2 and TM3, VDW representation. The figure was generated using molecular graphics package VMD (21).
FIGURE 9.
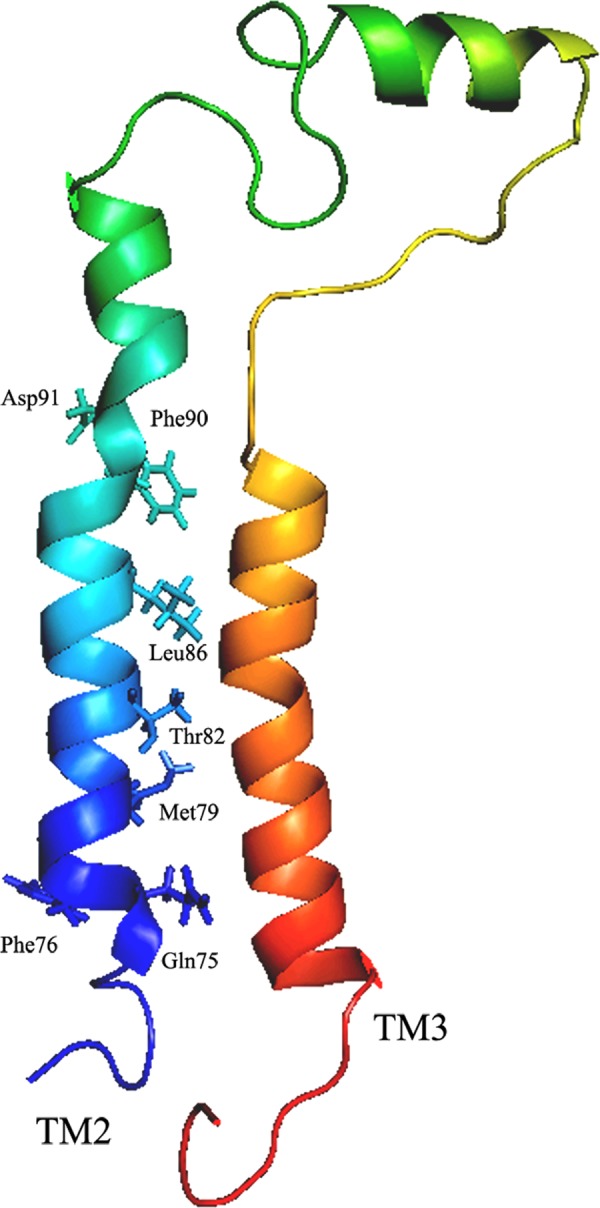
In silico rendering of the residues relevant for sodium transport. The relative orientation of the TM2 and TM3 in dimer model is depicted. The residues that show sodium sensitivities from TM2 are highlighted. Amino acids are represented by their three-letter codes and corresponding placement in the protein.
Conservation of TM2 Sodium-sensitive Residues in ASBTNM
Despite low sequence identity (24%) between hASBT and recently crystallized ASBTNM, sequence alignment indicates 60% overlap between TM2 of hASBT in a 7TM mammalian topology model and TM3 of ASBTNM (Fig. 10A). Both TMs span the plasma membrane in the same direction (intracellular to exofacial). Furthermore, the sodium-sensitive residues in TM2 of hASBT, Gln75/Phe76, Met79, Thr82, Leu86, are conserved in ASBTNM and reveal clear α-helical periodicity, facing TMs 5 and 8 (Fig. 10B). The ASBTNM crystal structure captured two bound Na+ ions, one of which interacts with five coordinating amino acids (Fig. 10C), including Gln77 (corresponding to residue Gln75 in hASBT). Combining these data, we propose that sodium-sensitive residues may play a role in sodium translocation either directly, by interacting with sodium, or indirectly, by perturbing the conformation of adjacent TM helices.
FIGURE 10.

