

Background: Sweet taste receptors are candidate nutrient sensors in adipose tissue.
Results: Sweet taste receptor ligands stimulate adipogenesis and suppress lipolysis; however, these effects do not require T1R2 and T1R3 despite their expression in adipose tissue.
Conclusion: Some artificial sweeteners regulate adipocyte differentiation and metabolism through a sweet taste receptor-independent mechanism.
Significance: Absorbed artificial sweeteners may regulate aspects of adipose tissue biology.
Keywords: Adipogenesis, Akt, G Protein-coupled Receptors (GPCR), Lipolysis, Metabolism, Acesulfame K, Saccharin, T1R2, T1R3
Abstract
G protein-coupled receptors mediate responses to a myriad of ligands, some of which regulate adipocyte differentiation and metabolism. The sweet taste receptors T1R2 and T1R3 are G protein-coupled receptors that function as carbohydrate sensors in taste buds, gut, and pancreas. Here we report that sweet taste receptors T1R2 and T1R3 are expressed throughout adipogenesis and in adipose tissues. Treatment of mouse and human precursor cells with artificial sweeteners, saccharin and acesulfame potassium, enhanced adipogenesis. Saccharin treatment of 3T3-L1 cells and primary mesenchymal stem cells rapidly stimulated phosphorylation of Akt and downstream targets with functions in adipogenesis such as cAMP-response element-binding protein and FOXO1; however, increased expression of peroxisome proliferator-activated receptor γ and CCAAT/enhancer-binding protein α was not observed until relatively late in differentiation. Saccharin-stimulated Akt phosphorylation at Thr-308 occurred within 5 min, was phosphatidylinositol 3-kinase-dependent, and occurred in the presence of high concentrations of insulin and dexamethasone; phosphorylation of Ser-473 occurred more gradually. Surprisingly, neither saccharin-stimulated adipogenesis nor Thr-308 phosphorylation was dependent on expression of T1R2 and/or T1R3, although Ser-473 phosphorylation was impaired in T1R2/T1R3 double knock-out precursors. In mature adipocytes, artificial sweetener treatment suppressed lipolysis even in the presence of forskolin, and lipolytic responses were correlated with phosphorylation of hormone-sensitive lipase. Suppression of lipolysis by saccharin in adipocytes was also independent of T1R2 and T1R3. These results suggest that some artificial sweeteners have previously uncharacterized metabolic effects on adipocyte differentiation and metabolism and that effects of artificial sweeteners on adipose tissue biology may be largely independent of the classical sweet taste receptors, T1R2 and T1R3.
Introduction
In conditions of persistent nutrient excess, adipocytes differentiate from mesenchymal precursor cells to provide additional reservoirs for lipid storage. These same nutritional conditions result in reciprocal regulation of anabolic and catabolic processes in mature adipocytes to promote triacylglycerol accumulation. In preadipocytes, adipogenic stimulation results in the activation of transcription factors PPARγ4 and C/EBPα, primary drivers of the adipogenic program that stimulate expression of terminal adipocyte genes such as FABP4 and GLUT4 (1). However, the upstream endogenous circulating, vascular or adipocyte-derived factors that are sensed as the key signals to indicate nutritional excess have not been fully elucidated. Nutritive signals themselves may serve this role as has been demonstrated by fatty acids acting through GPR43 and GPR120 to promote preadipocyte differentiation in vitro (2, 3). In adipocytes, energy availability is communicated indirectly through insulin concentrations and sympathetic tone (4) but is also sensed directly through G protein-coupled receptors. In adipose tissue, G protein-coupled receptors respond to short chain fatty acids, lactate, β-hydroxybutyrate, β-hydroxyoctanoate, and succinate to mediate effects on lipolysis (5–9). A hypothesis explored in this study is whether nutritive signals regulating adipocyte differentiation and metabolism are also mediated in part by sweet taste receptors.
The sweet taste receptor is generally believed to be an obligate heterodimer of the G protein-coupled receptors T1R2 and T1R3 (10, 11). Sugars and artificial sweeteners such as saccharin (Sacc) and acesulfame potassium (AceK) bind primarily to T1R2 (12), although direct binding to T1R3 has been described (13). Originally characterized in the tongue as a mediator of saccharin preference, these receptors have subsequently been described in the brain, bladder, pancreas, and gut (14–17) with metabolic roles defined in the latter two tissues. In the enteroendocrine cells of the small intestine, activation of sweet taste receptors promotes glucose uptake and release of incretin hormones such as glucagon-like peptide 1 (18, 19). T1R2/T1R3 also functions in the pancreas where its activation in β cells mediates stimulatory effects of sweeteners on glucose-induced insulin secretion (17, 20).
Numerous reports have demonstrated sweet taste receptor activation in response to artificial sweeteners in heterologous expression systems (10, 12, 21, 22); however, experiments in taste receptor knock-out (KO) animals suggest that an additional receptor(s) may be capable of binding and responding to sweet tastants (11, 23, 24). Additionally, binding of artificial sweeteners to the N-terminal domain of T1R2 or T1R3 in the absence of its dimerization partner suggests that these receptors may be capable of functioning independently (13, 25, 26). Although the input of T1R2/T1R3 may be important in the tongue and certain metabolic tissues, these studies indicate that there may be additional receptors sensitive to carbohydrates and sweeteners.
Here we report that T1R2 and T1R3 were constitutively expressed throughout adipogenesis of immortalized and primary cells and within adipose tissues. Treatment with artificial sweeteners such as saccharin and AceK induced adipogenesis of mouse and human precursors. Saccharin treatment also stimulated phosphorylation of Akt and its downstream effectors of preadipocyte differentiation. Surprisingly, T1R2 and T1R3 were dispensable for both saccharin-stimulated adipogenesis and phosphorylation on Thr-308, although activation of Ser-473 was at least partially dependent on sweet taste receptors. In mature adipocytes, exposure to artificial sweeteners suppressed both basal and stimulated lipolysis through a mechanism that was also independent of sweet taste receptors. Taken together, these data demonstrate unexpected roles for artificial sweeteners in adipocyte differentiation and metabolism and support the presence of additional “sweet” taste receptors.
MATERIALS AND METHODS
Cell Culture
3T3-L1 cells were differentiated as described previously (27). Briefly, cells 2 days after confluence (D0) were treated with DMEM containing 10% fetal bovine serum, 1 μm dexamethasone, 1 μg/ml insulin, and 0.5 mm methylisobutylxanthine or combinations thereof. Cells were fed every 2 days with insulin and fetal bovine serum (FBS) supplementation on D2 and FBS alone from D4 to the conclusion of the experiment. In general, artificial sweeteners were added to differentiation medium at induction and replaced with medium every 2 days. Saccharin, AceK, and sucralose were from Sigma-Aldrich. Adipogenesis was evaluated by Oil Red-O (Sigma-Aldrich) staining as described previously (28). LY294002, a phosphatidylinositol 3-kinase (PI3K) inhibitor, and U0216, a MEK1/2 inhibitor, were purchased from Cell Signaling Technology (Danvers, MA). U73122, a phospholipase Cβ inhibitor, was from Cayman Chemical (Ann Arbor, MI).
eMSC Isolation
eMSCs were isolated from controls, T1R2 KO, T1R3 KO, and T1R2/T1R3 double KO mice as described previously (29). Briefly, mouse ears were sterilized, minced, and incubated for 1 h in collagenase to obtain a cell suspension. Preconfluent cells were supplemented with 50 μg/ml FGF during the initial growth period (30) and maintained at 5% CO2 in DMEM/F-12 supplemented with 15% FBS. For differentiation, 1 μm dexamethasone, 5 μg/ml insulin, and 0.5 mm methylisobutylxanthine were added to maintenance medium for 2 days starting at D0. Insulin remained in medium for the first 4 days of adipogenesis. In general, cells were fed every 2 days until harvest.
Glucose Uptake
3T3-L1 adipocytes were serum-starved in HBSS (Invitrogen) with 0.5% BSA for 4 h. Cells were then incubated with 4 nm insulin in Krebs-Ringer HEPES buffer for 10 min before treating with 50 μm cytochalasin B to block background translocation. After 20 min of insulin treatment, 0.1 μCi/ml 2-deoxy[14C]glucose (PerkinElmer Life Sciences) was added. Ten min later (30-min post-insulin), adipocytes were placed on ice, washed, and lysed in 0.1% SDS. Uptake of 2-deoxyglucose was then quantified by scintillation counting.
Lipolysis
Lipolysis assays were conducted in differentiated adipocytes at least 8 days after induction of adipogenesis for 3T3-L1 cells or after 12 days for eMSCs and human SVCs. Secretion of glycerol and non-esterified fatty acid from cultured adipocytes into HBSS was determined with assay kits from Sigma-Aldrich (FG0100) and Wako Diagnostics (Richmond, VA; NEFA-HR(2)). Cells were treated for 2 h or the time indicated. Lipolysis in mouse explants was performed as described under “Animals.” Saccharin was used at 4.5 mm unless indicated otherwise.
cAMP
cAMP was measured by ELISA (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions.
Human SVC Isolation
SVCs were isolated from a subcutaneous fat depot of a 54-year-old diabetic patient in the same manner as eMSCs (29). Human SVCs were maintained and differentiated in the same manner as eMSCs. Human samples were obtained with the approval of the Institutional Review Board of the University of Michigan Medical School (HUM00060733).
