

Background: MLL fusion proteins, including Mll-Ell, induce overexpression of HoxA9 and HoxA10 in the bone marrow.
Results: Mll-Ell induces HoxA9-and HoxA10-dependent FGF2 transcription. Autocrine production of Fgf2 contributes to cytokine hypersensitivity in Mll-Ell expressing myeloid progenitor cells.
Conclusion: Mll-Ell induces Fgf2 production by myeloid progenitors and differentiating myeloid cells.
Significance: Proliferative effects of Fgf2 may influence the pathogenesis of leukemias with MLL gene translocations.
Keywords: Cell Proliferation, Fibroblast Growth Factor (FGF), Gene Transcription, Growth Factors, Leukemia, hox, mll
Abstract
The subset of acute myeloid leukemias (AML) with chromosomal translocations involving the MLL gene have a poor prognosis (referred to as 11q23-AML). The MLL fusion proteins that are expressed in 11q23-AML facilitate transcription of a set of HOX genes, including HOXA9 and HOXA10. Because Hox proteins are transcription factors, this suggests the possibility that Hox target genes mediate the adverse effects of MLL fusion proteins in leukemia. Identifying such Hox target genes might provide insights to the pathogenesis and treatment of 11q23-AML. In the current study we found that Mll-Ell (an MLL fusion protein) induced transcriptional activation of the FGF2 gene in a HoxA9- and HoxA10-dependent manner. FGF2 encodes fibroblast growth factor 2 (also referred to as basic fibroblast growth factor). Fgf2 influences proliferation and survival of hematopoietic stem cells and myeloid progenitor cells, and increased Fgf2-expression has been described in AMLs. We determined that expression of Mll-Ell in myeloid progenitor cells resulted in autocrine production of Fgf2 and Fgf2-dependent cytokine hypersensitivity. Therefore, our results implicated increased Fgf2 expression in progenitor proliferation and expansion in 11q23-AML. Because small molecule inhibitors of Fgf-receptors are in human clinical trials, this suggested a potential therapeutic approach to this treatment refractory leukemia.
Introduction
Clinical correlative studies defined a poor prognosis subset of acute myeloid leukemia (AML)2 with increased expression of HoxB3, HoxB4, HoxA9–11, and Meis1 in CD34+ bone marrow cells (1). In this leukemia subset, expression of these proteins was also aberrantly sustained in differentiating, CD34− myeloid cells. Further analysis determined that this pattern of gene expression was characteristic of AML with chromosomal translocations or partial tandem duplications involving the MLL gene (referred to as 11q23-AML), chromosomal translocation involving the MYST3 and CREBBP genes, and a poor prognosis subset of cytogenetically normal AML (2–7).
More than 20 different fusion partners have been described for the MLL gene in 11q23-AML (2). The expressed MLL fusion proteins include common N-terminal domains from MLL and C-terminal domains from the various different partner genes. Studies in murine models supported a functional role for MLL fusion proteins in Hox expression and leukemia. Engineered expression of various MLL fusion proteins in murine bone marrow cells increased Hox expression in a manner that was consistent with human 11q23-AML (1, 5, 8). MLL fusion proteins also expanded the hematopoietic stem cell (HSC) and myeloid progenitor cell bone marrow populations in vitro and induced a myeloproliferative neoplasm that progressed to AML over time in vivo (1, 5, 8). These results were similar with all of the MLL fusion proteins tested.
Other murine models supported a functional role for Hox proteins in leukemogenesis. Human and murine HOX genes are clustered in four groups (A-D) on four chromosomes (9, 10). HOX1–4 genes are most highly transcribed in HSC, and transcription of HOX7–11 is most active in committed progenitor cells (10). Consistent with this expression profile, engineered overexpression of HoxB3 or HoxB4 in murine bone marrow expanded the HSC population and led to an myeloproliferative neoplasm in vivo (11, 12). Overexpression of either HoxA9 or HoxA10 in murine bone marrow was characterized by expansion of the bipotential myeloid progenitor population (granulocyte/monocyte progenitors (GMP)) (13–17). In vivo overexpression of HoxA9 or HoxA10 resulted in an myeloproliferative neoplasm that evolved to AML in a manner similar to MLL fusion proteins (1, 17–19).
These studies suggested the possibility that Hox proteins were involved in expansion of the leukemia stem cell population in 11q23-AML. In various forms of AML this population has generally been defined as a GMP with HSC-like characteristics (20). However, the set of Hox target genes that mediated bone marrow progenitor expansion was not defined.
We used chromatin immunoprecipitation-based screening techniques to identify HoxA10 target genes that might be relevant to leukemogenesis (21–26). In these studies we identified the gene encoding fibroblast growth factor 2 (Fgf2, or basic Fgf) as a HoxA10 target gene (26). Fgf2 is produced by bone marrow stromal cells, hematopoietic progenitor cells, and mature phagocytes (27). Increased Fgf2 production by AML cells is described, and a search of publically available data bases specifically associated this with 11q23-AML (28, 29). Fgf2 plays a key role in expanding hematopoietic progenitor cell populations and is essential for hematopoietic and embryonic stem cell maintenance (30).
In myeloid progenitor cells, Fgf2 binding to Fgf-R1/R2 activates phosphoinositol 3-kinase (31, 32). Phosphoinositol 3-kinase activates Akt kinase, which inhibits glycogen synthase kinase 3β. Serine/threonine phosphorylation of β-catenin by glycogen synthase kinase 3β leads to β-catenin ubiquitination and degradation. Therefore, Fgf2-induced activation of phosphoinositol 3-kinase stabilizes β-catenin and enhances β-catenin activity (26).
We previously found that autocrine production of Fgf2 contributed to cytokine hypersensitivity of HoxA10-overexpressing myeloid progenitor cells in a phosphoinositol 3-kinase and β-catenin-dependent manner (26). The possibility that Fgf2 contributed to the pathogenesis of 11q23-AML was not previously explored.
HoxA9 and HoxA10 have highly conserved DNA binding homeodomains, suggesting the possibility of common target genes (33). However, HoxA9 and HoxA10 diverge outside of the homeodomain, suggesting the possibility of differential effects on some target genes. Consistent with the latter, we found that CYBB was a common target gene for HoxA10 and HoxA9 (the only previously identified common target gene) (34–37). CYBB encodes gp91phox; a rate-limiting component of the NADPH oxidase that generates H2O2/O3− during the innate immune response.
We found that HoxA10 interacted with and repressed a CYBB cis element in myeloid progenitor cells (34, 36). During myeloid differentiation, phosphorylation of conserved tyrosine residues in the DNA binding homeodomain (HD) of HoxA10 decreased binding affinity for CYBB (34, 36). HoxA9 activated CYBB transcription through the same cis element, but in this case phosphorylation of the conserved HD tyrosine residues of HoxA9 increased CYBB binding affinity during differentiation (37). These studies identified an antagonistic role for HoxA9 and HoxA10 during myeloid differentiation. This was consistent with previous studies indicating that HoxA10 was involved in a differentiation block in AML, but HoxA9 conferred a myeloid phenotype upon leukemia cells (13, 18).
However, in the current study we found that HoxA9 and HoxA10 activated common cis elements in the FGF2 promoter in myeloid progenitor cells and throughout myeloid differentiation. Expression of the leukemia-associated MLL fusion protein Mll-Ell also activated these two FGF2 cis elements in a HoxA9- and HoxA10-dependent manner. This resulted in autocrine production of Fgf2 by Mll-Ell-expressing myeloid progenitor cells and Fgf2-dependent cytokine hypersensitivity. This is a previously un-described and therapeutically targetable mechanism for MLL fusion proteins to influence expansion of myeloid progenitor cells.
MATERIALS AND METHODS
Plasmid Vectors
The cDNA for human HoxA10 was obtained from C. Largman (University of California, San Francisco) (38). The HoxA9 cDNA was generated by reverse transcription and PCR from U937 cells as described (37). These cDNAs were subcloned into the mammalian expression vector pcDNAamp (Invitrogen) and the murine retroviral vector pMSCVpuro (Clontech, Mountain View, CA), as described (21, 22). A vector with the Mll-Ell fusion protein was obtained from D. E. Zhang (University of California, San Diego, CA). HoxA10- and HoxA9-specific shRNA and scrambled control sequences were designed using the Promega website (Promega, Madison, WI) and subcloned into the pLKO.1puro vector (from Dr. Kathy Rundell, Northwestern University, Chicago). Several sequences were tested, and the most efficient were combined.
The FGF2 5′-flank was obtained from the genomic DNA of U937 cells by PCR. The sequence was compared with the published FGF2 5′-flank sequence (ENSEMBL database), and promoter fragments were subcloned into the pGL3-basic reporter vector (Promega) as described (26). Other constructs were generated with mutation of identified Hox binding sites in the FGF2 promoter. Reporter constructs were also generated using the pGL3-promoter vector (with a minimal promoter and reporter) and three copies of the proximal (−287 to −266 bp) or distal (−448 to −425 bp) Hox binding cis elements from the FGF2 promoter as described (26).