Sequence alignment and mapping of TM2 sodium sensitive residues onto the ASBTNM structure. A, sequence alignment of TM2 and its flanking residues of hASBT to ASBTNM using ClustalW. The alignment results were manually adjusted in JalView 2.8. The gray bars above and below the sequence pair indicate the amino acid range of each transmembrane domains. Residues that are identical in both sequences are highlighted in blue. B, ribbon representation of TM3 and TM8 structures of ASBTNM with residues shown to correspond to the sodium-sensitive residues in TM2 of hASBT. C, stick model of the amino acids that mediate sodium binding in the crystal structure. Amino acids are colored by elements: green for carbon, blue for nitrogen, and red for oxygen.
DISCUSSION
In the absence of a mammalian crystal structure, various biochemical approaches have been utilized to characterize hASBT at the molecular level. However, there is paucity of information on the role of TM2 and its constituent amino acids on the expression, structure, and function of hASBT. This study applied a targeted and systematic analysis of TM2 using the substituted cysteine accessibility method. Our findings suggest that TM2 may not be directly involved in bile acid substrate translocation, but we clearly demonstrate that several residues of TM2 are sensitive to changes in Na+ concentration; in particular, Asp91 may provide a Na+ binding/interaction site. Sodium-sensitive residues are positioned on one face of the membrane helix, indicating a putative Na+ translocation pathway. In silico simulations reveal high sodium distribution around residue Asp91, which further corroborates experimental findings.
TM2 has a high degree of sequence conservation among species (Fig. 1B), suggesting an important role in ASBT function and/or structure. It is noteworthy that a GXXP motif exists within TM2 that has been reported to provide a hinge-bending motion in the transmembrane regions of integral membrane proteins (32, 33). In fact, cysteine substitution of Pro80 resulted in a severe loss of function (Fig. 2C), suggesting a structural role for TM2 in hASBT function.
>60% of TM2 cysteine mutants had significantly impaired activity or loss of function (Fig. 2C). After normalizing uptake activity to each mutant's plasma membrane localization, we observed α-helical periodicity, i.e. every third or fourth residue (starting from Gln75) along TM2 had significantly diminished TCA uptake compared with control. Similar periodicity was noticed when TM2 mutants were examined for Na+ sensitivity (Fig. 4): Q75C, F76C, M79C, G83C, F84C, I85C, L86C, F90C, D91C, and I92C were significantly affected by lowered Na+ compared with C270A control. It should be noted that these changes in function did not result from altered total hASBT protein expression or plasma membrane localization of the mutant transporters (Fig. 2B), with V98C being the only exception.
Next, direct involvement of TM2 residues in bile acid substrate translocation was ruled out by solvent accessibility scanning using the positively charged, membrane-impermeable thiol modification agent MTSET (Fig. 3). Substituted cysteine accessibility method data should be interpreted with caution, as replacing any residue with a relatively hydrophobic cysteine may alter the protein microenvironment, especially if this residue is exposed to an aqueous environment. Nevertheless, absent solvent accessibility for any of the mutants tested, it can be concluded that TM2 may only play an indirect role in bile acid substrate transport.
Conservative and non-conservative mutations were utilized to evaluate the functional importance on a WT_ABST background of residues that were either sodium-sensitive or had loss of function. We found that residues Cys74, Gln75, Phe76, Pro80, Phe90, Asp91, Ile92, and Pro94 (mutated using WT_ASBT as the template) are critical for hASBT function (Fig. 5A). To further delineate the mechanism underlying altered transport activity of these residues, we performed detailed kinetic analysis. Mutants F76C, M79C, D91C, and I92C had decreased Jmax, yet only M79C appeared to have significantly altered TCA binding affinity (Km) compared with C270A control (Table 1). Even though TCA has affinity for these mutant transporters, they reveal significantly lowered turnover rates, perhaps by mutation-induced conformational changes leading to a perturbed transport cycle. We next hypothesized that reduced uptake rates could be caused by altered Na+ binding (affinity). M79C and L86C had significantly increased KNa+, accompanied by significant decreases in Jmax. Interestingly, L73C, F76C, I92C, and Q96C all demonstrate higher affinity to sodium than C270A yet significantly lower Jmax (Table 2), suggesting that these residues are involved in Na+ interactions. Mutating Asp91 to either glutamate or asparagine (conservative mutations) using WT_ASBT as the scaffold did not affect KNa+ or the Hill coefficient; however, mutation resulted in significantly lower Jmax values (Table 3). A Hill coefficient of 1 or lower for D91A and D91G suggests that mutations have affected Na+ binding to the transporter, whereas charge and size of the side chain at position 91 appears to be critical for interaction with Na+.