Immunoblot Analysis
Cell extracts were lysed in an SDS buffer (1% SDS, 12.7 mm EDTA, 60 mm Tris-HCl, pH 6.8) and heated to 95 °C. Lysates were then centrifuged to pellet cell debris and transferred to a fresh tube, and the protein concentration was quantified with a BCA assay (Thermo Scientific, Waltham, MA). 4× SDS loading buffer (4% SDS, 240 mm Tris-HCl, 40% glycerol, 0.05% bromphenol blue, 2.5% 2-mercaptoethanol) was added to a constant amount of protein before separation on Bis-Tris polyacrylamide gels (Invitrogen). For evaluation of adipogenesis markers, a constant volume of lysate was used. SDS-PAGE and immunoblotting were performed as described previously (28). Membranes were immunoblotted with antibodies from Cell Signaling Technology for C/EBPα (catalog number 2295), pAkt308 (catalog number 9271), pAkt473 (catalog number 9275 or 4060), pFOXO1 (catalog number 9461), pCREB (catalog number 9191), phosphorylated hormone-sensitive lipase (pHSL) (catalog number 4126), pERK (catalog number 9101), total Akt (catalog number 9272), total ERK1/2 (catalog number 4695), total FOXO1 (catalog number 9462), total hormone-sensitive lipase (HSL) (catalog number 4107), and total CREB (catalog number 9197). Laminin antibody was obtained from Novus Biologicals (Littleton, CO). PPARγ1/2 antibody was obtained from Millipore (Temecula, CA). FABP4 antibody was obtained from R&D Systems (Minneapolis, MN; MAB1143).
mRNA Quantification by RT-PCR
Total RNA was prepared from frozen tissue or cells using RNA Stat60 according to the manufacturer's protocol (Tel-Test, Inc., Friendsville TX). Total RNA was quantified and reverse transcribed with random hexamers (TaqMan Reverse Transcription kit, Applied Biosystems, Foster City, CA). Quantitative PCR was performed using the MyiQ real time PCR detection system with SYBR Green reagents (Bio-Rad). Reverse transcription, primer design, and quantitative PCR were performed as described previously (28). Primer sequences are as follows: T1R2: F, GTCCGCTGCACCAAGCA; R, GTTCGTCGAAGAAGAGCTGGTT; T1R3: F, CCAGGCAACCAGGTGCCAGTC; R, CGCCTTGCAGTCCACGCAGT; PPARγ: F, CCAGAGCATGGTGCCTTCGC; R, TTCCGAAGTTGGTGGGCCAGA; C/EBPα: F, TGGACAAGAACAGCAACGAG; R, TCACTGGTCAACTCCAGCAC; PREF1: F, CCTCCTGTTGCAGTATAACAGCG; R, GGTCATGTCAATCTTCTCGGG; WNT10b: F, ACGACATGGACTTCGGAGAGAAGT; R, CATTCTCGCCTGGATGTCCC; C/EBPβ: F, GGGACTTGATGCAATCCGG; R, AACCCCGCAGGAACATCTTT; C/EBPδ: F, CGCCGCAACCAGGAGAT; R, GCTGATGCAGCTTCTCGTTCT; FOXO1: F, GCTTTTGTCACATGCAGGT; R, CGCACAGAGCACTCCATAAA.
Animals
C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Procedures for this work were approved by the Committee on the Use and Care of Animals at the University of Michigan with daily care of animals overseen by the unit for laboratory animal medicine (PRO0001369). Animals were maintained on a 12-h light/dark cycle and fed standard chow ad libitum (LabDiet 5L0D, Purina, St. Louis, MO). Leprdb/db mice were obtained from The Jackson Laboratory. For saccharin effects on ex vivo lipolysis, 10-week-old male mice were injected with a sodium saccharin solution at 100 mg/kg. After 20 min, animals were euthanized with CO2, and epididymal adipose tissue depots were excised. Depots were then weighed and cut into ∼40-mg pieces, which were then cultured at 5% CO2 in HBSS for 4 h before quantifying glycerol in medium. T1R2 and T1R3 KO animals were created and analyzed by Charles Zuker, Columbia University (11). Double T1R2/T1R3 KO animals were generated by initially mating T1R2 KO and T1R3 KO mice to produce double heterozygote T1R2/T1R3. In turn, double heterozygote T1R2/T1R3 mice were mated to produce littermate double wild-type controls and double T1R2/T1R3 knock-out mice.
Calcium Imaging
3T3-L1 cells were incubated with Fluo4 (Invitrogen) in glass bottom dishes (MatTek, Ashland, MA) for 30 min before imaging by confocal microscopy (Olympus FV500 confocal microscope and Olympus IX-71). Sweeteners were delivered by pipette as 10× solutions and allowed to diffuse through dishes maintained at 37 °C while monitoring the green fluorescence channel. Experiments were conducted with the assistance of the Michigan Diabetes Research and Training Center Morphology and Image Analysis Core.
Unbiased Metabolomics
To prepare samples for untargeted metabolomics, metabolites were extracted from adipocytes according to the protocol of Lorenz et al. (31). Briefly, 500 μl of cold 8:1:1 methanol:chloroform:water were added to each well of a 6-well cell culture plate that had previously been aspirated of all medium and quenched by freezing with liquid nitrogen. The plate was then scraped with a cell scraper to release and lyse cells. The cell extract was transferred by pipette to a microcentrifuge tube, and residual cell debris was pelleted by centrifugation at 15,000 × g for 10 min. The supernatant was directly analyzed by LC-MS.
Cell extracts were analyzed by LC-MS using an Agilent 1200 HPLC coupled to an Agilent 6210 time-of-flight mass spectrometer. Chromatographic separation was performed via mixed mode anion exchange-hydrophilic interaction chromatography using a Phenomenex Luna NH2 3-μm column, 15 cm × 2-mm inner diameter. Mobile phase A for the separation was acetonitrile, and mobile phase B was 5 mm ammonium acetate in water adjusted to pH 9.9 with ammonium hydroxide. The gradient consisted of a 20-min linear ramp from 20 to 100% B followed by a 2-min hold at 100% B and a subsequent 13-min re-equilibration period at 20% B. The sample injection volume was 20 μl, and the flow rate was 0.25 ml/min. Detection was performed by electrospray ionization mass spectrometry in negative ion mode. MS parameters were as follows: gas temperature, 350 °C; drying gas, 10 liters/min; nebulizer, 20 p.s.i. gauge; capillary voltage, 3500 V; scan range, 50–1200 Da; internal reference mass correction, enabled.
Untargeted metabolomics data analysis was performed using Agilent Masshunter Qualitative Analysis and Mass Profiler Professional software. Features in the data were first detected using the Find by Feature algorithm in Masshunter Qualitative Analysis and then aligned between samples by accurate mass and retention time using Mass Profiler Professional. To minimize gaps in the data, recursive detection of aligned features was performed using the Find by Formula algorithm. Once a final list of features was generated, compounds were assigned putative identities by searching against the online Metlin database. In many cases, the Metlin search resulted in multiple possible matches for each feature within a 10-ppm mass error window. Metabolite matches were ranked in order of ascending mass error and among matches with equivalent mass error in order of ascending Metlin identification number. Following putative metabolite identification, statistical analysis and data reduction techniques, including principle component analysis were used to determine statistically different features between sample groups and to assess global differences in the metabolome between treatment conditions.
Saturation Transfer Difference NMR
Preparation of membranes and saturation transfer difference NMR was performed as described by Venkitakrishnan et al. (32) with some modifications. Briefly, WT and double KO (DKO) eMSCs were grown to confluence in 500-cm2 Corning dishes (Corning, NY) and suspended in EDTA. The cell suspension was pelleted at 800 × g and then lysed using a Polytron homogenizer (Kinematica, Lucerne, Switzerland). The resulting suspension was spun at low speed to collect cell debris followed by ultracentrifugation at 100,000 × g to isolate cell membranes. Following membrane isolation, saturation transfer difference NMR spectroscopy was performed as described previously (32).
RESULTS
Sweet Taste Receptors T1R2 and T1R3 Are Expressed throughout Adipogenesis in 3T3-L1 Cells and eMSCs
To evaluate the expression of taste receptors during adipogenesis, RNA was isolated at the indicated time points as 3T3-L1 preadipocytes were differentiated into mature adipocytes, and quantitative PCR analyses were performed (Fig. 1). Expression of both T1R2 and T1R3 peaked at day 2 of adipogenesis, returning to near preadipocyte levels by day 8 (Fig. 1A). We then examined sweet taste receptor expression in primary eMSCs as an independent adipogenic model. These cells are isolated from the mouse ear and are capable of chondrogenic, osteogenic, and adipogenic differentiation (29). We found that sweet taste receptor expression in this system was reminiscent of that in 3T3-L1 cells; T1R2 and T1R3 both peaked in expression at day 2 of adipogenesis before returning to preadipocyte levels at day 12 (Fig. 1B).
FIGURE 1.

Sweet taste receptors T1R2 and T1R3 are expressed constitutively throughout adipogenesis and in adipose tissues. A, T1R2 and T1R3 expression throughout 8 days of adipogenesis in 3T3-L1 cells. B, T1R2 and T1R3 expression over 12 days of adipogenesis in eMSCs. C, the indicated tissues were collected from control and 13–15-week-old female db/db mice maintained on a chow diet, and expression of T1R3 mRNA was evaluated by quantitative PCR. Data are expressed as mean ± S.D. (error bars) with n = 4–6/tissue. Analysis by two-way analysis of variance indicates a significant effect of genotype (p < 0.05). gWat, gonadal white adipose tissue; iWAT, inguinal white adipose tissue; pWAT, perirenal white adipose tissue.
To determine whether sweet taste receptors were present in adipose tissue in vivo, we evaluated T1R3 expression in adipose tissues and liver of control and db/db mice. Expression of T1R3 was different between tissues of control mice by as much as 2-fold (Fig. 1C). Based on a statistical interaction between genotypes, expression of T1R3 is generally reduced in obese animals. However, post hoc analyses revealed only trends within individual tissues due to substantial variation in expression. Taken together, these data indicate that sweet taste receptors are expressed throughout adipogenesis and in adipose tissues.