Oligonucleotides
Oligonucleotides were custom-synthesized by MWG Biotech (Piedmont, NC). These oligonucleotides represent Hox consensus sequences from the FGF2 promoter; −287 to −266 bp (5′-GAGAAAGTTGAGTTTAAACTTTTA-3′), −448 to −425 bp (5′-AATTTTAAAGTTTATGCCCCATT-3′), or Hox-consensus mutants of these sequences (26).
Myeloid Cell Line Culture
The human myelomonocytic leukemia cell line U937 (39) was obtained from A. Kraft (Hollings Cancer Center, Medical University of South Carolina, Charleston, SC). Cells were maintained and differentiated (with retinoic acid (RA) and dimethylformamide (DMF)) as described (39).
Primary Murine Bone Marrow Studies
Animal studies were performed according to a protocol approved by the Animal Care and Use Committees of Northwestern University and the Jesse Brown Veterans Administration Medical Center.
Bone marrow mononuclear cells were obtained from the femurs of WT or HoxA10−/− C57/BL6 mice (40). Bi-potential granulocyte/monocyte progenitor cells (GMPs) were cultured (2 × 105 cells/ml) for 48 h in DME media supplemented with 10% fetal calf serum, 1% pen-strep, 10 ng/ml murine GM-CSF (R & D Systems Inc., Minneapolis, MN), 10 ng/ml murine recombinant IL-3 (R & D Systems), and 100 ng/ml SCF (R & D Systems) followed by separation of CD34+ cells using the Miltenyi magnetic bead system (Miltenyi Biotechnology, Auburn, CA) (18, 23, 26). Some cells were CD34-separated after 24 h under these conditions followed by 24 h of differentiation in DME supplemented with 10% fetal calf serum, 1% pen-strep, 20 ng/ml G-CSF (R & D Systems) and 10 ng/ml IL3.
Retrovirus was generated with HoxA10/MSCV, HoxA9/MSCV or Mll-Ell/MSCV or control MSCV plasmid using the Phoenix cell packaging line according to manufacturer's instructions (Stratagene, La Jolla, CA). The average viral concentration was 107 pfu/ml. Bone marrow mononuclear cells were cultured for 24 h in 10 ng/ml GM-CSF, 10 ng/ml IL3, and 100 ng/ml SCF. Cells were transduced by incubation with retroviral supernatant supplemented with Polybrene (6 μg/ml) as described (18, 23, 26). Transduced cells were selected for 48 h in puromycin followed by CD34 selection and culture in GM-CSF, IL3, and SCF (as above). Transgene expression was confirmed by real time PCR and Western blot.
Quantitative Real Time PCR
RNA was isolated using TRIzol reagent (Invitrogen). Primers were designed with Applied Biosystems software. Real time PCR was performed with SYBR Green according to the “standard curve” method. Results were normalized to 18 S and to actin (for mRNA determination) or total input chromatin (for chromatin immunoprecipitation studies).
Chromatin Immunoprecipitation
Cells were incubated briefly in media supplemented with formaldehyde to generate DNA-protein cross-links. Cell lysates were sonicated to generate chromatin fragments with an average size of 200 bp (41). Lysates underwent one round of immunoprecipitation with antibodies to HoxA9, HoxA10, or irrelevant antibody (21, 25, 26). HoxA9 and HoxA10 antibodies are not cross-reactive and do not prevent DNA binding (N-20 and A-20 from Santa Cruz Biotechnology, Santa Cruz, CA). The irrelevant antibody is a purified antibody to glutathione S-transferase (anti-GST antibody, Santa Cruz Biotechnology).
Chromatin was amplified by real time PCR with sets of primers flanking the previously identified Hox binding cis elements in the FGF2 promoter or with primers representing irrelevant regions in the proximal 2.0 kb of FGF2 5′ flank as previously described (26).
Myeloid Cell Line Transfections and Assays
To generate stable U937 transfectant pools, cells were electroporated with equal amounts of a HoxA9, HoxA10, HoxA9 + HoxA10, or Mll-Ell expression vector (or empty control vector) plus a vector with a neomycin phosphotransferase cassette (pSRα) (30 μg each). Stable pools were selected in G418 (0.5 mg/ml), and aliquots were tested for transgene expression by real time PCR and Western blot.
For FGF2 promoter and cis element analysis, U937 cells were co-transfected with a construct with various sequences from the FGF2 5′-flank linked to a luciferase reporter (1.1 kb, 467 bp, 467 bp with mutation of the distal cis element, 424 bp, 297 bp, 297 bp with mutation of the proximal cis element, or 266 bp of FGF2-pGL3 or pGL3 control) (30 μg), a vector to express Mll-Ell (50 μg), HoxA9 (50 μg), HoxA10 (50 μg), HoxA9 + HoxA10 (25 μg each), or empty vector control (50 μg), and β-galactosidase reporter vector to control for transfection efficiency (CMVβ-gal).
In other experiments cells were co-transfected with an artificial promoter/reporter vector with three copies of the proximal (−287 to −266 bp) or distal (−448 to −425 bp) FGF2 cis elements (with the pGL3-p vector) (30 μg), a vector to express Mll-Ell, HoxA9, and/or HoxA10 (as above), and CMVβ-gal. In other experiments these reporter plasmids were co-transfected with vectors to overexpress HoxA9, HoxA10, or Mll-Ell with vectors to express HoxA10- or HoxA9-specific shRNAs (or scrambled control shRNA) and CMVβ-gal. Transfectants were analyzed with or without granulocyte differentiation with RA + DMF.
Western Blots
Cells were lysed by boiling in 2× SDS sample buffer. Lysate proteins (50 μg) were separated by SDS-PAGE (8% acrylamide) and transferred to nitrocellulose. Western blots were serially probed with antibodies to various proteins, including a loading control. Each experiment was repeated at least three times with different batches of lysate proteins. Representative blots are shown.
ELISA
Expression of Fgf2 in the media of cultured cells was determined using the Emax ImmunoAssay System according to manufacturer's instructions (Promega). Cells were maintained at a density of 0.6 × 106 cells/ml. Equivalent amounts of media were withdrawn at various time points for the assay (26). The assay is Fgf2-specific and does not cross-react with other Fgfs.
Cell Proliferation Assays
U937 stable transfectants with HoxA9, HoxA10, Mll-Ell, or empty control vector were deprived of fetal calf serum for 24 h followed by treatment with a dose titration of fetal calf serum (0.01–10%). Some cells were incubated with a specific blocking antibody for Fgf2 or control antibody (R & D Systems). Other cells were incubated with the Fgf-R1 inhibitor, PD173074 (Cayman Chemical, Ann Arbor, MI). Cell proliferation was determined by incorporation of [3H]thymidine (23, 26).
In other studies WT or HoxA10−/− murine bone marrow cells were transduced with a retroviral vector to overexpress HoxA9, HoxA10, and Mll-Ell or with empty MSCV vector, cultured in 10 ng/ml GM-CSF, 10 ng/ml IL3, 100 ng/ml SCF, deprived of cytokines for 24 h (in DME with 10% FCS), and stimulated for 24 h with a dose titration of GM-CSF (0.01–10 ng/ml + 5 ng/ml IL3). Some cells were incubated with Fgf2 blocking antibody (or control antibody) or Fgf-R1 inhibitor (or buffer control). Cell proliferation was determined as above (23, 26).
In Vitro DNA Binding Assays
Nuclear proteins were isolated by the method of Dignam et al. (42) as described (28). Oligonucleotides probes were prepared, and DNA binding was analyzed by electrophoretic mobility shift assay (EMSA) and DNA affinity purification techniques as described (18, 21–26). For EMSA, antibody to HoxA9 or HoxA10 (or irrelevant control antibody) was included in binding assays. For DNA affinity purification, Western blots of affinity-purified proteins were probed with antibodies to HoxA9, HoxA10, Ell, or irrelevant control antibody.
For all experiments at least three different batches of nuclear proteins were tested in two independent experiments. Nuclear protein integrity and equality of loading was determined in control EMSA with a probe representing a classical CCAAT box from the α-globin gene promoter.
Statistical Analysis
Statistical significance was determined by Student's t test and analysis of variance using SigmaPlot and SigmaStat software. Graphs are presented with error bars representing S.E. calculations. p values of ≤0.02 were considered statistically significant.
RESULTS
FGF2 Promoter Activity Is Increased in Mll-Ell-expressing Cells
We investigated the effect of Mll-Ell on the FGF2 promoter by co-transfecting U937 myeloid leukemia cells with a series of reporter vectors containing FGF2 promoter sequences and vectors to express Mll-Ell or empty control vector. Other cells were co-transfected with these reporter constructs and a vector to overexpress HoxA10 as a positive control. Based on the highly conserved DNA binding HD of HoxA9 and HoxA10, some cells were co-transfected with the reporter vectors and a HoxA9 expression vector.
We previously defined two FGF2 promoter cis elements that conformed to a derived Hox-DNA binding consensus sequence; that is, a proximal cis element between −297 and −266 bp and a distal cis element between −424 and −467 bp (from the transcription start site) (Fig. 1) (26). The FGF2-promoter constructs used in the current studies were designed around the cis elements, and some constructs included a mutation of the Hox-binding consensus sequences in the cis elements.