Analysis of sodium distribution profiles on our 7TM homology model revealed sodium binding regions in the N terminus (Asp9), EL1 loop (Asp120, Asp122), EL3 loop (Glu261), and TM2 (Asp91) regions. In agreement with experimental studies, MD simulations were able to capture the binding of sodium ions at Asp91 in one of the monomers. In view of the dimer model, this translates to sodium binding at two Asp91 sites, one for each monomer. The presence of two binding sites agrees with the 2:1 stoichiometric ratio. Although MD simulations were able to identify the possible initiation of the sodium translocation event, they could not capture the transfer of sodium ions from the extracellular to intracellular milieu. However, this information can be gathered from mapping the sodium-sensitive residues onto the dimer model. Seven additional cysteine mutants in TM2 (Q75C, F76C, M79C, G83C, L86C, F90C, and D91C) exhibited substantial sodium sensitivity, revealing a possible pathway for sodium translocation that faces the interior of the 7TM assembly. It is interesting to note that none of the residues face the membrane-bound side of the 7TM assembly, again in accordance with experimental data. The identification of two other key residues, Thr134 and Thr149, in TM3 that also exhibited sodium sensitivity strengthens the hypothesis of the predicted translocation pathway via the 7TM assembly. Thus, although the negative charge on Asp91 provides the focal point to initiate the uptake of sodium from the extracellular milieu, the subsequent translocation is governed by possible cation-π (Phe90, Phe76) and cation-polar residue (Met79 and Gln75 in TM2, Thr134 and Thr149 in TM3) interactions and supported by few non-polar residues (Gly83, Lys86) to complete the translocation pathway (Fig. 9).
Recently, a crystal structure of a putative bacterial homologue of ASBT from Neisseria meningitides, ASBTNM, was solved at 2.2 Å resolution (10). The crystal structure reveals a 10TM topology contrary to the systematically validated mammalian 7TM model used in this study. It has been argued that ASBTNM may not be a member of the SLC10A family (34) due to its relatively low amino acid identity (∼24%) compared with evolutionarily closer homologues (SLC10A3-6) that do not transport bile salts. Regardless, TM2 of hASBT shares ∼60% sequence similarity with its proposed counterpart (TM3) in ASBTNM (Fig. 10A), most notably the sodium-sensitive residues Gln75/Phe76, Met79, Thr82, and Leu83 are conserved in ASBTNM. In addition, the corresponding residues in ASBTNM line a discrete face along TM3 facing TM5 and TM8. The side chain of residue Gln77, corresponding to residue Gln75 in hASBT, was shown in the crystal structure to accommodate one of the two Na+ ions (Fig. 10C), in agreement with our hypothesis. Combining these observations, we conclude that the sodium translocation mechanism proposed here is conserved between hASBT and ASBTNM despite their remote evolutionary distance and highly divergent membrane topologies.
In conclusion, we report the central role of TM2 in hASBT function. Both experimental and computational analyses converge on the same understanding that residues of TM2, in particular Phe90/Asp91 and Gln75/Phe76, may interact with Na+ ions. Integrating the present data with our previous structure-function studies as well as structural information from its remote bacterial homolog, we speculate that the negative charge on Asp91 may initiate long-range electrostatic interactions with Na+ ions in cooperation residues from EL1 and EL3. Furthermore, the interface between TM2 and TM3 may form a Na+ translocation pathway. Because sodium-sensitive residues are conserved across species and are present in the alleged bacterial homolog ASBTNM, we conclude that the proposed sodium translocation mechanism may be universal to the SLC10A transporter family.