Saccharin Stimulates Adipogenesis of Mouse and Human Precursor Cells
To assess the effects of sweet taste receptor activity on adipogenesis, we utilized the artificial sweeteners Sacc and AceK as T1R2/T1R3 ligands. These agonists, as opposed to natural sugars, are useful in metabolic studies because they are not metabolized and are ∼500-fold sweeter than sucrose (33, 34). First, 3T3-L1 cells were treated throughout adipogenesis with a submaximal adipogenic mixture containing dexamethasone and insulin supplemented with increasing concentrations of saccharin. The addition of saccharin robustly stimulated adipogenesis in a concentration-dependent manner, resulting in increased lipid accumulation and FABP4 expression (Fig. 2A). FABP4 was then quantified over multiple experiments to empirically determine minimal saccharin concentrations necessary to enhance adipogenesis (Fig. 2B); this densitometric analysis indicated that 0.45 mm saccharin is the lowest concentration to significantly increase FABP4 accumulation in 3T3-L1 cells, whereas higher amounts resulted in a nearly 10-fold increase.
FIGURE 2.

Saccharin stimulates adipogenesis of mouse and human precursor cells. A, 3T3-L1 cells were differentiated with DI in the presence of the indicated concentrations of saccharin. Seven days after induction, cells were stained for neutral lipid with Oil Red-O (upper panel), and lysates were evaluated for expression of FABP4 (lower panel). ERK1/2 was used as a loading control. B, quantification of saccharin-stimulated FABP4 expression from four independent experiments in DI-treated 3T3-L1 cells. Data are expressed as mean ± S.D. (error bars). p < 0.01 is indicated with **, and p < 0.005 is indicated with ***. C, 3T3-L1 cells were differentiated with components of the MDI mixture as indicated in the presence or absence of 4.5 mm saccharin. Adipogenesis was then evaluated after 8 days by Western blotting for FABP4. D, eMSCs were incubated in the presence of MDI, DI, or FBS with or without 2 mm saccharin supplementation. At day 16 of differentiation, the degree of differentiation was evaluated with photomicrographs (upper panels) and by expression of FABP4 (lower panels). E, human SVCs were induced with MDI for 14 days in the absence or presence of 4.5 mm saccharin. Adipogenesis was assessed with photomicrographs and by expression of FABP4. M, methylisobutylxanthine; D, dexamethasone; I, insulin.
3T3-L1 cells induced with dexamethasone and insulin (DI) and treated with saccharin show enhanced adipogenesis, which could be due to a specific synergistic interaction of the signaling pathways activated by saccharin and DI. To test this, we differentiated 3T3-L1 cells with various combinations of the full methylisobutylxanthine, dexamethasone, and insulin (MDI) mixture, including each component individually or no induction at all (FBS), in the presence or absence of saccharin (Fig. 2C). Using this approach, we observed that saccharin was sufficient to induce adipogenesis under all tested conditions with mild effects even in the absence of MDI. These data suggest that saccharin does not require permissive activation of a specific adipogenic signaling pathway to stimulate differentiation.
To determine whether these effects of saccharin on adipogenesis are observed in other models of adipogenesis, we first evaluated multipotent, primary eMSCs and found that saccharin robustly stimulated lipid accumulation and expression of FABP4 (Fig. 2D). As observed with 3T3-L1 preadipocytes, artificial sweetener effects occurred independently of differentiation conditions as eMSCs induced with MDI, DI, or FBS all showed an enhancement of adipogenesis with saccharin treatment. We also tested the applicability of these findings to human precursors and observed that in SVCs isolated from human white adipose tissue saccharin markedly enhanced lipid accumulation and FABP4 expression following adipogenic induction with MDI (Fig. 2E). Taken together, these results indicate that adipogenesis of mouse and human precursors is stimulated by saccharin, suggesting that sweet taste receptors may sense nutritive signals and regulate preadipocyte differentiation.
AceK Stimulates Adipogenesis of Mouse and Human Precursor Cells
To test whether effects on adipogenesis are specific to saccharin, we repeated the previous experiments with another artificial sweetener and found that AceK also stimulated 3T3-L1 adipogenesis. Comparable with our results with saccharin, increased adipogenesis was observed at 4.5 mm AceK (Fig. 3A). Results similar to those with saccharin were also observed in eMSCs where AceK supplementation enhanced adipogenesis regardless of the differentiation induction conditions (Fig. 3B). Lastly, we repeated this experiment in human SVCs. In these primary cells, AceK stimulated lipid and FABP4 accumulation (Fig. 3C). These results suggest that proadipogenic effects may be broadly observed with different artificial sweeteners.
FIGURE 3.
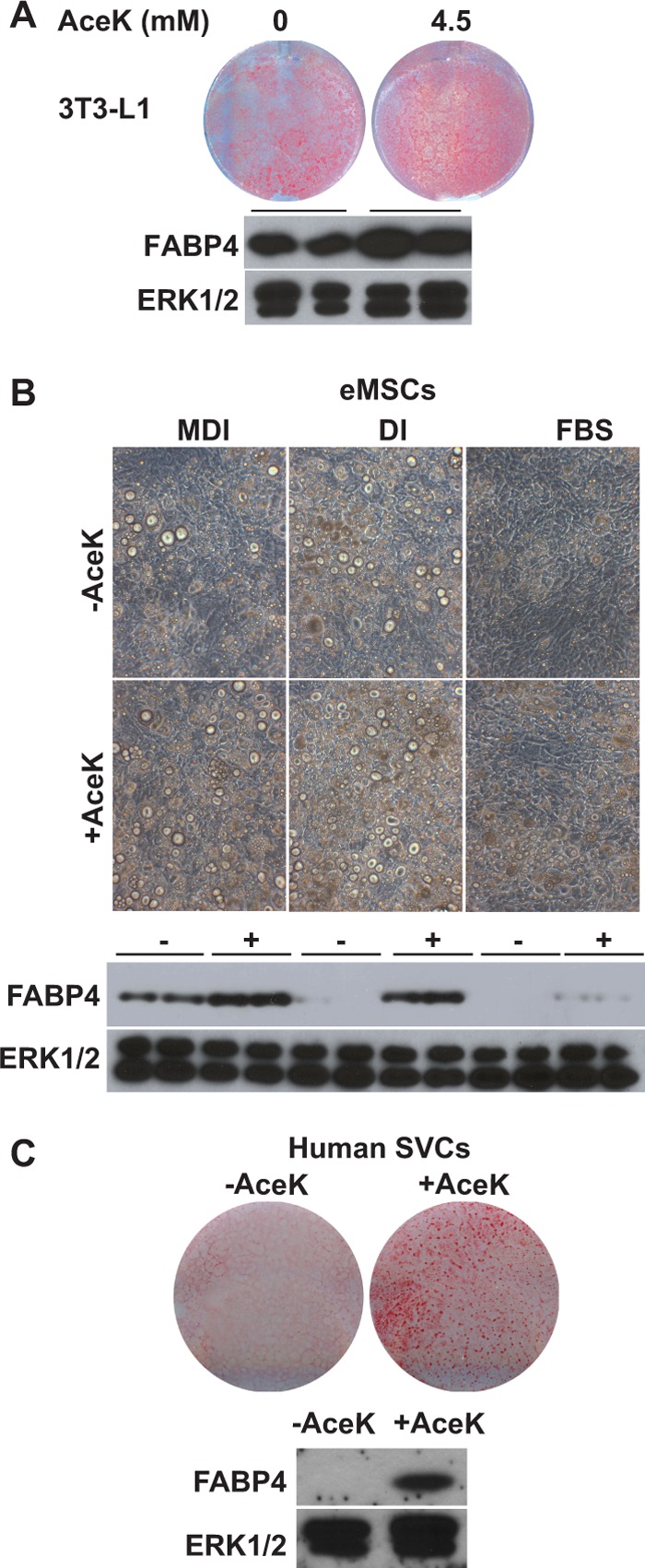
AceK stimulates adipogenesis of mouse and human precursor cells. A, 3T3-L1 cells were induced with DI and treated for 8 days with the indicated concentrations of AceK. After 8 days, cells were stained for neutral lipid with Oil Red-O. Lysates were evaluated for expression of FABP4. B, eMSCs were incubated in the presence of MDI, DI, or FBS with or without 2 mm AceK. After 16 days, the degree of differentiation was evaluated with photomicrographs (upper panels) and by expression of FABP4 (lower panels). C, human SVCs were induced with MDI for 14 days in the absence or presence of 4.5 mm AceK. Adipogenesis was determined with photomicrographs (upper panel) and by expression of FABP4 (lower panel).
Effects of Saccharin on Adipogenesis Are Dependent on Time and Duration
To further characterize sweetener-stimulated adipogenesis, we evaluated the temporal requirements of saccharin treatment to enhance adipogenesis. To do this, we treated differentiating 3T3-L1 cells with saccharin at varying time intervals (Fig. 4A). Cells treated with saccharin for the first 2 or 4 days of adipogenesis had elevated expression of PPARγ and FABP4 with 4 days of treatment showing a more pronounced effect on Oil Red-O staining. However, maximal effects on lipid accumulation and adipocyte gene expression were observed with the full 8 days of treatment. Saccharin treatment must begin within an early time window because saccharin treatment had no effect on adipogenesis when supplementation began on day 4. These observations suggest that, in addition to concentration (Figs. 2 and 3), both time and duration of saccharin treatment are important for effects on adipogenesis.
FIGURE 4.