FIGURE 1.

The proximal 500 bp of FGF2 promoter has consensus sequences for Hox-protein binding. Sequence analysis identified homology between the human (black) and murine (blue) FGF2 promoter sequences. Hox binding consensus sequences are shown in red, and the previously identified functional cis elements are in bold and underlined.
Because we previously found that HoxA9 and HoxA10 regulated CYBB transcription in a differentiation stage-specific manner, transfectants were assayed with or without differentiation to granulocytes with retinoic acid and dimethyl formamide (RA/DMF) (39).
We found that activity of reporter constructs with 467 bp (both cis elements) or 297 bp (proximal only) of FGF2 promoter was significantly increased in Mll-Ell transfectants in comparison to control (p < 0.001, n = 6) (Fig. 2A). This increase was significantly greater in transfectants with the 467-bp construct versus the 297-bp construct (388% ± 60% increase with Mll-Ell versus 235% ± 10% increase, p = 0.01, n = 6; Fig. 2A). Mutation of the single Hox binding consensus in the 297-bp construct abolished Mll-Ell-induced reporter activity (Fig. 2A). Mutation of the distal cis element in the 467-bp FGF2 promoter construct decreased Mll-Ell-induced promoter activity so that it was not significantly different from the wild type 297-bp construct (p = 0.8, n = 6).
FIGURE 2.

Activity of the FGF2 promoter is increased by expression of Mll-Ell or overexpression of HoxA9 in myeloid leukemia cells. A, Mll-Ell expression increases activity of the FGF2 promoter. U937 cells were co-transfected with a series of FGF2 promoter/reporter constructs and a vector to express Mll-Ell or control expression vector. Some FGF2 promoter/reporter constructs have mutations in Hox binding consensus sequences (i.e. mut-467 and mut-297). Transfectants were analyzed for reporter expression with or without granulocyte differentiation (with RA/DMF). Statistically significant differences in reporter activity are indicated by *, **, ***, #, or ##. B, overexpression of HoxA9 or HoxA10 increases activity of the FGF2 promoter in a non-differentiation stage-specific manner. U937 cells were co-transfected with the FGF2 promoter/reporter constructs (described above) and vectors to express HoxA9 or HoxA10 (or control vector). Transfectants were analyzed for reporter activity with or without differentiation. Statistically significant differences in reporter expression are indicated by *, **, ***, or #.
Although there was a trend to increased activity of these FGF2 reporter constructs in RA/DMF-differentiated transfectants versus control transfectants, this did not reach statistical significance with or without Mll-Ell (p ≥ 0.06, n = 6; Fig. 2A). This was consistent with the known production of Fgf2 by both myeloid progenitor cells and mature phagocytes.
We found the same pattern of increased reporter activity in U937 cells that were co-transfected with these FGF2 promoter constructs and vectors to overexpress HoxA9 or HoxA10 (Fig. 2B). There was no significant difference in the efficiency of activation of the FGF2 promoter constructs by HoxA9 versus HoxA10 (p > 0.8, n = 6). This was in contrast to the CYBB promoter, which was repressed by HoxA10 in myeloid progenitor cells but activated by HoxA9 during myeloid differentiation. Also in contrast to the CYBB promoter, HoxA9- or HoxA10-induced activity of the FGF2 promoter was only slightly increased by differentiation under each of the conditions studied (p ≥ 0.06, n = 6; Fig. 2B).
Activity of the empty control reporter construct was not altered by co-transfection with vectors to express Mll-Ell, HoxA9, or HoxA10 or differentiation with RA/DMF. This minimal activity was subtracted as background.
Mll-Ell Increases FGF2 Promoter Activity in a HoxA9- and HoxA10-dependent Manner
To verify that Mll-Ell influenced FGF2 transcription through the proximal and distal cis elements, U937 cells were co-transfected with a construct containing three copies of the distal or proximal FGF2 cis element linked to a minimal promoter and reporter (or control minimal promoter/reporter vector) and a vector to express Mll-Ell or empty expression vector. Some cells were co-transfected with these reporter constructs and a vector to overexpress HoxA10 as a positive control. To determine if there were differences in regulation of the two cis elements by HoxA9 versus HoxA10, some cells were transfected with the reporter constructs and a vector to overexpress HoxA9 or equivalent amounts of HoxA9 + HoxA10 (keeping the total amount of Hox vector constant). Because phosphorylation of conserved tyrosine (Tyr) residues in the HDs of HoxA9 or HoxA10 regulates CYBB cis element binding, some cells were co-transfected with the reporter constructs and vectors to express HD-Tyr mutant forms of HoxA9 or HoxA10 (35, 37). Transfectants were assayed with or without differentiation with RA/DMF.
We found that Mll-Ell significantly increased activity of both the proximal and distal FGF2 cis elements (p < 0.0001, n = 6; Fig. 3A). This effect of Mll-Ell was not significantly altered by differentiation, although there was a trend for increase (p ≥ 0.04, n = 6). Two FGF2 cis elements were equivalently activated by HoxA9, HoxA10, or HoxA9 + HoxA10 (p > 0.2, n = 6) (Fig. 3B). There was a trend to increased activity with differentiation that did not meet statistical significance for either the proximal or distal cis element with overexpression of these Hox proteins (p ≥ 0.07, n = 6) (Fig. 3B). Consistent with the latter results, WT HoxA9 or HoxA10 was only slightly more efficient in activating the cis elements in comparison to HD-Tyr mutant forms of these proteins (p ≥ 0.09, n = 6; Fig. 3B).
FIGURE 3.

Activity of two FGF2 cis elements is increased by expression of Mll-Ell or overexpression of HoxA9 in myeloid leukemia cells. A, activity of the proximal and distal FGF2 cis elements is increased by Mll-Ell expression. U937 cells were co-transfected with minimal promoter/reporter constructs with multiple copies of the proximal or distal FGF2 cis elements (or control vector) and a vector to express Mll-Ell or empty control vector. Transfectants were analyzed for reporter activity with or without differentiation (with RA/DMF). Statistically significant differences are indicated by * or **. B, HoxA9 and HoxA10 activate two FGF2 cis elements in an additive manner. U937 cells were co-transfected with the minimal promoter/reporter constructs with multiple copies of the proximal or distal Hox binding FGF2 cis element (or control vector) and vectors to overexpress HoxA9, HoxA10, ½ HoxA9 + ½ HoxA10, HoxA9 with mutation of a conserved HD-Tyr residue (Ymut-HoxA9), HoxA10 with mutation of a conserved HD-Tyr residue (Ymut-HoxA10), or control vector. Plasmid vectors were adjusted so that the total amount of Hox expression vector was the same in Hox-overexpressing experiments. Transfectants were analyzed for reporter activity with or without differentiation. Statistically significant differences with versus without Hox-overexpression are indicated by * and **.
These results indicated that Mll-Ell influenced two FGF2 cis elements that were activated by HoxA9 and/or HoxA10. To determine if activation of the cis elements by Mll-Ell required Mll-Ell-induced expression of HoxA9 or HoxA10, cells were co-transfected with the reporter constructs, a vector to express Mll-Ell (or vector control), and vectors to express HoxA9 or HoxA10-specific shRNAs (or scrambled control shRNAs). We found that knockdown of HoxA9, HoxA10, or both significantly decreased Mll-Ell-induced FGF2 cis element activity (p < 0.001, n = 6; Fig. 4A).
FIGURE 4.

Mll-Ell activates the FGF2 cis elements in a HoxA9- and HoxA10-dependent manner, and HoxA9 and HoxA10 are interchangeable for FGF2 activation. A, Mll-Ell increases the activity of the proximal and distal FGF2 cis elements in a HoxA9- and HoxA10-dependent manner. U937 cells were co-transfected with a minimal promoter/reporter construct with multiple copies of the proximal or distal FGF2 cis elements (described above), a vector to express Mll-Ell (or empty control vector), and vectors to express HoxA9 or HoxA10 specific shRNAs (or scrambled shRNA control vectors). Cells were analyzed for reporter activity. Statistically significant differences are indicated by *, **, ***, #, ##, or ###. B, HoxA9 overexpression compensates for HoxA10 knockdown for activation of the two FGF2 cis elements. U937 cells were co-transfected with the same minimal-promoter/reporter constructs as described above, a dose titration of HoxA9 expression vector (or empty control vector), and a vector to express a HoxA10-specific shRNA (or scrambled control vector). Statistically significant differences with versus without HoxA10-knockdown are indicated by *, **, ***, #, ##, or ###. C, HoxA10-overexpression compensates for HoxA9 knockdown for activation of the two FGF2 cis elements. The converse experiment was also performed, with the same proximal or distal FGF2 cis element containing artificial promoter-reporter constructs, a dose titration of HoxA10 expression vector, and a vector to express HoxA9-specific shRNAs (or relevant control vectors). Statistically significant differences with versus without HoxA9 knockdown are indicated by *, **, ***, #, ##, or ###. HoxA9 protein expression was decreased >70% using the HoxA9-specific shRNA vectors (see the inset Western blot (WB) of transfectant cell lysates probed with antibodies to HoxA9, HoxA10, Fgf2, or Gapdh loading control).