Acknowledgments
We thank Dr. Tatiana Claro da Silva for excellent advice during the course of this work. We also thank Dr. Robyn Moore for critical reading of the manuscript. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant OCI-1053575. We also acknowledge the use of computational facilities at computer aided drug design (CADD), University of Maryland Baltimore.
This work was supported, in whole or in part, by National Institutes of Health Grant DK061425 (NIDDK; to P. W. S.).
S. S. Mallajosyula, submitted for publication.
- (h)ASBT
- (human) apical sodium-dependent bile acid transporter
- MTSET
- [2-(trimethylammonium) ethyl]-methane-thiosulfonate
- TCA
- taurocholic acid
- TM
- transmembrane domain
- ASBTNM
- ASBT from N. meningitides
- MD
- molecular dynamics.
REFERENCES
- 1. Dawson P. A. (2011) Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb. Exp. Pharmacol. 201, 169–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dawson P. A., Lan T., Rao A. (2009) Bile acid transporters. J. Lipid Res. 50, 2340–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hofmann A. F., Hagey L. R. (2008) Bile acids. Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 65, 2461–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tremont S. J., Lee L. F., Huang H. C., Keller B. T., Banerjee S. C., Both S. R., Carpenter A. J., Wang C. C., Garland D. J., Huang W., Jones C., Koeller K. J., Kolodziej S. A., Li J., Manning R. E., Mahoney M. W., Miller R. E., Mischke D. A., Rath N. P., Fletcher T., Reinhard E. J., Tollefson M. B., Vernier W. F., Wagner G. M., Rapp S. R., Beaudry J., Glenn K., Regina K., Schuh J. R., Smith M. E., Trivedi J. S., Reitz D. B. (2005) Discovery of potent, nonsystemic apical sodium-codependent bile acid transporter inhibitors (Part 1). J. Med. Chem. 48, 5837–5852 [DOI] [PubMed] [Google Scholar]
- 5. Kitayama K. (2006) Novel non-systemic inhibitor of ileal apical Na+-dependent bile acid transporter reduces serum cholesterol levels in hamsters and monkeys. Eur. J. Pharmacol. 539, 89–98 [DOI] [PubMed] [Google Scholar]
- 6. Rais R., Fletcher S., Polli J. E. (2011) Synthesis and in vitro evaluation of gabapentin prodrugs that target the human apical sodium-dependent bile acid transporter (hASBT). J. Pharm. Sci. 100, 1184–1195 [DOI] [PubMed] [Google Scholar]
- 7. Balakrishnan A., Polli J. E. (2006) Apical sodium dependent bile acid transporter (ASBT, SLC10A2). A potential prodrug target. Mol. Pharm. 3, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weinman S. A., Carruth M. W., Dawson P. A. (1998) Bile acid uptake via the human apical sodium-bile acid cotransporter is electrogenic. J. Biol. Chem. 273, 34691–34695 [DOI] [PubMed] [Google Scholar]
- 9. Banerjee A., Swaan P. W. (2006) Membrane topology of human ASBT (SLC10A2) determined by dual label epitope insertion scanning mutagenesis. New evidence for seven transmembrane domains. Biochemistry 45, 943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu N. J., Iwata S., Cameron A. D., Drew D. (2011) Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 478, 408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. da Silva T. C. (2011) Transmembrane helix 1 contributes to substrate translocation and protein stability of bile acid transporter SLC10A2. J. Biol. Chem. 286, 27322–27332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hussainzada N., Claro Da Silva T., Swaan P. W. (2009) The cytosolic half of helix III forms the substrate exit route during permeation events of the sodium/bile acid cotransporter ASBT. Biochemistry 48, 8528–8539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang E. Y., Phelps M. A., Banerjee A., Khantwal C. M., Chang C., Helsper F., Swaan P. W. (2004) Topology scanning and putative three-dimensional structure of the extracellular binding domains of the apical sodium-dependent bile acid transporter (SLC10A2). Biochemistry 43, 11380–11392 [DOI] [PubMed] [Google Scholar]
- 14. Khantwal C. M., Swaan P. W. (2008) Cytosolic half of transmembrane domain IV of the human bile acid transporter hASBT (SLC10A2) forms part of the substrate translocation pathway. Biochemistry 47, 3606–3614 [DOI] [PubMed] [Google Scholar]