Saccharin stimulates expression of PPARγ and C/EBPα late during differentiation. A, 3T3-L1 cells were induced with DI and supplemented with 4.5 mm Sacc at the indicated time points. Cells were stained with Oil Red-O (upper panel), and lysates were collected for immunoblotting after 8 days (lower panels). C/EBPα (B) and PPARγ (C) expression was measured over a time course of adipogenesis in 3T3-L1 cells induced with DI and treated with vehicle, 4.5 mm Sacc, or 4.5 mm AceK for 4 days. Expression of C/EBPβ (D), C/EBPδ (E), and PREF1 (F) was measured over the first 5 days of differentiation in 3T3-L1 cells treated with Sacc or AceK. Significance was determined using Student's t test. Significant differences (p < 0.05) between vehicle and Sacc are denoted with *, and those between vehicle and AceK are denoted with #. Data are expressed as mean ± S.D. (error bars).
Saccharin Stimulates Expression of PPARγ and C/EBPα Late during Differentiation
To investigate mechanisms for enhancement of adipogenesis by artificial sweeteners, we next examined aspects of the transcriptional profile of cells that had been treated with DI ± saccharin or AceK for the first 4 days of adipogenesis. We observed that sweetener-stimulated PPARγ and C/EBPα expression did not differ from control cells until D5, after the removal of saccharin from the differentiation medium. By D8, expression of C/EBPα was increased ∼3-fold, and expression of PPARγ was elevated ∼2-fold in saccharin- or AceK-treated cells compared with DI alone (Fig. 4, B and C). These data suggest that sweeteners stimulate early events in adipogenesis (D0–D4) that are propagated following termination of sweetener treatment.
Because many essential elements of sweetener signaling occur in the first 2 or 4 days of adipogenesis (Fig. 4), we profiled important regulators of adipogenesis active in this time period. However, after profiling mRNA expression of numerous transcription factors and inhibitors of differentiation, including C/EBPβ (Fig. 4D), C/EBPδ (Fig. 4E), PREF1 (Fig. 4F), and FOXO1, EBF1, and EBF2 (data not shown), significant alterations in early events of adipogenesis were not observed. These data suggest that artificial sweeteners robustly stimulate adipogenesis in the first 4 days of adipogenesis, consistent with increased expression of PPARγ and C/EBPα by D5 and beyond; however, the mechanistic basis appears to be independent of the aspects of the program of adipogenesis evaluated.
Saccharin Acutely Activates Akt and ERK1/2 Signaling Pathways in Preadipocytes
We next examined signal transduction cascades that might mediate sweetener effects. In other contexts (e.g. taste cells, β cells, and enteroendocrine cells), sweet taste receptor activation produces intracellular Ca2+ transients (19, 20, 35). However, saccharin appears to work through an atypical signaling pathway in preadipocytes because we did not find evidence for sweetener-stimulated calcium flux in preadipocytes at or above concentrations that stimulate adipogenesis (data not shown). To evaluate potential mechanisms by which saccharin signals to stimulate preadipocyte differentiation, we screened known adipogenic signaling pathways (1). 3T3-L1 preadipocytes were serum-starved for 2 h and then treated with saccharin for 30 min, and lysates were screened by immunoblot for activated signaling proteins. Using this approach, we observed that saccharin treatment stimulated phosphorylation of Akt at Thr-308 and Ser-473 (Fig. 5A), modifications that are well characterized for their proadipogenic effects (36–39). Importantly, saccharin also stimulated phosphorylation of CREB and FOXO1, which are known downstream targets of Akt involved in promoting preadipocyte differentiation (36, 40, 41). Similar to the lack of calcium transients, this is the first known report of artificial sweeteners stimulating Akt signaling, which implicates novel pathways, novel receptors, or both, in transducing metabolic signals from artificial sweeteners.
FIGURE 5.

Saccharin activates Akt and ERK1/2 signaling pathways in preadipocytes. A, 3T3-L1 preadipocytes were serum-starved for 2 h in HBSS and then treated with 0.45 or 4.5 mm saccharin for 30 min before lysis and immunoblot analyses with the indicated antibodies. B, 3T3-L1 cells were serum-starved in HBSS for 2 h and pretreated with 50 μm LY294002 for 1 h. Cells were then treated with the indicated concentrations of saccharin in the absence or presence of LY294002 for 30 min. After lysis, samples were probed by immunoblot with the indicated antibodies. Insulin at 25 nm was included as a control. C, 3T3-L1 cells were serum-starved in HBSS for 2 h and pretreated with 1 μm U0216, 5 μm U73122, or 2 μm wortmannin for 1 h. Cells were then treated with 4.5 mm saccharin in the absence or presence of each inhibitor for 30 min before immunoblotting for the indicated proteins. D, 3T3-L1 preadipocytes were serum-starved in HBSS for 2 h and treated with 4.5 mm saccharin for the indicated time periods before lysis and immunoblotting. E, 3T3-L1 cells were stimulated with DI in the presence or absence of 4.5 mm saccharin. Lysates were prepared at the indicated time points and probed by immunoblot for the indicated proteins. T308, threonine 308; S473, serine 473; T Akt, total Akt; T CREB, total CREB; T FOXO1, total FOXO1; T ERK1/2, total ERK1/2; WM, wortmannin.
To further investigate this novel signal transduction mechanism in preadipocytes, we evaluated saccharin-stimulated signaling upstream of Akt phosphorylation by using PI3K, MEK, or phospholipase Cβ inhibitors. We observed that the PI3K inhibitors LY294002 (Fig. 5B) and wortmannin (Fig. 5C) reduced base-line phosphorylation of Akt as well as the ability of saccharin to stimulate Akt phosphorylation of Thr-308 or Ser-473. Inhibition of MEK signaling with U0216 had no effect on basal or saccharin-stimulated Akt phosphorylation, whereas inhibition of protein kinase C with U73122 suppressed basal Akt phosphorylation with a partial or full block of saccharin-stimulated Thr-308 or Ser-473 phosphorylation, respectively (Fig. 5C). These data suggest that this novel saccharin-stimulated Akt phosphorylation occurs in a PI3K-dependent manner.
We next explored the temporal properties of saccharin-stimulated Akt phosphorylation in 3T3-L1 cells. Here we observed that Akt Thr-308 phosphorylation was rapid, occurring within 5 min, and persisted for at least 1 h in serum-starved cells (Fig. 5D). Whereas maximal activation for Thr-308 occurred within 5 min, Ser-473 phosphorylation could only just be detected at that time point, but phosphorylation steadily increased throughout the time course for 60 min.
Finally, we evaluated the ability of saccharin to stimulate Akt phosphorylation in the context of adipogenesis when Akt is already strongly activated by high concentrations of insulin. Even in the presence of DI, 3T3-L1 preadipocytes demonstrated augmented phosphorylation at Thr-308 in the presence of saccharin (Fig. 5E). Elevated phosphorylation at Thr-308 was observed within 30 min of treatment and persisted to 6 h following induction. In contrast, saccharin caused a small increase in phosphorylation at Ser-473 only at 2 and 4 h. Interestingly, phosphorylation of ERK1/2 was also enhanced by saccharin in differentiating 3T3-L1 cells (Fig. 5E). However, this saccharin-stimulated ERK1/2 activity was not present in serum-starved cells (data not shown). Although maximal ERK1/2 phosphorylation at 30 min was not influenced by saccharin treatment, the activation of ERK was observed at 2 h and persisted to 16 h with some increase in basal phosphorylation observed out to 72 h postinduction. Although further experiments will be required to explain this difference in ERK activity between serum-starved and DI-stimulated conditions, a “priming” effect of growth factor or insulin/IGF-1 signaling appears to be necessary for saccharin-stimulated ERK1/2 phosphorylation.
T1R2 and T1R3 Are Not Required for Saccharin-stimulated Adipogenesis or Akt Phosphorylation
Although sweet taste receptors are the most obvious candidates for binding saccharin in adipose tissue, some of our observations such as saccharin-stimulated Akt signaling and a lack of calcium transients have not been consistent with the current literature on sweet taste receptor signaling in other contexts. To determine whether sweet taste receptors are the mediators of artificial sweetener effects in adipose tissue, we investigated the dependence of saccharin-stimulated adipogenesis and Akt phosphorylation on the presence of sweet taste receptors. We reasoned that if saccharin acts through T1R2/T1R3 then knock-out of either receptor would block the effects of artificial sweeteners on both adipogenesis and Akt phosphorylation. Therefore, we investigated these phenotypes in eMSCs derived from T1R2 or T1R3 knock-out mice beginning with T1R3-null eMSCs. Surprisingly, we observed that T1R3 KO did not prevent saccharin-stimulated lipid accumulation (Fig. 6A). Accordingly, when we also measured the ability of saccharin to stimulate Akt Thr-308 phosphorylation in the absence of T1R3, we observed no difference in the concentration dependence (Fig. 6B, upper panel) of Akt phosphorylation between genotypes. There was also no difference in kinetic responses to saccharin in the absence of T1R3 (Fig. 6A, lower panel) as phosphorylation of Thr-308 and Ser-473 occurred within 5 min of saccharin treatment regardless of genotype. Akt phosphorylation in response to insulin was also similar between genotypes.
FIGURE 6.