To determine if HoxA9 overexpression could compensate for loss of HoxA10 and vice versa, we co-transfected U937 cells with the proximal or distal FGF2 cis element-containing constructs, a vector to express specific shRNAs to HoxA10 or HoxA9 (or scrambled control shRNA), and a vector to overexpress HoxA9 or HoxA10 (or control vector). If HoxA9 and HoxA10 have unique effects on the cis elements, even large amounts of HoxA9 would not compensate for loss of HoxA10 and vice versa.
We found that knockdown of HoxA10 impaired the ability of overexpressed HoxA9 to activate these FGF2 cis elements (Fig. 4B). However, we were able to overcome the effect of HoxA10 knockdown with increasing amounts of HoxA9 (Fig. 4B). We found the same effect in transfectants with knockdown of HoxA9 and HoxA10 overexpression (Fig. 4C). This suggested that the mechanisms of FGF2 activation by HoxA9 and HoxA10 were not unique.
Activity of the minimal promoter-reporter control vector was not altered by Mll-Ell, overexpression or knockdown of HoxA9 or HoxA10, or differentiation of the transfectants. This minimal activity was subtracted as background. In control experiments we identified the amount of HoxA9- or HoxA10-specific shRNA vector that decreased the endogenous message by ∼70% (Fig. 4C and Ref 26). This amount was used in reporter gene assays.
Mll-Ell Increases Binding of HoxA9 and HoxA10 to the FGF2 Promoter
To determine if Mll-Ell increased binding of HoxA9 and/or HoxA10 to the FGF2 promoter, we first verified binding of HoxA9 to the cis elements. For these studies, chromatin was co-precipitated from U937 lysates with an antibody to HoxA9, HoxA10 (as a positive control), or irrelevant control antibody. Lysates were sonicated under conditions that generated chromatin fragments with an average size of 200 bp (25, 26). Chromatin was amplified by real time PCR with primers flanking the proximal or distal FGF2 cis element. We found in vivo HoxA9 binding to the FGF2 cis elements (Fig. 5A).
FIGURE 5.

Mll-Ell increases binding of HoxA9 and HoxA10 to the FGF2 promoter. A, HoxA9 binds to the previously identified HoxA10 binding FGF2 cis elements in vivo. Chromatin immunoprecipitation experiments were performed using U937 myeloid cells and an antibody (Ab) to HoxA9, HoxA10 (as a positive control), or irrelevant control antibody (negative control). Co-precipitating chromatin was amplified by real time PCR with primers flanking the FGF2 cis elements. Statistically significant differences in studies with control versus HoxA9 or HoxA10 antibody are indicated by * or **. B, Mll-Ell increased binding of HoxA9 and HoxA10 to the proximal or distal FGF2 cis elements in vitro. Nuclear proteins from U937 cells that were stably transfected with an Mll-Ell or empty control vector were incubated with a biotin-labeled, double-stranded oligonucleotide probe representing the proximal or distal FGF2 cis elements with or without mutation of the Hox binding consensus sequence. Proteins were collected by affinity of the probe to avidin-linked matrix, separated by SDS-PAGE, and identified by a Western blot (WB) with antibodies to HoxA9, HoxA10, or Ell. C, HoxA9 and HoxA10 bind to the same FGF2 cis elements in vitro by electrophoretic mobility shift assay. Nuclear proteins were isolated from U937 cells and incubated with a radio-labeled, double-stranded oligonucleotide probe representing the proximal FGF2 cis element. Bound proteins and free probe were separated by native gel electrophoresis. Some binding assays were preincubated with antibody to HoxA9, HoxA10, both, or irrelevant control (GST) antibody. The free probe and Hox-bound DNA-protein complex are indicated by arrows.
We also performed chromatin immunoprecipitation with U937 cell lysates sonicated to generate 1.0-kb chromatin fragments. Co-precipitating chromatin was analyzed with PCR primers to amplify overlapping 500-bp sequences in the proximal 2 kb of FGF2 5′-flank. No additional HoxA9 binding sites were identified (not shown).
We investigated the influence of Mll-Ell on HoxA9 and HoxA10 by DNA affinity purification assays. For these studies, nuclear proteins were isolated from U937 cells that were stably transfected with an Mll-Ell expression vector (or control vector) and incubated with double-stranded oligonucleotides representing the two FGF2 cis elements with or without mutation in the Hox binding consensus. DNA-bound proteins were collected by affinity of the biotin-labeled probe to an avidin-linked matrix, separated by SDS-PAGE, and identified by Western blot. We found that sequence-specific HoxA9 and HoxA10 binding to both cis elements was increased by Mll-Ell (Fig. 5B). However, we did not demonstrate binding of Mll-Ell to any of these probes (Fig. 5B).
We also investigated binding to the FGF2 cis elements by EMSA. For these studies, U937 nuclear proteins were incubated with a radiolabeled double-stranded oligonucleotide representing the proximal or distal FGF2 cis element. Nuclear proteins were preincubated with an antibody to HoxA9, HoxA10, both antibodies, or irrelevant antibody, and bound and free probes were separated by native PAGE.
We found that both FGF2 probes bound a low mobility protein complex that was partly disrupted by antibody to HoxA9 or HoxA10 and was completely disrupted by both antibodies together (Fig. 5C). Results are shown for the proximal cis element probe and were identical for the distal probe. We previously demonstrated that these probes generated a specific, low mobility complex with cross-competitive binding specificities with cis elements from other HoxA10 target genes (26).
Mll-Ell Induces Fgf2 Expression
We first investigated the effect of Mll-Ell on Fgf2 expression using U937 stable transfectants with an Mll-Ell versus control vector. We also investigated the relative effects of HoxA9 versus HoxA10 on Fgf2 using U937 cells stably overexpressing HoxA9, HoxA10, or HoxA9 + HoxA10 (½ each). Transfectants were analyzed with or without differentiation with RA/DMF.
We first investigated Fgf2 production by the transfectants using an Fgf2-specific ELISA to analyze cell-conditioned media. We found significantly more Fgf2 in the media of Mll-Ell-expressing U937 transfectants in comparison to control transfectants (p < 0.0001, n = 3), and this was only slightly increased by differentiation (p = 0.08, n = 3).
We also found significantly increased Fgf2 mRNA in Mll-Ell-expressing U937 cells relative to control transfectants by real time PCR (p < 0.0001, n = 6) (Fig. 6B). HoxA9 and HoxA10 mRNA was significantly increased in Mll-Ell-expressing cells (p < 0.002, n = 6; Fig. 6B). Although there was less HoxA10 mRNA versus HoxA9 mRNA in control cells (p < 0.01, n = 6), the increase in HoxA10 in cells expressing Mll-Ell was significantly greater than the increase in HoxA9 (568 ± 26% increase in HoxA10 with Mll-Ell versus 312 ± 25% increase in HoxA9, p = 0.001, n = 6; Fig. 6B). Expression of Mll-Ell was confirmed with a primer set that bridges the fusion. Western blots of lysate proteins from these cells demonstrated increased Fgf2, HoxA9, and HoxA10 protein in Mll-Ell-expressing cells in comparison to control cells (Fig. 6C).
FIGURE 6.

Fgf2 expression is increased in U937 cells expressing Mll-Ell or overexpressing HoxA9 or HoxA10 with or without differentiation. A, Fgf2 production by U937 cells is equivalently increased by expression of Mll-Ell or overexpression of HoxA9, HoxA10, or HoxA9 + HoxA10. U937 cells were stably transfected with vectors to express Mll-Ell or overexpress HoxA9, HoxA10, or HoxA9 + HoxA10 (1/2 of each) or with empty control vector. Media were harvested from the transfectants with or without RA/DMF differentiation and analyzed for Fgf2 by ELISA. Statistically significant differences in Fgf2 are indicated by * or **. B, Mll-Ell increases expression of Fgf2, HoxA9, and HoxA10 mRNA in U937 transfectants. U937 stable transfectants with Mll-Ell or empty control vector were analyzed for expression of HoxA9, HoxA10, and Fgf2 mRNA by real time PCR. Cells were analyzed with or without differentiation with RA/DMF. Statistically significant differences in mRNA expression are indicated by *, **, ***, or #. C, Mll-Ell increases expression of Fgf2, HoxA9, and HoxA10 proteins in U937 transfectants. Cell lysates from these stable transfectants were analyzed by Western blot (WB) with antibodies to HoxA9, HoxA10, Fgf2, Ell, and Gapdh (a loading control). D, overexpression of HoxA9, HoxA10, or HoxA9 + HoxA10 in U937 cells equivalently increases Fgf2 mRNA abundance. U937 cells were stably transfected with vectors to overexpress HoxA9, HoxA10, HoxA9 + HoxA10, or empty control vector and analyzed by real time PCR for expression of Fgf2, HoxA9, or HoxA10 mRNA. Cells were analyzed with or without RA/DMF-differentiation. Statistically significant differences in expression are indicated by *, **, ***, #, or ##. E, overexpression of HoxA9 in U937 cells increases Fgf2 protein expression. Cell lysates from these transfectants were analyzed by Western blots with antibodies to HoxA9, Fgf2, or Gapdh.