- 15. Moore R. H., Chothe P., Swaan P. W. (2013) Transmembrane domain V plays a stabilizing role in the function of human bile acid transporter SLC10A2. Biochemistry 52, 5117–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hussainzada N., Khandewal A., Swaan P. W. (2008) Conformational flexibility of helix VI is essential for substrate permeation of the human apical sodium-dependent bile acid transporter. Mol. Pharmacol. 73, 305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hussainzada N., Banerjee A., Swaan P. W. (2006) Transmembrane domain VII of the human apical sodium-dependent bile acid transporter ASBT (SLC10A2) lines the substrate translocation pathway. Mol. Pharmacol. 70, 1565–1574 [DOI] [PubMed] [Google Scholar]
- 18. González P. M., Hussainzada N., Swaan P. W., Mackerell A. D., Jr., Polli J. E. (2012) Putative irreversible inhibitors of the human sodium-dependent bile acid transporter (hASBT; SLC10A2) support the role of transmembrane domain 7 in substrate binding/translocation. Pharm Res. 29, 1821–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Banerjee A., Ray A., Chang C., Swaan P. W. (2005) Site-directed mutagenesis and use of bile acid-MTS conjugates to probe the role of cysteines in the human apical sodium-dependent bile acid transporter (SLC10A2). Biochemistry 44, 8908–8917 [DOI] [PubMed] [Google Scholar]
- 20. Ray A. (2006) Design of novel synthetic MTS conjugates of bile acids for site-directed sulfhydryl labeling of cysteine residues in bile acid binding and transporting proteins. Bioorg. Med. Chem. Lett. 16, 1473–1476 [DOI] [PubMed] [Google Scholar]
- 21. Humphrey W., Dalke A., Schulten K. (1996) VMD. Visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 22. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM. The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalé L., Skeel R., Bhandarkar M., Brunner R., Gursoy A., Krawetz N., Phillips J., Shinozaki A., Varadarajan K., Schulten, Klaus (1999) NAMD2. Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312 [Google Scholar]
- 24. Mackerell A. D., Jr., Bashford D., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., WiÃrkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 25. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) Extending the treatment of backbone energetics in protein force fields. Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 26. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R, W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 27. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A.. D., Jr., Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids. Validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald. An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 29. Ryckaert J.-P., Ciccotti G., Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints. Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 30. Hussainzada N., Da Silva T. C., Zhang E. Y., Swaan P. W. (2008) Conserved aspartic acid residues lining the extracellular loop 1 of sodium-coupled bile acid transporter ASBT Interact with Na+ and 7α-OH moieties on the ligand cholestane skeleton. J. Biol. Chem. 283, 20653–20663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loo T. W., Clarke D. M. (1993) Functional consequences of proline mutations in the predicted transmembrane domain of P-glycoprotein. J. Biol. Chem. 268, 3143–3149 [PubMed] [Google Scholar]
- 32. Cordes F. S., Bright J. N., Sansom M. S. (2002) Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323, 951–960 [DOI] [PubMed] [Google Scholar]
- 33. Gibbs N., Sessions R. B., Williams P. B., Dempsey C. E. (1997) Helix bending in alamethicin. Molecular dynamics simulations and amide hydrogen exchange in methanol. Biophys. J. 72, 2490–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lionarons D. A., Boyer J. L., Cai S. Y. (2012) Evolution of substrate specificity for the bile salt transporter ASBT (SLC10A2). J. Lipid Res. 53, 1535–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]