T1R3 and T1R2 are not required for saccharin-stimulated adipogenesis or Akt phosphorylation. A, WT and T1R3 KO eMSCs were differentiated in FBS supplemented with 0.45 mm saccharin. After 12 days, lipid accumulation was evaluated with Oil Red-O. B, WT and T1R3 KO eMSCs were serum-starved for 2 h in HBSS and treated for 30 min with increasing concentrations of saccharin (0, 0.02, 0.045, 0.2, 0.45, 2, and 4.5 mm) before collecting lysates for immunoblotting (upper panel). WT and T1R3 KO eMSCs were serum-starved for 2 h in HBSS before being treated with saccharin for the indicated time intervals (lower panel). C, WT and T1R2 KO eMSCs were differentiated in FBS supplemented with 0.45 mm saccharin. After 12 days, lipid accumulation was evaluated with Oil Red-O. D, WT and T1R2 KO eMSCs were serum-starved for 2 h in HBSS and treated for 30 min with increasing concentrations of saccharin (0, 0.02, 0.045, 0.2, 0.45, 2, and 4.5 mm) before lysis and immunoblotting (upper panel). WT and T1R2 KO eMSCs were maintained in calf serum (−) or treated with DI in the absence (−) or presence (+) of 4.5 mm saccharin for 30 min (lower panel). E, WT and T1R2/T1R3 KO (DKO) eMSCs were differentiated in FBS supplemented with 0.45 mm saccharin. After 12 days, lipid accumulation was evaluated with Oil Red-O. F, WT and DKO eMSCs were serum-starved for 2 h in HBSS and treated for 30 min with 4.5 mm saccharin before lysis and immunoblotting. T Akt, total Akt.
We next investigated saccharin activity in T1R2 KO eMSCs. As with T1R3, we observed that T1R2 was not required for saccharin-stimulated lipid accumulation (Fig. 6C). Although the T1R2 KO eMSC precursors were slightly desensitized to saccharin in terms of Akt Thr-308 phosphorylation, T1R2 was clearly not required for the effects of saccharin on Thr-308 phosphorylation (Fig. 6D, upper panel). In addition, we observed that saccharin-stimulated Thr-308 and Ser-473 phosphorylation in differentiating eMSCs was not affected by the absence of T1R2 (Fig. 6D, lower panel). These data suggest that neither T1R2 nor T1R3 is individually necessary for saccharin-mediated enhancement of adipogenesis or Akt phosphorylation in preadipocytes. In this case, it is plausible that T1R2 and T1R3 might be acting as homodimers to bind saccharin and transduce signals, or saccharin might be working through another receptor or mechanism.
Literature suggests that T1R2 and T1R3 are capable of individually responding to tastants rather than functioning as an obligate heterodimer (25, 42, 43). To directly address the possibility of T1R2 and T1R3 homodimer activity in preadipocytes, we produced T1R2/T1R3 DKO eMSCs. Using this DKO model, we observed that saccharin enhanced adipogenesis even in the absence of both T1R2 and T1R3 (Fig. 6E). These data suggest that, in this context, T1R2 and T1R3 do not function as homodimers to mediate effects of saccharin on differentiation. Interestingly, whereas DKO had no effect on saccharin-stimulated Akt phosphorylation at Thr-308, phosphorylation at Ser-473 was reduced (Fig. 6F). These results suggest that although stimulation of Akt phosphorylation on Ser-473 is not essential for saccharin-induced adipogenesis it is likely that artificial sweeteners influence at least some aspects of adipose biology through sweet taste receptors.
Artificial Sweeteners Suppress Adipocyte Lipolysis and Decrease Hormone-sensitive Lipase Phosphorylation
Although artificial sweeteners have proadipogenic activity, this activity appears to be independent of T1R2 and T1R3 in preadipocytes. Because the sweet taste receptors are expressed throughout differentiation and in mature adipocytes (Fig. 1), we then tested whether saccharin regulates adipocyte metabolism in a T1R2/T1R3-dependent manner. Fully differentiated 3T3-L1 adipocytes were treated with saccharin or AceK for 1 h. Cells were very rapidly washed with ammonium acetate and frozen with liquid nitrogen, and lysates were subjected to unbiased metabolomics analysis. After 1 h, 81 metabolites were significantly different with saccharin, and of these, 14 of 17 were also regulated by AceK (Fig. 7A; data not shown). All of the 14 co-regulated metabolites decreased upon saccharin or AceK treatment (Fig. 7B), including four fatty acids (in red). Many additional fatty acids were predicted to decline in response to saccharin (data not shown). These data suggest that artificial sweeteners regulate some aspect of adipocyte lipid metabolism.
FIGURE 7.

Artificial sweeteners suppress lipolysis. A, following 1-h Sacc or AceK treatment, 3T3-L1 adipocytes were flash frozen, and intracellular metabolites were evaluated on an LC-MS platform by the University of Michigan Molecular Phenotyping Core. Of the metabolites that were significantly altered by the treatment, 69 were unique to Sacc, three were unique to AceK, and 14 were shared between both sweeteners. B, distribution of metabolites altered by Sacc alone (blue) or Sacc and AceK (yellow) following 1-h treatment in 3T3-L1 adipocytes. Long-chain fatty acids decreased by both saccharin and AceK are shown in red. The significance level on the y axis extends to 10e−16. C, glucose uptake was measured in 3T3-L1 adipocytes as described under “Materials and Methods.” Significant differences (p < 0.05) between 0 and 4 nm insulin are denoted with #. D, incorporation of [13C]palmitate into dipalmitoyl diacylglycerol (DAG) with 1 or 4 h of 4.5 mm Sacc treatment in 3T3-L1 adipocytes. E, 3T3-L1 adipocytes were serum-starved for 2 h in HBSS before treatment with 4.5 mm Sacc, 4.5 mm mannitol, or 10 μm Fsk. Glycerol content of the medium was assayed at the indicated time points. F, non-esterified fatty acid (NEFA) (μm) was measured from assay medium after 60 min of 4.5 mm Sacc treatment. G, glycerol content of assay medium was measured following 1-h treatment with Sacc, AceK, or sucralose (Sucr) at 0.45 and 4.5 mm. H, epididymal white adipose tissue (eWAT) explants were collected from C57BL/6J mice 20 min following injection of Sacc. Glycerol content of the medium was then measured following a 4-h incubation with vehicle, Fsk, or Sacc. Significant differences from vehicle treatment p < 0.05, p < 0.01, and p < 0.005 are denoted with *, **, and ***, respectively. Data are expressed as mean ± S.D. (error bars). Significance was determined using Student's t test. 2DOG, 2-deoxyglucose.
To determine the basis for reduced intracellular fatty acids following saccharin treatment of adipocytes, we evaluated a number of metabolic processes that could be directly or indirectly involved. We observed that saccharin treatment had no effect on glucose uptake (Fig. 7C), conversion of [14C]glucose or [14C]acetate to triacylglycerol (data not shown), or uptake and incorporation of [13C]palmitate into lipids, including diacylglycerol (Fig. 7D). However, the effects of saccharin on lipolysis of 3T3-L1 adipocytes were striking with reduced basal secretion of glycerol (Fig. 7E) and non-esterified fatty acid (Fig. 7F) detected as early as 30 min. Importantly, this effect was not due to osmotic stress as lipolysis was not influenced by equimolar concentrations of mannitol (Fig. 7E).
To determine whether lipolysis was inhibited by artificial sweeteners that are structurally distinct from saccharin, we evaluated the effects of AceK and sucralose (Fig. 7G) and observed that, as with saccharin, these artificial sweeteners suppressed basal lipolysis in 3T3-L1 cells. Lastly, we evaluated the effects of saccharin on basal lipolysis in an ex vivo system and found that isolated pieces of epididymal adipose tissue incubated with saccharin for 4 h had reduced secretion of glycerol into culture medium. Interestingly, an intraperitoneal injection of saccharin was also sufficient to reduce glycerol release when the adipose tissue was excised 20 min postinjection and lipolysis was evaluated over the subsequent 4 h (Fig. 7H).
We then investigated mechanisms for lipolytic regulation by artificial sweeteners. We observed that the addition of saccharin to 3T3-L1 adipocytes suppressed phosphorylation of HSL (Fig. 8A). Interestingly, saccharin significantly blunted the stimulation of both glycerol release and HSL phosphorylation by forskolin (Fsk), an adenylyl cyclase activator. This indicates that saccharin suppresses both basal and Fsk-stimulated lipolysis. We next hypothesized that saccharin might act through Akt signaling as it does in preadipocytes (Fig. 5) to suppress lipolysis in a PI3K-dependent manner. However, treatment with LY294002 also failed to block saccharin-mediated suppression of basal lipolysis (Fig. 8B), consistent with our observation that saccharin stimulated phosphorylation of Akt Thr-308 in adipocytes but only after effects on lipolysis were already observed (data not shown). As cAMP is an important regulator of lipolytic activity upstream of HSL, we speculated that saccharin treatment might reduce cAMP concentrations in adipocytes. However, saccharin had no effect on cAMP concentrations (Fig. 8C), suggesting that artificial sweeteners may act downstream of PKA.
FIGURE 8.

T1R2/T1R3 are dispensable for saccharin-suppressed lipolysis. A, 3T3-L1 adipocytes were treated for 1 h with Sacc, Fsk, or both. Glycerol concentration in the medium was measured before collection of protein lysates from control and treated cells. Lysates were then probed for pHSL by Western blotting (left panel). HSL phosphorylation was then quantified by densitometry (right panel). B, 3T3-L1 adipocytes were serum-starved for 2 h and pretreated with LY294002 (LY) for 1 h before a 2-h Sacc treatment. Glycerol secretion was then measured in medium. C, 3T3-L1 adipocytes were serum-starved for 2 h and then treated for 45 min with Fsk or Sacc. cAMP concentration was quantified by ELISA. T1R3 KO (D), T1R2 KO (E), or DKO (F) eMSC adipocytes were treated with Sacc, Fsk, or both for 4 h before measuring glycerol accumulation in medium. Significant differences p < 0.05, p < 0.01, and p < 0.005 are denoted *, **, and ***, respectively. Data are expressed as mean ± S.D. (error bars). Significance was determined using Student's t test. tHSL, total HSL.