We found significantly increased Fgf2 in the media of HoxA9-overexpressing U937 cells versus control transfectants (p < 0.001, n = 3), and this was not substantially altered by differentiation (p = 0.2, n = 3; Fig. 6A). Overexpression of HoxA9, HoxA10, or HoxA9 + HoxA10 also significantly increased Fgf2 mRNA (p < 0.0001, n = 6). There was no significant difference in the amount of Fgf2 mRNA expression in transfectants overexpressing HoxA9 versus HoxA10 versus both with or without differentiation (p = 0.2, n = 6; Fig. 6D). Overexpression of HoxA9 versus HoxA10 was equivalent in these transfectants and approximately half as much in transfectants with both vectors (Fig. 6D). HoxA9 overexpression also increased Fgf2 protein in Western blot cells of lysates from U937 transfectants (Fig. 6E).
These studies suggested that Fgf2 was increased in myeloid progenitors and differentiating myeloid cells expressing Mll-Ell. However, U937 is a leukemia cell line that has abnormalities in proliferation and survival before introduction of oncoproteins. Therefore, we also investigated the effect of Mll-Ell on Fgf2 expression in primary murine bone marrow cells.
For these studies bone marrow cells from WT C57 Black 6 mice were transduced with an Mll-Ell retroviral expression vector (or empty control vector), cells were cultured in GM-CSF, IL3, and SCF, and CD34+ cells were separated. We referred to this as GMP conditions in these studies. Some cells were differentiated to granulocytes by treatment with G-CSF.
In other experiments bone marrow cells were transduced with retroviral vectors to express HoxA9, HoxA10, or HoxA9 + HoxA10 (or control vector). To determine if HoxA9 could substitute for HoxA10 for Fgf2 expression, bone marrow cells from HoxA10−/− mice were also transduced with these vectors. Serum Fgf2 is decreased in HoxA10−/− mice (26).
We found significantly increased Fgf2 in the media of Mll-Ell-expressing bone marrow cells in comparison to control vector-transduced cells (p < 0.0001, n = 3), and this was not altered by granulocyte differentiation (p = 0.6, n = 3) (Fig. 7A). We also found a significant increase in Fgf2, HoxA9, and HoxA10 mRNA in Mll-Ell-expressing bone marrow cells in comparison to control vector-transduced cells (p < 0.001, n = 6) (Fig. 7B). Expression of Fgf2 mRNA in Mll-Ell-expressing cells was not significantly altered by G-CSF-induced differentiation (p = 0.2, n = 6).
FIGURE 7.

Fgf2 expression is increased in primary murine myeloid progenitor cells expressing Mll-Ell or overexpressing HoxA9 or HoxA10 with or without granulocyte differentiation. A, expression of Mll-Ell or overexpression of HoxA9 or HoxA10 increased Fgf2 production by murine myeloid progenitor cells. Bone marrow was harvested from WT or HoxA10−/− mice and transduced with retroviral vectors to express Mll-Ell or overexpress HoxA9, HoxA10, HoxA9 + HoxA10, or empty control vector. Cells were cultured in GM-CSF, IL3, and SCF, and CD34+ cells were isolated (GMP conditions) or differentiated with G-CSF. Media were analyzed for Fgf2 by ELISA. Statistically significant differences in Fgf2 are indicated by *, **, or ***. B, Mll-Ell increases expression of Fgf2, HoxA9, and HoxA10 mRNA in primary murine bone marrow cells. The Mll-Ell-transduced cells (or control vector-transduced cells) described above were analyzed by real time PCR for Fgf2, HoxA9, HoxA10, or Mll-Ell fusion mRNA. Statistically significant differences in expression with Mll-Ell are indicated by *, **, ***, or #. C, Mll-Ell increases expression of Fgf2, HoxA9, and HoxA10 protein in primary murine bone marrow cells. These cells were also analyzed by Western blot (WB) with antibodies to Fgf2, HoxA9, HoxA10, Ell, or Gapdh. D, overexpression of HoxA9 increases Fgf2 mRNA expression in primary murine bone marrow cells. Primary murine bone marrow cells were transduced with vectors to express HoxA9, HoxA10, or HoxA9 + HoxA10 or with control vector and cultured under GMP or G-CSF differentiation conditions described above. Cells were analyzed by real time PCR for Fgf2, HoxA9, and HoxA10 mRNA expression. Statistically significant differences in expression are indicated by *, **, ***, #, or ##. E, HoxA9 overexpression increases Fgf2 protein expression in primary murine bone marrow cells. Cell lysate proteins from the transduced GMP cells, described above, were analyzed by Western blot with antibodies to Fgf2, HoxA9, or Gapdh. F, overexpression of HoxA9 rescues expression of Fgf2 in HoxA10−/− murine bone marrow cells. Primary HoxA10−/− murine bone marrow cells were transduced with vectors to express HoxA9, HoxA10, or HoxA9 + HoxA10 or with control vector and cultured under GMP or G-CSF differentiation conditions as described above. Cells were analyzed by real time PCR for Fgf2, HoxA9, and HoxA10 mRNA. Statistically significant differences in expression are indicated by *, **, ***, #, ##, or ###.
As noted for U937 transfectants, Mll-Ell resulted in a significantly greater increase in expression of HoxA10 versus HoxA9 (773 ± 49% increase in HoxA10 with Mll-Ell versus 372 ± 12% increase in HoxA9, p = 0.001, n = 6). Western blots demonstrated increased Fgf2, HoxA9, and HoxA10 protein in Mll-Ell-transduced murine bone marrow cells in comparison to control cells (Fig. 7C).
We found that overexpression of HoxA9 in murine bone marrow cells significantly increased Fgf2 in the media (p < 0.001, n = 3; Fig. 7A). HoxA9-induced increase in Fgf2 was not altered by G-CSF differentiation (p = 0.5, n = 3) and was not significantly different from the effect of expressing Mll-Ell or overexpressing HoxA10 or HoxA9 + HoxA10 (p = 0.2, n = 3; Fig. 7A). HoxA9 overexpression in HoxA10−/− myeloid progenitor cells also significantly increased Fgf2 production (p < 0.0001, n = 3; Fig. 7A). Fgf2 secretion by HoxA10−/− cells was equivalent with Mll-Ell expression or overexpression of HoxA9, HoxA10, or both (p = 0.4, n = 3; Fig. 7A).
Fgf2 mRNA was also significantly increased in HoxA9-overexpressing bone marrow cells relative to control cells (p < 0.0001, n = 6; (Fig. 7D). This increase was equivalent in cells expressing Mll-Ell or overexpressing HoxA9 versus HoxA10 versus both with or without G-CSF differentiation (p = 0.3, n = 6). In these studies HoxA9 and HoxA10 were equivalently overexpressed, and approximately half as much was expressed in cells with both vectors (Fig. 7D). Western blots of cell lysate proteins demonstrated increased Fgf2 protein with HoxA9 overexpression in comparison to control cells (Fig. 7E).
We found decreased Fgf2 mRNA expression in HoxA10−/− bone marrow cells in comparison to control cells (Fig. 7F) (26). Fgf2 expression in HoxA10−/− cells was equivalently increased by expression of Mll-Ell or overexpression of HoxA10, HoxA9, or HoxA9 + HoxA10 (p = 0.8, n = 6) (Fig. 7F). HoxA10 knock-out did not influence HoxA9 mRNA expression (Fig. 7F). We also demonstrated similar overexpression of HoxA9 and HoxA10 in these cells and approximately half the overexpression with both vectors (Fig. 7F).
Mll-Ell Induces Fgf2-dependent Cytokine Hypersensitivity
To investigate the role of autocrine Fgf2-production in cytokine-induced proliferation of Mll-Ell-expressing cells, U937 transfectants with Mll-Ell or control vector were treated with a dose titration of FCS. Some cells were also treated with an Fgf2 blocking antibody (or control antibody) or a specific Fgf-R1 inhibitor (PD173074). We found that FCS-induced proliferation was significantly greater in Mll-Ell-expressing U937 cells in comparison to control cells at all doses tested (p < 0.001, n = 3; Fig. 8A). Cytokine hypersensitivity of Mll-Ell transfectants was reversed by treatment with Fgf2 blocking antibody or Fgf-R1 inhibitor (Fig. 8A). The specificities of this antibody and inhibitor were previously validated (26).
FIGURE 8.

Mll-Ell induces Fgf2-dependent cytokine hypersensitivity in myeloid progenitor cells. A, Mll-Ell induces Fgf2-dependent cytokine hypersensitivity in U937 myeloid cells. U937 cells were stably transfected with a vector to express Mll-Ell or control vector and analyzed for proliferation in response to a dose titration of FCS (by incorporation of [3H]thymidine). Some cells were treated with a blocking antibody to Fgf2 or an Fgf-R1-inhibitor. Statistically significant differences in [3H]thymidine uptake at the same dose of FCS are indicated by an asterisk. B, Mll-Ell increases Fgf2-dependent GM-CSF hypersensitivity in primary murine myeloid progenitor cells. WT murine bone marrow cells were transduced with a vector to express Mll-Ell or empty control vector and analyzed under GMP conditions for proliferation in response to a dose titration of GM-CSF (by incorporation of [3H]thymidine). Some cells were treated with an Fgf2 blocking antibody or Fgf-R1 inhibitor. Statistically significant differences in [3H]thymidine uptake in Mll-Ell-expressing cells with versus without Fgf2 blocking antibody or Fgf-R1 inhibition are indicated by an asterisk. C, Mll-Ell increases β-catenin protein in an Fgf2-dependent manner in primary murine myeloid progenitor cells. Some transduced cells were analyzed by Western blots (WB) with antibodies to β-catenin, HoxA9, HoxA10, or Gapdh.
We also investigated the functional significance of autocrine Fgf2 production in Mll-Ell-expressing murine bone marrow cells. For these studies cells were transduced with a retroviral vector to express Mll-Ell or control vector, cultured under GMP conditions, and assayed for proliferation in response to a dose titration of GM-CSF. We found that GM-CSF-induced proliferation was significantly greater in Mll-Ell-expressing versus control cells at all cytokine doses (p < 0.001, n = 3; i.e. GM-CSF hypersensitivity). GM-CSF-induced proliferation of Mll-Ell-expressing cells was significantly decreased by treatment with Fgf2 blocking antibody or Fgf-R1 inhibition with PD173074 (p < 0.01, n = 3) (Fig. 8B).
Fgf2 binding to Fgf-R1/R2 results in stabilization of β-catenin protein (26). We tested the hypothesis that autocrine production of Fgf2 in Mll-Ell-expressing cells also stabilized β-catenin protein using the transduced murine bone marrow cells described above. Cells were treated with an Fgf2 blocking antibody (or control antibody), and Western blots of cell lysates were probed for β-catenin, HoxA9, HoxA10, Ell (to identify the fusion protein), or Gapdh (a loading control). We found an increase in β-catenin protein in Mll-Ell-expressing bone marrow cells that was abrogated by Fgf2 blocking antibody (Fig. 8C). Expression of β-catenin mRNA was not increased in Mll-Ell-expressing cells (not shown).
We also investigated the impact of increased production of Fgf2 in HoxA9-overexpressing U937 stable transfectants by proliferation assays similar to studies above. We found that HoxA9 overexpression induced cytokine hypersensitivity in these cells (Fig. 9A) similar to overexpressed HoxA10 (18, 26). HoxA9-induced cytokine hypersensitivity was significantly decreased by treatment with Fgf2 blocking antibody or Fgf-R1 inhibitor (p < 0.01, n = 3; Fig. 9A).
FIGURE 9.

HoxA9 overexpression induces Fgf2-dependent cytokine hypersensitivity in myeloid progenitor cells. A, overexpression of HoxA9 induces Fgf2-dependent cytokine hypersensitivity in U937 myeloid leukemia cells. Cells stably overexpressing HoxA9 or transfected with an empty control vector were analyzed for proliferation in response to a dose titration of FCS (by incorporation of [3H]thymidine). Some cells were treated with a blocking antibody (Ab) to Fgf2 or Fgf-R1 inhibitor. Statistically significant differences in [3H]thymidine uptake at the same dose of FCS are indicated by an asterisk. B, HoxA9 increases β-catenin protein expression in an Fgf2-dependent manner. U937 cells that stably overexpressed HoxA9, HoxA10, or HoxA9 + HoxA10 were analyzed by Western blot with antibody to β-catenin or Gapdh with or without Fgf2 blocking antibody. C, overexpression of HoxA9 in murine bone marrow myeloid progenitor cells induces Fgf2-dependent GM-CSF hypersensitivity. WT murine bone marrow cells were transduced with a vector to overexpress HoxA9 or HoxA10 or with empty control vector. Cells were analyzed for proliferation in response to a dose titration of GM-CSF with or without an Fgf2 blocking antibody or Fgf-R1 inhibitor. Statistically significant differences in [3H]thymidine incorporation at the same dose of GM-CSF are indicated by asterisks (* or **). D, HoxA9 rescues GM-CSF-induced proliferation in HoxA10−/− myeloid progenitor cells. HoxA10−/− murine bone marrow cells were transduced with a vector to overexpress HoxA9 or HoxA10 or with empty control vector. Cells were analyzed for proliferation in response to a dose titration of GM-CSF with or without an Fgf2 blocking antibody. Empty vector-transduced WT cells were a control in this study. Statistically significant differences in proliferation at comparable GM-CSF doses are indicated by asterisks (* or **).
We analyzed U937 stable transfectants overexpressing HoxA9, HoxA10 (positive control), or HoxA9 + HoxA10 for β-catenin protein expression by Western blot. We found that β-catenin protein was increased in cells overexpressing HoxA9, HoxA10, or HoxA9 + HoxA10 in comparison to control transfectants, but this was decreased by treatment with Fgf2 blocking antibody (Fig. 9B). Expression of β-catenin mRNA was not increased in HoxA9- or HoxA10-overexpressing cells (not shown), consistent with Fgf2-induced stabilization of β-catenin protein.
We also investigated the roles of HoxA9 and Fgf2 in cytokine hypersensitivity using transduced murine bone marrow cells as described above. We found that HoxA9-overexpressing murine myeloid progenitor cells exhibited significantly enhanced GM-CSF-induced proliferation at a low cytokine dose relative to control cells (p < 0.01, n = 3; Fig. 9C). We also found that treatment of HoxA9-overexpressing myeloid progenitor cells with Fgf2 blocking antibody or Fgf-R1 inhibitor significantly decreased GM-CSF induced proliferation (p < 0.01, n = 3) (Fig. 9C). HoxA10 vector and control vector-transduced murine myeloid progenitor cells were positive and negative controls for these studies.
To determine if HoxA9 could rescue proliferation defects in HoxA10−/− cells, HoxA10−/− murine myeloid progenitor cells were transduced with vectors to express HoxA9 or HoxA10 (as a positive control) or with empty retroviral vector (as a negative control) and treated with a dose titration of GM-CSF. We found that GM-CSF-induced proliferation of HoxA10−/− cells was impaired in comparison to WT cells at all GM-CSF doses (p < 0.01, n = 3; Fig. 9D). This was reversed, and cytokine hypersensitivity resulted, with overexpression of HoxA9 (Fig. 9D). HoxA9-induced cytokine hypersensitivity was decreased by treatment with Fgf2 blocking antibody.
DISCUSSION
Increased serum levels of Fgf2 are observed in some leukemia subjects, and increased Fgf2 mRNA expression is specifically found with 11q23-AML (28, 29). Consistent with this, we determined that expression of an 11q23-AML-associated MLL fusion protein (Mll-Ell) increased FGF2 transcription in a HoxA9- and HoxA10-dependent manner. This resulted in autocrine production of active Fgf2 by Mll-Ell-expressing cells and Fgf2-dependent cytokine hypersensitivity. Fgf2 is implicated in growth and survival of bone marrow progenitor cells (27, 28). One mechanism for this may be Fgf2-induced, phosphoinositol 3-kinase-dependent stabilization of β-catenin with consequent activation of β-catenin target genes that are involved cell proliferation/survival (c-myc, cyclinD1, survivin) (26). In the current studies we found that Mll-Ell-induced, HoxA9/A10-dependent autocrine production of Fgf2 by myeloid progenitor cells resulted in hypersensitivity to cytokines that share this pathway with Fgf2, such as GM-CSF.
The genes encoding Cdx4 and HoxA10 are also β-catenin target genes, and HOXA9 and HOXA10 are Cdx4 target genes in myeloid progenitor cells (25, 43). Therefore, the current studies define a positive feedback loop that leads from MLL fusion proteins to increased expression of HoxA9 and HoxA10 to autocrine production of Fgf2 and from Fgf2 back to increased expression of HoxA9 and HoxA10 through β-catenin and Cdx4 (Fig. 10). This feedback loop is functionally illustrated by the decrease in expression of HoxA9 or HoxA10 in Mll-Ell-transduced primary murine myeloid progenitor cells upon treatment with Fgf2 blocking antibody (Fig. 8C).
FIGURE 10.