T1R2/T1R3 Are Not Required for Suppression of Basal or Fsk-stimulated Adipocyte Lipolysis by Saccharin
Although T1R2 and T1R3 are likely binding candidates for artificial sweeteners, data in preadipocytes suggest that these receptors might not be necessary. We tested for saccharin-stimulated T1R2/T1R3 activity by performing lipolysis assays in T1R2 and T1R3 KO eMSCs. Similar to our observations in preadipocytes, loss of T1R3 (Fig. 8D) or T1R2 (Fig. 8E) failed to block saccharin effects on either basal or forskolin-stimulated lipolysis, and no significant differences were observed between genotypes. Furthermore, we tested effects of saccharin in the absence of both T1R2 and T1R3 to rule out the possibility of homodimer activity. However, as observed in preadipocytes, saccharin suppressed basal and forskolin-induced lipolysis even in the absence of both receptors (Fig. 8F). Taken together, these results suggest that saccharin reduces lipolysis independently of T1R2/T1R3 sweet taste receptors and through a PKA-mediated mechanism downstream of cAMP.
Saccharin Binds to Membranes of T1R2/T1R3 Double KO Adipocyte Progenitors
Thus far, our data have indicated that saccharin has effects on preadipocyte differentiation and adipocyte metabolism in the absence of sweet taste receptor expression. This suggests that either additional receptors are capable of binding and mediating effects of saccharin under these conditions or saccharin directly initiates intracellular signaling downstream of receptors. To distinguish between these two possibilities, we assessed saccharin binding by control and DKO preadipocyte membranes by saturation transfer difference NMR. Using this approach, we observed little difference in interactions between saccharin and either control or DKO preadipocyte membranes (Fig. 9A). Similar results were observed with an independent artificial sweetener, neotame, that has been well characterized within this assay (Fig. 9B and Ref. 32). Given that saccharin was capable of binding to the cell membrane of DKO preadipocytes, this suggests that receptors in addition to T1R2 and T1R3 mediate signal transduction of artificial sweeteners.
FIGURE 9.
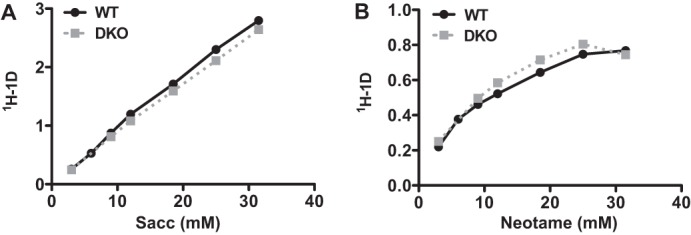
Saccharin and neotame bind similarly to membranes of control and T1R2/T1R3 DKO eMSC progenitors. WT and DKO membranes were incubated with the indicated concentrations of saccharin (A) or neotame (B), and binding of artificial sweeteners to membranes was evaluated by saturation transfer difference NMR.
DISCUSSION
One strategy currently utilized to curtail the obesity epidemic is extensive use of non-nutritive sweeteners. Such artificial sweeteners, including saccharin, AceK, aspartame, sucralose, and neotame, have little to no caloric value but are hundreds of times sweeter than sucrose. However, some studies suggest that high level consumers of artificial sweeteners may actually be at greater risk for overweight and obesity (44). Although not all studies agree with this assessment (45–47), the hypothesis that artificial sweeteners “uncouple” anticipated caloric density perceived by the tongue from the actual calories ingested has gained a distinct foothold. Some argue that this uncoupling leads to increased insulin secretion, food intake, and weight gain (48, 49), although there are little data that conclusively implicate artificial sweeteners in either weight gain or weight loss in humans (50).
Potential mechanisms for how artificial sweeteners may influence energy balance have been complicated by reports identifying functional sweet taste receptors in tissues outside the tongue. Our results demonstrate that artificial sweeteners such as saccharin and AceK can regulate adipocyte differentiation and metabolism. The active concentrations in preadipocytes and adipocytes are higher than would generally be observed in humans (51–54) as bolus oral doses of the recommended maximum daily intake of saccharin results in peak plasma concentrations of ∼75 μm (54); however, this is nonetheless an important proof of principle for unexpected effects of artificial sweetener use and raises the possibility that absorbed sweeteners may influence aspects of adipose tissue biology given sufficient “dose and duration” of exposure. However, further studies will be required to test this possibility.
Our data indicate that saccharin increases adipogenesis and represses adipocyte lipolysis in the absence of T1R2/T1R3, suggesting a novel saccharin receptor and/or mechanism of action. Some groups have hypothesized that T1R2/T1R3 does not function as an obligate heterodimer (42, 43), but rather each component functions independently as a homodimer, consistent with the N-terminal domains of both T1R2 and T1R3 containing ligand-binding sites and changing conformation in response to artificial sweeteners (13). The homodimerization model has found support from Shibata and co-workers (55), who have investigated sweet taste receptor activity in cultured 3T3-L1 and adipose tissue stromal cells. In their model, T1R3, but not T1R2, is required for inhibition of adipogenesis by sweet taste receptors. The authors show results conflicting with our own in that sodium saccharin and sucralose treatment inhibits adipogenesis and reduces expression of PPARγ and C/EBPα. Although we observed a mild inhibition of adipogenesis with sucralose (data not shown), in our hands, saccharin, sodium saccharin, and AceK consistently stimulated adipogenesis of mouse and human precursors. Masubuchi et al. (55) also showed that inhibition of adipogenesis is partially blocked with knockdown of T1R3 using shRNA; however, we did not observe impaired differentiation in primary eMSCs derived from mice lacking T1R2, T1R3, or both receptors (Fig. 6). Although slight differences in experimental models might be the cause of our disparate results, our work suggests that receptor homodimerization is not sufficient to mediate saccharin activity in preadipocytes. This is further supported by quantitative data demonstrating that saccharin and neotame bind precursor membranes in the absence of both T1R2 and T1R3 (Fig. 9). These data support a model in which tastants bind to an uncharacterized “sweet” receptor.
Further complexity to sweet taste receptor signaling is indicated by the analysis of gustatory nerves: T1R3 KO differentially effects the chorda tympani, a facial nerve innervating the anterior two-thirds of the tongue, versus the glossopharyngeal nerve, which innervates the posterior third of the tongue (42). Whole-nerve recordings indicate that sweet tastants are detectable in T1R3 KO mice, and reductions in sensitivity can vary greatly between the chorda tympani and glossopharyngeal nerves. In particular, the glossopharyngeal nerve of T1R3 KO mice has only a mildly blunted response to AceK. In the case of “savory” taste mediated by T1R1/T1R3, both nerves maintain robust responses to savory tastants in the absence of T1R3 despite behavioral data showing a reduction in preference for savory tastants in these animals.
Although NMR analysis suggests that saccharin activity in preadipocytes was not T1R2/T1R3-mediated (Fig. 9), the possibility remains that artificial sweeteners have direct intracellular activity. This has been suggested in rat liver where previous reports have indicated that saccharin might act directly on adenylyl cyclase (56). Additional reports in rat adipocytes have shown saccharin-stimulated increases (57) or decreases (58) in adenylate cyclase activity and lipolysis, varying with whether isolated adipocytes were directly treated with saccharin or rats were previously fed a high saccharin diet. Although our data do not show any clear indications of adenylyl cyclase or cAMP regulation by saccharin, it is possible that saccharin regulates local or compartmentalized cAMP populations that cannot be evaluated by ELISA. Such local cAMP regulation could be a mechanism for the reduction in HSL phosphorylation in both the presence and absence of forskolin. In addition, it is possible that saccharin could function through regulation of the HSL phosphatase. In line with a model in which phosphatases are regulated by artificial sweeteners, a role for inhibition of PP2A phosphatase activity in preadipocytes in response to saccharin would be consistent with elevated phosphorylation of Akt Thr-308 phosphorylation even in the presence of DI (59). Similarly, the need for priming phosphorylation on ERK with DI and serum as well as the sustained pERK signal through several hours (Fig. 5E) could be explained if saccharin inhibited the dual specificity ERK phosphatases (60).
An additional possibility for how artificial sweeteners mediate effects in preadipocytes and adipocytes is through bitter taste receptors with which saccharin and AceK interact in the tongue to cause the aversive “metallic” aftertaste at low mm concentrations (61, 62). To date, expression of one bitter taste receptor in an adipogenic system has been published, Tas2R46 in human mesenchymal stem cells (63). However, saccharin does not appear to activate hTas2R46 in vitro (64, 65), arguing against this bitter taste receptor mediating the effects observed in this study. Although to our knowledge expression has not been evaluated during adipogenesis, bitter taste receptors Tas2R43 and Tas2R44 are activated in cultured cells by concentrations of saccharin and AceK (62) similar to concentrations that regulate adipocyte differentiation and metabolism in this study. Slack et al. (66) also observed that concentrations of saccharin (ED50 of 0.61 mm) and AceK (ED50 of 0.49 mm) required to activate bitter taste receptors are compatible with our work. If saccharin and AceK were working through bitter taste receptors, we might predict that a bitter taste receptor activator would mimic their effects on adipose biology. Consistent with this notion, quinine, which activates nine bitter taste receptors (64), robustly stimulates adipocyte differentiation (data not shown) and represses adipocyte lipolysis (67). We have not directly tested the involvement of specific members of the bitter taste receptor family as these experiments are hampered by low receptor expression and complex and overlapping pharmacology among many receptors (64).
Perhaps the most intriguing question remaining from this study is the physiological role, if any, of sweet taste receptors in adipose tissue. Although we are the second group to describe them (55), there has been no consensus on the function of these receptors. We have screened numerous pathways for sensitivity to sweetener treatment, with Akt and ERK phosphorylation increased in preadipocytes and PKA signaling increased in adipocytes. However, many other candidate pathways remain untested. In muscle, T1R1 and T1R3 regulate autophagy and mammalian target of rapamycin complex activity (68). Although we have not found evidence that saccharin regulates autophagy (data not shown), the specificity of Ser-473 phosphorylation to T1R2/T1R3 DKO may represent a possible connection between taste receptors and TORC2 activity in preadipocytes. It is also possible that these receptors have significant functionality in vivo that is not observed in cultured cells. Taken together, these data unveil novel roles for artificial sweeteners in adipose biology and suggest that sweet taste receptors may represent a broader and more complex group of receptors than is currently appreciated.
Acknowledgments
This work utilized Animal Phenotyping, Morphology and Imaging, and Metabolomics Core Services supported by National Institutes of Health Grants P30-DK089503, P30-DK020572, and U24 DK097153.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-DK062876 and R01-DK095705 (to O. A. M.) and T32-GM007315 (to B. R. S.), a Cellular and Molecular Biology Program training grant, and T32-HD007505, a Training Program in Organogenesis grant (to B. R. S. and S. D. P.). This work was also supported by a Fulbright scholar's award (to O. A. M.).
- PPAR
- peroxisome proliferator-activated receptor
- CREB
- cAMP-response element-binding protein
- Sacc
- saccharin
- AceK
- acesulfame potassium
- eMSC
- ear mesenchymal stem cell
- HBSS
- Hanks' balanced salt solution
- SVC
- stromal vascular cell
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HSL
- hormone-sensitive lipase
- p
- phosphorylated
- F
- forward
- R
- reverse
- DKO
- double KO
- DI
- dexamethasone and insulin
- MDI
- methylisobutylxanthine, dexamethasone, and insulin
- Fsk
- forskolin
- pHSL
- phosphorylated hormone-sensitive lipase.
REFERENCES
- 1. Rosen E. D., MacDougald O. A. (2006) Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 7, 885–896 [DOI] [PubMed] [Google Scholar]
- 2. Hong Y. H., Nishimura Y., Hishikawa D., Tsuzuki H., Miyahara H., Gotoh C., Choi K. C., Feng D. D., Chen C., Lee H. G., Katoh K., Roh S. G., Sasaki S. (2005) Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 146, 5092–5099 [DOI] [PubMed] [Google Scholar]
- 3. Gotoh C., Hong Y. H., Iga T., Hishikawa D., Suzuki Y., Song S. H., Choi K. C., Adachi T., Hirasawa A., Tsujimoto G., Sasaki S., Roh S. G. (2007) The regulation of adipogenesis through GPR120. Biochem. Biophys. Res. Commun. 354, 591–597 [DOI] [PubMed] [Google Scholar]
- 4. Saltiel A. R., Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 5. Liu C., Wu J., Zhu J., Kuei C., Yu J., Shelton J., Sutton S. W., Li X., Yun S. J., Mirzadegan T., Mazur C., Kamme F., Lovenberg T. W. (2009) Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 284, 2811–2822 [DOI] [PubMed] [Google Scholar]
- 6. Taggart A. K., Kero J., Gan X., Cai T. Q., Cheng K., Ippolito M., Ren N., Kaplan R., Wu K., Wu T. J., Jin L., Liaw C., Chen R., Richman J., Connolly D., Offermanns S., Wright S. D., Waters M. G. (2005) d-β-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 280, 26649–26652 [DOI] [PubMed] [Google Scholar]
- 7. Ren N., Kaplan R., Hernandez M., Cheng K., Jin L., Taggart A. K., Zhu A. Y., Gan X., Wright S. D., Cai T. Q. (2009) Phenolic acids suppress adipocyte lipolysis via activation of the nicotinic acid receptor GPR109A (HM74a/PUMA-G). J. Lipid Res. 50, 908–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahmed K., Tunaru S., Offermanns S. (2009) GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol. Sci. 30, 557–562 [DOI] [PubMed] [Google Scholar]
- 9. Duncan R. E., Sarkadi-Nagy E., Jaworski K., Ahmadian M., Sul H. S. (2008) Identification and functional characterization of adipose-specific phospholipase A2 (AdPLA). J. Biol. Chem. 283, 25428–25436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nelson G., Hoon M. A., Chandrashekar J., Zhang Y., Ryba N. J., Zuker C. S. (2001) Mammalian sweet taste receptors. Cell 106, 381–390 [DOI] [PubMed] [Google Scholar]
- 11. Zhao G. Q., Zhang Y., Hoon M. A., Chandrashekar J., Erlenbach I., Ryba N. J., Zuker C. S. (2003) The receptors for mammalian sweet and umami taste. Cell 115, 255–266 [DOI] [PubMed] [Google Scholar]
- 12. Xu H., Staszewski L., Tang H., Adler E., Zoller M., Li X. (2004) Different functional roles of T1R subunits in the heteromeric taste receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 14258–14263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nie Y., Vigues S., Hobbs J. R., Conn G. L., Munger S. D. (2005) Distinct contributions of T1R2 and T1R3 taste receptor subunits to the detection of sweet stimuli. Curr. Biol. 15, 1948–1952 [DOI] [PubMed] [Google Scholar]
- 14. Elliott R. A., Kapoor S., Tincello D. G. (2011) Expression and distribution of the sweet taste receptor isoforms T1R2 and T1R3 in human and rat bladders. J. Urol. 186, 2455–2462 [DOI] [PubMed] [Google Scholar]
- 15. Ren X., Zhou L., Terwilliger R., Newton S. S., de Araujo I. E. (2009) Sweet taste signaling functions as a hypothalamic glucose sensor. Front. Integr. Neurosci. 3, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dyer J., Salmon K. S., Zibrik L., Shirazi-Beechey S. P. (2005) Expression of sweet taste receptors of the T1R family in the intestinal tract and enteroendocrine cells. Biochem. Soc. Trans. 33, 302–305 [DOI] [PubMed] [Google Scholar]
- 17. Nakagawa Y., Nagasawa M., Yamada S., Hara A., Mogami H., Nikolaev V. O., Lohse M. J., Shigemura N., Ninomiya Y., Kojima I. (2009) Sweet taste receptor expressed in pancreatic β-cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS One 4, e5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jang H. J., Kokrashvili Z., Theodorakis M. J., Carlson O. D., Kim B. J., Zhou J., Kim H. H., Xu X., Chan S. L., Juhaszova M., Bernier M., Mosinger B., Margolskee R. F., Egan J. M. (2007) Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc. Natl. Acad. Sci. U.S.A. 104, 15069–15074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mace O. J., Affleck J., Patel N., Kellett G. L. (2007) Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J. Physiol. 582, 379–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kyriazis G. A., Soundarapandian M. M., Tyrberg B. (2012) Sweet taste receptor signaling in β cells mediates fructose-induced potentiation of glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 109, E524–E532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang P., Cui M., Zhao B., Liu Z., Snyder L. A., Benard L. M., Osman R., Margolskee R. F., Max M. (2005) Lactisole interacts with the transmembrane domains of human T1R3 to inhibit sweet taste. J. Biol. Chem. 280, 15238–15246 [DOI] [PubMed] [Google Scholar]
- 22. Masuda K., Koizumi A., Nakajima K., Tanaka T., Abe K., Misaka T., Ishiguro M. (2012) Characterization of the modes of binding between human sweet taste receptor and low-molecular-weight sweet compounds. PLoS One 7, e35380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Treesukosol Y., Blonde G. D., Spector A. C. (2009) T1R2 and T1R3 subunits are individually unnecessary for normal affective licking responses to Polycose: implications for saccharide taste receptors in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R855–R865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zukerman S., Glendinning J. I., Margolskee R. F., Sclafani A. (2009) T1R3 taste receptor is critical for sucrose but not Polycose taste. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R866–R876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nie Y., Hobbs J. R., Vigues S., Olson W. J., Conn G. L., Munger S. D. (2006) Expression and purification of functional ligand-binding domains of T1R3 taste receptors. Chem. Senses 31, 505–513 [DOI] [PubMed] [Google Scholar]
- 26. Maîtrepierre E., Sigoillot M., Le Pessot L., Briand L. (2012) Recombinant expression, in vitro refolding, and biophysical characterization of the N-terminal domain of T1R3 taste receptor. Protein Expr. Purif. 83, 75–83 [DOI] [PubMed] [Google Scholar]
- 27. Hemati N., Ross S. E., Erickson R. L., Groblewski G. E., MacDougald O. A. (1997) Signaling pathways through which insulin regulates CCAAT/enhancer binding protein a (C/EBPα) phosphorylation and gene expression in 3T3-L1 adipocytes: correlation with GLUT4 gene expression. J. Biol. Chem. 272, 25913–25919 [DOI] [PubMed] [Google Scholar]
- 28. Cawthorn W. P., Bree A. J., Yao Y., Du B., Hemati N., Martinez-Santibañez G., MacDougald O. A. (2012) Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 50, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rim J. S., Mynatt R. L., Gawronska-Kozak B. (2005) Mesenchymal stem cells from the outer ear: a novel adult stem cell model system for the study of adipogenesis. FASEB J. 19, 1205–1207 [DOI] [PubMed] [Google Scholar]