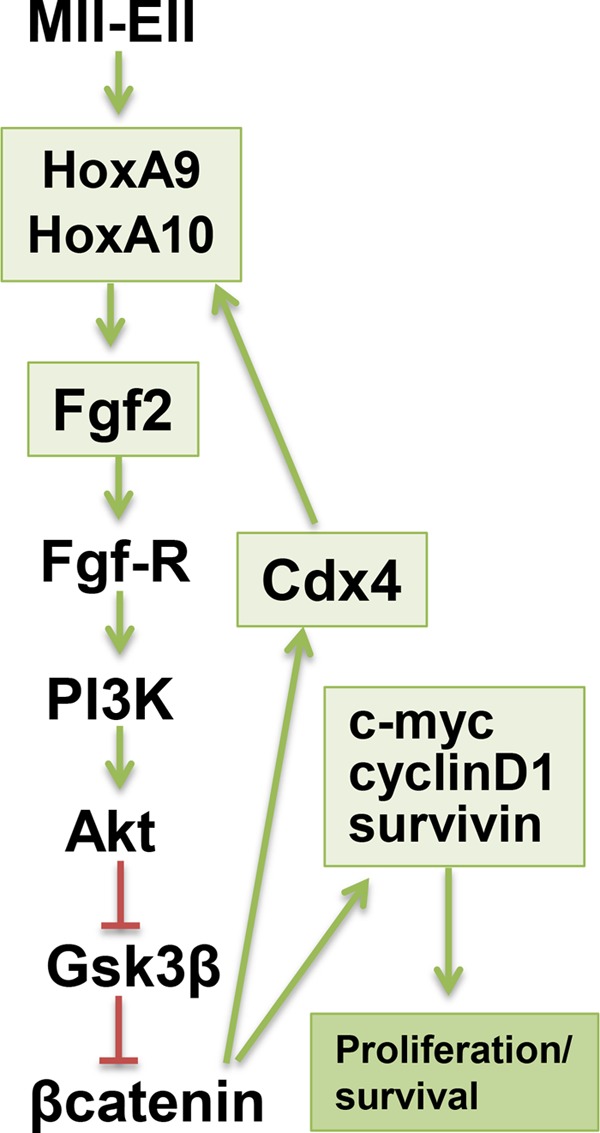
MLL fusion proteins induce a HoxA9- and HoxA10-dependent positive feedback loop involving Fgf2, β-catenin, and Cdx4. Shown is a schematic representation of the effects of MLL fusion proteins on HoxA9 and HoxA10, Fgf2, and downstream events. Activation is indicated by a green arrow, and inhibition is indicated by red lines. Events involving induction of gene transcription are enclosed in green boxes.
Increased β-catenin activity in leukemia stem cells is an indicator of poor prognosis, although mechanisms for this increase are not well defined (44). Perhaps consistent with this, Hox-overexpressing leukemias are relatively treatment refractory (45). Therefore, our studies suggest that targeting Fgf2 and/or cognate pathways might be a rational therapeutic approach to the Hox-overexpressing subset of AML, including 11q23 leukemias. Because Fgf-R1 inhibitor molecules are in clinical trials for solid tumors, this may rapidly become feasible and relevant.
Hox proteins are implicated in a variety of processes during definitive hematopoiesis, and proteins encoded by adjacent genes are presumed to have some functional redundancy. For example, knock-out of either HoxB3 or HoxB4 induces HSC dysfunction, and overexpression of either HoxA9 or HoxA10 expands the bone marrow GMP population in vitro or in vivo (15, 17). However, there are no studies that determine if this is due to regulation of common target gene sets by adjacent Hox proteins or regulation of different genes that control common or functionally redundant pathways. In the current study we found that HoxA9 and Hoxa10 contribute to myeloid progenitor expansion by activating transcription of the FGF2 gene. This is the first report of a common HoxA9 and HoxA10 target gene for which these proteins are functionally redundant.
In contrast, we previously identified CYBB as a common HoxA9 and HoxA10 target gene for which these proteins are antagonists (34, 37). CYBB encodes a component of the phagocyte NADPH oxidase, and CYBB transcription contributes to acquisition of phagocyte functional competence during differentiation (34). We defined a CYBB cis element that was repressed by HoxA10 in myeloid progenitor cells but activated by HoxA9 during phagocyte differentiation (34, 37). In the current studies we found that activation of FGF2 transcription by HoxA9 or HoxA10 was not significantly altered during granulocyte differentiation.
We determined that differentiation state-specific binding of HoxA9 and HoxA10 to the CYBB cis element was regulated by phosphorylation of conserved tyrosine residues in the DNA binding HDs in the two proteins (34–37). In contrast, phosphorylation of conserved HD-Tyr residues had an insignificant effect on HoxA9- or HoxA10-induced FGF2 transcription, consistent with the modest influence of differentiation on Fgf2 expression. Therefore, HoxA9 and HoxA10 were antagonists for phagocyte functional competence during myelopoiesis but redundant for production of Fgf2 during this process. This would be consistent with studies associating HoxA10 with differentiation block in AML and HoxA9 with myeloid phenotype of leukemia cells.
Fgf2 may be also be involved in cell motility through activation of non-canonical Fgf2-signaling by association of Fgf2/Fgf-R1 with αvβ3 integrin (46). A possible contribution of HoxA9 or HoxA10 to this process is an area of ongoing investigation in the laboratory. In addition, ITGB3 (encoding β3 integrin) is a HoxA10 target gene, and we found increased αvβ3-dependent adhesion and signaling in HoxA10-overexpressing myeloid cells (23).
We have identified additional common HoxA9 and HoxA10 target genes that are the subject of ongoing investigations in the laboratory. Some of these genes are activated by HoxA9 and HoxA10 in a redundant manner similar to their roles in regulation of FGF2. Other genes are activated by one of these proteins and repressed by the other in a differentiation stage-specific manner similar to regulation of CYBB. Therefore, the issue of Hox redundancy versus antagonism may be more complex than initially assumed. This has implications for understanding Hox-regulated molecular events that are relevant to the pathogenesis of 11q23-AML. This is a topic of translational relevance and ongoing investigations.
This work was supported, in whole or in part, by National Institutes of Health Grants HL87717 (to E. A. E.), CA155566 (to L. C. P.), and CA77816 (to L. C. P.). This work was also supported by Veterans Affairs Merit Reviews (to E. A. E. and L. C. P.), the Mander Foundation (to E. A. E.), and the Director's Research Fund of the Robert H. Lurie Comprehensive Cancer Center at Northwestern University (to E. A. E.).
- AML
- acute myeloid leukemia
- HSC
- hematopoietic stem cell
- Fgf2
- fibroblast growth factor 2
- HD
- homeodomain
- RA
- retinoic acid
- DMF
- dimethylformamide
- GMP
- granulocyte/monocyte progenitor cell
- SCF
- stem cell factor.
REFERENCES
- 1. Kawagoe H., Humphries R. K., Blair A., Sutherland H. J., Hogge D. E. (1999) Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 13, 687–698 [DOI] [PubMed] [Google Scholar]
- 2. Armstrong S. A., Staunton J. E., Silverman L. B., Pieters R., den Boer M. L., Minden M. D., Sallan S. E., Lander E. S., Golub T. R., Korsmeyer S. J. (2002) MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 30, 41–47 [DOI] [PubMed] [Google Scholar]
- 3. Guenther M. G., Jenner R. G., Chevalier B., Nakamura T., Croce C. M., Canaani E., Young R. A. (2005) Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 102, 8603–8608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Milne T. A., Briggs S. D., Brock H. W., Martin M. E., Gibbs D., Allis C. D., Hess J. L. (2002) MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 5. Ernst P., Mabon M., Davidson A. J., Zon L. I., Korsmeyer S. J. (2004) An Mll-dependent Hox program drives hematopoietic progenitor expansion. Curr. Biol. 14, 2063–2069 [DOI] [PubMed] [Google Scholar]
- 6. Camós M., Esteve J., Jares P., Colomer D., Rozman M., Villamor N., Costa D., Carrió A., Nomdedéu J., Montserrat E., Campo E. (2006) Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res. 66, 6947–6954 [DOI] [PubMed] [Google Scholar]
- 7. Roche J., Zeng C., Barón A., Gadgil S., Gemmill R. M., Tigaud I., Thomas X., Drabkin H. A. (2004) Hox expression in AML identified a distinct subset of patients with intermediate cytogenetics. Hox expression in AML identifies a distinct subset of patients with intermediate cytogenetics. Leukemia 18, 1059–1063 [DOI] [PubMed] [Google Scholar]
- 8. Li Z., Luo R. T., Mi S., Sun M., Chen P., Bao J., Neilly M. B., Jayathilaka N., Johnson D. S., Wang L., Lavau C., Zhang Y., Tseng C., Zhang X., Wang J., Yu J., Yang H., Wang S. M., Rowley J. D., Chen J., Thirman M. J. (2009) Consistent deregulation of gene expression between human and murine MLL rearrangement leukemias. Cancer Res. 69, 1109–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Acampora D., D'Esposito M., Faiella A., Pannese M., Migliaccio E., Morelli F., Stornaiuolo A., Nigro V., Simeone A., Boncinelli E. (1989) The human HOX gene family. Nucleic Acids Res. 17, 10385–10402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sauvageau G., Lansdorp P. M., Eaves C. J., Hogge D. E., Dragowska W. H., Reid D. S., Largman C. (1994) Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl. Acad. Sci. U.S.A. 91, 12223–12227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thorsteinsdottir U., Kroon E., Jerome L., Blasi F., Sauvageau G. (2001) Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol. Cell. Biol. 21, 224–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sauvageau G., Thorsteinsdottir U., Eaves C. J., Lawrence H. J., Largman C., Lansdorp P. M., Humphries R. K. (1995) Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 9, 1753–1765 [DOI] [PubMed] [Google Scholar]