- 30. Mori H., Prestwich T. C., Reid M. A., Longo K. A., Gerin I., Cawthorn W. P., Susulic V. S., Krishnan V., Greenfield A., Macdougald O. A. (2012) Secreted frizzled-related protein 5 suppresses adipocyte mitochondrial metabolism through WNT inhibition. J. Clin. Investig. 122, 2405–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lorenz M. A., Burant C. F., Kennedy R. T. (2011) Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem. 83, 3406–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Venkitakrishnan R. P., Benard O., Max M., Markley J. L., Assadi-Porter F. M. (2012) Use of NMR saturation transfer difference spectroscopy to study ligand binding to membrane proteins. Methods Mol. Biol. 914, 47–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sweatman T. W., Renwick A. G. (1979) Saccharin metabolism and tumorigenicity. Science 205, 1019–1020 [DOI] [PubMed] [Google Scholar]
- 34. Renwick A. G. (1986) The metabolism of intense sweeteners. Xenobiotica 16, 1057–1071 [DOI] [PubMed] [Google Scholar]
- 35. Liu D., Liman E. R. (2003) Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc. Natl. Acad. Sci. U.S.A. 100, 15160–15165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakae J., Kitamura T., Kitamura Y., Biggs W. H., 3rd, Arden K. C., Accili D. (2003) The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 4, 119–129 [DOI] [PubMed] [Google Scholar]
- 37. Menghini R., Marchetti V., Cardellini M., Hribal M. L., Mauriello A., Lauro D., Sbraccia P., Lauro R., Federici M. (2005) Phosphorylation of GATA2 by Akt increases adipose tissue differentiation and reduces adipose tissue-related inflammation: a novel pathway linking obesity to atherosclerosis. Circulation 111, 1946–1953 [DOI] [PubMed] [Google Scholar]
- 38. Kim S. P., Ha J. M., Yun S. J., Kim E. K., Chung S. W., Hong K. W., Kim C. D., Bae S. S. (2010) Transcriptional activation of peroxisome proliferator-activated receptor-gamma requires activation of both protein kinase A and Akt during adipocyte differentiation. Biochem. Biophys. Res. Commun. 399, 55–59 [DOI] [PubMed] [Google Scholar]
- 39. Magun R., Burgering B. M., Coffer P. J., Pardasani D., Lin Y., Chabot J., Sorisky A. (1996) Expression of a constitutively activated form of protein kinase B (c-Akt) in 3T3-L1 preadipose cells causes spontaneous differentiation. Endocrinology 137, 3590–3593 [DOI] [PubMed] [Google Scholar]
- 40. Klemm D. J., Leitner J. W., Watson P., Nesterova A., Reusch J. E., Goalstone M. L., Draznin B. (2001) Insulin-induced adipocyte differentiation. Activation of CREB rescues adipogenesis from the arrest caused by inhibition of prenylation. J. Biol. Chem. 276, 28430–28435 [DOI] [PubMed] [Google Scholar]
- 41. Cypess A. M., Zhang H., Schulz T. J., Huang T. L., Espinoza D. O., Kristiansen K., Unterman T. G., Tseng Y. H. (2011) Insulin/IGF-I regulation of necdin and brown adipocyte differentiation via CREB- and FoxO1-associated pathways. Endocrinology 152, 3680–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Damak S., Rong M., Yasumatsu K., Kokrashvili Z., Varadarajan V., Zou S., Jiang P., Ninomiya Y., Margolskee R. F. (2003) Detection of sweet and umami taste in the absence of taste receptor T1r3. Science 301, 850–853 [DOI] [PubMed] [Google Scholar]
- 43. Geraedts M. C., Takahashi T., Vigues S., Markwardt M. L., Nkobena A., Cockerham R. E., Hajnal A., Dotson C. D., Rizzo M. A., Munger S. D. (2012) Transformation of postingestive glucose responses after deletion of sweet taste receptor subunits or gastric bypass surgery. Am. J. Physiol. Endocrinol. Metab. 303, E464–E474 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44. Dergance J. M., Mouton C. P., Lichtenstein M. J., Hazuda H. P. (2005) Potential mediators of ethnic differences in physical activity in older Mexican Americans and European Americans: results from the San Antonio Longitudinal Study of Aging. J. Am. Geriatr. Soc. 53, 1240–1247 [DOI] [PubMed] [Google Scholar]
- 45. Rodin J. (1990) Comparative effects of fructose, aspartame, glucose, and water preloads on calorie and macronutrient intake. Am. J. Clin. Nutr. 51, 428–435 [DOI] [PubMed] [Google Scholar]
- 46. Porikos K. P., Koopmans H. S. (1988) The effect of non-nutritive sweeteners on body weight in rats. Appetite 11, Suppl. 1, 12–15 [PubMed] [Google Scholar]
- 47. Blackburn G. L., Kanders B. S., Lavin P. T., Keller S. D., Whatley J. (1997) The effect of aspartame as part of a multidisciplinary weight-control program on short- and long-term control of body weight. Am. J. Clin. Nutr. 65, 409–418 [DOI] [PubMed] [Google Scholar]
- 48. Swithers S. E., Martin A. A., Davidson T. L. (2010) High-intensity sweeteners and energy balance. Physiol. Behav. 100, 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Swithers S. E., Davidson T. L. (2008) A role for sweet taste: calorie predictive relations in energy regulation by rats. Behav. Neurosci. 122, 161–173 [DOI] [PubMed] [Google Scholar]
- 50. Anderson G. H., Foreyt J., Sigman-Grant M., Allison D. B. (2012) The use of low-calorie sweeteners by adults: impact on weight management. J. Nutr. 142, 1163S–1169S [DOI] [PubMed] [Google Scholar]
- 51. Sweatman T. W., Renwick A. G., Burgess C. D. (1981) The pharmacokinetics of saccharin in man. Xenobiotica 11, 531–540 [DOI] [PubMed] [Google Scholar]
- 52. Colburn W. A., Bekersky I., Blumenthal H. P. (1981) Dietary saccharin kinetics. Clin. Pharmacol. Ther. 30, 558–563 [DOI] [PubMed] [Google Scholar]
- 53. Colburn W. A., Bekersky I., Blumenthal H. P. (1981) A preliminary report on the pharmacokinetics of saccharin in man: single oral dose administration. J. Clin. Pharmacol. 21, 147–151 [DOI] [PubMed] [Google Scholar]
- 54. Pantarotto C., Salmona M., Garattini S. (1981) Plasma kinetics and urinary elimination of saccharin in man. Toxicol. Lett. 9, 367–371 [DOI] [PubMed] [Google Scholar]
- 55. Masubuchi Y., Nakagawa Y., Ma J., Sasaki T., Kitamura T., Yamamoto Y., Kurose H., Kojima I., Shibata H. (2013) A novel regulatory function of sweet taste-sensing receptor in adipogenic differentiation of 3T3-L1 cells. PLoS One 8, e54500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Striem B. J., Naim M., Zehavi U., Ronen T. (1990) Saccharin induces changes in adenylate cyclase activity in liver and muscle membranes in rats. Life Sci. 46, 803–810 [DOI] [PubMed] [Google Scholar]
- 57. Dib K., Oget I., Wrisez F., El Jamali A., Aguie-Aguie G., Correze C., Lambert B. (1996) Effects of sodium saccharin diet on fat-cell lipolysis: evidence for increased function of the adenylyl cyclase catalyst. Int. J. Obes. Relat. Metab. Disord. 20, 15–20 [PubMed] [Google Scholar]
- 58. Dib K., Wrisez F., el Jamali A., Lambert B., Correze C. (1997) Sodium saccharin inhibits adenylyl cyclase activity in non-taste cells. Cell. Signal. 9, 431–438 [DOI] [PubMed] [Google Scholar]
- 59. Andrabi S., Gjoerup O. V., Kean J. A., Roberts T. M., Schaffhausen B. (2007) Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 104, 19011–19016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang C. Y., Tan T. H. (2012) DUSPs, to MAP kinases and beyond. Cell Biosci. 2, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bartoshuk L. M. (1979) Bitter taste of saccharin related to the genetic ability to taste the bitter substance 6-n-propylthiouracil. Science 205, 934–935 [DOI] [PubMed] [Google Scholar]
- 62. Kuhn C., Bufe B., Winnig M., Hofmann T., Frank O., Behrens M., Lewtschenko T., Slack J. P., Ward C. D., Meyerhof W. (2004) Bitter taste receptors for saccharin and acesulfame K. J. Neurosci. 24, 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lund T. C., Kobs A. J., Kramer A., Nyquist M., Kuroki M. T., Osborn J., Lidke D. S., Low-Nam S. T., Blazar B. R., Tolar J. (2013) Bone marrow stromal and vascular smooth muscle cells have chemosensory capacity via bitter taste receptor expression. PLoS One 8, e58945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meyerhof W., Batram C., Kuhn C., Brockhoff A., Chudoba E., Bufe B., Appendino G., Behrens M. (2010) The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Senses 35, 157–170 [DOI] [PubMed] [Google Scholar]
- 65. Brockhoff A., Behrens M., Massarotti A., Appendino G., Meyerhof W. (2007) Broad tuning of the human bitter taste receptor hTAS2R46 to various sesquiterpene lactones, clerodane and labdane diterpenoids, strychnine, and denatonium. J. Agric. Food Chem. 55, 6236–6243 [DOI] [PubMed] [Google Scholar]
- 66. Slack J. P., Brockhoff A., Batram C., Menzel S., Sonnabend C., Born S., Galindo M. M., Kohl S., Thalmann S., Ostopovici-Halip L., Simons C. T., Ungureanu I., Duineveld K., Bologa C. G., Behrens M., Furrer S., Oprea T. I., Meyerhof W. (2010) Modulation of bitter taste perception by a small molecule hTAS2R antagonist. Curr. Biol. 20, 1104–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Markus H. B., Ball E. G. (1969) Inhibition of lipolytic processes in rat adipose tissue by antimalaria drugs. Biochim. Biophys. Acta 187, 486–491 [DOI] [PubMed] [Google Scholar]
- 68. Wauson E. M., Zaganjor E., Lee A. Y., Guerra M. L., Ghosh A. B., Bookout A. L., Chambers C. P., Jivan A., McGlynn K., Hutchison M. R., Deberardinis R. J., Cobb M. H. (2012) The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol. Cell 47, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]