- 13. Calvo K. R., Sykes D. B., Pasillas M., Kamps M. P. (2000) Hoxa9 immortalizes a granulocyte-macrophage colony-stimulating factor-dependent promyelocyte capable of biphenotypic differentiation to neutrophils or macrophages, independent of enforced meis expression. Mol. Cell. Biol. 20, 3274–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lawrence H. J., Helgason C. D., Sauvageau G., Fong S., Izon D. J., Humphries R. K., Largman C. (1997) Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood 89, 1922–1930 [PubMed] [Google Scholar]
- 15. Buske C., Feuring-Buske M., Antonchuk J., Rosten P., Hogge D. E., Eaves C. J., Humphries R. K. (2001) Overexpression of HOXA10 perturbs human lymphomyelopoiesis in vitro and in vivo. Blood 97, 2286–2292 [DOI] [PubMed] [Google Scholar]
- 16. Björnsson J. M., Andersson E., Lundström P., Larsson N., Xu X., Repetowska E., Humphries R. K., Karlsson S. (2001) Proliferation of primitive myeloid progenitors can be reversibly induced by HOXA10. Blood 98, 3301–3308 [DOI] [PubMed] [Google Scholar]
- 17. Thorsteinsdottir U., Mamo A., Kroon E., Jerome L., Bijl J., Lawrence H. J., Humphries K., Sauvageau G. (2002) Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 99, 121–129 [DOI] [PubMed] [Google Scholar]
- 18. Wang H., Lindsey S., Konieczna I., Bei L., Horvath E., Huang W., Saberwal G., Eklund E. A. (2009) Constitutively active SHP2 cooperates with HoxA10 overexpression to induce acute myeloid leukemia. J. Biol. Chem. 284, 2549–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kroon E., Krosl J., Thorsteinsdottir U., Baban S., Buchberg A. M., Sauvageau G. (1998) Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 17, 3714–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goardon N., Marchi E., Atzberger A., Quek L., Schuh A., Soneji S., Woll P., Mead A., Alford K. A., Rout R., Chaudhury S., Gilkes A., Knapper S., Beldjord K., Begum S., Rose S., Geddes N., Griffiths M., Standen G., Sternberg A., Cavenagh J., Hunter H., Bowen D., Killick S., Robinson L., Price A., Macintyre E., Virgo P., Burnett A., Craddock C., Enver T., Jacobsen S. E., Porcher C., Vyas P. (2011) Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 19, 138–152 [DOI] [PubMed] [Google Scholar]
- 21. Wang H., Lu Y., Huang W., Papoutsakis E. T., Fuhrken P., Eklund E. A. (2007) HoxA10 activates transcription of the gene encoding mitogen-activated protein kinase phosphatase 2 (Mkp2) in myeloid cells. J. Biol. Chem. 282, 16164–16176 [DOI] [PubMed] [Google Scholar]
- 22. Bei L., Lu Y., Bellis S. L., Zhou W., Horvath E., Eklund E. A. (2007) Identification of a HoxA10 activation domain necessary for transcription of the gene encoding β3 integrin during myeloid differentiation. J. Biol. Chem. 282, 16846–16859 [DOI] [PubMed] [Google Scholar]
- 23. Shah C. A., Wang H., Bei L., Platanias L. C., Eklund E. A. (2011) HoxA10 regulates transcription of the gene encoding transforming growth factor β2 (TGFB2) in myeloid cells. J. Biol. Chem. 286, 3161–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang H., Bei L., Shah C. A., Horvath E., Eklund E. A. (2011) HoxA10 influences protein ubiquitination by activating transcription of ARIH2; the gene encoding Triad1. J. Biol. Chem. 286, 16832–16845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bei L., Huang W., Wang H., Shah C., Horvath E., Eklund E. (2011) HoxA10 activates CDX4 transcription and Cdx4 activates HOXA10 transcription in myeloid cells. J. Biol. Chem. 286, 19047–19064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shah C. A., Bei L., Wang H., Platanias L. C., Eklund E. A. (2012) HoxA10 protein regulates transcription of gene encoding fibroblast growth factor 2 (FGF2) in myeloid cells. J. Biol. Chem. 287, 18230–18248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoon S. Y., Tefferi A., Li C. Y. (2000) Cellular distribution of platelet-derived growth factor, transforming growth factor-β, basic fibroblast growth factor, and their receptors in normal bone marrow. Acta Haematol. 104, 151–157 [DOI] [PubMed] [Google Scholar]
- 28. Aguayo A., Kantarjian H., Manshouri T., Gidel C., Estey E., Thomas D., Koller C., Estrov Z., O'Brien S., Keating M., Freireich E., Albitar M. (2000) Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 96, 2240–2245 [PubMed] [Google Scholar]
- 29. Rhodes D. R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A. M. (2004) ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilson E. L., Rifkin D. B., Kelly F., Hannocks M. J., Gabrilove J. L. (1991) Basic fibroblast growth factor stimulates myelopoiesis in long-term human bone marrow cultures. Blood 77, 954–960 [PubMed] [Google Scholar]
- 31. Holnthoner W., Pillinger M., Groger M., Wolff K., Ashton A. W., Albanese C., Neumeister P., Pestell R. G., Petzelbauer P. (2002) Fibroblast growth factor-2 induces Lef/Tcf-dependent transcription in human endothelial cells. J. Biol. Chem. 277, 45847–45853 [DOI] [PubMed] [Google Scholar]
- 32. Shimizu T., Kagawa T., Inoue T., Nonaka A., Takada S., Aburatani H., Taga T. (2008) Stabilized β-catenin functions through TCF/LEF proteins and the Notch/RBP-Jκ complex to promote proliferation and suppress differentiation of neural precursor cells. Mol. Cell. Biol. 28, 7427–7441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eklund E. (2011) The role of Hox proteins in leukemogenesis. Insights into key regulatory events in hematopoiesis. Crit. Rev. Oncog. 16, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eklund E. A., Jalava A., Kakar R. (2000) Tyrosine phosphorylation decreases HoxA10 DNA-binding and transcriptional repression during IFNγ differentiation in myeloid cell lines. J. Biol. Chem. 275, 20117–20126 [DOI] [PubMed] [Google Scholar]
- 35. Lindsey S., Zhu C., Lu Y. F., Eklund E. A. (2005) HoxA10 represses transcription of the gene encoding p67PHOX in phagocytic cells. J. Immunol. 175, 5269–5279 [DOI] [PubMed] [Google Scholar]
- 36. Lindsey S., Huang W., Wang H., Horvath E., Zhu C., Eklund E. A. (2007) Activation of SHP2 protein-tyrosine phosphatase increases HoxA10-induced repression of the genes encoding gp91PHOX and p67PHOX. J. Biol. Chem. 282, 2237–2249 [DOI] [PubMed] [Google Scholar]
- 37. Bei L., Lu Y., Eklund E. A. (2005) HoxA9 activates transcription of the gene encoding gp91PHOX during myeloid differentiation. J. Biol. Chem. 280, 12359–12370 [DOI] [PubMed] [Google Scholar]
- 38. Lowney P., Corral J., Detmer K., LeBeau M. M., Deaven L., Lawrence H. J., Largman C. (1991) A human Hox 1 homeobox gene exhibits myeloid-specific expression of alternative transcripts in human hematopoietic cells. Nucleic Acids Res. 19, 3443–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larrick J. W., Fischer D. G., Anderson S. J., Koren H. S. (1980) Characterization of a human macrophage-like cell line stimulated in vitro. A model of macrophage functions. J. Immunol. 125, 6–12 [PubMed] [Google Scholar]
- 40. Satokata I., Benson G., Maas R. (1995) Sexually dimorphic sterility phenotypes in Hoxa10-deficient mice. Nature 374, 460–463 [DOI] [PubMed] [Google Scholar]
- 41. Oberley M. J., Farnham P. J. (2003) Probing chromatin immunoprecipitates with CpG-island microarrays to identify genomic sites occupied by DNA-binding proteins. Methods Enzymol. 371, 577–596 [DOI] [PubMed] [Google Scholar]
- 42. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bei L., Shah C., Wang H., Huang W., Roy R., Eklund E. A. (2012) β-Catenin activates the HOXA10 and CDX4 genes in myeloid progenitor cells. J. Biol. Chem. 287, 39589–39601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simon M., Grandage V. L., Linch D. C., Khwaja A. (2005) Constitutive activation of the Wnt/β-catenin signalling pathway in acute myeloid leukaemia. Oncogene 24, 2410–2420 [DOI] [PubMed] [Google Scholar]
- 45. Schwind S., Marcucci G., Kohlschmidt J., Radmacher M. D., Mrózek K., Maharry K., Becker H., Metzeler K. H., Whitman S. P., Wu Y. Z., Powell B. L., Baer M. R., Kolitz J. E., Carroll A. J., Larson R. A., Caligiuri M. A., Bloomfield C. D. (2011) Low expression of MN1 associates with better treatment response in older patients with de novo cytogenetically normal acute myeloid leukemia. Blood 118, 4188–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Murakami M., Elfenbein A., Simons M. (2008) Non-canonical fibroblast growth factor signaling in angiogenesis. Cardiovasc. Res. 78, 223–231 [DOI] [PubMed] [Google Scholar]